Thérapie génique ex vivo de la dystrophie musculaire de
Duchenne à l’aide de cellules souches pluripotentes
induites
Mémoire
Chantale Maltais
Maîtrise en Biologie cellulaire et moléculaire
Maître ès sciences (M.Sc.)
Résumé
La dystrophie musculaire de Duchenne (DMD) est une myopathie héréditaire due à l'absence de dystrophine. Parmi les thérapies possibles, la greffe autologue de myoblastes dérivés de cellules souches pluripotentes induites (hiPSCs) provenant du patient dystrophique, préalablement corrigés génétiquement, est envisageable. Lors de la première partie de ma recherche, j'ai transplanté des hiPSCs de patient DMD différenciés en myoblastes chez la souris Rag/mdx. Ces cellules avaient été corrigées génétiquement à l'aide d'un vecteur lentiviral codant pour la micro-dystrophine, une dystrophine tronquée, mais toujours fonctionnelle. Mes résultats ont démontré l'expression de cette micro-dystrophine dans certaines fibres hybrides. Cependant, le protocole de différenciation des hiPSCs en myoblastes doit être amélioré. La deuxième partie de mon projet consistait donc à induire la myogenèse à l’aide de protéines recombinantes. Pour cela, des facteurs de transcription régulateurs de la myogenèse, fusionnés à un peptide de pénétration cellulaire, ont été produits et purifiés d’un système bactérien. Leur pénétration dans des cellules mésenchymateuses a été observée in vitro et leurs effets sur les cellules sont en cours d’étude. Lorsque ces approches thérapeutiques seront mises au point, elles pourraient être appliquées cliniquement pour traiter des patients dystrophiques.
Abstract
Duchenne muscular dystrophy (DMD) is a hereditary myopathy due to the absence of dystrophin. Among the possible therapies, there is the autologous transplantation of genetically corrected myoblasts derived from human induced pluripotent stem cells (hiPSCs) of a dystrophic patient. In the first part of my research project, I have transplanted myoblasts differentiated from iPSCs of a DMD patient in the Rag/mdx mouse. These cells had been previously genetically corrected with a lentiviral vector coding for micro-dystrophin, a functional truncated version of dystrophin. The results demonstrated the expression of this micro-dystrophin in some of the hybrid fibers. However, in order to increase the graft success, the protocol of differentiation of hiPSCs in myoblasts must be improved. The second part of my project was the induction of myogenesis from hiPSCs using recombinant proteins. To accomplish this, myogenic transcription factors fused with a cell penetrating peptide were produced and purified from the bacterial system. Their capacity to enter into mesenchymal-like cells in vitro was observed and their effects on the cells are currently under study. Once optimized, these therapeutic approaches could be clinically applied to treat dystrophic patients.
Table des matières
Résumé ... III Abstract ... V Table des matières ... VII Liste des figures ... IX Liste des tableaux ... XI Liste des abréviations ... XIII Remerciements ... XVII
Chapitre 1 : Introduction... 1
1.1 Tissu musculaire squelettique ... 1
1.1.1 Myogenèse ... 1
1.1.2 Muscle squelettique adulte ... 4
1.1.2.1 Anatomie et physiologie ... 4
1.1.2.2 Réparation des tissus musculaires ... 8
1.2 La dystrophie musculaire de Duchenne ... 10
1.2.1 Historique ... 10
1.2.2 Description de la maladie ... 10
1.2.3 Gène de la dystrophine et mutations ... 12
1.2.4 Dystrophine ... 13
1.2.4.1 Isoformes ... 13
1.2.4.2 Dp427 musculaire ... 14
1.2.5 Le complexe de glycoprotéines associé à la dystrophine ... 15
1.3 Soins et traitements de la DMD ... 17 1.3.1 Diagnostique ... 17 1.3.2 Traitements et thérapies ... 18 1.3.2.1 Traitements pharmacologiques ... 18 1.3.2.2 Thérapie génique ... 20 1.3.2.3 Thérapie cellulaire... 22 1.4 Modèles animaux ... 25 1.4.1 Modèles murins ... 25 1.4.2 Modèles canins ... 26 1.4.3 Modèle félin ... 26
1.5 Cellules souches humaines ... 27
1.5.1 Cellules souches embryonnaires ... 27
1.5.1.1 Historique et législation ... 27
1.5.1.2 Isolation et culture ... 28
1.5.1.3 Caractéristiques et mécanisme de pluripotence ... 30
2.2.1.1 Vecteur lentiviral codant pour la micro-dystrophine (Le.MCK-µDysV5) ... 37
2.2.1.2 Vecteur adénoviral codant pour MyoD (Ad.CAG-MyoD) ... 38
2.2.2 Culture cellulaire ... 38
2.2.2.1 Les myoblastes humains normaux ... 39
2.2.2.2 La lignée d’hiPSCs dystrophiques ... 40
2.2.2.3 Différenciation en cellules mésenchymateuses ... 40
2.2.2.4 Correction génique à l’aide du vecteur Le.MCK-µDysV5 ... 41
2.2.2.5 Différenciation en myoblastes à l’aide du vecteur Ad.CAG-MyoD ... 41
2.2.3 Détection de l’expression de la micro-dystrophine-V5 in vitro ... 41
2.2.4 Greffe des cellules dans les muscles de souris Rag/mdx ... 43
2.2.5 Analyses immunohistologiques ... 44
2.3 Résultats ... 45
2.3.1 Détection de l’expression de µDysV5 in vitro ... 45
2.3.2 Immunomarquages ... 45
2.3.3 Évaluation du nombre des fibres positives aux immunomarquages ... 47
2.4 Discussion ... 49
Chapitre 3. Induction de la myogenèse à l’aide de facteurs de transcription... 53
3.1 Objectif ... 53
3.2 Matériels et méthodes ... 55
3.2.1 Construction des vecteurs d’expression ... 55
3.2.2 Production des protéines recombinantes ... 60
3.2.2.1 Transformation des bactéries thermo-compétentes ... 60
3.2.2.2 Culture des bactéries transformées ... 61
3.2.2.3 Induction de la production des protéines recombinantes ... 61
3.2.2.4 Détection des protéines par immunobuvardage de type Western ... 62
3.2.2.5 Lyse des cellules bactériennes ... 62
3.2.2.6 Purification des protéines recombinantes ... 62
3.2.2.7 Dessalage et renaturation des protéines recombinantes ... 63
3.2.3 Essais in vitro de 6xHis-Tat-MyoD ... 64
3.2.3.1 Culture des cellules mésenchymateuses ... 64
3.2.3.2 Transduction des protéines recombinantes dans les cellules mésenchymateuses 64 3.2.3.3 Détection des protéines ... 64
3.3 Résultats ... 67
3.3.1 Construction des vecteurs d’expression ... 67
3.3.2 Production et purification des protéines recombinantes ... 67
3.3.3 Détection des protéines recombinantes dans les cellules transduites ... 70
3.4 Discussion ... 71
Chapitre 4 : Conclusion ... 73
Liste des figures
Figure 1. Facteurs impliqués lors de la myogenèse embryonnaire. ... 2
Figure 2. Développement du muscle squelettique adulte. ... 3
Figure 3. Anatomie du muscle squelettique d’un point de vue macroscopique et microscopique. ... 6
Figure 4. Liaison des myofibrilles au sarcolemme via les costamères. ... 7
Figure 5. Facteurs impliqués lors de la réparation musculaire. ... 9
Figure 6. Manœuvre de Gower ... 11
Figure 7. Isoformes de la dystrophine. ... 14
Figure 8. Dystrophine et le complexe de glycoprotéines associé. ... 17
Figure 9. Versions de la dystrophine. ... 20
Figure 10. Étapes d'une greffe de myoblastes allogéniques. ... 23
Figure 11. Isolation des cellules souches embryonnaires et mise en culture. ... 29
Figure 12. Vecteur Le.MCK-µDysV5. ... 37
Figure 13. Différents types cellulaires greffés. ... 39
Figure 14. Immunomarquages des cryosections consécutives de muscles greffés à l'aide de myoblastes humains normaux. ... 45
Figure 15. Immunomarquages des cryosections consécutives de muscles greffés à l'aide de MSCs corrigées génétiquement à l’aide du vecteur Le.MCK-µDysV5 et différenciées en myoblastes. ... 46
Figure 16. Immunomarquages des cryosections consécutives de muscles greffés à l'aide de MSCs corrigées génétiquement à l’aide du vecteur Le.MCK-µDysV5, mais non différenciées en myoblastes. ... 47
Figure 17. Succès de greffe évalué par le compte de fibres positives aux immunomarquages. ... 48
Figure 18. Représentation schématique du plasmide pET-16b. ... 56
Figure 19. Mécanisme de l'induction à l'IPTG. ... 57
Figure 20. Immunobuvardage de type Western de l'induction de la production des protéines recombinantes. ... 67
Figure 21. SDS-PAGE de la purification de 6x[His]-Tat-MyoD. ... 68
Figure 22. SDS-PAGE de la purification de 6x[His]-Tat-Pax3. ... 69
Figure 23. SDS-PAGE de la purification de 6x[His]-Tat-Pax7. ... 69
Figure 24. Immunobuvardage de type Western pour la détection de la protéine 6x[His]-Tat-MyoD intracellulaire. ... 70
Liste des tableaux
