THESE
THESE
En vue de l'obtention du
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Physiopathologie
JURY
Dr. Jocelyn CERALINE Rapporteur Dr. Alexandre DE LA TAILLE Rapporteur Dr. Jacques PORTOUKALIAN Rapporteur Pr. Jean François ARNAL Examinateur Dr. Olivier CUVILLIER Directeur de thèse Pr. Bernard MALAVAUD Directeur de thèse
Ecole doctorale : Biologie, Santé et Biotechnologies
Unité de recherche : Institut de Pharmacologie et de Biologie Structurale - CNRS UMR 5089 Directeur(s) de Thèse : Dr. Olivier CUVILLIER et Pr. Bernard MALAVAUD
Rapporteurs : (s'ils ne font pas partie des membres du jury)
Présentée et soutenue par Audrey DAYON Le 22 Juillet 2008
Titre : RÔLE DE LA SPHINGOSINE KINASE-1 DANS LA SURVIE ET LA PROGRESSION DES CELLULES TUMORALES PROSTATIQUES LNCaP VERS L'ANDROGÉNO-INDÉPENDANCE
Ce travail de thèse a fait l’objet de plusieurs communications écrites et orales :
Manuscrit soumis pour publication :
Dayon A, Pirot N, Doumerc N, Mazerolles C, Nogueira L, Golzio M, Teissié J, Serre G, Rischmann P, Malavaud B, Cuvillier O
‘Sphingosine kinase-1 is a regulator of androgen-dependent prostate cancer cell growth and survival’
Cancer Res.
Présentations par poster :
Dayon A, Pirot N, Doumerc N, Mazerolles C, Noguera L, Golzio M, Teissié J, Serre G, Rischmann P, Malavaud B, Cuvillier O
‘Role of sphingosine kinase-1 in survival and progression of LNCaP to androgen refractory state’
7th Young Scientist Forum, 32th FEBS Congress, Vienne, Autriche, Juillet 2007
Dayon A, Pirot N, Doumerc N, Mazerolles C, Nogueira L, Golzio M, Teissie J, Serre G, Rischmann P, Malavaud B, Cuvillier O
‘Role for sphingosine kinase-1 in survival and progression of LNCaP prostate cancer cells to androgen independence’
23rd Annual Congress European Association of Urology, Milan, Italie, Mars 2008 Eur. Urology Supplements, 7 (2008) 312 - Meeting Abstract: 966
Communications orales :
‘Sphingosine kinase et androgènes’
14ème Journée de l’Association pour la Recherche sur les Tumeurs de la Prostate, Paris, France, Décembre 2005
‘Role of sphingosine kinase-1 in survival and progression of LNCaP to androgen refractory state’
Résumé
Le cancer de la prostate est le cancer le plus fréquent chez l'homme de plus de 50 ans et la deuxième cause de décès par cancer. Comme les cellules tumorales prostatiques ont besoin d'hormones mâles - les androgènes - afin de proliférer et de survivre, la privation androgénique est un traitement de référence dans la prise en charge du cancer de la prostate. Malheureusement, la réponse au traitement hormonal n'est que transitoire et la majorité des patients progressent vers un statut androgéno-résistant conduisant à la mort. Les mécanismes moléculaires de cet échappement sont encore mal connus. Au laboratoire, nous avons montré que la sphingosine kinase-1 (SphK1) est surexprimée dans le tissu tumoral prostatique par rapport au tissu sain adjacent, et que son activité enzymatique augmente avec l'agressivité tumorale. La SphK1 - de nature oncogénique - est responsable de la production de sphingosine 1-phosphate, un sphingolipide qualifié de promoteur de tumeur car impliqué dans la prolifération cellulaire, l'angiogenèse et la résistance à l'apoptose. Dans notre travail de thèse, nous avons exploré le rôle potentiel de la SphK1 dans la régulation androgéno-dépendante de la prolifération et la survie des cellules tumorales prostatiques.
