INTOLÉRANCE À L’EFFORT EN
HYPERTENSION ARTÉRIELLE
PULMONAIRE :
Implication des déterminants périphériques
Thèse
Simon Malenfant
Doctorat en kinésiologie
Philosophiæ doctor (Ph.D.)
Québec, Canada
© Simon Malenfant 2016
INTOLÉRANCE À L’EFFORT EN
HYPERTENSION ARTÉRIELLE
PULMONAIRE :
Implication des déterminants périphériques
Thèse
Simon Malenfant
Sous la direction de :
Steeve Provencher, directeur de recherche
Sébastien Bonnet, codirecteur de recherche
Résumé
L’hypertension artérielle pulmonaire (HTAP) est une maladie caractérisée par l’augmentation progressive des résistances vasculaires pulmonaires causant une augmentation de la pression artérielle pulmonaire qui mène au décès prématuré des patients. Malgré une amélioration rapide ces dernières années des traitements spécifiques, les patients souffrant d’HTAP demeurent dyspnéiques et intolérants à l’effort. L’atteinte vasculaire pulmonaire est actuellement irréversible. Elle est également la source de plusieurs anomalies au niveau des systèmes cardiovasculaires, ventilatoires et musculaires constituant les principaux déterminants physiologiques de la capacité à l’effort des patients. Cette thèse a investigué différentes facettes de la tolérance à l’effort en HTAP : les différents mécanismes ayant un impact sur l’apport musculaire en oxygène, l’altération des voies de signalisation cellulaire impliquées dans l’angiogenèse musculaire et les mécanismes ayant un impact sur la régulation du débit sanguin et l’oxygénation cérébrale en HTAP. Nous avons premièrement documenté une diminution de l’apport en oxygène aux muscles squelettiques à l’effort des patients en relation avec une diminution de la densité capillaire musculaire. Ce défaut d’angiogenèse corrélait d’ailleurs avec la capacité à l’effort des sujets. Par la suite, nous avons étudié les voies de signalisations cellulaires de l’angiogenèse musculaire. Ces résultats ont permis de démontrer une diminution de l’expression de miR-126, unique aux patients HTAP, qui était responsable de la diminution de la densité capillaire et qui contribuait à leur intolérance à l’effort. De plus, il était possible de moduler in vivo l’expression de miR-126. L’expérimentation in vivo, à l’aide d’un modèle murin d’HTAP, a permis de rétablir l’expression de miR-126, d’augmenter la microcirculation musculaire et d’améliorer la tolérance à l’effort des animaux, ce qui met en lumière le potentiel thérapeutique de l’angiogenèse musculaire pour améliorer la capacité à l’effort en HTAP. Notre dernier projet a démontré que les patients HTAP présentaient une diminution de débit sanguin cérébral. Ce projet a également démontré que les changements de pression artérielle sont moins bien amortis par les vaisseaux cérébraux des patients et que leurs vaisseaux cérébraux étaient moins réactifs aux changements de CO2. Les patients présentaient aussi une augmentation de la sensibilité des chémorécepteurs centraux qui contribuait à augmenter leur ventilation au repos, mais aussi à l’exercice. Finalement, à l’effort, nous avons démontré que le débit sanguin cérébral des patients HTAP était principalement influencé par la pression artérielle alors que chez les sujets sains, le débit sanguin cérébral était influencé principalement par la PETCO2. Nous avons également démontré que les patients HTAP présentaient une diminution progressive de leur oxygénation cérébrale, qui corrélait avec leur capacité à l’effort. Les résultats obtenus au cours de ce doctorat démontrent bien que la capacité à l’effort en HTAP est aussi déterminée par plusieurs anomalies physiopathologiques périphériques.
Abstract
Pulmonary arterial hypertension (PAH) is characterized by a progressive increase in pulmonary vascular resistance ultimately leading to patients’ premature death. The most recent available specific therapies have considerably improved the long-term prognosis of PAH patients. However, the vast majority still displays persistent and significant exercise intolerance despite being optimally treated. Persistent and irreversible pulmonary vascular damage causes several cardiovascular, respiratory and muscular impairments, which constitute the main physiological determinants of exercise capacity. We wanted to investigate the integrative physiological mechanisms that impair exercise tolerance in PAH patients. We first documented that capillary rarefaction within skeletal muscle influences exercise tolerance and quadriceps muscle function at least partly through impaired muscle oxygen perfusion to working muscle myocytes. We then assessed that miR-126 down-regulation accounted for this reduced muscular capillarity, which contributed to PAH exercise intolerance. We showed that in vivo down-regulation of miR-126 expression in the skeletal muscle of otherwise healthy rats is associated with microcirculation impairment, and ultimately exercise intolerance, making miR-126 an attractive therapeutic target. Finally, we demonstrated that PAH patients have a lower cerebral blood flow (CBF) at rest and during exercise related to alterations in the pressure-flow relationship resulting in inadequate buffering of blood pressure changes, an increased central chemoreceptor sensibility leading to excessive ventilation and hyperventilation, an abnormal cerebrovascular reactivity to CO2 contributing to lower CBF for any CO2 levels and potentially a loss in cerebral microvessels. We also demonstrated that PAH patients exhibited marked impairments in cerebral oxygenation, which correlated with their exercise capacity. Collectively, we provide evidence that CBF regulation and oxygenation is compromised in PAH patients. The results obtained throughout this doctoral training provide support to an integrative physiological comprehension of exercise intolerance in PAH. A better understanding of those mechanisms might lead to new therapeutic targets that will ultimately lead to a refinement in specific therapies and increase in exercise tolerance and quality of life of patients.
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des tableaux ... ix
Liste des figures ... x
Liste des équations ... xi
Abréviations ... xii
Remerciements ... xvi
Avant-propos ... xviii
CHAPITRE 1 – Introduction générale ... 1
CHAPITRE 2 – Systèmes cardiovasculaire, respiratoire et cérébral ... 4
LE SYSTÈME CARDIOVASCULAIRE ... 5
Constituant macroscopique du cœur ... 5
Chambres cardiaques ... 6
Valves cardiaques ... 6
Le système circulatoire ... 7
Système artériel, capillaire et veineux ... 7
Baroréflexe artériel ... 8
LA FORMATION DE NOUVEAUX VAISSEAUX ... 8
Les facteurs de croissance ... 9
Les micro-ARN ... 9
LE SYSTÈME RESPIRATOIRE ... 10
Voie et surface respiratoire ... 11
Vascularisation pulmonaire ... 11
SYSTÈME MUSCULAIRE ET CAPILLARITÉ ... 12
Les ergorécepteurs ... 13
LE SYSTÈME CÉRÉBROVASCULAIRE ... 13
MÉCANISMES PHYSIOLOGIQUES DÉTERMINANT LA RÉPONSE CÉRÉBROVASCULAIRE AU REPOS ... 15 La pression artérielle ... 15 L’autorégulation cérébrale ... 15 Approche myogénique ... 17 Approche neurologique ... 17 Le débit cardiaque ... 18
Le dioxyde de carbone et l’oxygène ... 18
CHAPITRE 3 – Les déterminants de la tolérance à l’effort chez l’adulte sain ... 20
LA RÉPONSE CARDIOVASCULAIRE AU REPOS ET À L’EFFORT ET SES DÉTERMINANTS PHYSIOLOGIQUES ... 21
Le débit cardiaque et ses déterminants ... 21
Adaptations cardiaques à l’effort et à l’entrainement soutenu ... 23
La pression artérielle au repos et à l’effort ... 23
La réponse des baroréflexes artériels à l’effort ... 24
LA RÉPONSE VENTILATOIRE AU REPOS ET À L’EFFORT ET SES DÉTERMINANTS PHYSIOLOGIQUES ... 25
Les pressions partielles de l’oxygène et du dioxyde de carbone ... 25
Mécanismes physiologiques régissant le contenu en oxygène ... 26
Mécanismes physiologiques régissant le transport du dioxyde de carbone ... 28