Tableau 1. Séquences des amorces utilisées. ... 58 Tableau 2. Paramètres pour l'amplification par PCR des gènes MyoD, Pax3 et Pax7. ... 59 Tableau 3. Températures de production des protéines recombinantes dans les bactéries E. coli BL21 (DE3). ... 61
Liste des abréviations
2′OMe = 2′-O-méthyl-phosphorothioate ADN = acide déoxyribonucléique ADNc = ADN complémentaire ARN = acide ribonucléique ARNm = ARN messager ATP = adénosine triphosphate BCA = acide bicinchonique
bFGF = basic fibroblast growth factor BMP 4 = bone morphogenetic protein 4
CAG = cytomegalovirus early enhancer/chicken β-actin CH= calponin homology
CKCS-MD = cavalier King Charles spaniels muscular dystrophy cm = centimètre
CPP = cell-penetrating peptide DAPI = 4',6'-diamidino-2-phénylindole
DGC = complexe de glycoprotéines associé à la dystrophine DMD = Dystrophie Musculaire de Duchenne
DMEM = Dulbecco's modified Eagle medium DTT = dithiothreitol
ESCs = cellules souches embryonnaires Eya 1/2 = eyes-absent homologs 1 et 2 FBS = sérum de veau foetal
FGF = fibroblast growth factor g = force relative de centrifugation
GRMD = Golden Retriever Muscular Dystrophy HBSS = Hank’s balanced salts solution
hESCs = ESCs humaines
HFMD = hypertrophy feline muscular dystrophy HGF = hepatocyte growth factor
HRP = Horseradish peroxydase hiPSCs= iPSCs humaines IGF = insulin growth factor IgG = immunoglobuline G IL-6 = interleukine-6
IPTG = isopropyl-β-D-thiogalactopyranoside iPSCs = cellules souches induites à la pluripotence kb = kilobase
Mdx = muscular dystrophy X-linked MEF = mouse embryonic fibroblasts mg = miligramme
mm = milimètre ml = mililitre mM = milimolaire
MI = multiplicité d’infection
MRFs = facteurs régulateurs de la myogénèse MSCs = cellules mésenchymateuses
Myf5 = facteur myogénique 5 MyHC = myosin heavy chain
MyoD = facteur de différenciation myogénique MyoG = myogénine
NaCl = chlorure de sodium
NAPDH = nicotinamide adénine dinucléotide phosphate nNos = neural nitric oxide synthase
ONA = oligonucléotide antisens
Pax 3/7 = paired box transcription factors 3 et 7 pb = paire de bases
PBS = phosphate buffer solution
PCR = réaction en chaîne par polymérase PDGF = platelet-derived growth factor
PMO = morpholino phosphorodiamidate oligonucléotide PMSF = phenylmethylsulfonylfluoride
P/S = pénicilline et streptomycine rpm = rotations par minute SDS = sodium dodécyl sulfate
SDS-PAGE = sodium dodecyl sulfate polyacrylamide gel electrophoresis Shh = Sonic hedgehog
Six1/4 = sine oculis–related homeobox 1 et 4
SSEA-3/4 = antigènes de surface spécifiques au stade embryonnaire 3 et 4 TA = Tibialis anterior
Tat = trans-acting activator of transcription TGF- = transforming growth factor beta TNF = tumor necrosis factor
Tris-HCl = Tris(hydroxymethyl)aminomethane hydrochloride µDys = micro-dystrophine
µDysV5 = micro-dystrophine avec un tag V5 µl = microlitre
µg = microgramme µm = micrometre V = Volts
Ces travaux ont été effectués dans l’espoir de contribuer au développement d’un traitement curatif contre la dystrophie musculaire de Duchenne.
Remerciements
En premier lieu, je tiens à remercier le Dr. Jacques P. Tremblay de m’avoir accueillie dans son laboratoire. Il a su me transmettre ses connaissances et sa passion pour la recherche, et m’a permis d’évoluer dans un environnement innovateur. J’ai eu la chance de travailler sur de multiples projets et ainsi d’apprendre une grande variété de techniques.
De plus, je suis reconnaissante envers tous les membres de l’équipe de recherche pour m’avoir conseillée et pour tous les bons moments passés au laboratoire. Un merci spécial à Joël Rousseau et Catherine Gérard qui ont su répondre à mes innombrables questions, surtout au début de ma maîtrise, et me soutenir lorsque j’étais dans le creux de la vague.
J’aimerais aussi remercier particulièrement Carl Lebel, qui m’a montré les premiers pas en thérapie génique, et William-Édouard Gravel, qui est toujours de bonne humeur et prêt à aider.
Finalement, je tiens à remercier ma famille et mes amis, spécialement ma mère et mon beau-père. Sans leurs encouragements, leur soutien et leur amour, je ne serais pas la moitié de la personne que je suis aujourd’hui.
Chapitre 1 : Introduction
Ce mémoire porte sur le développement d’une thérapie de la dystrophie musculaire de Duchenne (DMD) basée sur l’utilisation de cellules souches pluripotentes induites humaines (hiPSCs). Des thèmes seront d'abord introduits, comme le tissu musculaire squelettique, la DMD et ses traitements et thérapies, les modèles animaux existants et les cellules souches en médecine régénérative. Par la suite, deux parties de mon projet de recherche seront décrites, d’une part la thérapie génique ex vivo de la DMD à l'aide d'hiPSCs et d'un vecteur lentiviral codant pour la micro-dystrophine, et d’autre part l’induction de la myogenèse à l’aide de facteurs de transcription.
1.1. Tissu musculaire squelettique
Les tissus musculaires squelettiques ont pour fonction principale de permettre le mouvement volontaire, le maintien de la posture et la production de chaleur. Ils représentent près de 40 % du corps humain et leur formation débute lors de l’embryogenèse.
1.1.1. Myogenèse
Lors de l’embryogenèse, des gradients de morphogènes, comme l'acide rétinoïque, le fibroblast
growth factor (FGF) et Wnt, ainsi que des changements dans l’expression des gènes, vont provoquer
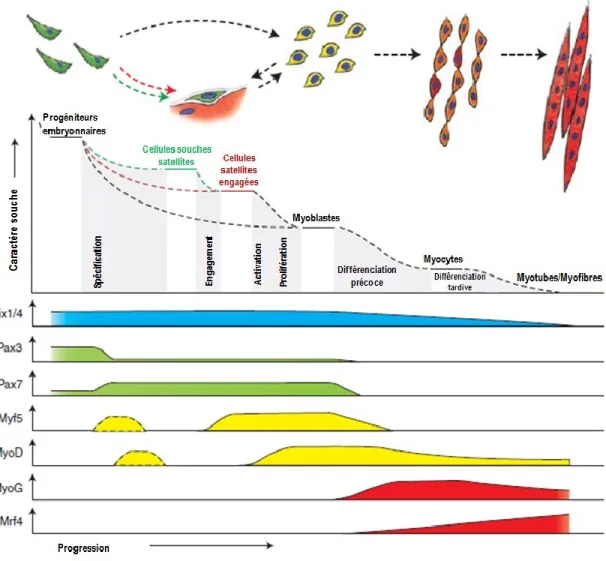
la formation des somites à partir du mésoderme para-axial. Lorsque les cellules quittent la partie caudale, un gradient d’acide rétinoïque permet leur polarisation pour former le dermomyotome dorsal et le sclérotome ventral. Des signaux venant des tissus avoisinants, tels que Wnts, Sonic Hedgehog (Shh) et Noggin, activent l’expression des facteurs de régulation myogéniques (MRFs) primaires, alors que le bone morphogenetic protein 4 (BMP4) inhibe l’engagement des cellules dans la voie myogénique, tel que démontré par la Figure 1 [1, 2].
Figure 1. Facteurs impliqués lors de la myogenèse embryonnaire.
Les muscles squelettiques proviennent du dermomyotome, lui-même dérivé du compartiment dorsal du somite. Image adaptée de Gilbert, 2013 [2].
Le compartiment dorsal du somite deviendra le dermomyotome, dont les muscles squelettiques (sauf ceux de la tête) sont dérivés. Les protéines sine oculis–related homeobox 1 et 4 (Six1 et Six4) lient les eyes-absent homologs 1 et 2 (Eya1 et Eya2) et les transloquent au noyau. Ainsi, ils agiront comme cofacteurs pour activer les gènes cibles de Six1/4, c'est-à-dire paired box transcription
factor 3 (Pax3), facteur de différenciation myogénique (MyoD), Mrf4, et myogénine (MyoG). Les
cellules du dermomyotome sont marquées par l’expression des paired box transcription factors 3 et 7 (Pax3 et Pax7) et par une faible expression du facteur myogénique 5 (Myf5). Pax3 est requis pour la migration des précurseurs myogéniques des somites et active l’expression précoce et transitoire de Myf5 et MyoD [3]. Pax7 sera nécessaire pour la spécification en cellules satellites [4]. Une partie du dermomyotome va donc se développer pour former le myotome. À ce stade, les cellules sont dites engagées dans la voie musculaire, puisqu’elles expriment MyoD et Myf5, deux marqueurs de la spécification terminale des cellules musculaires. Les cellules qui les expriment se nomment des myoblastes [5]. Les myoblastes vont proliférer pour finalement se retirer du cycle cellulaire. Par la suite, ils expriment des MRFs tardifs, comme la MyoG et Mrf4. Les cellules deviennent alors des myocytes, qui vont s’aligner, puis fusionner pour former les fibres musculaires,
c’est-à-dire les cellules musculaires adultes qui expriment des protéines spécifiques au muscle, comme la myosin heavy chain (MyHC) et muscle creatin kinase (MCK) (Figure 2) [6].
Figure 2. Développement du muscle squelettique adulte.
Plusieurs facteurs de transcription régulent la myogenèse. Six1/4 et Pax3/7 sont des régulateurs de la spécification initiale, alors que Myf5 et MyoD engagent les cellules dans la voie myogénique. La fusion des myocytes en myotubes est assurée par l'expression des gènes de la différenciation terminale, soit MyoG et Mrf4. Figure adaptée de Bentzinger et al., 2012 [6].
et ont un rôle clé dans la régénération musculaire. Leur implication dans ce processus de réparation sera expliquée en détail à la section 1.1.2.2.
