Développement d’un ribozyme
spécifique à l’ARNm de Tau pour
contrer les Tauopathies
Mémoire
Laura Eyoum Jong
Maîtrise en neurobiologie
Maître ès sciences (M.Sc.)
Québec, Canada
Développement d’un ribozyme
spécifique à l’ARNm de Tau pour
contrer les Tauopathies
Mémoire
Laura Eyoum Jong
Sous la direction de :
Emmanuel Planel, directeur de recherche
Georges Lévesque, codirecteur de recherche
iii
Résumé
Une des causes principales de la Maladie d’Alzheimer et des autres Tauopathies est la présence d’enchevêtrements neurofibrillaires (ENF). Les ENF sont des agrégations intracellulaires de la protéine Tau hyperphosphorylée. Plusieurs études ont montré que les ENF sont associés à la pathogénèse des troubles neurodégénératifs et à la neurotoxicité. De plus, il a été démontré qu’une diminution du niveau de la protéine Tau permet de prévenir les déficits cognitifs dans les modèles murins. Basé sur ces études, notre hypothèse de recherche est que réduire le niveau total de Tau dans le cerveau, pourrait diminuer les ENF et retarder la pathologie. Notre objectif est de développer une molécule capable de cibler l’ARNm de Tau plutôt que la protéine Tau hyperphosphorylée. Ainsi, nous avons développé le SOFA-delta ribozyme, capable de cliver l’ARNm de Tau. Notre ribozyme a trois composantes soit le bloqueur, le biosenseur et l’effecteur. Nous avons développé des delta-ribozymes capable cibler les exons constitutifs et l’exon 10 (spécifique au Tau4R) de l’ARNm de Tau, ainsi nous pouvons cibler tous les isoformes de Tau. Dans ce projet, nous avons tout d’abord synthétisé le delta-ribozyme par clonage moléculaire et nous avons confirmé son effet grâce au clivage in vitro. Dans un second temps, nous avons transfecté le ribozyme dans des cellules neuronales, et nous avons caractérisé son effet sur l’ARNm de Tau par RT-PCR. Enfin, nous avons produit des virus AAV, infecté des cellules neuronales et caractérisé encore une fois l’effet du ribozyme sur l’ARNm de Tau par RT-PCR.
Cette approche thérapeutique est basée sur la spécificité et l’efficacité du SOFA-delta-ribozyme, qui identifie et coupe l’ARNm de Tau.
Ainsi, dans ce mémoire nous avons identifié le ribozyme 350 et 395 comme candidats potentiellement capable de cliver l’ARNm de Tau.
iv
Abstract
One of the main causes of Alzheimer’s disease and Tauopathies is the presence of neurofibrillary tangles (NFT). NFT consist in intracellular aggregation of the abnormally hyperphophorylated protein Tau. Several studies have shown that NFT are associated with the pathogenesis of neurodegenerative disorders and neurotoxicity. Moreover, it has been demonstrated that decreasing the level of Tau protein could prevent cognitive deficits in mouse models. Based on these studies, our hypothesis is that reducing the level of total Tau in the brain could decrease NFTs and delay the pathology. Our objective is to design a molecule that will target directly Tau mRNA instead of the hyperphosphorylated protein. We developed a modified delta-ribozyme, the SOFA (Specific On/Off Adaptor) ribozyme, which is able to cleave the Tau mRNA. Our ribozyme is composed of three components: the blocker, the biosensor, and the effector. We have designed delta-ribozymes that target constitutive exons of Tau mRNA as well as the exon 10 specific of
Tau 4R. Therefore, we can target all the Tau isoforms. In this project, we
first synthesized the delta-ribozyme by molecular cloning and we confirmed its effect by in vitro cleavage. Secondly, by transfecting it in neuronal cells we characterized its effects on Tau mRNA by RT-PCR. Finally, we produced AAV viruses and infected neuronal cells and
characterized its effects on Tau mRNA by RT-PCR. This therapeutic
approach is based on specificity and efficiency of the SOFA-ribozymes, which identify and cut the Tau mRNA. Thus, in this project we identified ribozymes 350 and 395 as potential good candidates able to cleave the Tau mRNA.
v
Table des matières
Résumé ...iii
Abstract ... iv
Listes des figures ... viii
Listes des tableaux ...x
Listes des abbréviations ... xi
Remerciements ... xiv
Introduction ... 1
I. Les Tauopathies ... 1
1.1. Statistiques ... 1
1.2. Regroupement des Tauopathies et caractéristiques de ces maladies ... 2
1.3. La Maladie d’Alzheimer ... 7
1.4. Le peptide -amyloïde ... 9
II. La protéine Tau ... 13
2.1. Le gène et la protéine de Tau ... 13
2.2. Modifications post-traductionnelles ... 15
2.3. Rôle de Tau et Neurotoxicité ... 18
2.4. Propagation dans la maladie d’Alzheimer ... 22
III. Les Ribozymes ... 24
3.1. Traitements ... 24
3.2. Généralités : les ribozymes naturels ... 27
3.3. Généralités : les ribozymes naturels ... 29
3.4. SOFA-Delta Ribozyme ... 32
IV. Hypothèse et objectifs ... 36
Chapitre 1 : matériel et méthodes ... 38
1. Bioinformatique ... 38 2. Clonage moléculaire ... 38 2.1. pAAV-U6-GFP ... 38 2.2. pBlueScript SK ... 39 2.3. pEGFP-C1-del-EGFP et pGBKT7... 40 2.4. Annealing ... 41
vi
2.5. Digestion, déphosphorylation et purification avec le gel
extraction kit du vecteur. ... 42
2.6. Digestion de la séquence du ribozyme ... 44
2.7. Ligation ... 44
2.8. Transformation bactérienne ... 46
2.9. Isoler et repiquer une colonie ... 47
2.10. Mini préparation ... 47
2.11. Dosage de l’ADN ... 48
2.12. Vérification du clonage et séquençage ... 48
3. Clivage in vitro ... 49
3.1. Digestion des plasmides ... 49
3.2. Transcription in vitro du ribozyme et de l’ARNm de Tau et clivage in vitro ... 50
3.3. Réaction de reverse transcriptase et DNAse I ... 52
3.4. PCR ... 54
4. Culture cellulaire ... 56
5. Transfection ... 57
5.1. Cotransfections des HEK-293 au Jet Prime® ... 57
5.2. Transfection des SH-SY5Y au CaCl2 ... 58
6. Extraction d’ARN et RT-PCR ... 59
6.1. Extraction d’ARN ... 59
6.2. Dosage et qualité de l’ARN... 60
6.3. Reverse Transcriptase ... 60
6.4. PCR ... 61
7. Production virale ... 62
7.1. Transfection dans les HEK-293AAV ... 62
7.2. Récolte et purification des virus ... 62
7.3. Infection des SK-N-MC ... 64
Chapitre 2 : Résultats ... 65
I. Objectif 1 : Synthèse du ribozyme capable de cibler l’ARNm de Tau et effectuer un clivage in vitro ... 65
1.1. Sélection des ribozymes ... 65
1.2. Clivage in vitro ... 66
vii
II. Objectif 2 : Valider et caractériser l’efficacité du ribozyme en
transfection transitoire ... 72
2.1. Transfections transitoires dans les HEK-293 ... 72
2.2. Transfections transitoires dans les SH-SY5Y ... 79
III. Objectif 3 : Valider et caractériser l’efficacité du ribozyme en infection ... 84
3.1. Infection des SK-N-MC ... 84
3.2. PCR et quantification ... 85
Chapitre 3 : Discussion ... 87
1. Sélectionner l’outil thérapeutique ... 87
2. Contexte cellulaire ... 89
3. Vision vers l’in vivo ... 92
Conclusions et perspectives ... 94
viii
Liste des figures
Figure 1. Classement des Tauopathies selon leur profil d’électrophorèse. ... 4
Figure 2. Inclusions de Tau suivant la Tauopathies dans une préparation de cerveau humain. ... 6
Figure 3. Marqueurs et propagation dans la Maladie d’Alzheimer. ... 9
Figure 4. Mécanisme de clivage de l’APP menant à la formation de plaques amyloïdes. ... 11
Figure 5. Le gène Tau et les différents isoformes du Tau humain. ... 14
Figure 6. Phosphorylation du plus long isoforme de la protéine Tau. ... 17
Figure 7. Tau dans des conditions normales et dans des conditions pathologiques. ... 20