La ventilation pulmonaire et alvéolaire ... 28
Le contrôle nerveux de la ventilation ... 29
Les échanges gazeux ... 30
LA RÉPONSE DU MUSCLE SQUELETTIQUE À L’EFFORT ... 30
Le métabolisme des différents types de fibre musculaire ... 31
La réponse des ergoréflexes à l’effort ... 31
L’impact de l’activité physique sur l’angiogenèse ... 32
LA TOLÉRANCE À L’EFFORT ... 33
La consommation maximale d’oxygène ... 33
Mécanisme régissant l’apport en oxygène à l’effort ... 36
Réponse du débit sanguin cérébral à l’exercice ... 37
CHAPITRE 4 – L’hypertension artérielle pulmonaire ... 39
L’HYPERTENSION PULMONAIRE ... 40
La classification clinique ... 41
L’HYPERTENSION ARTÉRIELLE PULMONAIRE ... 43
PRÉVALENCE ET INCIDENCE DE L’HYPERTENSION ARTÉRIELLE PULMONAIRE ... 44
PHYSIOPATHOLOGIE DE L’HYPERTENSION ARTÉRIELLE PULMONAIRE44 Lésions histologiques vasculaires en HTAP ... 45
Histologie caractéristique ... 45 Facteurs environnementaux ... 47 Hypoxie ... 47 Substances anorexigènes ... 48 La prise d’interféron ... 49 Anormalités génétiques ... 49 Mutations BMPR2 ... 49
Autres mutations génétiques ... 51
Débalancement prolifération/apoptose ... 52
Instabilité dans l’angiogenèse pulmonaire en HTAP – le rôle de l’angiopoïétine ... 52
Le rôle des microARN ... 52
Dommage à l’ADN ... 54
DYSFONCTION VENTRICULAIRE DROITE ... 54
L’hypertrophie adaptative ... 55
Défaut contractile ... 56
Fibrose myocardique ... 57
DIAGNOSTIC DE L’HYPERTENSION ARTÉRIELLE PULMONAIRE ... 59
Mesures diagnostiques non invasives ... 60
Mesure diagnostique invasive ... 61
Les biomarqueurs sériques en HTAP ... 62
Diagnostic final d’HTAP ... 62
ÉVALUATION DE LA CAPACITÉ FONCTIONNELLE ... 63
PRONOSTIC ET SURVIE DE l’HTAP ... 65
OPTIONS THÉRAPEUTIQUES ... 66
Recommandations générales ... 66
Bloqueurs des canaux calciques (BCC) et anticoagulants ... 67
Activité physique et réhabilitation cardio-pulmonaire ... 69
Traitements pharmacologiques spécifiques à l’HTAP ... 71
Dérivés de la prostacycline ... 72
Antagoniste des récepteurs de l’endothéline-1 ... 75
Inhibiteurs de la phosphodiestérase de type 5 ... 76
Combinaisons thérapeutiques ... 78
Atrioseptostomie et transplantation pulmonaire ... 79
Perceptives thérapeutiques ... 80
L’utilisation de ß-bloqueurs en HTAP ... 80
Cibler les facteurs de croissance en HTAP, de l’espoir à la désillusion ... 81
Thérapie cellulaire ... 82
Dénervation sympathique de l’artère pulmonaire ... 83
LES DÉTERMINANTS DE L’INTOLÉRANCE À L’EFFORT EN HTAP ... 84
Le système cardiovasculaire ... 85
Limitations cardiaques observables au test à l’effort progressif maximal ... 85
Augmentation de l’activité nerveuse sympathique ... 86
Le système ventilatoire ... 86
La fonction pulmonaire en HTAP ... 87
Les muscles respiratoires ... 87
La ventilation au repos et à l’effort ... 88
Le système musculaire squelettique ... 89
Le système cérébrovasculaire ... 90
OBJECTIFS ET HYPOTHÈSES DE RECHERCHE ... 92
Objectif de recherche – Chapitre 5 ... 92
Hypothèses spécifiques Chapitre 5 ... 92
Objectif de recherche – Chapitre 6 ... 93
Hypothèses spécifiques Chapitre 6 ... 93
Objectif de recherche – Chapitre 7 ... 94
Hypothèses spécifiques Chapitre 7 ... 94
CHAPITRE 5 – Impaired skeletal muscle oxygenation and exercise tolerance in pulmonary hypertension ... 95
CHAPITRE 6 – Impaired angiogenesis and peripheral muscle microcirculation loss contributes to exercise intolerance in pulmonary arterial hypertension ... 135
CHAPITRE 7 – Compromised cerebrovascular regulation and oxygenation in pulmonary
arterial hypertension ... 185
CHAPITRE 8 – Discussion et conclusion ... 234
PRINCIPAUX RÉSULTATS ET DISCUSSION ... 235
Diminution de l’apport musculaire en oxygène en HTAP ... 235
Limitations du Near-infrared spectroscopy (NIRS) ... 237
Limitation de la mesure de l’hémodynamie centrale (Nexfin) ... 238
Perceptive ... 239
Présence d’un défaut angiogénique musculaire intrinsèque en HTAP ... 240
Perceptive ... 242
L’apport sanguin et de l’oxygénation cérébrale en HTAP ... 243
Limitations Doppler transcrânien ... 246
Perspective ... 248
CONCLUSION ... 250
BIBLIOGRAPHIE ... 251
ANNEXE 1 – Skeletal muscle proteomic signature and metabolic impairment in pulmonary hypertension ... 311
Liste des tableaux
Tableau 1. Valeur de référence V̇O2 max sur vélo ergocycle ... 36
Tableau 2. Classification clinique des différentes formes d’hypertension pulmonaire ... 42 Tableau 3. Classement de la capacité fonctionnelle des patients HTP selon l’Organisation
mondiale de la Santé ... 64
Liste des figures
Figure 1. Cavités cardiaques ... 5
Figure 2. Composition structurelle d’une artère ... 7
Figure 3. Mécanismes d’actions de miR-126 ... 10
Figure 4. Artères cérébrales ... 14
Figure 5. Concept de l’autorégulation cérébrale statique ... 17
Figure 6. Relation linéaire entre le débit cardiaque et la vitesse d’écoulement du sang à l’intérieur de l’artère cérébrale moyenne ... 18
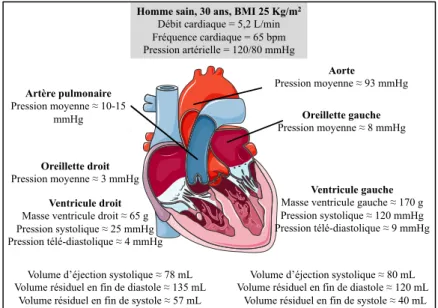
Figure 7. Exemple pour un adulte sain des différentes valeurs de pressions et de volumes cardiaque ... 22
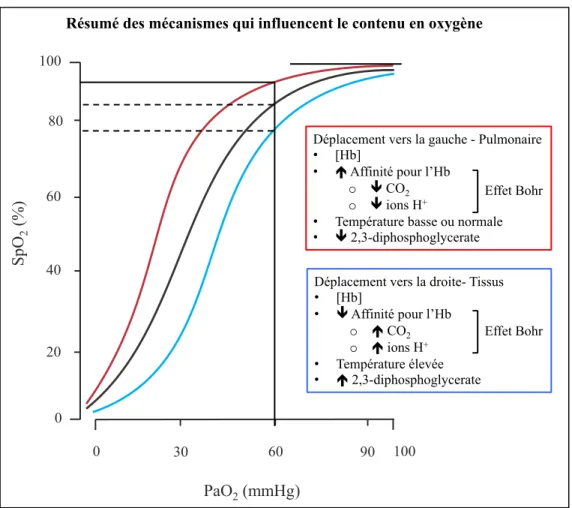
Figure 8. Pressions partielles de l’oxygène et du dioxyde de carbone ... 26
Figure 9. Courbe de dissociation de l’hémoglobine ... 27
Figure 10. Modèle intégré des différents mécanismes physiologiques cardiaque, pulmonaire et circulatoire et la réponse à l’exercice ... 34
Figure 11. Revue des différentes voies de signalisations cellulaires impliquées dans le remodelage vasculaire en HTAP ... 46
Liste des équations
Équation 1. Débit cardiaque ... 21
Équation 2. Volume d’éjection systolique ... 22
Équation 3. Fraction d’éjection systolique ... 22
Équation 4. Pression artérielle moyenne ... 24
Équation 5. Ventilation minute ... 28
Équation 6. Déterminants de la consommation d’oxygène (Principe de Fick) ... 33
Abréviations
CO2 : dioxyde de carboneVEGF : Vascular Endothelial Growth Factor FGF : Fibroblast Growth Factor
PDGF : platelet-derived growth factor ANG-1 : Angiopoïétine 1
ANG-2 : Angiopoïétine 2 miR : microARN
Spred1 : Srouty-related EVH1 domain-containing protein 1 PI3K : phosphoinositol-3 kinase
Egfl7 : EGF-like domain 7
PI3KR2 : phosphoinositol-3 kinase regulatory subunit 2 AKT : Protein kinase B
RAF1 : proto-oncogene serine/threonine-protein kinase ERK : extracellular-signal-regulated kinases
PAPm : pression artérielle pulmonaire moyenne RVP : résistance vasculaire pulmonaire
IIa : fibre musculaire de type IIa IIx : fibre musculaire de type IIx H+ : ions hydrogène
PAM : pression artérielle moyenne PA : pression artérielle
MCAv : vitesse du débit sanguin à l’intérieur de l’artère cérébrale moyenne (traduit de l’anglais : middle cerebral artery velocity)
PaCO2 : pression partielle artérielle en dioxyde de carbone PaO2 : pression partielle artérielle en oxygène
PatmO2 : pression partielle atmosphérique en oxygène PAO2 : pression partielle alvéolaire en oxygène
PACO2 : pression partielle alvéolaire en dioxyde de carbone PvO2 : pression partielle veineuse en oxygène
PvCO2 : pression partielle veineuse en dioxyde de carbone SpO2 : saturation systémique
mmHg : millimètre de mercure DC : débit cardiaque
FC : fréquence cardiaque
VES : volume d’éjection systolique
V̇O2 max : consommation maximale d’oxygène VTD : volume télédiastolique
VTS : volume télésystolique FES : fraction d’éjection systolique Hb : hémoglobine
H2CO3 : acide carbonique (HCO3-) : ion carbonate
FiO2 : fraction d’oxygène inspirée
FiCO2 : fraction de dioxyde de carbone inspirée FeO2 : fraction expirée en oxygène
V̇E : ventilation minute V̇T : Volume courant FR : fréquence respiratoire CI : capacité inspiratoire V̇A : ventilation alvéolaire V̇D : espace mort physiologique
V̇D/V̇T : ratio espace mort physiologique/volume courant V̇A/Q : ratio ventilation perfusion