1.1.2. Muscle squelettique adulte
1.1.2.1. Anatomie et physiologie
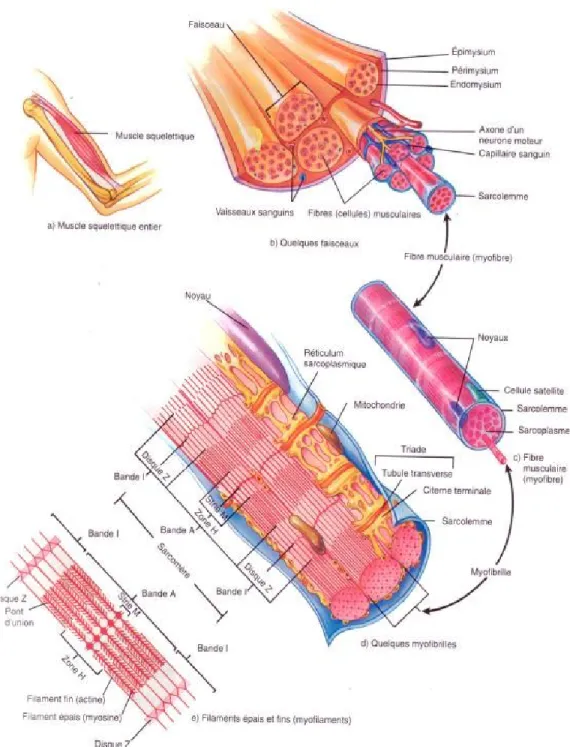
Les cellules du muscle squelettiques sont appelées fibres musculaires. Elles sont multinucléées et possèdent une forme cylindrique d'un diamètre variant de 10 à 100 micromètres (µm) et dont la longueur peut atteindre jusqu'à 30 centimètres (cm) chez l'humain. Elles sont formées par la fusion de myoblastes, des cellules précurseurs mononucléées [7].
Les fibres possèdent un sarcoplasme qui contient plusieurs organites, dont de multiples mitochondries, le réticulum sarcoplasmique ainsi que de nombreux noyaux cellulaires, qui sont retrouvés en périphérie de la fibre, sous la membrane plasmique. Cette dernière est appelée le sarcolemme et fait face à la lame basale, une matrice de tissu conjonctif, principalement constituée de fibronectine pour permettre la migration de cellules mononucléées lors de la réparation musculaire. Cette lame basale est située sous la lame réticulaire, et font tous deux partie de l’endomysium. Diverses cellules s’y retrouvent, comme des fibroblastes et des macrophages. De plus, les cellules satellites, qui sont les cellules souches musculaires adultes, sont situées dans la lame basale [8].
Les fibres musculaires sont formées de myofibrilles, les éléments contractiles du muscle. Il s'agit d'organites complexes faits de groupes de filaments d'actine et de myosine. Ces filaments sont organisés en sarcomère, l'unité fonctionnelle du muscle, et portent des stries qui sont alignées avec celles des myofibrilles voisines. C'est l'alternance régulière des bandes A et I qui donne l'apparence striée du muscle [7, 8].
Le regroupement de plusieurs fibres musculaires entourées de leur endomysium est un faisceau musculaire, lui-même étant recouvert de tissu conjonctif dense régulier appelé le périmysium. Ce dernier comprend de multiples vaisseaux sanguins nécessaires pour l'irrigation du muscle. Plusieurs faisceaux regroupés ensemble sont recouverts par l'épimysium, une couche de tissu conjonctif dense irrégulier. Elle est impliquée dans la transmission de la force mécanique et l’élasticité du muscle, en
plus de servir de réservoir de facteurs de croissance et d’eau pour le développement et la régénération musculaire (Figure 3) [8, 10].
Figure 3. Anatomie du muscle squelettique d’un point de vue macroscopique et microscopique.
Les muscles squelettiques sont faits de myofibrilles composées de sarcomères contenant des filaments d'actine et de myosine. Les myofibrilles sont regroupées en fibres musculaires. Chaque fibre est entourée d'endomysium. Un ensemble de fibres est entouré du périmysium et s'appelle un faisceau. Plusieurs faisceaux sont enveloppés par l’épimysium. Image tirée de Tortora et al., 1994 [8].
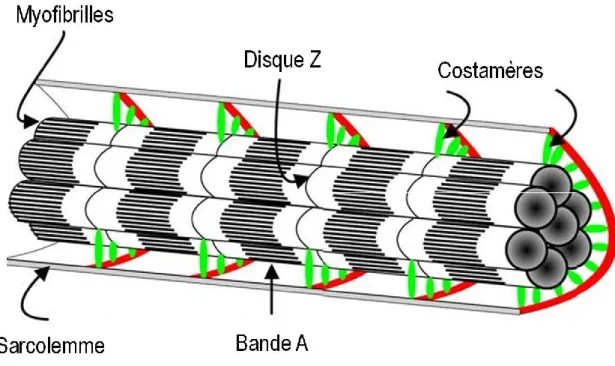
La contraction musculaire est provoquée par le glissement des filaments d'actine le long des filaments de myosine, un processus qui est dépendant de l’adénosine triphosphate (ATP) et du calcium. La régulation du transport des ions calcium est assurée par le réticulum sarcoplasmique, qui entoure chaque myofibrille. Les myofibrilles périphériques sont connectées à la membrane plasmique via les costamères, c’est-à-dire des complexes protéiques situés sous la membrane dont le rôle est de permettre la transmission des forces contractiles d’une fibre à l’autre. Les costamères sont des régions enrichies en plusieurs protéines, dont deux complexes importants, soit les complexes dystrophine-glycoprotéines et intégrine-vinculine-taline (Figure 4). Ces derniers empêchent donc les micro-ruptures de la membrane en synchronisant les contractions des fibres musculaires [7].
Figure 4. Liaison des myofibrilles au sarcolemme via les costamères.
Les costamères sont riches en protéines. On y retrouve une concentration de complexes dystrophine-glycoprotéines et intégrine-vinculine-taline. Image adaptée de Ervasti, 2003 [9].
1.1.2.2. Réparation des tissus musculaires
Les fibres musculaires entrent dans un processus de réparation en réponse à des bris qui peuvent être causés par des stress mécanique, ischémique, thermique ou pharmacologique. La réparation musculaire s'effectue en deux phases, soit la phase dégénérative et la phase régénérative.
La phase dégénérative est caractérisée par une nécrose. Elle survient immédiatement après la rupture du sarcolemme de la fibre, ce qui provoque une entrée de calcium intracellulaire, et la destruction d'organites, comme les mitochondries, les ribosomes et les myofibrilles. Cette rupture provoque aussi une sortie des protéines musculaires vers le système sanguin, telles la créatine kinase et la troponine, ainsi que des molécules biologiquement actives.
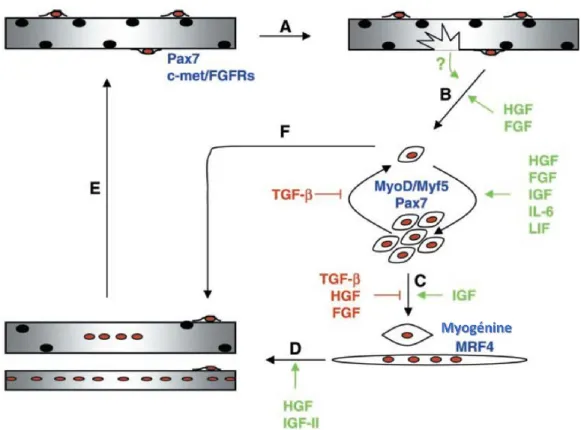
La phase régénérative est divisée en trois étapes : la réaction inflammatoire, l’activation et différenciation des cellules satellites, et finalement la maturation des nouvelles fibres et le remodelage des fibres régénérées. Tout d’abord, une réaction inflammatoire est enclenchée, au cours de laquelle des macrophages et neutrophiles sont attirés aux sites endommagés pour phagocyter les débris cellulaires [11]. Ces cellules ainsi que la matrice extracellulaire sécrètent des facteurs de croissance qui activent les cellules satellites, comme le FGF, le transforming growth
factor beta (TGF-), l'insulin growth factor (IGF), l'hepatocyte growth factor (HGF), le tumor necrosis
factor (TNF), l'interleukine-6 (IL-6) et le leukemia inhibitory factor (LIF) (Figure 5). Les cellules
satellites sont normalement quiescentes et ont un faible taux de synthèse de macromolécules et une activité métabolique minimale. Elles se protègent bien des dommages, via des facteurs et des enzymes qu’elles produisent, en plus d’utiliser la glycolyse anaérobique, dans le but de limiter l’accumulation d’oxygène réactif. Elles ont la capacité de se régénérer elles-mêmes et de se différencier [12, 13]. Elles produisent des molécules qui régulent les cascades de signalisation pour leur entrée dans le cycle cellulaire. Si nécessaire, elles peuvent être rapidement activées puisqu’elles possèdent des récepteurs et des enzymes pour répondre aux molécules de signalisation lors d’un bris de fibres musculaires. De plus, les cellules satellites transcrivent les facteurs myogéniques, mais ceux-ci sont inactifs grâce à des corépresseurs ou des mécanismes post-transcriptionnels. Ces facteurs peuvent donc être rapidement libérés lors de l’activation des cellules satellites [14]. Ainsi, suite à leur activation, le niveau d'expression de Pax7 diminue alors que celui de MyoD et Myf5 augmente dans les cellules satellites. Leur division asymétrique et la différenciation des cellules qui
en découle résultent en des myoblastes qui vont fusionner aux fibres endommagées, ou former de nouvelles fibres [7]. Une petite portion des cellules ne se différencie pas et retourne à un état de quiescence pour préserver le pool de cellules satellites [15]. Les fibres régénérées et nouvellement formées sont petites et centro-nucléées. Leur croissance et maturation, ainsi que la migration des noyaux vers la périphérie des fibres, correspond à la troisième phase de la régénération, et dépend de plusieurs facteurs, comme la nature du bris ainsi que le rétablissement des jonctions neuromusculaires et des vaisseaux sanguins [11].