Figure 8. Mécanisme SN2 de l’auto-clivage des ribozymes du 2nd groupe. ... 29
Figure 9. Structures secondaires et structures cristallines du Hammerhead ribozyme et du Hairpin ribozyme. ... 30
Figure 10. Structure secondaire et structure cristalline du VS ribozyme. ... 31
Figure 11. Mécanisme SN2 de l’auto-clivage de ribozyme delta. ... 33
Figure 12. Structure du SOFA-Delta-Ribozyme. ... 34
Figure 13. Vecteur d’expression utilisé pour le clonage moléculaire de la séquence du ribozyme. ... 39
Figure 14. Vecteur d’expression utilisé pour le clonage moléculaire de la séquence du ribozyme. ... 40
Figure 15. Vecteur d’expression utilisé pour la transfection de Tau endogène et pour la transcription de l’ARNm de Tau in vitro... 41
Figure 16. Schéma explicatif du clonage moléculaire dans pAAV-U6-GFP. ... 45
Figure 17. Schéma explicatif de la digestion des plasmides pour le clivage in vitro. ... 50
Figure 18. Schéma explicatif des étapes de transcription et du clivage in vitro dans le même tube. ... 51
Figure 19. Schéma explicatif de l’étape de reverse transcription dans le même tube. 53 Figure 20. Schéma explicatif de l’étape DNAse I dans le même tube. ... 53
Figure 21. Schéma explicatif de la PCR du clivage in vitro (exemple avec les ribozymes 350 ou 395). ... 56
Figure 22. Clivage in vitro de l’ARNm de Tau par les ribozymes 350, 395 et 638. ... 68
Figure 23. Transfection des SH-SY5Y avec les ribozymes transcrits in vitro. ... 70
Figure 24. ARNm de Tau normalisé sur GAPDH en fonction des différentes conditions de transfection avec les ribozymes transcrits in vitro dans les SH-SY5Y. ... 71
Figure 25. Photographie de la transfection des HEK-293 au Jet Prime post 48 heures avec le vecteur pAAV-U6-GFP/rz. ... 72
Figure 26. Qualité de l’ARN total de l’extraction d’ARN provenant de la transfection dans les HEK 293. ... 74
Figure 27. Cotransfection des HEK-293 au Jet Prime avec le plasmide de Tau et le plasmide du ribozyme pour un ratio 1 : 5 (Tau : Rz). ... 76
Figure 28. Cotransfection des HEK-293 au Jet Prime avec le plasmide de Tau et le plasmide du ribozyme pour un ratio 1 : 10 (Tau : Rz). ... 77
Figure 29. ARNm de Tau normalisé sur GAPDH en fonction des différentes conditions de transfection avec les ribozymes dans les HEK 293. ... 78
Figure 30. Photographie de la transfection des SH-SY5Y au CaCl2 post 48 heures avec le vecteur pAAV-U6-GFP/rz. ... 79
Figure 31. Transfection des SH-SY5Y au CaCl2 avec le plasmide du ribozyme. ... 80
Figure 32. Transfection des SH-SY5Y au CaCl2 avec le plasmide du ribozyme. ... 81
Figure 33. ARNm de Tau normalisé sur GAPDH en fonction des différentes conditions de transfection avec les ribozymes dans les SH-SY5Y. ... 82
ix
Figure 35. ARNm de Tau normalisé sur GAPDH en fonction des différentes conditions de transfection dans les SH-SY5Y. ... 83 Figure 36. Photographie de la transduction dans les SK-N-MC post 48 heures avec les virus contenant le ribozyme. ... 85 Figure 37. ARNm de Tau normalisé sur GAPDH en fonction des différentes conditions d’infection dans les SK-N-MC. ... 86
x
Liste des tableaux
Tableau 1. Symptômes et âge d’apparition des Tauopathies. ... 3
Tableau 2. Enzymes de restriction utilisées. ... 43
Tableau 3. Recette de la ligation du vecteur avec l’insert. ... 45
Tableau 4. Oligonucléotides utilisés pour le clivage in vitro. ... 54
Tableau 5. Oligonucléotides utilisés en fonction des types cellulaires. ... 61
xi
Liste des abréviations
AAV Adénovirus associé A Peptide amyloide A Peptide amyloide 40 A Peptide amyloide 42 AD Alzheimer disease
ADAM10 Metalloproteinase domain-containing protein 10
ADN Acide désoxyribonucléique ADNc ADN complémentaire AGD Argyrophilic grain disease
AICD APP intracellular domain ALS Amyotrophic lateral sclerosis
AMPA -N-amino-3-hydroxy-5-methyl-4-isoxazolepropionic APOE Apolipoprotéine E
APP Protéine précurseur du peptide bêta-amyloïde ARN Acide ribonucléique
ARNm ARN messager ARN P Ribonucléase P ARNt ARN de transfert
BACE -site amyloid precursor protein cleaving enzyme
BGS Bovine growth serum
BL Bloqueur BS Biosenseur
CamKIIa Calcium/calmodulin-dependent protein kinase type II
CBD Dégénérescence cortico-basale CDC2 Cell division cycle protein 2
CDK5 Cyclin-dependent kinase
CMV Cytomégalovirus CTF Fragment C-terminal CSF Cerebrospinal fluid
DS Down syndrome
DMEM Dulbecco’s modified Eagle’s medium
DMSO Diméthyl sulfoxide DNAse Désoxyribonucléase
ENF Enchevêtrements neurofibrillaires FTD Démence fronto-temporale FYN Tyrosine protéine kinase G RCF
GAPDH Glyceraldehyde 3’-phosphate dehydrogenase
GFP Green fluorescent protein
GTP Guanosine triphosphate GSK3 Glycogen synthase kinase 3
H1 Haplotype 1 H2 Haplotype 2
HDV Hepatitis delta virus
HEK Humain embryonic kidney
kDa Kilodalton
LTD Long-term depression
LTP Long-term potentialisation
xii MTs Microtubules N2a Neuro2a NFT Neurofibrillary tangles NMDA N-méthyl-D-aspartate nt Nucléotide pb Paire de bases
PBS Phosphate Buffered Saline
PCR Polymerase chain reaction
PEP Postencephalitic parkinsonism
PEG Polyéthylène Glycol
PHF Paired helocoidals filaments
PiD Pick disease
PP Protéine phosphatase PS1 Présilinine 1
PSD95 Postsynaptic density protein 95
PSP Paralysie progressive supranucléaire PSEN Présiniline
qPCR PCR quantitative en temps réel RCF Relative centrifugal force
RNAse Ribonucléase
RPM Révolutions par minute RT Reverse transcriptase
RZ Ribozyme
sAPP Fragment soluble d’APP clivé par l’-sécrétase sAPP Fragment soluble d’APP clivé par la -sécrétase siARN Petit ARN interférent
SOFA Specific On/Off adapter
SNC Système nerveux central SNP Sytème nerveux périphérique SH-SY5Y Neuroblastome humain SV40 Simian virus 40
TAE Tris Acetate EDTA Tau Tubulin associated unit
Tau3R Tau three repeats Tau 4R Tau four repeats TD Tangle-only dementia
Tm Température d’hybridation TrisHCl Tris Hydrochloride
μg Microgramme μL Microlitres
VIH Virus d’immunodéficience humaine VS Varkud Satellite
xiii
A Mamie… Na si mende dimbea oa, tomtom…
Mbua é ti tè, ké bonji é si ma busa.
xiv
Remerciements
2 ans finalement cela passe comme 2 semaines. Et on n’a pas le temps de dire au revoir, de dire bonjour, de voir l’hiver ou l’été (surtout l’été). Je voudrais prendre le temps ici de dire merci à toutes les personnes qui m’ont soutenu de près ou de loin dans ce projet. Dans le projet de ma vie finalement.
Merci au Dr Emmanuel Planel, mon directeur de recherche, sans qui jamais je n’aurais eu l’opportunité de travailler dans son laboratoire. Merci, car vous m’avez donné ma chance. Merci également à toute l’équipe : Françoise Morin, Serena Petry, Maud Gratuze, Andréanne Turgeon (une amie remarquable), Isabelle Guisle et Ophélie Lerdu.
Merci au Dr Georges Lévesque, mon co-directeur de recherche, qui a toujours eu beaucoup de patience pour toutes les fois où j’ai cogné à sa porte pleine de doutes. Merci pour vos longues explications, votre calme implacable, votre temps surtout ! Merci aussi à l’équipe : Chantal Godin et Pascal Smith.
Un merci spécial à ma collègue et amie Anne Blois (MF), membre de l’équipe du Dr Lévesque, qui a réussi à donner à mon cœur durant ces 2 années passées à Québec, ce qui lui manquait le plus : de la chaleur ! Merci, car avec une goutte de Brésil tous les jours je me sentais un peu chez moi, au Cameroun.
Merci au Dr Madeleine Carreau pour son esprit critique, ces conseils avisés, toutes les sucreries et pour l’évaluation de mon mémoire.