V̇E/V̇O2 : ventilation nécessaire pour consommer un litre d’oxygène V̇E/V̇CO2 : ventilation nécessaire pour excréter un litre de CO2 P(A-a)O2 : différence de tension alvéolo-capillaire en oxygène PETCO2 : pression en fin d’expiration en dioxyde de carbone
P(a-ET)CO2 : différence de tension en CO2 artérielle et en fin d’expiration ATP : adénosine triphosphate
Diff(a-v)O2 : différence de contenu artério-veineuse HTP : hypertension pulmonaire
HTAP : hypertension artérielle pulmonaire
HTAP-SSc : hypertension artérielle pulmonaire associée à une sclérodermie iHTAP : hypertension artérielle pulmonaire idiopathique
PAPo : pression artérielle pulmonaire d’occlusion Alk-1 : active-like kinase I
ENG : endoglin
Smad : mother against decapentaplegic homolog BMPR2 : bone morphogenetic protein receptor II CAV-1 : caveolin-1
KCNK3 : gène encodant le potassium channel subfamily K, member 3 VIH : virus de l’immunodéficience humaine
WU : Wood Units
HTAP-SSc : HTAP associée avec une sclérodermie CaO2 : contenu artériel en oxygène
NFATc2 : nuclear factor of activated T-cell WES : whole-exome sequencing
Tie2: endothlium-specific tyrosine kinase-2 ROCK : kinase Rho
STAT3-PIM1 : signal transducer and activator of transcription 3-Moloney murine leukemia
virus
c-Src : sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog FAK : focal adhesion kinase
PARP-1 : poly(ADP-ribose) polymerase-1 BRD4 : Bromodomain-containing protein 4 β-bloqueurs : beta-bloqueurs
MCH : myosin heavy chain
HIF-1α : hypoxia-inducible factor 1α ROS : reactive oxygen species
Axe mef2/Hand2 : Islet1/myocyte enhencer factor 2 (mef2)/Hand2 Spred1 : Strouty-related EVH1 domain containing protein 1 MAPK : mitogen-activated protein kinase
ECG : électrocardiogramme
DLCO : diffusion pulmonaire du monoxyde de carbone TAPSE : tricuspid annular systolic excursion
BNP : brain natriuretic peptide pro-BNP: N-terminal fragment (NT) OMS: Organisation Mondiale de la Santé NYHA : New York Heart Association TM6 : test de marche de 6 minutes
MPOC : maladie pulmonaire obstructive chronique BCC : Bloqueurs des canaux calciques
AMP : l’adénosine monophosphate ET-1 : endothéline-1
GMPc : guanosine monophosphate cyclique PDE-5 : phosphodiestérase de type 5
TK: tyrosine kinase
PCE : précurseurs cellulaires endothéliales
VEMS: volume expiratoire maximal en une seconde CVF: capacité vitale forcée
Ratio VEMS/CVF : indice de Tiffeneau
Ratio V̇O2/FC : ratio de la consommation d’oxygène sur la fréquence cardiaque PETO2 : pression partielle en oxygène en fin d’expiration
V̇E/V̇O2 : Équivalent ventilatoire pour l'oxygène
V̇E/V̇CO2 : Équivalent ventilatoire pour le dioxyde de carbone TFA : transfer function analysis
Do not go gentle into that good night, Old age should burn and rave at close of the day Rage, rage against the dying of the light Dylan Thomas, Welsh poet (1914-1953)
Remerciements
À mon directeur de recherche, Dr Steeve Provencher. Merci de m’avoir accueilli et de
m’avoir partagé ton savoir sur l’HTAP, merci aussi d’avoir dit oui au projet HTAP-CBF !! Tu as été une source de soutien, d’inspiration, de transformation et de dépassement de soi. Malgré un emploi du temps chargé, tu as toujours pris le temps pour une rencontre pour être à l’écoute autant de mes problèmes professionnelles que mes angoisses !
À mon co-directeur de recherche, Dr Sébastien Bonnet. Tes conseils dans le domaine de
la recherche sont toujours à propos. J’ai énormément appris sur le fonctionnement et les aléas du processus de création scientifique avec toi.
À mes collègues, Didier Saey et François Maltais. Vous avez été des inspirations en début
de parcours en recherche. J’ai vraiment apprécié vous côtoyer au cours des dernières années.
À mon ancien collègue, Vincent Mainguy. C’est avec toi que j’ai eu mon premier contact
avec l’HTAP. Tu as mis la table pour mes projets, j’ai simplement eu à reprendre le flambeau et continuer à le faire progresser, merci à toi.
À Luce Bouffard et Éric Nadreau. Votre savoir-faire et votre grand professionnalisme
m’ont permis d’apprendre énormément dans l’approche patient et la réalisation de test à l’effort.
À ami et collègue, François Potus. Frère d’armes. Sans tout le talent que tu as déployé en
laboratoire, sans ta force de travail, cette thèse aurait été fortement différente. À tous nos soupers, à toutes nos virées, moi je dis chapeau ! Le futur est plein de promesses pour toi, j’en suis persuadé.
À Sandra, Ève, Valérie et à chaque membre du Groupe d’hypertension artérielle pulmonaire. C’est grâce à vos enseignements que j’ai pu avoir l’air de savoir ce que je
faisais avec une pipette.
À mon collègue, Patrice Brassard. Tu as dit oui sans hésitation quand je t’ai présenté mon
idée de projet en HTAP. Tu m’as accueilli à bras ouverts dans ton laboratoire et sans toi mon dernier projet n’aurait tout simplement pas été possible, point.
À Myriam, Olivier et Audrey. Myriam et Olivier, c’est grâce à vous que j’ai appris les
différentes techniques de mesure et d’analyse dans le domaine de la physiologie cérébrovasculaire. Audrey, ton aide en laboratoire a été très précieuse. Ton désire d’apprendre te de bien faire les choses sont des qualités essentiels, continue de les entretenir.
À mes parents, Michelle et Sylvain. Après un début de parcours scolaire difficile, parsemé
d’embuche, de tracas, de jours de semaine d’année difficile, un seul mot me vient en tête pour honorer vos combats et votre détermination à faire de moi un adulte complet, travaillant et déterminé, un mot qui résume modestement le tout : Voilà.
À mes frères, Éric et Mathieu et ma sœur, Marie-Hélène. Notre clan est fort et uni. Je
puise toujours dans cette force pour avancer et faire cheminer mes différents projets, recherches ou autres.
À Sœur Julie. Après mes parents, tu as cru en moi. J’ai mesuré plus tard tout l’impact que tu
as eu sur moi.
À mes amis Gab, Jay et Yoann. Pour votre présence, votre inspiration silencieuse. Je m’y
suis servi. Gab, sans toi, la suite des choses n’aurait pas été possible. Merci encore.
À vous, chers patients. J’ai eu la chance de rencontrer personnellement chacun de vous. J’en
sors profondément marqué et inspiré. Vous faites preuve d’une résilience sans frontière qui m’a motivé pour les six dernières années. Pour reprendre les mots de Vincent, restez positifs, le futur est beau pour vous.
Finalement, j’aimerais conclure en remerciant les Fonds de Recherche du Québec – Santé ainsi que le département de kinésiologie, le département de médecine et le centre de recherche de l’IUCPQ pour l’octroi de bourses de recherche. J’ai pu consacrer entièrement mon temps à la recherche grâce à leur aide financière.
Avant-propos
Cette thèse traite des différents mécanismes de l’intolérance à l’effort en hypertension artérielle pulmonaire (HTAP). La présente thèse débute avec une introduction générale qui présente l’HTAP, les défis actuels des patients et en décrit l’objectif général (Chapitre 1). Ce chapitre fait également le lien avec les chapitres qui suivent. Ainsi, une description générale de la physiologique des différents systèmes cardiovasculaire, respiratoire musculaire et cérébrovasculaire chez l’adulte sain sert d’introduction (Chapitre 2 et 3). Une description complète de l’HTAP est ensuite présentée (Chapitre 4). Ce chapitre présente les différents groupes d’hypertension pulmonaire, avec une attention particulière pour l’HTAP. Cette partie inclut également les différentes modalités pharmacologiques et non pharmacologiques de traitement de l’HTAP. Pour terminer, ce chapitre traite des différents déterminants de l’intolérance à l’effort en HTAP. Cette partie reprend les différents systèmes présentés dans les chapitres 2 et 3 et illustre les différences dans la réponse physiologique à l’effort des patients HTAP comparativement aux sujets sains.