Figure 5. Facteurs impliqués lors de la réparation musculaire.
Lors d'un bris musculaire, des facteurs sont sécrétés par la matrice extracellulaire et les cellules inflammatoires, activant ainsi les cellules satellites qui se mettent alors à exprimer MyoD et Myf5. Ces dernières vont donc proliférer et se différencier, et ainsi exprimer les gènes tardifs, soit MRF4 et
1.2. La dystrophie musculaire de Duchenne
1.2.1. Historique
La dystrophie musculaire de Duchenne, ou DMD, est une myopathie qui a été décrite pour la première fois en 1852 par Edward Meryon. Toutefois, c'est au physiologiste français Guillaume Benjamin Amand Duchenne à qui on doit de considérables caractérisations cliniques en 1861, alors qu'il croyait que la maladie était d'origine cérébrale. Toutefois, en 1868, il s'est aperçu que la maladie était plutôt d’origine musculaire. Il la nomma alors « paralysie musculaire pseudo-hypertrophique ou paralysie myo-sclérosique », mais elle est maintenant largement connue sous le nom de dystrophie musculaire de Duchenne. Il fut le premier à proposer des critères diagnostiques bien précis et il inventa l'appareil à biopsie, qui a été largement utile pour décrire les processus microscopiques de la maladie [16].
1.2.2. Description de la maladie
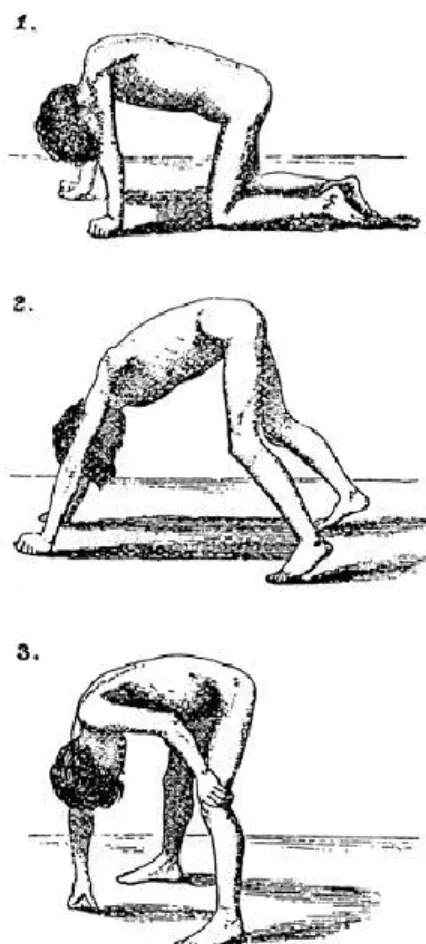
La DMD est une myopathie héréditaire récessive liée au chromosome X, ayant une incidence d'un cas sur 3 500 naissances mâles. Il s'agit de la forme de dystrophie la plus sévère, étant létale. Elle est caractérisée par une perte progressive de force musculaire dès l'âge de 2 ou 3 ans. Durant ce stade de l'enfance, les symptômes sont plutôt rares, mais des signes cliniques, comme l'élévation du taux de créatine kinase sérique et la nécrose des tissus musculaires, sont des preuves du début de la dégénérescence musculaire. Vers l'âge de 4 ans, il est possible d'observer une démarche anormale, une pseudo-hypertrophie des mollets, ainsi qu'une difficulté à monter les escaliers. On note l'apparition des signes de Gower vers l'âge de 5 à 7 ans (Figure 6). Il s'agit d'une manœuvre effectuée par l'enfant pour se relever du sol. Les muscles de ses jambes et de ses hanches étant devenus trop faibles pour cette tâche, il doit se servir de tout son corps pour se relever [16].
Figure 6. Manœuvre de Gower
La faiblesse musculaire des patients DMD les force à utiliser tous les muscles de leur corps pour se relever du sol. Cette méthode est appelée Manœuvre de Gower [17].
Vers l'âge de 12 ans, les patients se retrouvent confinés dans un fauteuil roulant puisque les muscles de leurs membres ont perdu trop de force pour les soutenir. La majorité des patients développent alors une scoliose. Des difficultés respiratoires surviennent peu après, les forçant à recourir à un système de respiration assistée. L'espérance de vie des patients varie entre 20 et 30 ans et les principales causes de décès sont l'insuffisance respiratoire ou cardiaque [18].
De plus, les patients atteints ont un taux sérique de créatine kinase très élevé dès la naissance, en raison des dommages continuels que subissent leurs tissus musculaires. En effet, les muscles des patients sont très fragiles puisqu'une protéine très importante, la dystrophine, est absente. Au fil du temps, le tissu musculaire est remplacé par du tissu adipeux et conjonctif, jusqu'à ce qu'il y ait perte de fonction musculaire [20].
1.2.3. Gène de la dystrophine et mutations
Le gène de la dystrophine responsable de la maladie a été découvert par Kunkel et al. en 1985 [21]. Il est situé sur le bras court du chromosome X au locus Xp21.2. Cette découverte est attribuable à l'étude de quelques patientes atteintes de la DMD ayant une translocation autosomale du chromosome X, et une inactivation de l’autre chromosome X [22, 23].
Le gène possède environ 2.4 mégabases (Mb) d'acides désoxyribonucléiques (ADN), ce qui en fait un des plus grands gènes connu chez l’humain, représentant à lui seul près de 1 % du chromosome X. Il code pour un acide ribonucléique messager (ARNm) de 14 kilobases (kb) de 79 exons, ayant des introns inhabituellement longs. Ainsi, seulement 0,6 % du gène représente la partie codante.
Dans 70 % des cas, les patients ont des réarrangements de larges sections d'ADN, soit une délétion ou duplication d’un ou plusieurs exons. Ces mutations sont retrouvées généralement près de deux sites en particulier : près de l’extrémité 5’ du gène et vers le centre, là où il y a de très longs introns (plus de 100 kb). Dans 30 % des cas, il s’agit de mutations ponctuelles ou de micro-délétions impliquant un petit nombre de nucléotides. Chez le tiers des patients DMD, les mutations sont spontanées et n’ont pas d'antécédent familial [16].
La gravité des symptômes de la DMD n’a pas de lien avec la longueur de la mutation. Généralement, les mutations produisent une erreur de cadre de lecture lors de la traduction de l’ARNm. Le ribosome rencontrera un codon stop prématuré et la protéine ne sera pas produite.
Dans les cas où la mutation dans le gène de la dystrophine ne causerait pas de codon stop prématuré, une protéine tronquée est produite. Ces patients sont atteints de la dystrophie musculaire de Becker. La sévérité des symptômes des patients Becker dépend de l’ampleur de la délétion et du
site de la mutation. Si c'est dans un endroit de liaisons (domaine N-terminal ou riche en cystéines), la dystrophine ne pourra plus se lier à son complexe glycoprotéique associé (DGC) ou à l’actine, et un phénotype de DMD est obtenu [24]. Toutefois, si la protéine garde une certaine fonctionnalité, le phénotype de la maladie sera atténué. La dystrophie de Becker se caractérise donc par des symptômes beaucoup moins sévères que ceux de la DMD et la progression de la maladie est plus lente [16, 24].
1.2.4. Dystrophine
1.2.4.1. Isoformes
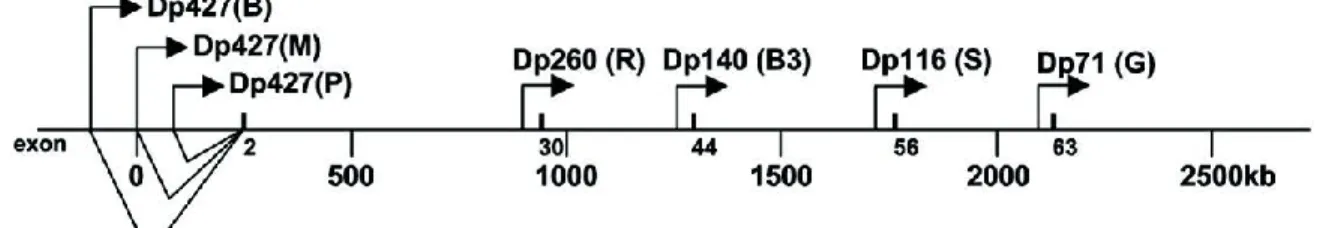
Il existe plusieurs isoformes de la dystrophine (Figure 7). La transcription du gène de la dystrophine pleine longueur (Dp427) est sous le contrôle de trois promoteurs principaux indépendants, selon le tissu :
1) Cerveau (B); 2) Muscle (M); 3) Purkinje (P).
Il existe aussi quatre isoformes courtes, régulées par quatre promoteurs internes qui sont eux aussi tissus spécifiques :
1) Rétinienne (R) = Dp260; 2) Cerveau 3 (B3) = Dp140;
3) Cellules de Schwann (S) = Dp116; 4) Général (G) = Dp71.
Figure 7. Isoformes de la dystrophine.
Il existe sept isoformes de la dystrophine, soit trois longues et quatre courtes. Leur transcription dépend des tissus. Figure adaptée de Blake et al., 2002 [25].
Les fonctions des isoformes sont toujours mal connues, mais l'absence de l'isoforme neurale expliquerait potentiellement le retard mental parfois observé chez le tiers des patients DMD [16, 25].