Merci également au Dr Frédéric Bretzner pour l’évaluation de mon mémoire. Merci à Mélody Mazon de m’avoir rappelé qu’il fallait toujours rester révoltée et merci de m’avoir appris la meilleure blague au monde (cf fourmis). Merci à Nicolas Josset de nous avoir supportées (Anne et moi) durant ces 2 ans et de m’avoir énormément aidée pour mon mémoire. Merci à vous deux pour votre si belle amitié. On se revoit à Houston.
Merci à Maxou et Nounou (mes deux plus grands fans) de faire de ma vie un grand fou rire. Merci à Magali, Sandra et Ivaha (mes sœurs) de m’avoir écrit ou appelé presque tous les jours pendant ces 2 ans. Merci à Céline et Johan de rester mes partenaires du crime depuis bientôt 7 ans pour l’une et 25 ans pour l’autre ! Merci à Kency et Patou pour votre amitié inébranlable. Merci à Eden, mon refuge…
Mes amis, ma famille que serais-je sans vous? Vous avez tous commencé cette aventure avec moi sans jamais douter, pendant ces deux années vous avez cru en moi, et vous m'avez donné l'éternité quand le temps semblait trop court, l'amour quand le temps était trop long...
Tant de personnes à remercier en si peu de mots.
Merci surtout, à mon papa qui m'a appris à remonter sur le vélo quand je tombais pour la 1ère fois. À ma maman qui m'a toujours réconfortée depuis les toutes 1ères fois où je dormais dans son lit, petite, jusqu’à maintenant (où j’y dors toujours quand j’en ai l’occasion).
Sans vous jamais je n'aurais appris le respect, la persévérance, la gentillesse, le courage et, surtout l'amour ! Sans vous je ne serai rien. Vous m’avez tout appris, vous m’avez tout donné. Bref merci pour tout.
1
Introduction
I. Les Tauopathies
1.1. Statistiques
La Maladie d’Alzheimer et la Maladie de Parkinson font parties des démences les plus connues dans le monde. Ces démences sont non seulement particulièrement répandues mais elles présentent aussi un risque majeur de santé publique puisqu’on ne leur connait encore aucun traitement curatif.
Les démences regroupent plus de cinquante maladies distinctes, définies par une diminution totale ou partielle de plusieurs fonctions cognitives (telles que la mémoire, le langage, les mouvements etc.) qui affectent la
vie quotidienne et ont des conséquences désastreuses sur l’autonomiedes
patients (http://www.alzheimer.ca). C’est sur un groupe spécifique de pathologies que nous allons nous concentrer ici : Les Tauopathies, maladies définies par l’accumulation de la protéine Tau dans le cerveau. Toutes les démences ne sont pas forcément des Tauopathies, mais la grande majorité des Tauopathies sont des démences. Ces pathologies rassemblent de nombreuses maladies neurodégénératives : la maladie d’Alzheimer, les démences fronto-temporales (FTD), la dégénérescence cortico-basale (CBD), la paralysie supranucléaire progressive (PSP), la Maladie de Pick (PiD). Les symptômes associés à ces différentes pathologies sont en revanche très divers. Ceux-ci peuvent aller d’un trouble moteur (cf CBD), à un trouble de l’humeur et du comportement (cf PiD), ou encore à un trouble de la mémoire (cf Maladie d’Alzheimer).
2
Aujourd’hui, les démences sont un problème majeur de santé publique. Les personnes les plus touchées par les démences sont les personnes âgées (dans la plupart des cas autour de 65ans). Les femmes étant plus affectées que les hommes probablement dû à leur plus longue espérance de vie (respectivement 83 ans et 73 ans).
Aujourd’hui, un nombre record de patients souffrant de démences est atteint. En 2012, 747 000 personnes vivaient avec une démence au Canada, et avec la prolongation de l’espérance de vie, 1.4 millions de
personnes seront probablement affectées d’ici 15ans
(http://www.societealzheimerdequebec.com). En tout et pour tout
Alzheimer Society Canada, comptabilise plus d’1.1 millions d’individus
touchés directement ou indirectement par les démences
(http://www.alzheimer.ca). Il en va de même pour le reste du monde avec 35.6 millions de personnes atteintes de démences en 2010 et tout comme au Canada, il est attendu que ce chiffre sera doublé d’ici 20 ans. Par conséquent, les coûts des soins pour les démences en 2010 s’élèvent à 604 milliards de dollars (américains) dans le monde entier (https://www.statcan.gc.ca). Il est donc urgent de trouver une solution pour réduire l’incidence et le coût de ces pathologies.
1.2. Regroupement des Tauopathies et caractéristiques de ces maladies
La forme de démence la plus répanduerestant la maladie d’Alzheimer,
de nombreuses autres Tauopathies sont souvent confondues avec cette dernière. Une question est donc posée: A quel stade reconnait-on une Tauopathie et comment se distinguent-elles les unes des autres ?
Les Tauopathies sont des maladies neurodégénératives impliquant l’hyperphosphorylation et l’accumulation de la protéine Tau dans le
3
cerveau (Avila, Lucas, Perez, & Hernandez, 2004). Les Tauopathies diffèrent selon la région où la protéine s’accumule, sa forme, les lésions causées, et les symptômes associés. De nombreux autres critères permettent la distinction entre les Tauopathies. Néanmoins, nous allons exposer les différentes caractéristiques de quelques Tauopathies en fonction de leurs spécificités symptomatiques.
Pathologies Symptômes Age d’apparition Maladie d’Alzheimer Perte de mémoire, aphasie,
apraxie, trouble du comportement <65 ans sans mutation >65 ans avec mutation Dégénérescence Cortico-Basal (CBD) Aphasie, apraxie, dysfonctionnement moteur (rigidité, mouvement involontaire) 50 à 70 ans Paralysie Progressive Supranucléaire (PSP) Problème d’équilibre, aphasie, trouble du comportement 60 ans
Maladie de Pick Aphasie, trouble du comportement (anxiété, dépression, changement
d’humeur)
40 à 60ans
Démence fronto-temporale avec syndrome parkisonien lié au
chromosome (FTDP-17)
Trouble du comportement, perte des fonctions
exécutives
>65ans
Down Syndrome Retard dans le développement physique et
cognitif
Dès la naissance
Tableau 1. Symptômes et âge d’apparition des tauopathies (http://www.alzheimer.ca)
4
Outre ces caractéristiques indispensables à l’identification initiale des Tauopathies, l’épissage alternatif de la protéine Tau permet aussi la distinction entre chaque pathologie. En effet, il a été démontré que certaines isoformes de Tau sont plus présentes dans les agrégats de Tauopathies que dans d’autres (Sergeant, Wattez, & Delacourte, 1999). Ce critère de différentiation des Tauopathies est en effet primordial puisqu’il permet de classer ces pathologies dans 4 groupes différents, selon la prévalence d’une isoforme de Tau par rapport à une autre. Les Tauopathies de la classe I (Maladie d’Alzheimer, Syndrome de Down, FTDP-17 etc.) possèdent les 6 isoformes. La classe II (CBD, PSP etc.) connait de façon majoritaire les isoformes Tau4R. Tandis que la classe III (Maladie de Pick) présente uniquement les isoformes Tau3R (Sergeant et al., 1999).
Figure 1. Classement des Tauopathies selon leur profil d’électrophorèse.
La classe 1 comporte un triplet avec Tau à 60kDa, 64kDa et 69kDa. La classe 2 comporte un doublet avec Tau à 64kDa et 69kDa. La classe 3 comporte un doublet avec Tau à 60kDa et 64kDa. Et la classe 4 comporte Tau à 60kDa (Adaptée de Buée et al. 2000).
AD, DS, PEP, ALS, FTDP-17 CBD, PSP, FTDP-17 Pick’s Disease, FTDP-17 Myotonic dystrophy Of type I or II
5
En plus des symptômes cliniques et des isoformes de Tau, les Tauopathies peuvent aussi être différenciées selon les cellules dans lesquelles se trouvent les inclusions de Tau et la forme de ces lésions. Ici un exemple flagrant de la différence des inclusions se trouvent dans la maladie de Pick. Ces inclusions, appelées corps de Pick, se développent dans les cellules nerveuses corticales et sous-fauxcorticales du lobe fronto-temporal (Brion, Plas, & Jeanneau, 1991). Les corps de Pick se retrouvent également dans les cellules nerveuses granulaires du dentée gyrus de l’hippocampe (Buée-Scherrer et al., 1996). Les inclusions sont des agrégats de Tau comme dans toutes les Tauopathies et provoquent un renflement des neurones.
Ces inclusions sont différentes dans la maladie d’Alzheimer, puisque ces dernières se forment dans les cellules nerveuses pyramidales de l’hippocampe et du cortex enthorhinal (Braak & Braak, 1985). Elles se retrouvent aussi dans l’amygdale ou dans le locus coeruleus (Buée, Bussière, Buée-Scherrer, Delacourte, & Hof, 2000).