Le premier article de cette thèse (Chapitre 5) s’intitule Impaired Skeletal Muscle
Oxygenation and Exercise Intolerance in Pulmonary Hypertension. L’objectif était de tester
l’hypothèse que l’apport en oxygène musculaire chez les patients HTAP était affecté, comparativement à des sujets sains. Nous souhaitions également explorer les différents mécanismes contribuant à limiter cet apport, mais aussi mesurer son impact sur l’intolérance à l’effort des patients. Cette étude a démontré que la diminution de la densité capillaire à l’intérieur du muscle squelettique des patients HTAP influence, du moins en partie, la capacité à l’effort et la force musculaire par une diminution de l’apport en oxygène. Cette étude a été réalisée sous la supervision du Dr Steeve Provencher, avec la précieuse collaboration des Drs Didier Saey et Sébastien Bonnet. Les Drs Vincent Mainguy, Mathieu Malenfant, Évelyne Leblanc, François Potus et Mme Fernanda Ribeiro ont participé à la collecte de données. J’ai rédigé le protocole du projet. J’ai également recruté les patients, mené les expérimentations et analysé les différents résultats. J’ai rédigé le manuscrit et effectué sa soumission et la révision subséquente des commentaires des réviseurs, sous la supervision du Dr Provencher. L’article a été soumis pour publication dans le Medicine and
Science in Sport and Exercise en janvier 2015 et a été accepté pour publication dans en avril
2015.
Le deuxième article de cette thèse (Chapitre 6) s’intitule Impaired Angiogenesis and
Peripheral Muscle Microcirculation Loss Contribute to Exercise Intolerance in Pulmonary Arterial Hypertension. L’objectif de ce projet était de déterminer les mécanismes de
signalisations cellulaires impliqués dans l’altération de l’angiogenèse musculaire chez les patients HTAP. Cette étude a donc démontré une diminution de l’expression de miR-126, unique aux patients HTAP. Cette diminution de miR-126 entrainait une diminution de l’angiogenèse musculaire, contribuant également à leur intolérance à l’effort. De plus, il était
possible de moduler in vivo l’expression de miR-126. L’expérimentation in vivo, à l’aide d’un modèle murin d’HTAP, a permis de rétablir l’expression de miR-126, de restaurer la microcirculation musculaire et d’améliorer la tolérance à l’effort des animaux. Ces résultats ont donc confirmé le potentiel thérapeutique de l’angiogenèse musculaire pour potentiellement améliorer la capacité à l’effort en HTAP. Cette étude a été réalisée sous la supervision des Drs Sébastien Bonnet et Steeve Provencher. J’ai recruté les patients, réalisé les tests à l’effort (test à l’effort maximal sur vélo et test d’endurance musculaire) et contribué à analyse les différents résultats avec M. François Potus. Ce dernier a mené les extractions cellulaires et la mesure de l’expression de miR-126 sur les biopsies musculaires. Il a également mené les manipulations in vitro et in vivo. J’ai participé également aux expérimentations in vivo. Vincent Mainguy a contribué au recrutement des patients et à l’élaboration du test à l’effort maximal sur vélo. Colin Graydon a aidé à l’extraction cellulaire et à la mesure de l’expression de miR-126. Ève Tremblay et Sandra Breuils-Bonnet ont élaboré les protocoles de préparation des biopsies musculaires lors du prélèvement en laboratoire et de leurs conservations subséquentes. Fernanda Ribeiro m’a assisté dans l’élaboration du protocole du test d’endurance musculaire. Alexandra Porlier a participé à la mesure de l’expression de miR-126 chez les patients atteints de la maladie pulmonaire obstructive chronique (MPOC). François Maltais a mis à notre disposition des échantillons de tissus musculaires de patients MPOC et a également contribué intellectuellement à la révision du manuscrit. J’ai rédigé le manuscrit avec François Potus et supervisé sa soumission et la révision subséquente des commentaires des réviseurs, sous la supervision des Drs Bonnet et Provencher. Sur cet article, je suis donc co-premier auteur avec M. Potus. L’article a été soumis pour publication en février 2014 à l’American Journal of Respiratory and Critical
Care Medicine et accepté pour publication en juin 2014.
Le troisième article de cette thèse (Chapitre 7) s’intitule Compromised Cerebrovascular
Regulation and Oxygenation in Pulmonary Arterial Hypertension. L’objectif de ce projet
était de déterminer si l’HTAP est caractérisée par des anomalies dans la régulation de la perfusion cérébrale pouvant ainsi contribuer à limiter l’apport sanguin et en oxygène cérébral et ainsi être un mécanisme supplémentaire venant limiter la capacité à l’effort des patients. Nous avons démontré que les changements de pression artérielle sont moins bien amortis par les vaisseaux cérébraux des patients et que leurs vaisseaux cérébraux étaient moins réactifs aux changements de CO2. Les patients présentaient aussi une augmentation de la sensibilité des chémorécepteurs centraux qui contribuait à augmenter leur ventilation au repos, mais aussi à l’exercice. Finalement, à l’effort, nous avons démontré que le débit sanguin cérébral des patients HTAP était principalement influencé par la pression artérielle, contrairement aux sujets sains pour qui le débit sanguin cérébral était influencé principalement par la PETCO2. Nous avons également démontré que les patients HTAP présentaient une diminution progressive de leur oxygénation cérébrale, qui corrélait avec leur capacité à l’effort. J’ai rédigé le protocole de recherche sous la supervision des Drs Patrice Brassard et Steeve Provencher. J’ai déterminé l’ordre des différentes expérimentations en laboratoire et recruté les participants. J’ai eu la chance d’apprendre les différentes techniques expérimentales de mesure de la régulation de la perfusion cérébrale avec Mme Myriam Paquette et M. Olivier Le Blanc en collaborant à leur projet de recherche. Ces derniers m’ont ensuite aidé à réaliser
les mêmes expérimentations pour mon projet. J’ai ensuite mené l’ensemble des analyses de données. Mme Audrey Chouinard m’a également assisté lors des différentes expérimentations en laboratoire avec les participants. Dre Valérie Nadeau a mis au point les immunohistochimies pour la mesure de la densité capillaire cérébrale sur les échantillons de cerveaux humains. M. Philip D. Allan a corrigé les artéfacts sur les différents fichiers bruts de mesure de la relation pression artérielle-débit sanguin cérébral. Le Dr Yu-Chieh Tzeng a élaboré le programme utilisé pour la mesure de la précédente relation et nous l’a généreusement prêté. Il a également continué intellectuellement à la révision du manuscrit. Dr Sébastien Simard a déterminé la batterie de tests neurocognitifs, m’a formé pour leur administration et en a supervisé leur déroulement. Dr Sébastien Bonnet a contribué à l’élaboration du protocole de mesure de la densité capillaire cérébrale et a continué intellectuellement à la révision du manuscrit. J’ai rédigé l’article sous la supervision des Drs Patrice Brassard et Steeve Provencher. L’article a été soumis pour publication en mai 2016. Les projets présentés dans le cadre de cette thèse représentent mes principales réalisations dans le domaine de la recherche clinique en HTAP au cours de mon doctorat. J’ai également réalisé un projet de recherche évaluant les anomalies de signalisation cellulaire à l’intérieur du muscle squelettique en HTAP. Nous avons démontré, à l’aide d’une analyse protéomique, que le métabolisme mitochondrial était anormal en HTAP. La microscopie électronique permettait d’observer une physionomie mitochondriale différente comparativement à des sujets sains. De plus, les anomalies de la fonction mitochondriale contribuaient à l’intolérance à l’effort des patients. Cette étude a été réalisée à l’aide de biopsies musculaires fraichement prélevées chez des sujets sains et HTAP. Ces échantillons ont permis d’extraire plus de 900 protéines. De ce nombre, 9 protéines principalement impliquées dans la phosphorylation oxydative étaient sous-exprimées tandis que 10 protéines principalement impliquées dans la glycolyse et la structure musculaire étaient sur-exprimées. J’ai recruté les patients et mené les tests à l’effort. J’ai réalisé les immunohistochimies. M. François Potus a dosé l’expression du citrate synthase et a mesuré la densité des mitochondries. Dr Frédéric Fournier a réalisé l’ensemble des mesures in silico. Mmes Sandra Breuils-Bonnet, Aude Pflieger et Ève Tremblay ont préparé les biopsies musculaires pour leur acheminement à la plateforme protéomique. M. Benjamin Nehmé a réalisé l’ensemble des manipulations techniques de l’extraction des protéines jusqu’à la génération d’un fichier brut d’analyse. Dr Arnaud Droit est responsable de cette plateforme et a contribué intellectuellement au manuscrit. J’ai rédigé le manuscrit avec M. François Potus, sous la supervision des Drs Bonnet et Provencher. J’ai ensuite supervisé les révisions et la réponse aux différents réviseurs. L’article a été soumis pour publication dans le Journal of Molecular Medicine en octobre 2014 et a été accepté pour publication en décembre 2014. Cet article constitue l’annexe 1 de cette thèse.