Dans un premier temps, nous montrons que la privation en androgènes, dans la lignée prostatique hormono-dépendante LNCaP, entraîne une inhibition rapide et transitoire de l'activité SphK1 qui est corrélée à une forte inhibition de la prolifération cellulaire. Ces données ont été confirmées in vivo, sur un modèle de xénogreffe orthotopique de cellules LNCaP/GFP sur souris SCID, puisque la perte du volume tumoral induite par la castration est associée à une profonde inhibition de l'activité SphK1 dans le tissu prostatique. Nous démontrons que cette perte des capacités prolifératives peut être surmonté par la surexpression du gène codant pour la SphK1. L'addition de dihydrotestostérone (DHT) stimule l'activité SphK1 et permet de ré-induire la prolifération cellulaire vraisemblablement via un mécanisme PI3Kinase-dépendant. Par ailleurs, l'inhibition pharmacologique de la SphK1 inhibe de façon très significative les effets prolifératifs de la DHT.
Dans un deuxième temps, nous avons démontré l'implication de la voie SphK1/S1P dans la progression des cellules LNCaP vers le statut androgéno-résistant. Lors de la privation prolongée en androgènes nous observons une augmentation progressive de l'activité et de l'expression protéique de la SphK1 associées à la transdifférenciation neuroendocrine des cellules qui est caractérisée par un changement de morphologie et l'augmentation de la sécrétion du NSE. Ces phénomènes sont de type adaptatif puisque la réintroduction des androgènes induit une complète réversion du phénomène.
Ces travaux impliquant la SphK1 dans la transition vers l'hormono-indépendance suggèrent que l’inhibition pharmacologique de la SphK1 pourrait représenter une stratégie viable pour prévenir ou retarder la progression vers l’androgéno-indépendance.
Abstract
Prostate cancer is the most frequently diagnosed cancers in men and is the second leading cause of death from cancer. As prostate cancer cell proliferation is regulated and stimulated by androgens, strategies aimed at reducing the production of androgens and/or effects are the standard of care in the management of patients with recurrent or advanced disease. Notwithstanding that androgen deprivation therapy (ADT) is initially effective, ultimately all patients invariably become resistant to hormonal manipulation. It is not clear how prostate cancer cells make the transition from being dependent to being androgen-independent after hormone ablation therapy. We have shown in the Lab that sphingosine kinase-1 (SphK1) enzymatic activity and expression are markedly increased in tumor samples from prostate cancer patients (as compared with normal counterparts) correlating with other markers such as PSA level, tumor grade as well as with the clinical outcome after prostatectomy. SphK1 is an oncogenic enzyme, which produces sphingosine 1-phosphate, a lipid mediator lipid mediator playing a major regulatory role in tumor cell growth, survival, invasion, and angiogenesis. We have explored the potential role of SphK1 in the regulation of androgen-dependent cell growth and survival in the hormono-sensitive LNCaP prostate cancer cell model. We provide the first evidence that androgen privation induces a differential effect on SphK1 activity in the hormono-sensitive LNCaP cell model.
Short-term androgen removal induced a rapid and transient SphK1 inhibition that was associated with a pronounced reduced cell growth. This effect was confirmed in an orthotopically LNCaP model established in SCID mice, where the loss of tumor volume after surgical castration correlated with a marked SphK1 inhibition in the prostatic tissues. Supporting the critical role of SphK1 inhibition in the rapid effect of androgen depletion, the overexpression of SphK1 could impaired the cell growth decrease. Similarly, the addition of dihydrotestosterone (DHT) to androgen deprived LNCaP cells re-established cell proliferation, throught a PI3K/Akt dependent stimulation of SphK1, and pharmacological inhibition of SphK1 could markedly impede the effects of DHT.
Conversely, long-term removal of androgen support in LNCaP cells resulted in a progressive increase in SphK1 expression and activity throughout the progression to androgen-independence state, which was characterized by the acquisition of a neuroendocrine (NE)-like cell phenotype. Fascinatingly, the reversability of the NE phenotype by exposure to normal medium was linked with a pronounced inhibition of SphK1 activity.
These results suggest that SphK1 activation upon chronic androgen privation may serve as a compensatory mechanism allowing prostate cancer cells to survive in androgen-depleted environment, giving support to its inhibition as a potential therapeutic strategy to delay/prevent the transition to androgen-independent prostate cancer.