6
Figure 2. Inclusions de Tau suivant la Tauopathies dans une préparation de cerveau humain.
Maladie d’Alzheimer (AD) et Tangle-only dementia (TD) : enchevêtrements neurofibrillaires et neuropil threads; Argyrophilic grain disease (AGD) : grains argyrophiliques ; Paralysie Progressive
Supranucléaire (PSP) : enchevêtrements neurofibrillaires globuleux et
neuropil threads ; Dégénérescence cortico-basale (CBD) : petits enchevêtrements neurofibrillaires et nombreux neuropil threads; Maladie de Pick (PiD) : corps de Pick. (Clavaguera et al., 2013).
Les causes des Tauopathies sont encore mal connues et la cause
principale reste inconnue. Cependant plusieurs facteurs
environnementaux et génétiques peuvent aussi influencer la prédisposition à ces maladies. On compte par exemple 80 mutations pour
le gène MAPT(http://alzforum.com), ces mutations sont le plus souvent
des mutations faux sens qui changent l’épissage alternatif de Tau favorisant ainsi une isoforme par rapport à une autre. C’est le cas, par exemple, de la mutation G272V, qui diminue la proportion des isoformes Tau4R (Götz et al., 2001). Les maladies résultantes des mutations faux
sens observéessont la FTDP-17, la CBD ou la PSP (Rademakers, Cruts, &
7
détachement des microtubules de la protéine et ainsi favoriser l’agrégation de la protéine, comme la mutation P301L (Hong, 1998). Mutation qui a d’ailleurs permis la création des souris transgéniques
P301L présentant une pathologie de type FTDP-17(http://alzforum.com).
Toutes ces caractéristiques (symptômes cliniques, inclusions de Tau, mutations etc.) permettent la différentiation et la classification des Tauopathies. Le vieillissement, quant à lui, constitue un risque important de développer la maladie.
1.3. La Maladie d’Alzheimer
La Maladie d’Alzheimer est une des Tauopathies les mieux décrites, mais à ce jour aucun traitement curatif n’a été découvert. Celle-ci se définit par une perte de la mémoire à court terme, un trouble du langage appelé aphasie, un problème de reconnaissance des objets ou des
visages, et un trouble moteur appelé apraxie(http://www.alzheimer.ca).
Il existe 2 formes de la maladie : la forme familiale (5% des cas) et la forme sporadique (95% des cas) (Wu et al., 2012).
La forme sporadique se déclenche généralement chez les individus âgés de plus de 65 ans. Plusieurs facteurs sont des facteurs de risque en ce qui concerne cette forme de la Maladie d’Alzheimer, notamment l’obésité, une pression artérielle élevée ou encore le diabète (J. A. Luchsinger & Mayeux, 2004; José a Luchsinger & Mayeux, 2004; Jose a Luchsinger, Tang, Shea, & Mayeux, 2004). Il a aussi été démontré que les fumeurs ont deux fois plus de risque d’être atteint de la maladie d’Alzheimer (Cataldo, Prochaska, & Glantz, 2010). De plus, une mutation de l’allèle 4 du gène de l’apolipoprotéine E (APOE) serait un facteur de risque important (Corder et al., 1993). D’autres facteurs seraient quant à eux bénéfiques,
8
comme l’exercice physique régulier (Adlard, 2005), les activités mentales ou une éducation élevée (Stern, 2012).
La forme familiale touche une faible proportion (5%) desmoins de
65 ans (Wu et al., 2012). Les mutations sont observées sur les gènes d’APP, PSEN1 et PSEN2 (présiniline 1 et présiniline 2) qui sont étroitement liées avec la formation du peptide A (Citron et al., 1997; Rogaev et al., 1995).
La perte progressive des cellules nerveuses dans cette maladie se caractérise par une atrophie débutant dans les lobes temporaux et pariétaux et s’étendant jusqu’au cortex frontal (cf figure 3). Ainsi, plusieurs stades de la pathologie peuvent être observés. Le stade léger se traduit par une atrophie du cortex entorhinal et de l’hippocampe, responsable des fonctions cognitives liées à la mémoire, notamment la mémoire épisodique. Lors du stade modéré et avancé, on observe une réduction des lobes temporaux et pariétaux, mais aussi une réduction du lobe frontal. Une atrophie de ces régions entraine divers problèmes tels que des troubles de lecture, une désorientation ou de l’anxiété voir de la colère. Enfin, le stade sévère se caractérise par une atrophie du cortex frontal, ce qui amène à des problèmes moteurs mais aussi des troubles
de la parole(http://www.agenebio.com).
Au niveaucellulaire, il existe deux marqueurs connus de la maladie
d’Alzheimer (cf figure 3) : Les plaques séniles de la protéine -amyloïde (plaque A) (Glenner & Wong, 1984) et les enchevêtrements neurofibrillaires de la protéine Tau (ENFs) (Brion et al., 1991). Le dépôt, la propagation et l’accumulation de ces deux protéines sont les principales causes des symptômes de la maladie d’Alzheimer.
9
Figure 3. Marqueurs et propagation dans la Maladie d’Alzheimer.
(a) Les plaques amyloïdes retrouvées dans la partie extracellulaire et les enchevêtrements neurofibrillaires retrouvées dans les neurones. (b) La propagation des plaques amyloïdes et des enchevêtrements neurofibrillaires des lobes pariétaux et temporaux au cortex frontal suivant le niveau de sévérité (d’après Masters et al., 2015).
1.4. Le peptide -amyloïde
Les plaques séniles furent découvertes par le Dr Alois Alzheimer en 1907. La protéine -amyloïde, qui est le constituant principal de ces plaques séniles, est issue du précurseur amyloïde APP (Amyloid precursor
protein). En effet, APP est une protéine transmembranaire qui se retrouve
dans de nombreux tissus (Polanco et al., 2018). Cette protéine possède plusieurs isoformes, mais certaines formes (APP695, APP770, APP751) sont prédominantes dans les neurones (Del Turco et al., 2016). Étant une protéine transmembranaire, sa partie N-terminale, possédant un domaine de liaison héparine et un domaine de liaison métallique, se situe du côté extracellulaire. Tandis que sa partie C-terminale, assez courte, se trouve du côté intracellulaire (Zhou et al., 2011). APP est une protéine qui
10
jouerait un rôle dans la différentiation cellulaire (Bergmans et al., 2010), la migration neuronale (Young-Pearse et al., 2007), l’adhésion cellulaire (Breen, Bruce, & Anderton, 1991) et la formation synaptique (Priller et al., 2006). Il existe deux processus (cf figure 4) par lequel APP est clivé : la voie non-amyloïdogénique, stimulé par l’activité synaptique, et la voie amyloïdogénique, productrice de peptides A.
La voie non-amyloïdogénique dépend de l’-sécrétase et d’ADAM10 (metalloproteinase domain-containing protein 10). En effet, ces enzymes permettent la coupure d’APP et entrainent la libération de la forme soluble APP du coté extracellulaire (Prox et al., 2013). La -sécrétase coupe par la suite la forme d’APP restée ancrée dans la membrane (autrement dit la partie C-terminale), et provoque la libération de l’AICD (APP intracellular
domain), qui lui-même à son tour sera coupé et induira la formation du
peptide p3 (Takagi-Niidome et al., 2015). Tous les fragments d’APP générés lors de la voie non-amyloïdogénique ont démontré jouer des rôles bénéfiques que ce soit dans la neurogénèse ou la plasticité synaptique (Zhou et al., 2011). Cette voie est donc très importante pour le bon fonctionnement du cerveau. Elle pourrait dès lors avoir un avantage certain pour contrer la maladie d’Alzheimer. La voie amyloïdogénique se définit, quant à elle, aussi par une coupure d’APP. Les parties N-terminal d’APP (extracellulaire) n’ayant pas subi de coupure par l’-secretase, peuvent subir une coupure par la -secretase (BACE) (Vassar et al., 2014). Il existe deux -sécrétases différentes appelées BACE1 et BACE2. Ces enzymes vont couper APP du coté N-terminal libérant un APP soluble. La partie C-terminal restante ancrée dans la membrane, appelée -CTF, sera clivée par la -sécrétase. Ce qui va induire la production d’AICD et de deux formes de protéines amyloïde- : l’A48 et l’A49 (Takami et al. 2009). C’est par le clivage de l’A48 par la -secretase, que sera produit l’A42, forme d’A qui aura une tendance à l’agrégation et
11
provoquera la formation de plaques amyloïdes (Haass & Selkoe, 2007). Bien que d’autres formes du peptide existent, c’est en effet la surproduction du peptide A42 qui entrainera la neurotoxicité et l’accumulation des plaques amyloïdes dans la Maladie d’Alzheimer.