L’hypertension artérielle pulmonaire (HTAP) est une maladie rare, sévère, pour laquelle il n’existe aucun traitement. Elle est caractérisée par une augmentation des résistances vasculaires pulmonaires et d’une augmentation de la pression artérielle pulmonaire. Ces deux déterminants sont liés à une augmentation de la prolifération cellulaire de la paroi des petites artères pulmonaires. La maladie évolue inévitablement vers l’insuffisance cardiaque droite, principalement responsable de la mort des patients. Ces anomalies participent grandement à l’importante diminution de la tolérance à l’effort de ces derniers.
La première description anatomique remonte à plus de 100 ans, tandis que le terme hypertension pulmonaire (HTP) a été adopté en 1951 [1, 2]. La vente sans prescription de l’aminorex, un anorexigène, fut à la base de la survenue d’une première épidémie d’HTAP en Suisse en 1967 et attira l’attention sur cette affection rare. Quelques années plus tard, l’Organisation Mondiale de la Santé (OMS) organisa en 1973 à Genève une première réunion pour clarifier ce concept [3]. Les principaux résultats de cette rencontre furent de recommander une classification clinique et anatomo-pathologique distincte et, sur le plan épidémiologique, la mise en place d’un registre prospectif pour mieux connaitre la survie, la prévalence et l’incidence de la maladie. Depuis cette toute première conférence, quatre autres conférences internationales ont eu lieu. Celle tenue en France en 1998 a permis la mise en place de la classification des différents types d’HTP en cinq grandes catégories selon leur présentation clinique [4, 5]. Elle est toujours utilisée aujourd’hui d’ailleurs. Le terme hypertension pulmonaire primaire a été remplacé par le terme hypertension artérielle pulmonaire suite à cette même conférence [6]. La classification des différents types d’HTP a également été révisée aux conférences de 2008 aux États-Unis et de 2013 en France. Ainsi, le travail commun de plusieurs équipes de recherches cliniques et fondamentales a permis diverses avancées dans la compréhension en HTAP de la physiopathologie, de la génétique et de la pharmacologie, débouchant sur des progrès majeurs dans la prise en charge de ces patients au cours des 40 dernières années.
Aujourd’hui encore, les causes exactes de l’HTP demeurent inconnues. L’intolérance à l’effort demeure l’un des premiers symptômes cliniques ressentis par les patients. Malgré divers progrès pharmacologiques au cours des dernières années, l’intolérance à l’effort n’est pas complètement réversible. Ainsi, cette absence de guérison amène les patients à adopter
leur quotidien en conséquence, de façon à vivre le mieux possible en maintenant leur état stable. Cette caractéristique fait en sorte que la qualité de vie des patients est affectée par la maladie.
Cette thèse a comme objectif général de décrire de nouveaux mécanismes impliqués dans la diminution de la tolérance à l’effort des patients. Les causes de l’intolérance à l’effort sont multiples. Elles touchent les systèmes cardiovasculaire, ventilatoire et musculaire. La compréhension de ces mécanismes s’avère primordiale afin de raffiner les interventions susceptibles d’améliorer la capacité des patients. Ainsi, comme nouveaux mécanismes, nous avons premièrement documenté une diminution de l’apport en oxygène aux muscles squelettiques à l’effort des patients en relation avec une diminution de la densité capillaire musculaire et l’intolérance à l’effort [7] (Chapitre 5). Nous avons ensuite documenté une diminution de l’expression du microARN (miR)-126 dans les cellules endothéliales isolé à partir de biopsies musculaires, responsable de la diminution de la densité capillaire et qui contribuait à leur intolérance à l’effort [8]. La diminution de l’expression de ce miR était unique aux patients HTAP (Chapitre 6). Le dernier projet présenté dans cette thèse s’est attardé aux différents mécanismes régulant le débit sanguin cérébral. Nous avons démontré donc que les patients HTAP présentaient une diminution de débit sanguin cérébral, mais également que les changements de pression artérielle sont moins bien amortis par les vaisseaux cérébraux des patients et que leurs vaisseaux cérébraux étaient moins réactifs aux changements de dioxyde de carbone (CO2). Les patients présentaient aussi une augmentation de la sensibilité des chémorécepteurs centraux qui contribuait à augmenter leur ventilation au repos, mais aussi à l’exercice. Finalement, à l’effort, nous avons démontré que le débit sanguin cérébral des patients HTAP était principalement influencé par la pression artérielle alors que chez les sujets sains, le débit sanguin cérébral était influencé principalement par la pression partielle en CO2. Nous avons également démontré que les patients HTAP présentaient une diminution progressive de leur oxygénation cérébrale, qui corrélait avec leur capacité à l’effort (Chapitre 7).
CHAPITRE 2 – Systèmes cardiovasculaire,
LE SYSTÈME CARDIOVASCULAIRE
Le système cardiovasculaire est un continuum fermé qui inclut une pompe, le cœur, et un système de conduit, les vaisseaux sanguins. Le système cardiovasculaire procure un environnement pour le transport des nutriments vers les tissus périphériques, ainsi que l’excrétion des déchets métaboliques.
Constituant macroscopique du cœur
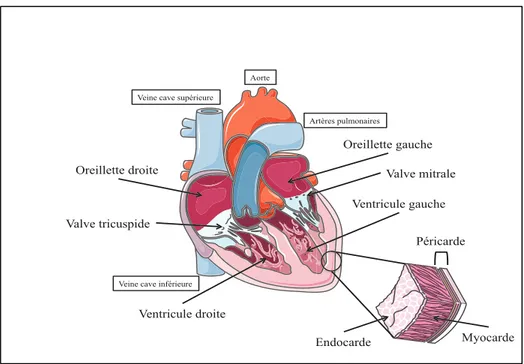
Le cœur agit comme une pompe séparée par une cloison médiale, les septums auriculaire et ventriculaire. Les oreillettes reçoivent le sang qui arrive au cœur soit par les veines caves ou les veines pulmonaires. Ces dernières ont une paroi plus fine. Les ventricules reçoivent le sang en provenance des oreillettes pour ensuite l’expulser vers la circulation pulmonaire ou systémique. Le ventricule droit pompe le sang à l’intérieur de la circulation pulmonaire. De son côté, le ventricule gauche pompe le sang dans la circulation systémique. Sa paroi est plus épaisse que celle du ventricule droit, car il développe plus de force comparativement au ventricule droit, qui est génétiquement adapté pour pomper le sang dans un système circulatoire à basse pression. La figure 1 présente les différentes composantes anatomiques du cœur.
Figure 1. Cavités cardiaques
Oreillette droite Oreillette gauche Ventricule gauche Ventricule droite Péricarde Myocarde Endocarde Valve tricuspide Valve mitrale
Veine cave supérieure
Veine cave inférieure
Aorte
Chambres cardiaques
En position anatomique, le bord droit du cœur est formé par l’oreillette droite, recevant le sang désoxygéné provenant de trois vaisseaux. Les deux veines caves inférieure et supérieure drainent le sang de la circulation systémique tandis que le sinus coronaire draine le sang de la circulation coronaire. Le sang passe ensuite dans le ventricule droit qui constitue la plus grande partie de la face antérieure et diaphragmatique du cœur. Le retour du sang oxygéné de la circulation pulmonaire se fait par les veines pulmonaires droites et gauches à l’intérieur de l’oreillette gauche, qui forme la base de la face postérieure du cœur. Le dernier passage du sang avant d’atteindre la circulation systémique par l’aorte se fait dans le ventricule gauche, qui occupe la face antérieure, diaphragmatique et pulmonaire gauche et forme l’apex du cœur.
Valves cardiaques
La valve tricuspide est située entre l’oreillette et le ventricule droit. Elle est formée de trois cuspides fermement ancrées à un anneau fibreux qui stabilise et permet de maintenir l’ouverture de la valve. La valve mitrale est située entre l’oreillette et le ventricule gauche. Elle est formée de deux cuspides également ancrées au même type d’anneaux fibreux récemment décrit. Les bords libres de chaque cuspide sont attachés à des cordages tendineux provenant des muscles papillaires. Au cours du remplissage des ventricules, les cuspides basculent dans ces derniers. À la puissante contraction des ventricules, les muscles papillaires se contractent pour maintenir les valves bien étanches et ainsi prévenir le retournement des cuspides dans les oreillettes.
L’entrée du tronc pulmonaire est occupée par la valve pulmonaire, qui est constituée de trois valvules semi-lunaires (gauche, droite, antérieur). Les bords libres se projettent dans la lumière du tronc pulmonaire. Le bord supérieur de chaque valvule présente une portion épaisse et des rebords plus fins. La valve forme un sinus en forme de poche après la contraction ventriculaire permettant une fermeture étanche de la valve pulmonaire au moment du remplissage des ventricules. L’orifice entre le ventricule gauche et l’aorte ascendante est fermé par la valve aortique. Sa structure, semblable à celle de la valve pulmonaire, présente trois valvules semi-lunaires qui forment les sinus aortiques droit, gauche et postérieur. Les artères coronaires droite et gauche naissent des sinus droit et gauche. Ainsi, lorsque la valve est fermée et que le sang remplit les sinus aortiques, il est automatiquement poussé dans les
artères coronaires, qui se remplissent donc principalement au moment de la diastole cardiaque.