1.2.4.2. Dp427 musculaire
L’isoforme musculaire de la dystrophine (Dp427(M)) a un poids moléculaire de 427 kiloDaltons (kDa), et est constituée de 3685 acides aminés [16, 26]. Sa séquence est hautement conservée chez l’homme et la souris, et est riche en hélice-alpha, impliquant un rôle structurel. Elle est présente dans tous les tissus musculaires (squelettiques, cardiaques et lisses) à environ 0.002 % des protéines totales [27].
La dystrophine est composée de quatre domaines principaux. Son extrémité N-terminale contient une paire de modules CH, pour calponin homology, et sert de domaine de liaison à l'actine. Le deuxième domaine est composé de 24 répétitions en tandem de style triple hélice et quatre domaines charnières, ce qui lui confère sa flexibilité. Ce domaine a une certaine homologie avec les multiples répétitions de la spectrine. Le troisième domaine contient un module WW qui se lie à la bêta-dystroglycane, une protéine du complexe associé à la dystrophine. Ce module est suivi par une séquence riche en cystéines, permettant une liaison calcium-dépendante de la calmoduline. Le quatrième domaine, le C-terminal, ne présente pas d'homologie avec aucun gène connu et est spécifique à la dystrophine. Il s’agit d’un domaine superhélice alpha qui permet la liaison à la dystrobrévine [16, 28].
La dystrophine est située sur la surface cytoplasmique du sarcolemme et sert d’ancre pour le cytosquelette à la matrice extracellulaire via le DGC [29]. Elle assure ainsi l’intégrité du sarcolemme et la transduction de la force le long de la fibre lors des contractions musculaires. Elle empêcherait ainsi les micro-ruptures et les fuites d’ions. Chez le patient DMD, la nécrose cellulaire progressive serait due à des protéases calcium-dépendantes. La dystrophine étant absente, les micro-ruptures du sarcolemme créeraient des fuites d’ions qui augmenteraient la nécrose [26]. La dystrophine possède aussi un rôle important lors de l’échafaudage du DGC, mais les mécanismes moléculaires impliqués sont incertains.
L’absence de la dystrophine provoque l’instabilité du sarcolemme des fibres musculaires. Les contractions musculaires des patients dystrophiques provoquent donc des ruptures membranaires entrainant une succession de cycles de dégénérescence et régénérescence des myofibrilles. Ces cycles continus épuisent le réservoir de cellules satellites qui deviennent sénescentes à force de se diviser continuellement. Ceci entraîne la dégénération musculaire progressive caractéristique de la maladie.
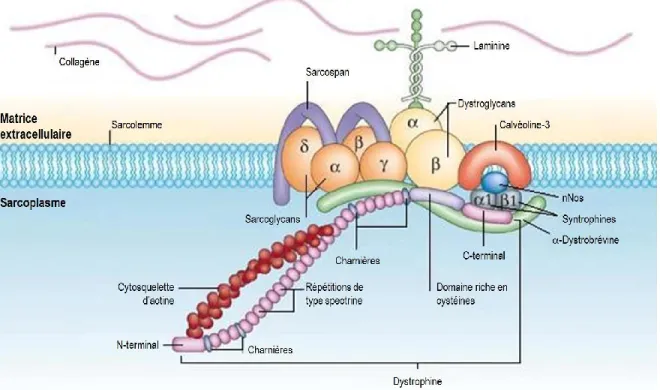
1.2.5. Le complexe de glycoprotéines associé à la dystrophine
Le complexe de glycoprotéines associé à la dystrophine (DGC) compte plusieurs éléments, dont la distribution varie selon la localisation ou le tissu : la laminine-2, les dystroglycans (α et β), les sarcoglycans (α, β, δ, ε, γ et δ ), la sarcospan, la dystrobrévine, les syntrophines (α1, β1, β2, γ1 et γ2), la neural nitric oxyde synthase (nNos), la microtubule associated serine/threonine kinase 205
Kd (MAST205), la syncoiline, et la calvéoline-3. Chez les vertébrés, au niveau musculaire, son rôle
est de maintenir la structure du sarcolemme et des jonctions neuromusculaires en liant le cytosquelette à la matrice extracellulaire [30]. De plus, il serait aussi impliqué dans la transmission synaptique et le regroupement de récepteurs de l’acétylcholine aux jonctions neuromusculaires. Il possède aussi un rôle lors de la signalisation, puisqu'il interagit avec des molécules de signalisation, comme nNos, les ions calcium, la calmoduline, la protéine kinase A, Grb2, Ras,
Parmi les éléments du DGC, des mutations dans les gènes codant pour les sarcoglycans, dystroglycans, dystrobrévines, la laminine et la calvéoline-3 sont connues pour être létales ou causant des myopathies. En particulier, nNos est importante pour la production d'oxyde nitrique, qui augmente la circulation sanguine localement selon les besoins lors d’un exercice physique. Chez les patients dystrophiques, nNos est délocalisé du sarcolemme et se retrouve au cytosol, mais conserve son activité enzymatique. Ainsi, il augmenterait la constriction des vaisseaux sanguins, ce qui causerait un stress ischémique [32], en plus d’augmenter l’activité de la nicotinamide adénine dinucléotide phosphate (NADPH) oxydase, l’inflammation et la dégradation de protéines, ainsi que de diminuer l’activation des cellules satellites [33]. En raison de cela, les muscles des patients dystrophiques seraient plus fragiles, subiraient un plus grand stress oxydatif et ischémique, et auraient une plus faible capacité régénérative.
Figure 8. Dystrophine et le complexe de glycoprotéines associé.
La dystrophine relie le cytosquelette d’actine par son extrémité N-terminale à la matrice extracellulaire via le complexe de glycoprotéines associé à la dystrophine (DGC). Image adaptée de Davies et al., 2006 [34].
1.3. Soins et traitements de la DMD
1.3.1. Diagnostique
Les premiers symptômes de la maladie sont visibles vers l’âge de 3 ans. Une prise de sang pour l’analyse du taux de créatine kinase sérique permettra de déterminer l’importance des bris
1.3.2. Traitements et thérapies
Il n’existe à ce jour aucun traitement curatif pour la DMD. Cependant, des traitements ou soins permettant de ralentir ou d’atténuer certains symptômes de la maladie sont disponibles, telles la physiothérapie, l'utilisation d'orthèses et la chirurgie pour réparer les scolioses dues à l'affaiblissement des muscles dorsaux. Rendu aux stades tardifs de la maladie, il est nécessaire d'avoir recours à la ventilation artificielle. Toutefois, plusieurs stratégies thérapeutiques sont envisageables et peuvent être classifiées comme suit : pharmacologique, génique et cellulaire [18].
1.3.2.1. Traitements pharmacologiques
Les traitements pharmacologiques de base ont généralement pour but de diminuer l’inflammation ou d’augmenter la prolifération des cellules musculaires. C’est le cas des corticostéroides, comme la Prednisone et le Déflazacort, qui peuvent ralentir la perte de fonction musculaire de 6 à 24 mois [35]. Une avenue thérapeutique possible consiste à surexprimer l’utrophine, un gène orthologue à la dystrophine, pouvant compenser fonctionnellement le manque de cette dernière. Chez une personne normale, l’utrophine est située aux jonctions neuromusculaires et myotendineuses. Par contre, chez le patient dystrophique et lors du développement embryonnaire, elle se localise au sarcolemme. Il a été démontré par Tinsley et al. qu’une version tronquée de l’utrophine pouvait réduire le phénotype DMD tout en restaurant la localisation d’une partie du DGC [36]. Toutefois, Li et al. ont établi que même la version pleine longueur de l’utrophine ne permet pas de recruter nNos au sarcolemme des fibres musculaires [37]. Ces études indiquent que la surexpression d'utrophine chez les patients DMD pourrait améliorer le phénotype de la maladie, mais ceux-ci souffriraient toujours d'une myopathie, puisque l’absence de nNos au sacrolemme cause un stress ischémique, comme mentionné précédemment [26, 37]. Une petite molécule qui s’annonçait prometteuse, la BMN195, aurait dû pouvoir augmenter le niveau d’utrophine, comme démontré dans les essais pré-cliniques. Cependant, les essais cliniques de phase I n’ont pas montré d’élévation de l’utrophine [38]. De son côté, la SMT C1100 aurait augmenté de deux fois le taux d’utrophine chez des patients DMD et une étude clinique de phase II est en cours.
Il existe aussi des approches visant à restaurer l’expression de la dystrophine. C’est le cas des molécules permettant d’ignorer un codon stop prématuré, telles la gentamicine et l’ataluren. La
gentamicine a obtenu des résultats variables lors des essais cliniques, allant de 0 à 15 % d’expression de dystrophine chez les patients. Aurino et al. ont démontré que l’ataluren, anciennement connu sous le nom de PTC124, avait des effets similaires à la gentamicine, sans les conséquences adverses. Lors de l’essai clinique de phase IIa, 61 % des patients ont démontré une augmentation d’expression de dystrophine [39, 40]. Cette molécule est présentement en essai clinique de phase III.
L'analyse des gènes de patients atteints de la dystrophie musculaire de Becker ont permis de conclure qu'une version tronquée de la protéine pouvait lui conférer une certaine fonctionnalité [41]. La technique du saut d'exon est basée sur cette connaissance. En effet, des oligonucléotides antisens (ONA) sont synthétisés de façon à être chimiquement protégés contre la dégradation enzymatique et être complémentaires à une séquence particulière. Ainsi, l'ONA se lie sur la séquence cible de l'ARN pré-messager, empêchant l'épissage d'un ou plusieurs exons. La protéine produite est tronquée, mais fonctionnelle. Les premiers essais cliniques faits avec deux types d’ONA, le 2′-O-méthyl-phosphorothioate (2′OMe) et le morpholino phosphorodiamidate oligonucléotide (PMO), ont donné des résultats variables, allant jusqu’à 55 % de fibres positives pour la dystrophine chez un patient [42]. L’essai clinique de phase III utilisant la technique du saut d'exon vient d'être arrêté subitement. En effet, l'étude clinique du Drisapersen faite par GlaxoSmithKline et Prosensa s'annonçait prometteuse, mais l'analyse des tests de marche de 6 minutes faite après 48 semaines de traitement ne démontre aucune amélioration significative entre les patients traités avec l'ONA et ceux ayant reçu le placebo [43].