Abréviations
AC Adénylate Cyclase
Acy-1 Aminoacylase-1
AMACR Alpha-Methylacyl-CoA Racemase AMPc Adénosine Monophosphate cyclique
ARA70 Androgen Receptor Associated coregulator 70 ARE Androgen Responsive Element
ARNm Acide Ribonucléique messager ATP Adénosine Triphosphate Bcl-2 B-Cell Lymphoma-2 BSA Bovine Serum Albumin
Caspase Cysteinyl aspartate-specific proteinase CDK Cyclin-Dependent Kinase
CDKi Cyclin-Dependent Kinase inhibitor CgA Chromogranine A
COX-2 Cyclooxygénase-2
CREB C-AMP Responsive Element Binding protein DHS Dihydrosphingosine
DHT Dihydrotestostérone
EDG Endothelial Differentiation Gene EGF Epidermal Growth Factor
EGFR Epidermal Growth Factor Receptor REβ Récepteur des œstrogènes β
ERK Extracellular signal-Regulated protein Kinase FGF Fibroblast Growth Factor
FAK Focal Adhesion Kinase HGF Hepatocyte Growth Factor HSP Heat-Shock Protein
IRM Imagerie par Résonance Magnétique IGF-1 Insulin Growth Factor-1
IL-1β Interleukine-1β IL-6 Interleukine-6 ISEL In Situ End Labeling JNK c-Jun Kinase
KO Knock-Out
LDL Low Density Lipoprotein
LHRH Luteinizing Hormone-Releasing Hormone LH Luteinizing Hormone
LPA Lysophosphatidic Acid
mAchR muscarinic Acetylcholine Receptor MAPK Mitogen-Activated Protein Kinase
MEK Mitogen-Activated Protein Kinase Kinase
MNAR Modulator of Nongenomic Activity of estrogen Receptor NAO Nonyl Acridine Orange
NB Northern Blot
NE Neuroendocrine
NES Nuclear Export Sequence NGF Nerve Growth Factor
NT Neurotensine
NLS Nuclear Localisation Sequence NSE Neuron-Specific Enolase PARP Poly(ADP-Ribose)Polymerase PCDH-PC Protocadherin-PC
PDGF Platelet-Derivated Growth Factor
PECAM-1 Platelet Endothelial Cell Adhesion Molecule-1 PI3K Phosphoinositide 3-Kinase
PKA Protéine Kinase A PKC Protéine Kinase C PLC Phospholipase C
PMSF Phenylmethylsulfonyl Fluoride PSA Prostate-Specific Antigen
PTHrP Parathyroid Hormone-related Protein
PTEN Phosphatase and Tensin homolog deleted on chromosome Ten PTP1B Protein Tyrosine Phosphatase 1B
RA Récepteur des Androgènes
RAm Récepteur des Androgènes membranaire
Rb Rétinoblastome
RCPG Récepteur Couplé aux Protéines G RPMI Roswell Park Memorial Institute SCID Severe Combined ImmunoDeficiency SDS Sodium Dodecyl Sulfate
SH3 Src Homology 3 domain
SHBG Sex-Hormone Binding Globulin
SHBGR Sex-Hormone Binding Globulin Receptor siRNA small interfering Ribonucleic acid
SKIP Sphingosine kinase Interacting Protein SphK Sphingosine Kinase
S1P Sphingosine 1-Phosphate T-BSA Testostérone-BSA
TGF-β Transforming Growth Factor-β TNF-α Tumor Necrosis Factor-α
TPA 12-O-Tetradecanoyl-Phorbol-13-Acetate t-PA tissue Plasminogen Activator
TRAF2 TNF Receptor Associated Factor-2
TRPM-2 Transient Receptor Potential cation channel M2
TUNEL Terminal deoxynucleotide transferase-mediated dUDP Nick End Labeling VEGF Vascular Endothelial Growth Factor
Sommaire
REVUE GÉNÉRALE 1
Chapitre I : Le Cancer de la prostate 1
1. La prostate 1
2. L'épidémiologie 2
3. L'anatomopathologie 4
a- la localisation 4
b- l'extension 4
c- le grade tumoral : la classification de Gleason 4
4. Le bilan d'extension : la classification TNM 5
5. Les traitements 6
6. L'hormonothérapie 7
a- la production des androgènes 7
b- les différents traitements hormonaux 8
i. castration cxhirurgicale 8
ii. castration chimique 8
Clin d'œil : le PSA 10
Chapitre II : De la cellule prostatique androgéno-dépendante au cancer 11 hormono-réfractaire
1. Les modes d'action des androgènes 11