Figure 4. Mécanisme de clivage de l’APP menant à la formation de plaques amyloïdes.
Voie non-amyloïdogénique impliquant l’-sécrétase et générant l’AICD et
APP soluble. Voie amyloïdogénique impliquant la -sécrétase et générant
l’AICD, l’APP soluble ainsi que les peptides A formant les plaques
amyloïdes (d’après Bachurin, Bovina, & Ustyugov, 2017).
Le peptide A (et plus particulièrement A42) provoque plusieurs effets neurotoxiques sur les cellules nerveuses : une hyperactivité neuronale liée à un influx de Ca2+ (Busche et al., 2008), une dérégulation
des mitochondries (Rhein et al., 2009) et une dérégulation des fonctions synaptiques. En condition normale, la LTP (long-term potentialisation) est régulée, entre autres, par les récepteurs NMDA (N-methyl-D-aspartate) et AMPA (alpha-N-amino-3-hydroxy-5-methyl-4-isoxazolepropionic). Cependant, en condition pathologique, la protéine A se lierait de façon directe à ces récepteurs, et induirait une diminution de leur activité par un mécanisme qui demeure encore inconnu. Ce qui provoquerait une réduction de la LTP en faveur de la LTD (long-term depression) (Selkoe,
12
2008). Aussi, il semblerait qu’A interagisse par un processus indirect avec la protéine PSD95 (Postsynaptic density protein 95), nécessaire dans l’assemblage des récepteurs NMDA et AMPA, provoquant la diminution de PSD95 (Pham et al., 2010) et engendrant par la suite une réduction des récepteurs NMDA et AMPA ou induisant une endocytose de ces derniers. Les mécanismes liés à la neurotoxicité du peptide A dans le cerveau sont encore mal compris et restent à investiguer.
La forme du peptide A qui est neurotoxique est aussi à déterminer. En effet, les plaques amyloïdes sont aussi retrouvées dans les cerveaux sains (Giannakopoulos et al., 2003), et ne sont parfois que le symptome d’un vieillissement du cerveau et non d’une pathologie.
De plus, les souris APPPS1 (transgène PS1 et transgène d’APP muté (http://alzforum.com)) formeraient bien des plaques amyloïdes mais on n’observerait pas de mort neuronale dans les début de la pathologie (Rupp, Wegenast-Braun, Radde, Calhoun, & Jucker, 2011). Il existe néanmoins de nombreuses formes de ce peptide soluble ou insoluble qui pourraient jouer ce rôle neurotoxique.
Un autre marqueur de la maladie d’Alzheimer est la protéine Tau. Cette protéine s’accumule dans les neurones contrairement au peptide A qui s’accumule à l’extérieur des neurones. La formation d’enchevêtrement neurofibrillaire de la protéine Tau hyperphosphorylé (ENFs) pourrait être un des facteurs entrainant une neurotoxicité et possiblement l’apparition de la maladie d’Alzheimer.
13
II. La protéine Tau
2.1. Le gène et la protéine de Tau
Découverte en 1975 par le Dr. Weingarten Tau est une protéine faisant partie de la famille des MAPs (microtubule-associated protein) (Weingarten, Lockwood, Hwo, & Kirschner, 1975). Cette famille de protéine permet la régulation des microtubules. On y retrouve par exemple MAP2 (dans les cellules nerveuses) ou MAP4 (dans tout type cellulaire). Le gène de Tau se situe sur le chromosome 17 à la position
17q21.1. (Buée et al., 2000). Il existe deux haplotypes différents : H1 et
H2 du gène de Tau. Un haplotype est un groupe d’allèles sur le même chromosome qui sont transmis ensemble lors de la méiose. En effet, il a été démontré qu’il y aurait une prévalence de l’haplotype H1 chez les personnes atteintes de la PSP, CBD et de la maladie de Parkinson (Ezquerra et al., 1999 ; Baker et al., 1999). Cet haplotype serait donc un facteur de risque pour certaines Tauopathies. Le gène de Tau, quant à lui, est constitué de 16 exons qui seront épissés alternativement (Wang & Mandelkow, 2016). Cet épissage va mener à la formation de plusieurs transcrits primaires différents, 6 en tout, et donc à la formation de 6 isoformes de Tau (Goedert, Spillantini, Jakes, Rutherford, & Crowther, 1989) (cf figure 5). Les exons 1, 4, 5, 7, 9, 11, 12 et 13 sont constitutifs. Tandis que les exons 2, 3 et 10, spécifiques au cerveau humain, sont épissés alternativement. Les transcrits possédant l’exon 8 ne sont pas retrouvés dans le SNC humain. L’exon 4A permet la formation de la Big Tau, cette forme de Tau faisant à peu près 100kDa (Nunez & Fischer, 1997), se retrouve généralement dans le système nerveux périphérique. L’exon 1, quant à lui, fait partie du promoteur, il n’est donc pas traduit (Buée et al., 2000).
14
Une étape cruciale de l’épissage alternatif est l’inclusion ou l’exclusion de l’exon 10. L’inclusion de l’exon 10 induit l’ajout d’un domaine de liaison aux microtubules. En effet, lorsque l’exon 10 est épissé, donc exclus, il reste 3 séquences répétées sur la partie C-terminal de la protéine permettent la liaison aux microtubules, produisant ainsi un isoforme appelé Tau3R (3 repeats regions). Lorsque l’exon 10 est inclus, il y a 4 séquences répétées sur la partie C-terminal de la protéine, induisant l’isoforme Tau4R (Lee, Cowan, & Kirschner, 1988). Les exons 2 et 3 permettent aussi la formation d’isoformes différentes, suivant la présence ou l’absence de ces inserts, ces isoformes seront nommées Tau0N, Tau1N ou Tau2N (N correspondant à N-terminal). L’exon 3 est uniquement inséré lorsque l’exon 2 est aussi présent (Wang & Mandelkow, 2016).
Cet épissage alternatif produit ainsi 6 isoformes de Tau variant entre 352 et 441 acides aminés.
Figure 5. Le gène Tau et les différents isoformes du Tau humain.
Épissage alternatif du gène de Tau produisant 6 isoformes de Tau : suivant que les exons 2 et 3 sont insérés, les isoformes Tau2N ou Tau1N sont produites, l’exclusion de ces exons induit l’isoforme Tau0N ; lorsque l’exon 10 est inséré l’isoforme Tau4R est produite, l’exclusion de cet exon induit l’isoforme Tau3R ; lorsqu’aucun des exons 2, 3 et 10 sont insérés l’isoforme Tau0N3R est formée (Adapté de Wang & Mandelkow, 2016).
15
Chez l’humain adulte, la proportion de Tau4R par rapport au Tau3R est égale, c’est-à-dire que le ratio est de 1 : 1 (Goedert & Jakes, 1990). Néanmoins, il y un débalancement de ce ratio dans certaines Tauopathies. La protéine de Tau varie entre 45kDa et 65kDa selon l’ARNm de Tau exprimé. Comme évoqué précédemment (cf figure 3), les différentes Tauopathies appartiennent à des classes différentes suivant l’isoforme exprimée dans leurs inclusions (Sergeant et al., 1999).
La protéine Tau est généralement cytoplasmique mais elle peut aussi être retrouvée dans le nucléole, celle-ci pourrait jouer un rôle de protection de l’ADN contre le stress (Violet et al., 2014). Cette protéine est formée de deux domaines distincts : le domaine de projection N-terminal et le domaine de liaison aux microtubules C-N-terminal. Ces deux domaines sont séparés d’une région riche en proline. La fonction de la protéine Tau est diverse, cependant ces deux domaines définissent son rôle au sein de la cellule. La partie N-terminal aurait un rôle d’espaceur entre les microtubules axonaux (Chen, Kanai, Cowan, & Hirokawa, 1992), alors que la partie C-terminale servirait à se lier aux microtubules grâce aux séquences répétées (Buée et al., 2000). Ce qui favoriserait par la suite la polymérisation et la stabilité des microtubules.