D’autres méthodes sont basées sur la correction du gène ou de son expression, soit par l’utilisation de méganucléases, de zinc finger nucléases ou de systèmes de transposons [35]. Le principe est que le cadre de lecture peut être réparé en induisant des micro-délétions ou micro-insertions dans le gène de la dystrophine. Chapdelaine et al. ont démontré que les méganucléases pouvaient cibler une séquence spécifique, induire une micro-délétion et restaurer le cadre de lecture du gène in vitro et in vivo [44].
comme le saut d’exon, pourrait être bénéfique dans le cas d’une thérapie de la DMD. Des essais cliniques sont en cours [45].
1.3.2.2. Thérapie génique
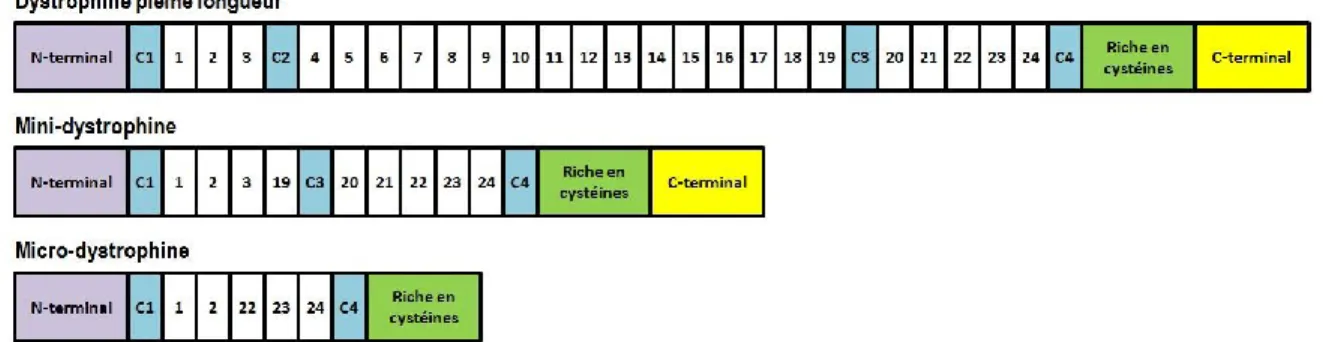
La thérapie génique consiste à introduire une copie fonctionnelle du gène de la dystrophine dans les fibres musculaires du patient. La très grande taille de la dystrophine est un facteur limitant lors du développement de thérapies. Le groupe de Chamberlain a donc créé des versions tronquées, mais toujours fonctionnelles, de la dystrophine afin de diminuer la taille de la séquence codante. Il a été démontré que ces versions, malgré des délétions de plus de 50 % de la séquence codante, possédaient toujours une fonctionnalité ressemblant à celle de la dystrophine pleine longueur [46]. En effet, les micro- et mini-dystrophines créées se localisent au sarcolemme et permettent l’échafaudage du DGC, à l’exception de nNOs. En effet, pour un échafaudage adéquat de nNos, les exons 42 à 45 de la dystrophine, codant pour les domaines homologues à la spectrine 16 et 17, sont requis [47]. Les versions tronquées ont l’avantage d’être plus faciles à insérer dans les cellules de par leur plus petite taille (Figure 9).
Figure 9. Versions de la dystrophine.
L’ADNc de la dystrophine pleine longueur fait près de 11 kb. La mini-dystrophine a subi une délétion allant de la deuxième charnière à la répétition de type spectrine 18 (ΔC2-R18) et a une taille d'environ 6 kb. La micro-dystrophine a une délétion allant de la deuxième charnière à la répétition de type spectrine 21 (ΔC2-R21), et le c-terminal est manquant.
La complication de la thérapie génique réside en la livraison difficile du gène d’intérêt, puisque les tissus musculaires sont les plus abondants du corps humain et composés de fibres qui ne se divisent pas, elles-mêmes entourées d'épaisses couches de tissus conjonctifs. Les principaux vecteurs utilisés sont les plasmides et les vecteurs viraux [48]. Les plasmides peuvent être injectés directement, administrés par pression hydrodynamique ou par électroporation. Toutefois, puisqu'une longue expression du transgène est désirée, il est préférable d'avoir recours à une méthode résultant en l'intégration du transgène dans le génome du patient. Pour ces raisons, les vecteurs viraux non intégratifs, comme les vecteurs adénoviraux ou dérivés de virus adéno-associés (VAA) sont à écarter. Les vecteurs lentiviraux sont ceux de prédilections, dû à leur capacité de transduire des cellules en division ou non, d'intégrer le génome de l’hôte ainsi que par la longue expression du transgène. De plus, il leur est possible de contenir les micro- et mini-dystrophines.
Le transfert de gènes utilisant les vecteurs viraux a causé un grand enthousiasme au début des années 1990, mais a connu plusieurs embardées depuis. En effet, quelques patients participants à des essais cliniques ont connu des fins tragiques suite aux traitements à l'aide de ces vecteurs. C'est le cas notamment de Jesse Gelsinger, en 1999, décédé 98 heures après avoir reçu une injection d'un vecteur adénoviral de type 5 qui a engendré une sévère réponse immunitaire [49, 50]. En 2002, un essai clinique utilisait un vecteur rétroviral afin de traiter le déficit immunitaire combiné sévère lié à l’X. Malgré que cela était considéré très peu probable chez l’humain, une mutagenèse d’insertion s’est produite, résultant en un patient souffrant de leucémie lymphocytaire aiguë, ce qui lui a été fatal [51]. En 2007, un autre patient, cette fois traité pour l'arthrite, est décédé suite au traitement à l'aide d’un VAA [52]. Ces décès démontrent les limitations des études faites chez l'animal afin de prédire les réactions humaines, la toxicité potentielle des vecteurs viraux non-réplicatifs, ainsi que la variabilité de la réponse d'un patient à l'autre.
Un certain succès pour le traitement de la DMD dans les modèles animaux à l’aide de vecteurs viraux a été obtenu. Toutefois, les équipes de recherche se sont heurtées à divers problèmes, comme la toxicité, le rejet immunitaire ou la courte durée d’expression du transgène. Un essai
1.3.2.3. Thérapie cellulaire
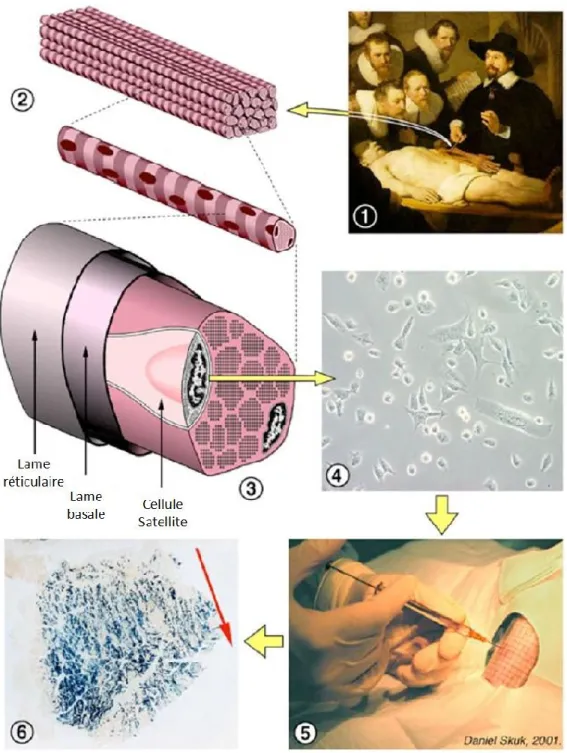
De son côté, la thérapie cellulaire consiste à greffer des cellules exprimant une dystrophine fonctionnelle, dans les muscles des patients dystrophiques. Ces cellules peuvent provenir d’un donneur sain (greffe allogénique) ou de cellules souches ou progénitrices, venant du patient lui-même (greffe autologue), corrigées préalablement ex vivo. La Figure 10 démontre le principe de la thérapie cellulaire, où les cellules saines ou corrigées génétiquement sont micro-injectées de façon intramusculaire afin de fusionner avec les cellules de l’hôte, et ainsi former des fibres hybrides. Le noyau de la cellule normale permet l’expression de la dystrophine sur quelques centaines de microns de la fibre hybride [54, 55].
Figure 10. Étapes d'une greffe de myoblastes allogéniques.
(1) Une biopsie de tissus musculaire d'un donneur sain est effectuée pour obtenir des fibres musculaires (2) qui seront digérées de façon enzymatique (3). Ces cellules, une fois mises en culture
Des essais cliniques utilisant la greffe de myoblastes ont été réalisés et ont eu des résultats variables. Les premiers essais cliniques utilisant cette méthode comportaient l'utilisation de la cyclosporine A comme traitement immunosuppresseur. Toutefois, il a été démontré que la dose utilisée était trop faible pour empêcher une réaction immunitaire [57]. Il a aussi été démontré par Huard et al. que des myoblastes allogéniques greffés à une souris immunosupprimée seulement avec la cyclosporine A ou un sérum anti-lymphocyte causaient une réaction immunitaire résultant en un rejet de la greffe [58]. Depuis, le tacrolimus est devenu le traitement de prédilection et les résultats de greffe se sont améliorés. Notre laboratoire a d’ailleurs réussi un essai clinique de Phase I chez neuf patients au cours duquel une greffe de myoblastes provenant d'un donneur sain immunocompatible a été effectuée. Jusqu'à 26 % des fibres musculaires de la zone traitée exprimaient la dystrophine [59].