Le système circulatoire
Système artériel, capillaire et veineux
À la sortie du cœur, le sang entre dans la circulation vasculaire, qui est composée de plusieurs types de vaisseaux. Les artères transportent le sang de l’extérieur du cœur vers de plus petits embranchements, les artérioles. Ces dernières sont ensuite reliées aux capillaires qui permettent les différents échanges avec les tissus. Le sang entre ensuite dans la circulation veineuse pour converger vers les plus grandes veines et revenir au cœur.
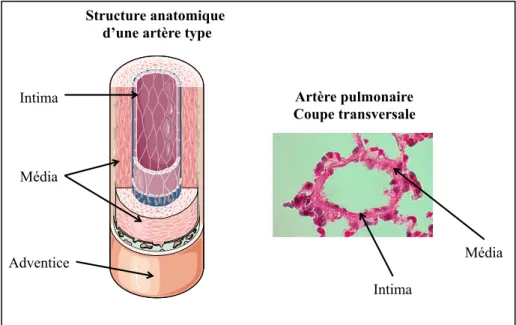
Figure 2. Composition structurelle d’une artère
Les différents vaisseaux du système vasculaire diffèrent par la structure anatomique de leur paroi. La figure 2 présente les différentes composantes de la structure d’une artère. La paroi des artères et des artérioles sont composé de l’adventice, de la média et de l’intima. Chaque constituant de la paroi artérielle possède une signature anatomique particulière qui permet à l’artère d’absorber les hautes pressions qu’elle subit. La composition anatomique des veines et veinules est similaire aux artères. Par contre, elles sont plus minces que ces dernières. Certaines veines, comme celles des jambes contiennent une série de valves unidirectionnelles qui assistent le retour veineux vers le cœur.
Intima Média Adventice Artère pulmonaire Coupe transversale Intima Média Structure anatomique d’une artère type
L’adventice est situé en périphérie et est constitué de tissus conjonctifs, de collagène et d’élastine, jouant un rôle de soutien. Elle reçoit également les éléments fonctionnels de l’artère telle que les terminaisons nerveuses à l’origine de la vasomotricité et le vasa vasorum qui irrigue la paroi en elle-même. La média est composée principalement de fibres musculaires lisses, mais aussi de fibre de collagène et d’élastine. Cette couche peut être influencée par le contrôle neuronal sympathique et parasympathique, le contrôle hormonal, mais aussi par des médiateurs vasoactifs locaux tels que le pH et les niveaux d’oxygène et de dioxyde de carbone (CO2). L’intima est en contact direct avec le sang. Elle est constituée de cellules endothéliales et d’une couche sous endothéliale. Elle permet (ou bloque) le passage de différentes substances transportées par le sang tout en produisant différentes substances vasoactives. Les capillaires forment une architecture dense de microvaisseaux que l’on retrouve en grande quantité dans chaque tissu. Leur paroi est très mince, ce qui facilite l’apport en nutriment aux différentes cellules et l’évacuation des déchets métaboliques. Ils sont constitués principalement de cellules endothéliales, mais aussi de fibres de collagène et d’élastine qui aident au maintien de leur forme.
Baroréflexe artériel
Le baroréflexe artériel est un mécanisme homéostatique important qui vise à maintenir la pression artérielle relativement constante. Ce système repose sur un type de neurone spécifique, nommé barorécepteur, situé entre autres dans l’arc aortique et le sinus carotidien. Ces fibres nerveuses informent en continu le système nerveux central sur les changements de pression artérielle. Ainsi, au repos, une augmentation de la pression artérielle est accompagnée d’une bradycardie réflexe et une diminution de la pression artérielle est accompagnée d’une tachycardie réflexe et une vasoconstriction [9].
LA FORMATION DE NOUVEAUX VAISSEAUX
Les nouveaux vaisseaux sanguins se forment selon deux types de formation qui impliquent différents facteurs de croissance. Au niveau embryonnaire, les facteurs de croissance endothéliaux angioblastiques permettent la formation d’un réseau vasculaire primitif (vasculogenèse). À la naissance, d’autres facteurs de croissance s’ajoutent à ceux déjà actifs. Ainsi, le processus d’angiogenèse prend le dessus sur la vasculogenèse, quoique cette dernière reste quand même active [10].
Les facteurs de croissance
L’angiogenèse débute par une déstabilisation des différents constituants cellulaires de la paroi vasculaire du vaisseau-mère afin qu’un nouveau vaisseau s’y intègre. Cette étape implique différents facteurs de croissance. Ainsi, l’action angiogénique du vascular
endothelial growth factor (VEGF) et du fibroblast growth factor (FGF) commence par la
stimulation de la production de protéase ou d’activateurs de plasminogène par les cellules endothéliales [11]. Ces derniers induisent la dégradation de la membrane basale du vaisseau-mère, permettant à différentes cellules d’envahir la matrice cellulaire environnante. Ces cellules migratoires vont ensuite proliférer et se différencier en cellules endothéliales pour former un nouveau vaisseau. Après sa formation, le nouveau vaisseau est constitué d’une seule couche de cellules, sans même un lumen. Les nouvelles cellules endothéliales vont alors sécréter différents facteurs de croissance comme le platelet-derived growth factor (PDGF) ou l’angiopoïétine 1 (ANG-1) pour attirer différentes cellules comme les pericytes et les cellules musculaires lisses, permettant de stabiliser le lumen et la structure du nouveau vaisseau [12]. La génération d’un nouveau vaisseau ou même l’élimination d’un vaisseau existant nécessite la déstabilisation du vaisseau existant, également engendré par différents facteurs de croissance comme l’angiopoïétine 2 (ANG-2) [10].
Les micro-ARN
Les miRs sont des modulateurs de l’expression des gènes d’une importance capitale. Ce sont de courts brins d’ARN non-codant, composés de 21 à 24 nucléotides. L’activité des miRs oscille constamment entre répression et activation de l’expression des gènes en fonction du cycle cellulaire [13]. Ces derniers régulent l’expression d’un gène de deux façons principalement, soit en agissant comme répresseur post-transcriptionnel ou en déstabilisant l’ARN messager [14]. Un profil de l’expression de différents miRs pouvant contribuer à l’angiogenèse a été effectué dans les cellules endothéliales ou dans les cellules souches d’embryons entres autres, révélant une expression très élevée de plusieurs miRs, dont le miR-126 [15], qui est un miR spécifique aux cellules endothéliales. miR-miR-126 est un régulateur clé de l’angiogenèse et de l’intégrité vasculaire dans les cellules endothéliales. Un inhibition de son expression dans des embryons de poissons-zèbres ou dans des embryons de souris induit des malformations vasculaires et des saignements embryologiques importants [15], en plus d’une capacité angiogénique moins importante après la naissance [16]. Les deux cibles de
miR-126 sont le Srouty-related EVH1 domain-containing protein 1 (Spred1) et la sous-unité de PI3K, le PI3KR2. Comme Spred1 et PI3KR2 agissent comme des régulateurs négatifs de la cascade signalétique de VEGF, miR-126 favorise donc l’angiogenèse [15-17] (Figure 3). miR-126 adopte la forme d’une tige-boucle et est encodé par l’intron 7 du gène Egfl7
(EGF-like domain 7) [18] (Figure 3).
Figure 3. Mécanismes d’actions de miR-126, adaptés de Fish et al. [15] et de Wang et al. [16]. L’augmentation de l’expression de miR-126 inhibe l’expression de Spred1 et
de PI3KR2, deux inhibiteurs de la voie de signalisation angiogénique de VEGF. Les abréviations non décrites dans le texte sont : PI3KR2 : phosphoinositol-3 kinase regulatory subunit 2; AKT: Protein kinase B; RAF1: proto-oncogene
serine/threonine-protein kinase; ERK: extracellular-signal-regulated kinases.
LE SYSTÈME RESPIRATOIRE
Le système respiratoire est en contact permanent avec l’environnement extérieur pour permettre les échanges gazeux entre la circulation sanguine et l’air ambiant. Les mouvements continus de l’appareil respiratoire (les muscles de la cage thoracique, du diaphragme, et de l’abdomen, la plèvre pariétale et viscérale et le tissu élastique des poumons) permettent un passage continu de l’air dans les poumons. La contraction des muscles inspiratoires entraine l’air extérieur (21% O2, 0,04% CO2, balance N2) dans les voies respiratoires jusqu’aux
VEGF Endothélium RAF1 ERK PI3K% AKT%
Angiogenèse et intégrité vasculaire
SPRED1 PI3KR2 miR-126 miR-126 Egfl 7 Egfl 7 VEGFR-2
alvéoles. Le retour passif de l’appareil respiratoire à sa position de repos entraine l’expiration de l’air (15,5% O2, 4% CO2, balance N2) vers l’environnement extérieur.