2.2. Modifications post-traductionnelles
La protéine Tau subit plusieurs modifications post-traductionnelles qui peuvent influencer sa liaison aux microtubules. La phosphorylation a été démontrée comme modification cruciale pour la protéine Tau (Butler & Shelanski, 1986). En effet, il existe 85 sites putatifs de phosphorylation sur le plus long isoforme de Tau, soit 20% des acides aminés qui sont
16
phosphorylés (Stoothoff & Johnson, 2005). Ces acides aminés étant le plus souvent des Sérines, Thréonines et Tyrosine. Plusieurs tyrosines kinases permettent cette phosphorylation. Notamment les kinases telles que FYN (proto-oncogène tyrosine protéine kinase), Cdk5
(cyclin-dependent kinase 5) ou Cdc2 (cyclin-(cyclin-dependent protein kinase Cdk1/Cdc2). Une des kinases les plus présentes qui phosphorylent Tau
est GSK3 (Lovestone et al., 1994). Les protéines phosphatases, dont PP2B ou PP2C, sont des enzymes qui permettent la déphosphorylation de Tau. La protéine phosphatase PP2A est l’une des plus importantes phosphatases, celle-ci assure 70% de la déphosphorylation de la protéine Tau (Goedert & Jakes, 1990). Il a été démontré que la phosphorylation de la protéine Tau peut être modifiée lors de l’apparition de pathologie. En effet, il y aurait une hyperphosphorylation de Tau (site phosphorylé en condition physiologique) ou une phosphorylation anormale (site non phosphorylé en condition non physiologique) dans certaines Tauopathies. Nous savons par exemple qu’il existe une diminution de l’activité de PP2A chez les personnes atteintes de la maladie d’Alzheimer (Gong, Singh, Grundke-Iqbal, & Iqbal, 1993). Une diminution qui pourrait être due à l’hypothermie. En effet, il a été démontré que la phosphorylation de Tau est exacerbée dans des conditions d’hypothermie (par exemple lors de l’hibernation des rongeurs) probablement dû à la réduction de l’activité de PP2A (Planel et al., 2007). Un autre exemple de la dérégulation de la phosphorylation de Tau dans des cas pathologiques est la phosphorylation anormale de la Serine 262 qui se trouve dans une région répétée se liant aux microtubules. Ce qui entraine une diminution de l’affinité de la protéine aux microtubules (Biernat, Gustke, Drewes, Mandelkow, & Mandelkow, 1993). De même, la phosphorylation anormale de la Serine 214 (Schneider, Biernat, Von Bergen, Mandelkow, & Mandelkow, 1999) et l’hyperphosphorylation de la Thréonine 231 entraine une réduction de l’affinité aux microtubules (Lu, Wulf, Zhou, Davies, & Lu, 1999). Enfin, il
17
a été démontré que la phosphorylation anormale de la Serine 422 de la partie C-terminale empêche la dégradation de Tau par la Caspase 3 (Reynolds et al., 2006).
L’hyperphosphorylation de Tau ou la phosphorylation anormale cause ainsi divers problèmes tels que la réduction de l’affinité aux microtubules ou l’impossibilité de dégrader la protéine Tau. Ceci pourrait mener ultimement à l’augmentation des agrégats neurotoxiques.
Figure 6. Phosphorylation du plus long isoforme de la protéine Tau.
Les flèches jaunes sont les épitopes reconnus par des anticorps précis. En vert la phosphorylation normale, en rouge la phosphorylation anormale (site non phosphorylé en condition normale) et en bleu l’hyperphosphorylation (d’après Munoz et al. 2013).
Toutefois, la protéine Tau subit aussi d’autres modifications post-traductionnelles. La O-GlcNAcylation, par exemple, entrerait en compétition avec la phosphorylation, car elle ajouterait un glucide sur la chaine latérale des Sérines et Thréonines, empêchant ainsi la phosphorylation. Cependant, ce phénomène est moins fréquent chez les individus atteints de la maladie d’Alzheimer (Liu, Iqbal, Grundke-Iqbal,
18
Hart, & Gong, 2004). La N-glycosylation, quant à elle, induirait plutôt l’hyperphosphorylation, et donc l’agrégation de Tau (Liu, Zaidi, Iqbal, Grundke-Iqbal, & Gong, 2002).
L’acétylation serait aussi une modification post-traductionnelle importante puisqu’elle aurait un double rôle dans les modifications de Tau. L’acétylation de la Lysine 353 ou 321 par exemple aiderait à la dégradation de Tau et serait présent dans un cerveau sain, alors que l’acétylation de la Lysine 280 empêcherait la dégradation de Tau et serait présent plutôt chez des personnes atteintes de Tauopathies (Maladie d’Alzheimer, Maladie de Pick ou PSP) (Min et al., 2010).
Mise à part la phosphorylation, l’acétylation ou la glycosylation de Tau, il existe d’autres modifications post-traductionnelles qui influencent cette protéine : méthylation, troncation, ubiquitination ou sumoylation (Buée et al., 2000). Toutes ces modifications peuvent avoir un effet sur l’agrégation de Tau, sa liaison aux microtubules, le repliement ou la dégradation de cette protéine.
2.3. Rôle de Tau et Neurotoxicité
Comme nous l’avons évoqué précédemment, la protéine Tau jouerait un rôle essentiel en lien avec les microtubules. Dans un cerveau sain, il a déjà été démontré que la protéine Tau se lie aux microtubules (Weingarten et al., 1975) grâce à ces régions répétées en C-terminal et permet ainsi leur stabilisation et leur polymérisation. Cette protéine est le plus souvent retrouvée dans la partie axonale du neurone ou elle peut jouer son rôle de protéine stabilisatrice. Cependant, elle pourrait aussi interférer dans le transport axonal (von Bergen et al., 2000). En effet, la dynéine et la kinésine sont deux protéines motrices du transport axonal qui se lient aussi aux microtubules pour effectuer le transport d’organelles
19
ou de vésicules vers le corps cellulaire (dynéine – transport rétrograde) ou vers la terminaison axonale (kinésine – transport antérograde). Tau rentre ainsi en compétition avec ces deux protéines motrices quant à la liaison aux microtubules et peut, de ce fait, empêcher le transport d’organelles et de protéines (Ross, Shuman, Holzbaur, & Goldman, 2008). Ce changement dans le transport axonal a un effet sur le transport d’organelles et notamment sur le transport des mitochondries (Stamer, Vogel, Thies, Mandelkow, & Mandelkow, 2002).
Un autre rôle important de Tau serait la protection de l’ADN dans le nucléole dans des conditions de stress cellulaire. Il a été démontré in vitro (Violet et al., 2014) sous certaines conditions (stress) qu’il y aurait une surexpression de cette protéine, et qu’elle servirait ainsi à préserver l’intégrité de l’ADN et l’ARN présent dans le nucléole. En plus d’être retrouvée dans les axones et dans le nucléole, on observerait aussi la présence de Tau dans les dendrites (Ittner et al., 2010). Le rôle dans cette partie du neurone reste encore à déterminer, mais elle pourrait avoir un lien avec la transmission synaptique.
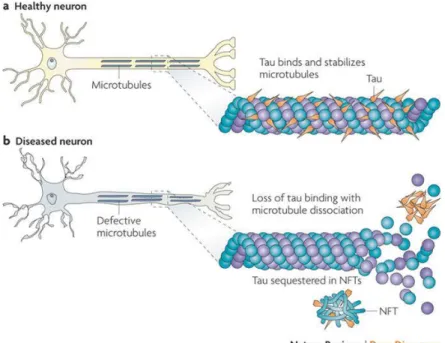
Dans des conditions pathologiques (Tauopathies), la protéine Tau liée aux microtubules par les régions répétées sur le domaine C-terminal se détache de ceux-ci. En effet, il y aurait une perte d’affinité, peut-être dû à une hyperphosphorylation ou à d’autres modifications post-traductionnelles. Ce détachement provoquerait une déstabilisation des microtubules. Mais aussi une dérégulation du transport axonal vue (LaPointe et al., 2009).
20
Figure 7. Tau dans des conditions normales et dans des conditions pathologiques.
(a) Tau se lie aux microtubules et permet leur stabilisation et leur polymérisation dans des neurones sains. (b) Tau hyperphosphorylé se détache des microtubules, s’agrège et entraine une dissociation des microtubules dans des neurones malades (d’après Brunden, Trojanowski, & Lee, 2009)
Suite à cela, Tau formera des monomères qui vont devenir des
oligomères.Ce phénomene est spontané, il nécessite uniquement la
présence du PHF6 hexapeptide dans une des régions répétées qui se lie aux microtubules (von Bergen et al., 2000). En plus des oligomères, plusieurs études montrent qu’il existe des Ghost Tangles. Ces formes de la protéine serait à la fois les précurseurs des enchevêtrements neurofibrillaires et seraient l’espèce qui subsisterait lorsque le neurone
aurait été dégradé(Maeda et al., 2007). Enfin, les oligomères vont former
des fibrils de Tau, appelés PHFs (paired helocoidals filaments) dans le cas de la maladie d’Alzheimer. Ces fibrils n’ont pas toujours la même forme selon les Tauopathies. Ils peuvent a leur tour former des enchevêtrements neurofibrillaires (Guzmán-Martinez, Farías, & Maccioni, 2013). Ce sont ces
21
ENFs qui sont retrouvés dans la maladie d’Alzheimer ou dans d’autres Tauopathies et qui sont devenus des marqueurs de Tauopathies.