a- la voie génomique classique 11
i. description du mécanisme 11
ii. régulation de la prolifération : cycle cellulaire et facteurs de 13 croissance
iii. régulation de la survie : inactivation de l'apoptose 14 b- les effets rapides des androgènes : la voie non classique 14 i. implication de récepteur des androgènes "classique" 15
ii. implication du récepteur de SHBG 18
2. Les effets de la privation androgénique 22
a- la prolifération et le cycle cellulaire 22
b- l'apoptose 22
c- l'angiogenèse et les vaisseaux sanguins 26
d- la différenciation neuroendocrine 26
3. Les mécanismes impliqués dans l'hormono-indépendance tumorale 27
a- la sélection clonale 27
b- l'activation du récepteur des androgènes 28
i. activation dépendante de la fixation du ligand 28 ii. activation indépendante de la fixation du ligand 30 c- l'inactivation de l'apoptose : implication de Bcl-2 33 d- la différenciation neuroendocrine des cellules d'adénocarcinome 34 prostatique
i. mise en évidence de la différenciation neuroendocrine 34 ii. comparaison des cellules NE versus NE-like 35 iii. mécanismes impliqués dans la différenciation neuroendocrine 36 iv. corrélation différenciation neuroendocrine et agressivité tumorale 37
Chapitre III : La voie sphingosine kinase-1/S1P dans le cancer 40
1. La sphingosine kinase 41
a- la mise en évidence : purification et clonage 42
b- les propriétés structurales 42
c- les propriétés biochimiques 44
d- la localisation 45
i. distribution tissulaire 45
ii. localisation cellulaire 45
e- la régulation 46
i. régulation par la translocation membranaire 48
ii. régulation par la protéine kinase C 49
iii. régulation par la phospholipase D 49
iv. régulation par les interactions protéine-protéine 50
v. régulation par le calcium 51
vi. régulation par la transcription/traduction 52
2. Les modes d'action de la voie SphK1/S1P 53 a- la voie intracellulaire : la S1P comme second messager 53 b- la voie extracellulaire : la S1P comme ligand pour des récepteurs
membranaires 53
3. La voie SphK1/S1P dans le cancer 58
a- l'implication dans la transformation, la tumorigenèse et la prolifération 58 b- l'implication dans la survie et la résistance aux traitements anti-tumoraux 59 c- l'implication dans l'angiogenèse et l'inflammation 60
Clin d'œil : les sphingolipides 63
PROBLÉMATIQUE 65
MATÉRIELS ET MÉTHODES 67
1. Le matériel biologique 67
a- les lignées et la culture cellulaire 67
b- les animaux 68
2. Les conditions expérimentales 68
a- l'ensemencement 68
b- la privation androgénique 69
c- la dihydrotestostérone 69
d- les inhibiteurs 69
3. Les protocoles expérimentaux 71
a- l'etude de la viabilité cellulaire : le test du MTT 71
b- la quantification des protéines 71
c- l'analyse des protéines par western-blot 71
d- la dosage de l'activité enzymatique de la SphK1 75
e- la quantification du PSA et du NSE 77
f- l'expérimentation in vivo 77
g- l'immunohistochimie 79
DISCUSSION ET PERSPECTIVES 111
RÉFÉRENCES BIBLIOGRAPHIQUES 115
Chapitre I : Le cancer de la prostate
1. La prostate
La prostate est une glande de l’appareil génital masculin qui a la forme et la taille d’une châtaigne et pèse 15 à 25 g à l’âge adulte. Située en avant du rectum, la prostate entoure le col de la vessie et la partie initiale de l’urètre, le canal qui permet l’évacuation des urines et du sperme (figure 1a). Elle comprend trois zones principales: la zone centrale, la zone de transition et la zone périphérique [McNeal JE., 1988a] (figure 1b).