Voie et surface respiratoire
L’entrée de l’air dans les poumons se fait par les voies de conduction respiratoire qui ont pour rôle, en plus de conduire l’air inspiré vers les alvéoles, de la nettoyer, de l’humidifier et de la réchauffer. Par exemple, la cavité nasale et le nasopharynx sont richement vascularisés pour réchauffer et humidifier l’air inspiré. L’oropharynx et le laryngopharynx constituent l’entrée respective des systèmes digestif et respiratoire. À l’entrée du larynx se trouve l’épiglotte, un cartilage en forme de feuille attachée par sa tige au versant postérieur du cartilage thyroïde, qui agit comme un clapet empêchant les aliments d’être aspirés à l’intérieur du système respiratoire. Après le larynx, les voies de conduction se différencient successivement de la trachée aux bronchioles respiratoires.
La plus petite unité pulmonaire visible est le lobule, délimité par des septa interlobulaires de tissus conjonctifs et la plèvre viscérale. En son centre, le lobule possède un paquet bronchovasculaire central qui comprend une bronchiole et une artériole. Les veinules pulmonaires se trouvent en périphérie du lobule. Chaque lobule contient les acini, qui comprennent une multitude de bronchioles respiratoires, les sacs alvéolaires et les alvéoles. Dans l’acinus, les échanges gazeux surviennent à l’intérieur des alvéoles, qui forment une très mince membrane à l’interface de l’air et de la circulation sanguine. Les alvéoles sont enveloppés de capillaires alvéolaires dont la paroi est également mince pour faciliter les échanges gazeux.
Vascularisation pulmonaire
L’apport sanguin aux poumons est double : pulmonaire et systémique. La circulation sanguine pulmonaire est un système de basse pression qui dépend du cœur droit et ayant pour rôle d’oxygéner le sang. La pression artérielle pulmonaire moyenne (PAPm) saine se maintient à l’intérieur d’un intervalle de 14 à 17 mmHg [19], tandis que la résistance vasculaire pulmonaire (RVP) demeure sous 240 dyne.s.cm-5. Avec l’avancée en âge, la PAPm augmente très légèrement, mais restent sous les seuils pathologiques de 25 mmHg [19, 20]. À l’effort, la PAPm augmente légèrement tandis que la RVP diminue [20]. La circulation
systémique est un système de haute pression (pression moyenne entre 100-120 mmHg) qui dépend du cœur gauche et ayant pour but de distribuer le sang oxygéné à tous les organes. Les artères bronchiques font partie de cette circulation et proviennent soit de l’aorte descendante ou des artères intercostales.
Il existe des anastomoses entre le réseau pulmonaire et bronchique. Cette communication ajoute un petit volume de sang désoxygéné provenant de la circulation veineuse bronchique à l’intérieur de la circulation veineuse pulmonaire qui contient le sang fraichement oxygéné. Cette anastomose est considérée comme faisant partie du shunt anatomique. Il existe également des anastomoses artérioveineuses intrapulmonaires, qui dévient le sang désoxygéné vers les veines pulmonaires sans être réoxygéné par les alvéoles. Le recrutement de ces communications artérioveineuses intrapulmonaires semble modulé par l’hypoxie alvéolaire pour maximiser les échanges gazeux en condition hypoxique [21], quoique son apport physiologique est encore source de débat.
SYSTÈME MUSCULAIRE ET CAPILLARITÉ
Le système musculaire est composé de trois types de muscle : les muscles squelettiques qui réalisent les mouvements volontaires, les muscles lisses qui réalisent les mouvements involontaires et le muscle cardiaque qui constitue la majeure partie du cœur. Une fibre musculaire isolée, elle-même constituée de myofibrilles (contenant les éléments contractiles), forme en réalité la cellule musculaire. Il existe deux types de fibres musculaires, les fibres de type lentes (slow-twitch ; type I) et les fibres de type rapides (fast-twitch ; type II). Les fibres de type II sont divisées en deux sous-divisions, les fibres de type IIa (IIa) et les fibres de type IIx (IIx).
La proportion pour les différents types de fibres musculaires varie principalement en fonction du sexe, du niveau d’entrainement, du type de muscle et de la présence ou non d’une pathologie. Chez l’adulte sain, une biopsie du vaste latérale révèle un décompte de fibre de type I de 36% chez l’homme et de 44% chez la femme tandis que le décompte de fibre de type IIa est de 41% chez l’homme et seulement de 34% chez la femme [22]. De plus, pour l’ensemble des fibres musculaires mesurées par Staron et al, l’aire de surface sectionnelle était plus élevée chez l’homme comparativement à la femme [22]. Les athlètes d’endurance
présentent une proportion plus élevé de fibre de type I comparativement aux sujets sains sédentaires, tous sexes confondus [23, 24]. Chez les athlètes entrainés en résistance musculaire, c’est une proportion plus élevé de fibre de type IIa et IIx comparativement aux sujets sains sédentaires, tous sexes confondus [25, 26], indiquant donc que la typologie musculaire est aussi dépendante du type d’activité physique qui est réalisé en plus du niveau d’entrainement des sujets.
Les ergorécepteurs
Le muscle squelettique contient deux type d’ergorécepteurs : les métaborécepteurs et les mécanorécepteurs [27, 28]. Les métaborécepteurs sont sensibles aux changements métaboliques à l’intérieur du muscle. Ils sont stimulés par les métabolites générés par la contraction musculaire (lactates, ions hydrogène (H+), bradykinines, prostaglandines, phosphates) et répondent en envoyant un signal nerveux à la commande centrale via les fibres nerveuses musculaires afférentes du groupe IV principalement [29]. Les mécanorécepteurs (par ex. organes tendineux de Golgi et fuseaux neuromusculaires) sont sensibles aux changements physiques musculaires. Ils sont plus sensibles à la fréquence successive de contractions et d’étirements musculaires qu’à la force de contraction proprement dite. La stimulation des mécanorécepteurs peut activer ou inhiber une contraction musculaire. Ces derniers envoient également un signal nerveux à la commande centrale via les fibres nerveuses musculaires afférentes, mais principalement du groupe III [30]. Il est important de garder en tête que l’activation des fibres afférentes des groupes III/IV se fait en même temps, mais diffère en terme de processus de réponse (par ex. entré en action des impulsions nerveuses, intensité des impulsions, stimulus déclenchant les impulsions) [31].
LE SYSTÈME CÉRÉBROVASCULAIRE
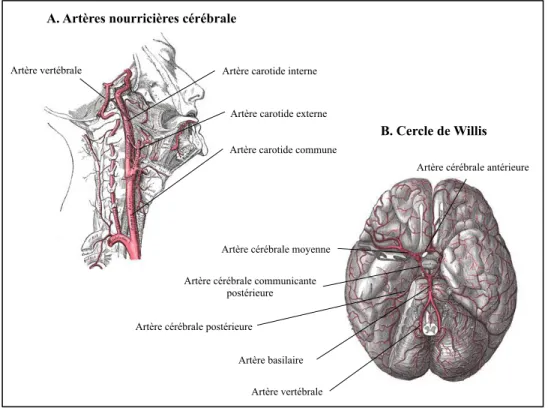
La figure 4 illustre les principales artères nourricières cérébrales. Les artères vertébrales et carotides internes sont tortueuses à la hauteur de leur entrée intracrânienne réciproque, ce qui a pour effet d’augmenter la turbulence du sang à l’intérieur de ces vaisseaux menant à une diminution de la pression artérielle [32]. Ce mécanisme protège la circulation sanguine cérébrale des augmentations rapides de tension artérielle.
Figure 4. Artères cérébrales. A. L’artère vertébrale droite provient de la portion
ascendante de l’artère subclavière. Elle chemine vers le cerveau à travers les foramens transverses des six premières vertèbres cervicales, entrent la cavité crânienne par le foramen magnum et rejoint l’artère vertébrale gauche pour former l’artère basilaire. Chaque carotide interne entre la cavité intracrânienne par les canaux carotidiens. B. L’apport sanguin cérébral provient des artères vertébrales droite et gauche, ainsi que des artères carotides internes et externes droite et gauche qui s’anastomosent dans la cavité crânienne pour former le cercle artériel, le Cercle de Willis.
L’imagerie par résonance magnétique en phase de contraste permet de quantifier la perfusion cérébrale jusqu’aux petites artères d’un diamètre de 1,5 mm. Zarrinkoob et al ont donc été en mesure de déterminer que la perfusion cérébrale totale était en moyenne 717 (123) mL/min chez l’adulte sain et qu’elle était légèrement supérieure chez la femme (724 (131) mL/min) comparativement à l’homme (709 (114) mL/min), sans que cette différence ne soit significativement différente [33]. Le pourcentage de l’apport sanguin cérébral provenant des carotides internes correspondait à 72% tandis que 28% de l’apport sanguin cérébral provenait des artères vertébrales.