La protéine Tau et le peptide A sont étroitement liés en ce qui concerne la neurotoxicité. En effet, la protéine Tau peut cibler l’enzyme FYN, qui va phosphoryler le récepteur NMDA à la sous-unité NR2B et provoquer le recrutement de la protéine PSD95. PSD95 va induire la formation d’un complexe via lequel le peptide A va entrainer l’excitotoxicité neuronale (Ittner et al., 2010),
De même, l’oligomère A56 peut se lier au récepteur NMDA, provoquant une entrée importante de Ca2+ intracellulaire. Le Ca2+ présent dans la
cellule va permettre l’activation de l’enzyme CaMKII
(calcium/calmodulin-dependent protein kinase type II subunit alpha), enzyme capable de phosphoryler la protéine Tau (Amar et al., 2017). Ce qui peut engendrer divers problèmes dans la neuropathologie de la maladie.
A quoi est due la neurotoxicité ? Il a longtemps été pensé que les enchevêtrements neurofibrillaires ainsi que les plaques séniles dans la maladie d’Alzheimer seraient responsables de la neurotoxicité. Cependant, comme vu précédemment, les plaques séniles ne sont pas forcément responsable de la neurotoxicité. Qu’en est il pour les enchevêtrements neurofibrillaires ? Il semblerait y avoir une corrélation positive entre l’accumulation des enchevêtrements neurofibrillaires et la mort neuronale (Duyckaerts et al., 1997). De plus il semblerait aussi y avoir une amélioration cognitive liée à la baisse des ENFs (Noble et al., 2005). Néanmoins, plusieurs études montrent aujourd’hui que les ENFs ne sont pas forcement responsables de la neurotoxicité. Les autres formes de la protéine sont désormais à investiguer. En effet, il a été prouvé que
22
chez des Drosophiles possédant des caractéristiques de Tauopathies, il n’a pas été observé de ENFs malgré une neurodégénérescence (Wittmann et al., 2001). Aussi, des souris exprimant elles aussi une pathologie de type tauopathie, ont démontré une dégénéresence neuronale dans les premiers stades de la maladie sans pour autant qu’il y ait présence de ENFs. Ceux-ci seraient apparus plus tard dans le développement de la maladie (Andorfer, 2005). Reste à savoir pourquoi si les ENFs ne sont pas toxiques, Tau formerait-il ces enchevêtrements ? Il a été montré que des neurones peuvent subsister pendant des décénies avec des ENFs (Morsch, Simon, & Coleman, 1999). De plus en plus d’études se tournent vers d’autres formes de Tau en ce qui concerne la neurotoxicité tels que les oligomères de Tau ou les monomères de Tau solubles. En effet, une étude intéressante a montré chez des souris rTg4510 (présentant la mutation P301L) qu’il était possible de diminuer la quantité de Tau totale et de réduire la forme soluble de Tau grace au bleu de méthylène. Ceci améliorant les fonctions cognitives notamment la mémoire, sans pour autant réduire le Tau insoluble (O’Leary et al., 2010). Ainsi toutes ces formes de Tau présentent des preuves en faveur de la toxicité comme en défaveur.
2.4. Propagation dans la maladie d’Alzheimer
Les enchevêtrements neurofibrillaires ne permettent pas la propagation de la maladie car ils ne peuvent traverser la membrane plasmique. Les oligomères, les filaments ou les monomères de Tau qui pourraient être neurotoxiques, par contre, ont été retrouvés dans la partie extracellulaire.
De plus en plus d’évidences tendent vers l’hypothèse selon laquelle les Tauopathies et la maladie d’Alzheimer plus précisément seraient des
23
maladies à Prion. C’est-à-dire qu’il y aurait la propagation d’une forme infectieuse de la protéine Tau dans le cerveau, d’une cellule à une autre. Ce qui engendrerait la propagation de la maladie. En effet, dans la maladie d’Alzheimer, il a été démontré qu’en injectant des extraits de cerveau humain provenant d’individus atteints par différentes Tauopathies dans l’hippocampe et le cortex de souris transgéniques, des inclusions de Tau sont retrouvées à l’endroit de l’injection mais aussi et surtout dans des régions plus éloignées de l’injection (Clavaguera et al., 2014). La propagation de Tau suivrait les connections synaptiques (Ahmed et al., 2014). Les stades de Braak sont décrits comme suit : la pathologie Tau se développerait tout d’abord dans le cortex enthorinal ce qui correspondrait aux stades I et II ; elle se propagerait par la suite dans l’hippocampe, stades III et IV ; puis dans les derniers stades de la maladie, à savoir les stades V et VI, elle se retrouverait dans tout le cortex (Braak & Braak, 1991)(cf figure 3).Comment et pourquoi cette pathologie se propagerait en suivant ce schéma dans tous les cerveaux atteints ? La sécrétion de Tau serait un phénomène normal, puisque la protéine Tau est retrouvée dans le CSF (Cerebrospinal fluid) de cerveaux de patients atteints de la maladie d’Alzheimer mais aussi dans les cerveaux d’individus sains. Il n’en demeure pas moins que la sécrétion de Tau hyperphosphorylé semble plus élevée dans les cerveaux atteints de la maladie que dans les cerveaux sains (Hampel et al., 2004). Plusieurs hypothèses sont en expérimentation quant au mécanisme de propagation de la protéine.
La 1ère hypothèse proposée fut que la protéine Tau était libérée dû à la mort neuronale. Cependant des études ont prouvé que la sécrétion de Tau
est indépendante de la mort cellulaire (Kanmert et al., 2015). La secondee
hypothèse explique que la sécrétion de Tau pourrait se faire via un mécanisme impliquant les exosomes (Saman et al., 2012) ou que les cellules microgliales pourraient induire la propagation de Tau toujours
24
grâce à un mécanisme dépendant des exosomes (Asai et al., 2015). Une troisième hypothèse implique la propagation de Tau à travers des nanotubes qui relieraient les cellules entre elles (Tardivel et al., 2016). Enfin, une dernière hypothèse proposerait que Tau pourrait traverser la membrane plasmique sans passer par un mécanisme vésiculaire (Chai, Dage, & Citron, 2012).
Les espèces sécrétées pourraient provoquer un mauvais repliement de la protéine Tau. Ces espèces sont hypothétiquement des monomères, des oligomères ou des filaments de Tau (Jucker & Walker, 2011).
Comme nous avons pu le voir, les Tauopathies sont des pathologies assez diverses, posant un certain nombre de problèmes cognitifs. Dans le cas de la Maladie d’Alzheimer, les plaques amyloïdes et les enchevêtrements neurofibrillaires sont deux marqueurs de la pathologie qui engendrent supposément la neurotoxicité. Une problématique importante, dans le cas de cette pathologie, porte par conséquent sur le traitement de cette maladie.
III. Les Ribozymes 3.1. Traitements
De nos jours, il n’existe aucun traitement curatif de la Maladie d’Alzheimer.
Le traitement le plus utilisé, qui permet de réduire les symptômes associés à la progression de la maladie est la mémantine. Ce traitement serait un antagoniste du récepteur NMDA et bloquerait ce récepteur pour éviter un influx important de Ca2+ à l’intérieur de la cellule et donc une
25
Sachant que les peptides Abêta sont générés par deux enzymes : BACE et la -sécrétase ; une des premières cibles fut la -sécrétase. Des études ont montré qu’en inhibant cette enzyme il était possible de réduire les plaques amyloïdes (Dovey et al., 2001). Néanmoins, l’inhibition de cette enzyme implique l’inhibition de la voie amyloïdogénique mais aussi d’autres voies plus importantes. En effet, la -secretase clive le récepteur Notch1, clivage qui s’est montré essentiel dans le développement embryonnaire (De Strooper et al., 1999). L’inhibition de cette enzyme causerait ainsi de nombreux problèmes pour cette voie mais aussi pour d’autres voies importantes (Milano et al., 2004).
L’autre cible envisagée était donc naturellement BACE1. Il y aurait une augmentation de l’activité de cette enzyme chez les personnes atteintes de la maladie d’Alzheimer (Zetterberg et al., 2008), néanmoins on ne pourrait pas dire si cette augmentation est une cause ou une conséquence de la maladie. Des inhibiteurs de cette enzyme sont à l’essai. Des souris
knock-out BACE1 n’ont pas démontré de problèmes majeurs, néanmoins
elles ont démontré un problème de myéline dans le système nerveux périphérique (Sreedharan et al., 2008).
Étant donné qu’il est difficile de cibler directement les enzymes responsables de la production du peptide A, une autre approche fut de cibler le peptide A en tant que tel.