Il existe toutefois plusieurs facteurs limitant la thérapie cellulaire, comme la mort rapide des cellules greffées, le faible taux de migration à travers le muscle de l‘hôte et le rejet immunitaire [35]. Notre laboratoire travaille activement à améliorer ces différents aspects. Suite à leur transplantation, les myoblastes ne migrent que vers les fibres musculaires qui sont en régénération, parce qu’ils sont attirés par des facteurs chémo-attractants. Ainsi, les myoblastes transplantés auront tendance à fusionner près des sites d'injection avec les fibres qui ont été endommagées par l’injection. Certains critères ont d’ailleurs été établis afin d'augmenter le succès des greffes, soit des injections à haute densité de cellules, bien distribuées dans tout le muscle, avec des trajectoires d'injection près les unes des autres, de façon homogène sur la longueur de chaque trajectoire [60]. De plus, des études démontrent que la co-injection de facteurs de croissance, comme l'IGF et le bFGF, peut favoriser la migration des myoblastes [61]. Il a été montré que près du tiers des myoblastes mourraient moins de 24 heures suivant la greffe. Les mécanismes exacts de ce processus sont inconnus, mais une réaction inflammatoire pourrait y jouer un rôle [62]. De plus, les myoblastes provenant d'un donneur sain expriment des antigènes qui causeront une réaction immunitaire chez le patient. Sans traitement immunosuppresseur, la greffe sera rejetée [56].
Mendell et al. ont démontré, lors d’une étude clinique, que des patients dystrophiques traités à l’aide d’un VAA codant pour une mini-dystrophine pouvaient développer des anticorps contre un épitope de la mini-dystrophine. De plus, ils ont découvert que certains patients dystrophiques possédaient déjà des anticorps contre la dystrophine, et ce, même avant traitement visant à rétablir son expression.
Des lymphocytes T auto-réactifs spécifiques à la dystrophine ont aussi été observés chez un patient dystrophique ayant été traité à la gentamycine pour augmenter la lecture des codons stops prématurés (résultats non-publiés) [63]. Ces résultats mettent l’emphase sur la nécessité de prendre en compte la potentielle réaction immunitaire du patient contre la dystrophine, et ce, peu importe la manière dont elle est réintégrée chez le patient [64].
Il n'en demeure pas moins que le principal obstacle à ce type de thérapie est la quantité considérable de cellules requises pour traiter un muscle complet. Ainsi, le traitement de tous les muscles squelettiques d'un patient n'est pas envisageable à ce moment.
1.4. Modèles animaux
Quelques modèles animaux sont disponibles pour expérimentation : murin, canin, et félin. Chaque modèle présente des avantages et des inconvénients. Les modèles les plus couramment utilisés seront vus en détail.
1.4.1. Modèles murins
Il existe quelques modèles murins pour la DMD, notamment la souris mdx, pour X-linked muscular
dystrophy, les mdx2cv, mdx3cv, mdx4cv et mdx5cv, ainsi que la mdx52 [65]. La souris mdx est un
modèle spontané découvert en 1984 et qui possède une mutation non-sens à l’exon 23, résultant en une absence de dystrophine. Il s’agit du modèle animal le plus couramment utilisé. Cette souris a un phénotype ressemblant à celui de la DMD chez l’humain, mais en moins sévère. En effet, on note l’infiltration de neutrophiles et de macrophages, ainsi que des cycles de nécrose et de régénération, amenant éventuellement une dégénérescence musculaire progressive, mais insuffisante pour provoquer le même phénotype que chez l'humain dystrophique. Les capacités contractiles des muscles de ces souris sont altérées, leur force est diminuée et une augmentation plasmatique de la créatine kinase est notable [65].
musculaire mutante est produite (Dp415), mais n’est pas fonctionnelle. Ces lignées présentent aussi des variations de niveau d’expression des isoformes musculaire, nerveuse et hépatique [65, 66]. La souche mdx52 a été créée par recombinaison homologue, en désactivant le gène DMD à l’exon 52, causant une absence de dystrophine. Elle présente le même phénotype que la souris mdx régulière, sauf qu’elle n’exprime pas l’isoforme rétinienne (Dp260), ce qui amène quelques anormalités à ce niveau [65].
1.4.2. Modèles canins
Des mutations dans le gène de la DMD ont été identifiées chez le Golden Retriever (GRMD), Rottweiler, le German Short-Hair Pointer, et le Cavalier King Charles Spaniels (CKCS-MD) [65]. La dystrophie musculaire du Golden Retriever a été la plus caractérisée. L’absence de la dystrophine est causée par un codon stop prématuré dû à une mutation ponctuelle au site d’épissage de l’intron 6, ce qui cause le saut de l’exon 7. Le phénotype observé chez le modèle GRMD est semblable à celui de l’humain DMD, présentant une perte de force musculaire progressive ainsi qu’une atteinte cardio-respiratoire. Toutefois, ce modèle n’est pas optimal puisque la mutation se trouve dans un site non fréquemment muté chez l’humain [67].
Le modèle CKCS-MD possède un phénotype dystrophique sévère, comme le GRMD, mais il a une mutation dans un site d’épissage de l’exon 50, résultant en sa délétion. Ceci présente un avantage du point de vue thérapeutique, puisque la majorité des mutations du gène humain de la DMD sont entre les exons 45 à 55. Le CKCS-MD représente donc le modèle idéal pour tester les méthodes thérapeutiques comme le saut d’exon, les zinc finger nucléases et les transposons [68].
1.4.3. Modèle félin
L’HFMD, ou Hypertrophy Feline Muscular Dystrophy, n’exprime pas la dystrophine. Toutefois, son phénotype est particulier : hypertrophie de certains muscles (langue, cou, épaules), salivation excessive, problème de marche causant des sautillements de lapin, cardiomyopathie, hépatosplénomégalie et insuffisance rénale. De larges délétions sont présentes dans le gène de la DMD au niveau des promoteurs musculaire et Purkinje. En raison de la faible similarité des phénotypes entre l’HFMD et l’humain DMD, ce modèle n’est que très rarement utilisé [69].
1.5. Cellules souches humaines
Les cellules souches humaines possèdent des caractéristiques particulières qui les rendent très intéressantes aux yeux de la médecine régénérative. Elles ont la capacité de s'auto-renouveler indéfiniment in vitro, en plus de pouvoir se différencier en cellules plus spécialisées, pouvant ainsi réparer ou remplacer les cellules endommagées par une maladie, une lésion ou le vieillissement.
1.5.1. Cellules souches embryonnaires
Les cellules souches embryonnaires humaines (hESCs) sont dites totipotentes, puisqu’elles ont la capacité de se différencier dans les trois feuillets embryonnaires, soit le mésoderme, l’endoderme et l’ectoderme. De plus, elles possèdent la capacité d’auto-renouvellement. Les lignées d'hESCs déjà établies peuvent servir pour les études de différenciation spécifiques et le développement d'avenues thérapeutiques à l'aide des cellules qui en sont dérivées. Leur utilisation thérapeutique présente plusieurs limitations, comme le rejet immunitaire et les problèmes éthiques.
1.5.1.1. Historique et législation
C'est à partir de l'étude du développement des embryons, dès les années 1860 en Allemagne, que s'est fait la déduction que des cellules souches existent. Toutefois, ce n'est qu'en 1961 que deux Canadiens, les docteurs James Till et Ernest McCulloch, prouvent leur existence expérimentalement et publient leur découverte en définissant les caractéristiques précises de ces cellules. Ces chercheurs sont considérés comme les pères des cellules souches, puisque leurs expériences ont ouvert la voie à la recherche actuelle sur les cellules souches embryonnaires et adultes. Les premières ESCs murines ont été extraites d'un blastocyste, en 1981, au Royaume-Uni et aux États-Unis. Le premier mammifère, une brebis, fut cloné en Écosse en 1996 [70]. C'est en 1998 que la première lignée d'hESCs a été établie aux États-Unis par Thomson et al., à partir d'un embryon préimplantatoire [71].
Depuis 2005, les études faites sur des embryons humains sont soumises à la loi sur la reproduction assistée (Bill C6), principalement dans le but d'empêcher toute forme de rémunération que les femmes pourraient en tirer, ainsi que les manipulations génétiques, le clonage, les hybrides, les chimères ou toutes lubies scientifiques [73].
1.5.1.2. Isolation et culture
L'isolation de hESCs se fait normalement à partir d'un embryon surnuméraire qui provient d'une clinique de fertilisation. Celui-ci est alors fertilisé pour obtenir un zygote qui sera mis en culture pour permettre sa division cellulaire. Après 5 jours, les divisions cellulaires ont formé une sphère d'environ 200 cellules, appelée le blastocyste. Dans cet agrégat se retrouve la masse cellulaire interne, formée d'environ 35 cellules pluripotentes. Cette masse est extraite manuellement avant d'être remise en culture pour obtenir une lignée de hESCs (Figure 11) [72].
Figure 11. Isolation des cellules souches embryonnaires et mise en culture.