Artère carotide commune Artère carotide externe Artère carotide interne Artère vertébrale
Artère vertébrale Artère basilaire Artère cérébrale postérieure
Artère cérébrale communicante postérieure
Artère cérébrale moyenne
Artère cérébrale antérieure B. Cercle de Willis A. Artères nourricières cérébrale
MÉCANISMES PHYSIOLOGIQUES DÉTERMINANT LA RÉPONSE CÉRÉBROVASCULAIRE AU REPOS
Le cerveau compte pour un total de 2-3% de la masse corporelle, mais reçoit environ 15% du débit cardiaque total et consomme environ 20% de l’apport total en oxygène au repos. Face à une capacité de stockage énergétique limitée, le maintien d’une perfusion cérébrale adéquate est donc nécessaire pour assurer son haut niveau de fonctionnement énergétique. Plusieurs mécanismes physiologiques entrent donc en jeu en vue de moduler la perfusion cérébrale selon les besoins énergétiques exigés par une situation particulière, comme l’exercice, le sommeil ou pour faire face à un environnement particulier comme l’élévation en altitude. Cette dernière section du présent chapitre, consacrée à la physiologique cardio-respiratoire, musculaire et vasculaire chez l’adulte sain, centrera donc son attention sur les différents déterminants de la perfusion cérébrale.
La pression artérielle
La pression de perfusion cérébrale participe activement au maintien de la perfusion cérébrale et est déterminée par la différence entre la pression artérielle moyenne (PAM) et la pression intra-crânienne, qui est généralement estimée à 10 mmHg [32]. La formule décrivant la PAM est (pression artérielle (PA) systolique + 2PA diastolique)/3. Au repos, chez l’adulte sain, avec une PAM équivalente à 90 mmHg, la pression de perfusion cérébrale équivaut à 80 mmHg. Lorsque la pression artérielle augmente ou diminue, plusieurs mécanismes physiologiques entrent en jeu afin de maintenir la perfusion cérébrale relativement constante. Entre autre, lorsque la pression systémique augmente, il y a une vasoconstriction des artérioles cérébrales pour limiter l’augmentation du débit sanguin au cerveau, ce qui se traduit par une augmentation de la résistance vasculaire cérébrale. Inversement, lorsque la pression diminue, il y aura une vasodilatation des artérioles cérébrale dans le but de maintenir le débit sanguin, ce qui se traduit par une diminution de la résistance vasculaire cérébrale.
L’autorégulation cérébrale
L’autorégulation cérébrale est constituée d’une composante statique et dynamique. L’autorégulation cérébrale statique régule le débit sanguin cérébral selon les changements progressifs, graduels et en état stable de la pression cérébrale [34, 35]. L’autorégulation cérébrale dynamique réfère à la régulation aiguë de la perfusion cérébrale en réponse à un changement aigu de la PAM [35].
En 1959, Lassen détermina, à partir de sept études touchant la relation entre la pression artérielle et la perfusion cérébrale, qu’un plateau existait à partir duquel le débit sanguin cérébral restait relativement stable malgré une augmentation de la PAM [36]. Ces observations ont conduit à la définition classique de l’autorégulation cérébrale qui réfère à la capacité intrinsèque des vaisseaux sanguins cérébraux à maintenir la perfusion cérébrale relativement constante lorsque la PAM se situe entre 60 et 150 mmHg [34]. Cette étude pivot contient malgré tout certaines limitations, entre autres le fait que les sujets provenant des différentes études étaient soit porteurs d’une pathologie ou participaient à un protocole pharmacologique qui agissait sur la pression artérielle. De plus, ces résultats proviennent d’une comparaison intersujet, alors qu’une comparaison intrasujet est plus appropriée pour être en mesure d’observer comment la perfusion cérébrale s’adapte au changement de pressions artérielles.
La littérature actuelle indique que cette relation n’est pas aussi stable sur un aussi grand intervalle de PAM. Lucas et al ont démontré que la perfusion cérébrale et l’oxygénation cérébrale ne sont pas indépendantes des changements de pression artérielle [37]. Les travaux de Tan et de Tzeng ont également permis de démontrer que le plateau de l’autorégulation cérébrale est bien présent tout en étant plus petit, indiquant une relation PAM-perfusion cérébrale passive plus importante que le modèle initial de Lassen [38]. De plus, la perfusion cérébrale est plus efficace à contrecarrer une augmentation de la PAM qu’une diminution de cette dernière [39]. Les résultats de Tzeng et al ont également été corroborés à l’aide d’une récente méta-analyse [40]. Ainsi, la figure 5 résume le changement de paradigme qui s’impose aujourd’hui en passant d’une vision classique de l’autorégulation cérébrale à une vision plus moderne de cette dernière.
Figure 5. Concept de l’autorégulation cérébrale statique selon Lassen (gauche) [36] et
le concept moderne de l’autorégulation cérébrale statique (droite) [32, 38, 39].
Les mécanismes physiologiques derrière l’autorégulation cérébrale reste, encore aujourd’hui, relativement inconnus. Malgré tout, certaines théories ont été mises de l’avant pour l’expliquer.
Approche myogénique
Les muscles lisses des parois des artères cérébrales seraient sensibles aux changements de pression. Ces derniers font preuve de compliance pour s’adapter aux changements de PAM [41]. Lorsque des bloqueurs calciques sont donnés à des adultes sains pour diminuer la réponse myogénique du système vasculaire, les augmentations et diminutions de PAM artificiellement, produites à l’aide d’une chambre spécialement conçue, induisent des changements plus importants de perfusion cérébrale comparativement à la condition sans bloqueur calcique [42]. Ces résultats impliquent donc directement la composante myogénique des vaisseaux cérébraux comme l’un des mécanismes de l’autorégulation cérébrale.
Approche neurologique
Le système vasculaire cérébral est richement innervé par les fibres des systèmes nerveux sympathique et parasympathique. Les fibres du système nerveux parasympathique sont surtout présentes au niveau de l’espace Virchow-Robin [32]. La composante neurologique est aussi importante comme mécanisme de l’autorégulation cérébrale au repos et à l’effort, mais son apport est encore source de débat, principalement au repos [43]. L’activité nerveuse a un impact sur l’autorégulation cérébral. En bloquant l’activité des systèmes parasympathique et
sympathique, le débit sanguin cérébral augmente plus passivement avec la pression artérielle, dénotant un temps d’adaptation plus long des mécanismes intrinsèques des vaisseaux cérébraux pour s’adapter aux changements de pression artérielles [35].
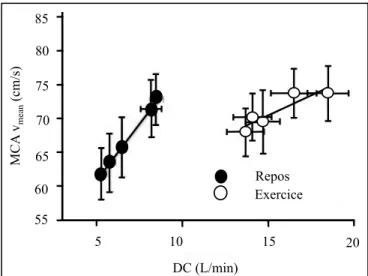
Le débit cardiaque
L’apport sanguin cérébral dépend également, comme pour tous les organes vitaux, du débit cardiaque (DC). Une relation linaire a d’ailleurs été déterminée entre le débit cardiaque et la perfusion cérébrale à l’aide de la vitesse de passage du sang dans l’artère cérébrale moyenne (MCAv), autant au repos qu’à l’exercice [44]. Cette relation linéaire est conservée malgré les changements qui peuvent survenir au niveau de l’autorégulation cérébrale et de la pression partielle artérielle en CO2 (PaCO2)[45]. La figure 6 illustre cette relation.
Figure 6. Relation linéaire entre le débit cardiaque (DC) et
la vitesse d’écoulement du sang à l’intérieur de l’artère cérébrale moyenne (MCA vmean). Adapté de Ogoh et al [44].
Le dioxyde de carbone et l’oxygène
Il est également bien établi que la PaCO2 influence grandement les vaisseaux sanguins cérébraux [46]. L’étroite relation qui existe entre la perfusion cérébrale et les changements de PaCO2 est essentielle pour la régulation et le maintien du pH, qui peut affecter la sensibilité des chémorécepteurs centraux et la ventilation [47]. En condition de normoxie, l’interaction entre la perfusion cérébrale et une augmentation de la PaCO2 induit une vasodilatation des artères cérébrales tandis qu’une diminution de la PaCO induit une
5 10 15 20 DC (L/min) Repos Exercice 55 60 65 70 75 80 85 MCA vmean (cm/s)
![Figure 3. Mécanismes d’actions de miR-126, adaptés de Fish et al. [15] et de Wang et al](https://thumb-eu.123doks.com/thumbv2/123doknet/6358950.167867/30.918.217.740.278.644/figure-mécanismes-actions-mir-adaptés-fish-al-wang.webp)
![Figure 5. Concept de l’autorégulation cérébrale statique selon Lassen (gauche) [36] et le concept moderne de l’autorégulation cérébrale statique (droite) [32, 38, 39]](https://thumb-eu.123doks.com/thumbv2/123doknet/6358950.167867/37.918.210.753.136.332/figure-concept-autorégulation-cérébrale-statique-autorégulation-cérébrale-statique.webp)
![Figure 10. Modèle intégré des différents mécanismes physiologiques cardiaque, pulmonaire et circulatoire et la réponse à l’exercice, adapté de [85]](https://thumb-eu.123doks.com/thumbv2/123doknet/6358950.167867/54.918.255.709.397.755/modèle-intégré-mécanismes-physiologiques-cardiaque-pulmonaire-circulatoire-réponse.webp)