Plusieurs autres stratégies ont été envisagées dès lors, à savoir : empêcher l’agrégation du peptide A (Gervais et al., 2007) ou induire la dégradation du peptide A (Jacobsen et al., 2008), cependant ces traitements qui ont atteints la phase clinique III n’ont pas eu l’efficacité escomptée.
Une stratégie, en vogue, serait l’immunothérapie. Plusieurs études démontrent qu’il serait possible d’injecter des agrégats du peptide A42, ceci provoquerait la synthèse d’anticorps capable de cibler les dépôts
26
amyloïdes (Morgan et al., 2000; Schenk et al., 1999). Le mécanisme d’action de cette thérapie n’est pas encore pleinement compris et plusieurs hypothèses restent ouvertes (recrutement mitochondrial, dégradation par les anticorps etc.). Plusieurs molécules sont à l’essai pour cette approche, notamment Bapineuzumab (de Elan and Wyeth) ou Solanezumab (d’Eli Lilly and Company). Néanmoins des cas de méningo encéphalite sont apparus lors de la phase II d’un essai clinique103.
Pour ce qui est des Tauopathies (maladie d’Alzheimer comprise), plusieurs médicaments sont prescrits mais ne servent qu’à atténuer les symptômes: le donepezil (pour FTD), le lévodopa (Tauopathies avec
déficience motrice), ou divers antipsychotiques(http://alzforum.com).
De nombreuses stratégies sont envisagées en ce qui concerne le traitement de la pathologie Tau. Une stratégie serait de cibler l’agrégation du Tau hyperphosphorylé en inhibant ce processus. Pourtant, plusieurs problèmes se présentent à nous : passer la barrière hématoencéphalique ou cibler uniquement l’interaction protéine-protéine de Tau sans pour autant cibler les autres interactions protéines-protéines (Bulic et al., 2009). Une autre stratégie serait l’inhibition des kinases impliquées dans la phosphorylation de Tau telles que GSK3 ou Cdk5. Cependant, ces kinases sont aussi impliquées dans d’autres mécanismes cellulaires important, leur inhibition pourrait provoquer des effets secondaires sérieux.
Enfin, le bleu de méthylène, comme évoqué précédemment, a démontré des effets encourageants. En effet, cette molécule a permis la réduction des agrégats de Tau in vitro. Néanmoins, il a démontré, pour l’instant, en phase clinique II uniquement une diminution de la progression de la maladie et non pas un traitement curatif (Wischik et al., 2015).
Ainsi, plusieurs approches thérapeutiques sont à l’essai concernant le peptide amyloïde ou concernant la protéine Tau.
27
Étant donné qu’il existe plusieurs causes de la pathogénicité en ce qui concerne les Tauopathies (modifications post-traductionnelles, agrégation de Tau, épissage alternatif, oligomères de Tau etc.) de nombreuses questions restent en suspens pour ce qui est de la cible de la neurotoxicité.
Nous avons choisi dans ce projet de développer une approche thérapeutique capable de cibler la quantité de Tau totale plutôt qu’une seule de ces causes. Pour ce faire, le but principal ici sera de viser l’ARNm de Tau grâce à une molécule développée dans les laboratoires du Dr Jean-Pierre Perrault et Dr Georges Lévesque : le SOFA-Delta-Ribozyme.
3.2. Généralités : les ribozymes naturels
En 1982, le Dr. Thomas Cech découvrait un ARN capable d’effectuer une réaction catalytique, chez le Tetrahymena Thermophila (Kruger et al., 1982), cet ARN-enzyme pouvait effectuer l’épissage de ces propres introns. Le Dr. Altman confirma cette découverte chez E.Coli et montra que la ribonucléase P (ARN P) était capable d’avoir une activité enzymatique (Guerrier-Takada, Gardiner, Marsh, Pace, & Altman, 1983). Ces deux découvertes leur ont valu le prix Nobel de Chimie en 1989. Cette molécule changeant complètement le dogme selon lequel une enzyme était obligatoirement une protéine. Celle-ci a par la suite été nommée ribozyme, soit ribonucléotide-enzyme. Il existe plusieurs exemples de ribozymes dans la nature. La grande majorité effectue des modifications catalytiques sur leur propre structure, mise à part la RNAse P qui permet en partie la synthèse d’un ARNt (ARN de transfert) (Guerrier-Takada et al., 1983). De plus, la plupart de ces ribozymes sont considérés comme metalloenzyme, ils ont besoin d’un cation, souvent le MgCl2+, pour
28
On distingue 2 groupes de ribozymes.
Le premier groupe comporte les ribozymes pouvant atteindre 3000 nucléotides. Il est composé de la RNAse P (Houser-Scott et al., 2012), et des introns du groupe I (Kruger et al., 1982) et II (Michel & Ferat, 1995). Ces ribozymes effectuent une transestérification et génèrent des produits 3’-hydroxyle et 5’-phosphate. En effet, les introns du groupe I et II permettent la maturation d’un ARNm en effectuant un auto-épissage des régions introniques. Les introns du groupe I s’aident d’une guanosine ou d’une GTP exogène (guanosine triphosphate) pour effectuer le mécanisme de transestérification tandis que les introns du groupe II effectuent une attaque nucléophile grâce à l’adénosine présente dans son propre ARNm (Doudna & Cech, 2002).
Le second groupe comporte des ribozymes de plus petites tailles, à peu près 200 nucléotides : le hammerhead, hairpin, Varkud Satellite (VS), et virus de l’hépatite delta ribozyme (HDV). Ces ribozymesTau effectuent
aussi une transestérification par un mécanisme SN2. En effet, le
phosphate subit une attaque nucléophile du 2’-oxygène qui se trouve sur le ribose, ceci provoque le clivage du lien phosphodiester. Il en résulte 2 produits : 2’-3’-cyclique phosphate et 5’-hydroxyle (Tanner, 1999).
29
Figure 8. Mécanisme SN2 de l’auto-clivage des ribozymes du second groupe.
Une base déprotone le 2’-hydroxyle, ce qui lui permet de faire une attaque nucléophile sur le phosphate et ainsi de provoquer le clivage, induisant les 2 produits (Adapté Doudna & Cech, 2002).
3.3. Généralités : les ribozymes naturels
Le premier ribozyme du second groupe ayant été découvert fut le
hammerhead ribozyme. Ce ribozyme fait partie de ce qu’on appelle les
ARN satellites. L’ARN satellite est un parasite de virus infectant les plantes, il se réplique à travers ce virus (Roossinck, Sleat, & Palukaitis, 1992). Le hammerhead ribozyme fut découvert pour la 1ère fois dans le virus ringspot qui infecte la feuille de tabac (Prody, Bakos, Buzayan, Schneider, & Bruening, 1986). Ce ribozyme, d’à peine 40 nucléotides, est le plus petit des ribozymes naturels. Il effectue un autoclivage de l’ARN satellite circulaire qui va par la suite mener à la réplication de ce dernier, il est capable de catalyser 1 molécule par minute (Scott, Horan, & Martick, 2013). La structure cristalline révèle qu’il est sous la forme d’un Y, composé de 3 hélices (I, II et III) reliées par une région conservée de 15 nucléotides. Les hélices I et II seraient les bras, tandis que l’hélice III le
30
corps du Y (Tanner, 1999). La réaction de clivage se passe après un triplet NUH (avec N n’importe quel nucléotide et H n’importe quel nucléotide sauf la guanosine) selon le mécanisme expliqué précédemment.
Le hairpin ribozyme fait aussi parti de l’ARN satellite parasitant le virus
ringspot de la feuille de tabac. Il a été nommé ainsi car il adopte la forme
d’une épingle à cheveux. Il est composé de 4 hélices (A, B, C et D) ce qui permet une meilleure stabilité de sa structure (Ferré-D’Amaré & Scott, 2010). L’attaque nucléophile se déroule sur le brin A, le phosphate attaqué est situé entre deux purines (G et A) importante pour la réaction de clivage (Rupert & Ferré-D’Amaré, 2001).
Ces deux ribozymes ont été adaptés pour cibler d’autres ARN que leur propre ARN, par exemple l’ARN du VIH (Sun et al., 1995).
a) b)
Figure 9. Structures secondaires et structures cristallines du Hammerhead ribozyme et du Hairpin ribozyme.
(a) Hammerhead ribozyme : Structure secondaire à gauche avec le site de clivage indiqué par une flèche ; Structure cristalline à droite avec le site de clivage indiqué en doré. (b) Hairpin ribozyme : Structure secondaire à gauche avec le site de clivage indiqué par une flèche ; Structure cristalline à droite avec le site de clivage indiqué en doré. (Adapté de Doudna & Cech, 2002).