Les techniques de culture des hESCs ont beaucoup évolué au fil des années. En premier lieu, des fibroblastes embryonnaires de souris (MEF), mitotiquement inactivés avec des rayons gamma ou la mitomycine-C, étaient requis pour la culture à l'aide d'un milieu riche en sérum. Le rôle des MEF est de favoriser l'adhésion des hESCs au pétri et de sécréter des protéines importantes pour le maintien de l'état de pluripotence et l'auto-renouvèlement [72, 75]. Cependant, recourir à des cellules murines amène le risque de xéno-contamination puisqu'elles sont reconnues comme potentiellement porteuses de divers pathogènes pouvant infecter l'humain, ce qui pourrait compromettre leur utilisation clinique ultérieure. Pour cette raison, des systèmes basés sur des cellules nourricières humaines ont été développés, notamment avec des fibroblastes humains adultes et fœtaux [76]. Toutefois, il a été remarqué que les cellules nourricières venant de souris n'expriment pas les mêmes taux de certains facteurs de croissance que celles provenant de l’humain, par exemple l'activine A et FGF-2. De plus, les différentes lignées de cellules nourricières humaines variaient entre elles [75]. Malgré les bénéfices apportés par les cellules nourricières humaines, il n'en reste pas moins qu'elles impliquent l'utilisation d'un milieu dont la composition est variable.
C'est ainsi que se sont développées différentes matrices, comme la laminine, la fibronectine ou le Matrigel, pouvant remplacer les cellules nourricières. Toutefois, un milieu supplémenté par une combinaison de différents facteurs de croissance (bFGF, TGF-β, Nodal, activine A, LIF, Noggin, et
platelet-derived growth factor (PDGF)) est requis afin de compenser le manque causé par l'absence
de cellules nourricières [77]. Parmi ces facteurs, la présence de bFGF est indispensable. Il s'est donc développé, en 2007, un milieu chimiquement défini, appelé le mTeSR [78]. L’utilisation du Matrigel, combinée au mTeSR, est maintenant la norme pour la culture d'hESCs. Leur avantage réside en la standardisation des techniques de culture ainsi que la réduction des variabilités du milieu et par la diminution de la charge de travail. En effet, l'utilisation de cellules nourricières était laborieuse, demandant d’être préparées la veille de la mise en culture des ESCs.
1.5.1.3. Caractéristiques et mécanisme de pluripotence
Les caractéristiques nécessaires à l'utilisation des hESCs en médecine régénérative correspondent à leur stade indifférencié. Une lignée d'hESCs doit être dérivée d'un embryon, capable d'être proliférée et de maintenir un état indifférencié pour une longue période de temps, en gardant un caryotype normal. Généralement, ces cellules conservent un caryotype normal sur une période de temps assez
étendue in vitro, mais il peut arriver que des conditions de stress rendent instables les chromosomes 12 et 17 [79, 80]. Il est donc important d’évaluer le caryotype fréquemment puisque ces cellules peuvent continuer d'exprimer des marqueurs de pluripotence tout en étant aneuploïdes [81]. La pluripotence est la capacité de développer les cellules des trois feuillets embryonnaires, c'est-à-dire l'endoderme, l'ectoderme, et le mésoderme. Elle peut être vérifiée par la formation de tératomes chez la souris immunodéficiente ayant reçu une greffe de ces cellules. Les tératomes sont un amas de cellules des trois feuillets embryonnaires. De plus, l'enzyme télomérase est fortement exprimée chez les hESCs. Celle-ci permet de répliquer l'extrémité des chromosomes, empêchant ainsi leur rétrécissement lors des divisions cellulaires [72].
Lorsque les hESCs sont mises en culture, elles poussent en colonies compactes ayant un contour rond et bien défini, et les cellules qui les forment possèdent un ratio noyau/cytoplasme élevé. Elles expriment différents marqueurs typiques de l'état embryonnaire, tels les antigènes de surface spécifiques au stade embryonnaire 3 et 4 (SSEA-3, SSEA-4), ainsi que TRA-1-60 et TRA-1-81. Elles sont aussi caractérisées par une forte expression d'alcaline phosphatase, une hydrolase de phosphate, ainsi que par des facteurs de transcription, comme Oct-4, Sox2, Nanog, Foxd3 et Rex1 [81].
Le mécanisme de pluripotence est un processus hautement régulé qui requiert l'implication de plusieurs gènes pour maintenir l'état indifférencié. Trois facteurs sont considérés comme les éléments clés régulateurs de la pluripotence, c'est-à-dire Oct4, Nanog et Sox2. Ces trois régulateurs partagent plusieurs gènes cibles et contrôlent la transcription de plusieurs gènes importants dans le processus de pluripotence, et ce, par des voies de signalisations distinctes ou non. Oct4 et Nanog sont abondamment exprimés dans les cellules pluripotentes et y sont spécifiques. L'absence de Oct4 entraîne la différenciation des hESCs en trophoblastes, alors que sa surexpression dirige les cellules vers la voie endodermique primitive et mésodermique [82]. Nanog inhibe la différenciation en cellules endodermiques primitives, alors que sa surexpression n'a pas de conséquences connues [83]. Sox2, pour sa part, est exprimé chez les cellules pluripotentes, les cellules germinales, ainsi que les
1.5.2. Cellules souches pluripotentes induites
Les caractéristiques des hESCs font en sorte qu'elles pourraient représenter un atout pour la médecine régénérative. Toutefois, leur provenance soulève des problèmes éthiques. De plus, il s'agit de cellules allogéniques, et leur utilisation clinique requerrait une immunosuppression du receveur. En 2006, Yamanaka et al. ont découvert les quatre facteurs de transcription nécessaires à la production de cellules souches pluripotentes induites (iPSCs) murines, c’est-à-dire Oct4, Klf4, Sox2 et c-Myc. Les cellules transduites à l'aide de rétrovirus codant pour ces quatre facteurs possèdent les caractéristiques des ESCs, c'est-à-dire la capacité d'auto-renouvellement et de différenciation dans les trois feuillets embryonnaires [86, 87]. En 2007, deux équipes de chercheurs ont réussi à dériver des iPSCs à partir de cellules somatiques humaines. Tout d'abord, le Dr. Yamanaka et son équipe ont poursuivi leur lancée en utilisant cette fois des lentivirus codant pour les quatre facteurs humains afin de transformer des fibroblastes en cellules souches humaines induites à la pluripotence (hiPSCs) [88]. L'autre équipe, celle du Dr. Thomson, a utilisé quatre lentivirus codant pour Oct4, Sox2, Nanog et Lin28. Malgré que les facteurs utilisés soient différents, les résultats obtenus étaient très similaires et permettaient l'obtention de cellules semblables aux hESCs [89]. Depuis, les recherches se poursuivent afin d'éliminer l'oncogène c-Myc ainsi que l'oncogène et suppresseur de tumeur Klf4, et éliminer l'utilisation de vecteurs viraux intégratifs, voir même remplacer ce type de vecteur totalement, par des plasmides, des protéines ou d'autres systèmes. Toutefois, l'élimination de c-Myc réduit l'efficacité de reprogrammation, déjà faible avec les quatre facteurs originaux. De plus, il faut déjà compter deux mois pour parvenir à une reprogrammation complète [89]. Des équipes travaillent donc à augmenter l'efficacité de reprogrammation à l'aide d'autres facteurs ou de petites molécules chimiques permettant de modifier la structure de la chromatine ou de moduler certaines voies de signalisation [90, 91]. Certaines recherches se concentrent sur des micro-ARN pour remplacer les facteurs ou améliorer l'efficacité de programmation [92]. La technique la plus courante de nos jours comprend l'utilisation du virus à ARN Sendaï, non intégratif, et efficace [93]. Plusieurs lignées d’hiPSCs ont été produites afin de modéliser diverses maladies pour en permettre l’étude. De plus, les hiPSCs représentent une avenue thérapeutique particulièrement intéressante puisqu’il est possible de créer des lignées de cellules spécifiques au patient, réduisant ainsi les risques de réactions immunitaires lors d’une greffe autologue. Il est donc nécessaire d’obtenir une
large quantité de cellules tissu-spécifiques, non tumorigéniques, capable de s’intégrer et de fonctionner chez l’hôte suite à une transplantation [94].
1.5.3. Myogenèse dirigée
Pour être utiles dans la médecine régénérative, les cellules souches doivent être différenciées spécifiquement au moment désiré. Les trois méthodes les plus couramment utilisées sont la formation de corps embryoïdes, l'utilisation d'un cocktail de facteurs de croissance spécifiques ainsi que la surexpression ou l’inhibition de gènes.
Plusieurs équipes ont tenté de mettre au point un protocole de différenciation myogénique efficace, permettant d'obtenir des myoblastes à partir d'hiPSCs, qui possèderaient les mêmes capacités de prolifération et de fusion que les myoblastes provenant d'une culture primaire [95, 96]. Toutefois, ces techniques, parfois complexes, ont connu des taux de succès variables in vitro, ou étaient peu reproductibles. Les cellules dérivées de ces protocoles ressemblent à des myoblastes, mais l'analyse de l'expression des gènes et l'observation de leurs capacités myogéniques révèlent qu'elles ne sont pas tout à fait de vrais myoblastes.
Darabi et al. ont démontré que l’expression inductible de Pax7 dans des iPSCs murines lors de la formation de corps embryoïdes résulte en des cellules pouvant être purifiées par l’expression du récepteur alpha du PDGF, un marqueur du mésoderme para-axial. La transplantation des cellules myogéniques précurseurs ainsi obtenues chez la souris dystrophique a résulté en un succès de greffe élevé et une augmentation de la contractilité des muscles [97].
Récemment, le groupe de Tanaka et al. a montré que l'expression inductible de MyoD dans des hiPSCs spécifiait les cellules dans la voie myogénique avec une efficacité de 70-90 %. L'analyse de l'expression des gènes des myoblastes résultants démontre une grande ressemblance avec des myoblastes humains venant d'une culture primaire. Toutefois, l'expression de Myf5 est manquante dans ces cellules, impliquant que le protocole utilisé emprunte une voie de différenciation différente