La prostate est constituée d’un stroma fibromusculaire et d’un épithélium glandulaire composé de trois types cellulaires (figure 2) :
- les cellules sécrétrices qui bordent la lumière;
- les cellules basales qui sont situées entre la membrane basale et le pôle basal des cellules sécrétrices;
- les cellules neuroendocrines qui sont peu nombreuses et présentes dans tous les compartiments glandulaires de la prostate.
Le stroma est principalement constitué de fibroblastes, de cellules musculaires lisses, de cellules endothéliales et de cellules dendritiques. La prostate est séparée des structures adjacentes par une bande de collagène appelée capsule prostatique.
Figure 1 : l'appareil génital masculin (a) [adapté de National Cancer Institute] et la prostate (b)
[De Marzo AM., 2007].
Figure 2 : l'épithélium glandulaire prostatique
[adapté de Bok RA., 2002.].
Le développement et le fonctionnement de la prostate sont dépendants d’une hormone appelée testostérone, qui est le principal androgène circulant chez l’homme. La testostérone est essentielle au développement des organes génitaux masculins et à l’acquisition des caractères sexuels secondaires à la puberté. Cette hormone est principalement synthétisée et sécrétée par les testicules et en moindre proportion par les glandes surrénales (10%). La principale fonction de la prostate est de sécréter et de stocker le liquide séminal, l’un des constituants du sperme.
2. L'épidémiologie
Le premier cas de cancer de la prostate a été décrit en 1853 par J. Adams, un chirurgien du "London Hospital" [Adams J., 1853]. Ce cancer est aujourd’hui un problème de santé publique majeur : il s'agit du deuxième cancer masculin le plus fréquent dans le monde avec plus de 670000 nouveaux cas recensés en 2002 [statistiques Globocan 2002, http:/www-dep.iarc.fr]. En France, où l’incidence a atteint 29434 nouveaux cas en 2002 (tableau 1), le cancer de la prostate est le troisième cancer le plus fréquent après le cancer du sein et du côlon. Chez l’homme, c’est le cancer le plus fréquent et la deuxième cause de mortalité par cancer (tableau 2) [statistiques Globocan 2002].
L’incidence du cancer de la prostate varie en fonction du pays, de l’origine ethnique et de l’âge des individus :
- variations géographiques et ethniques : il existe une très grande variation d’incidence selon les pays et l’origine ethnique. L’incidence la plus élevée dans le monde est observée aux États-Unis et la plus faible est observée en Chine, au Japon et en Inde. Aux États-Unis, l’incidence est plus élevée chez les afro-américains que chez les américains caucasiens. De plus, il a été démontré que les asiatiques vivant aux États-Unis ont un risque de cancer de la prostate plus élevé que leurs homologues qui vivent en Asie mais moindre que les afro-américains ou les afro-américains caucasiens [Muir CS., 1991]. Ces variations suggèrent l’implication de facteurs génétiques et environnementaux.
Cancer Incidence Mortalité
Sein 41957 11643
Côlon-rectum 34947 17097
Prostate 29434 9789
Bronches / poumon 27551 26225
Cancer Incidence Mortalité
Prostate 29434 9789
Bronches / poumon 23044 21760
Côlon-rectum 19229 9078
Tableau 1 : incidence et mortalité par cancer en France dans les deux sexes [Globocan 2002].
Tableau 2 : incidence et mortalité par cancer en France chez l'homme [Globocan 2002].
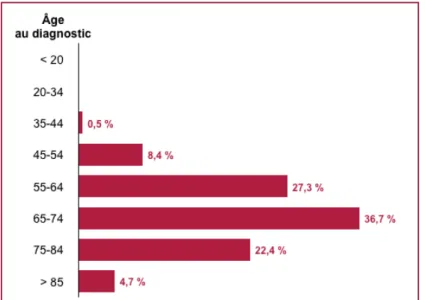
- variations avec l’âge : entre 2000 et 2004, l’âge médian du diagnostic du cancer de la prostate était de 68 ans. Ce cancer est rare avant 45 ans (0,5% des cas) et plus de 75% des nouveaux cas sont diagnostiqués après 55 ans [SEER "Surveillance Epidemiology and End Results" - National Cancer Institute] (figure 3).
L’âge et l’origine ethnique représentent des facteurs de risque pour le développement d’un cancer de la prostate. Un autre facteur de risque clairement identifié est l’existence d’antécédents familiaux de cancer de la prostate ou du sein.
- facteurs familiaux et héréditaires : les formes de cancer de la prostate dites "familiales" c’est-à-dire comportant au moins deux cas de cancer de la prostate dans la famille représentent environ 20% des cas [Valeri A., 1999]. Parmi ces formes familiales, certaines sont liées au hasard ou à l’exposition des membres de la famille à un carcinogène commun, alors que d’autres sont véritablement des formes héréditaires susceptibles d’être transmises au cours des générations successives. Ces dernières ne représentent que 3,6% des cas de cancer de la prostate. La transmission héréditaire peut se faire non seulement par un mode autosomique dominant mais aussi sur un mode lié au sexe via le chromosome X. Aujourd'hui, plusieurs loci pour des gènes de prédisposition aux formes héréditaires de cancer de la prostate ont pu être identifiés.
Par ailleurs, il a été montré que le risque de cancer de la prostate était 1,4 fois plus élevé chez les hommes apparentés au 1er et au 2e degré à une femme présentant un cancer du sein.
Figure 3 : l'incidence du nombre de cancers de la prostate en fonction de l'âge entre 2000 et 2004 aux Etats-Unis.
3. L'anatomopathologie
a- la localisation
La majorité des cancers de la prostate sont des adénocarcinomes c’est-à-dire des cancers de l’épithélium glandulaire et plus particulièrement des cellules sécrétrices. Dans 70% des cas, les cancers se développent dans la zone périphérique de la prostate. La zone centrale héberge 10% des cancers et 20% siègent dans la zone de transition [McNeal JE., 1988b]. Une des caractéristiques du cancer de la prostate est la multifocalité des lésions au sein d’une même glande.
b- l'extension
Après avoir franchi la capsule prostatique, la tumeur s’étend dans la graisse périprostatique puis vers les vésicules séminales par contiguïté. Les sites métastatiques les plus fréquents sont ganglionnaires et osseux. L’extension ganglionnaire est d’abord pelvienne puis rétropéritonéale. Les atteintes osseuses touchent préférentiellement le bassin, le rachis, les côtes et le sternum.
c- le grade tumoral : la classification de Gleason
La classification de Gleason est une classification anatomopathologique [Gleason DF., 1974] utilisée pour exprimer les résultats obtenus sur les biopsies prostatiques, les copeaux de résection endoscopique ou les pièces de prostatectomies. Le grade de Gleason comporte cinq sous-groupes notés de 1 (bien différencié) à 5 (indifférencié) selon l’architecture glandulaire de la tumeur (figure 4).
Étant donné que les tumeurs prostatiques ont en général une structure hétérogène, la différen- ciation est exprimée par le score de Gleason qui est la somme du grade des deux contingents tumoraux les plus représentés au sein de la tumeur étudiée. Le pronostic est d’autant plus défavorable que les tumeurs sont moins bien différenciées.
On distingue : - les tumeurs bien différenciées : score 2 à 4;
- les tumeurs moyennement différenciées : score 5 à 7; - les tumeurs peu ou pas différenciées : score 8 à 10.
4. Le bilan d’extension : la classification TNM
Le bilan d’extension permet d’évaluer le stade du cancer, le pronostic et de préciser les indications thérapeutiques. Plusieurs éléments permettent d’établir le bilan d’extension : - le toucher rectal qui apprécie l’extension locale;
- les examens d’imagerie qui évaluent l’extension locale et régionale par IRM (Imagerie par Résonance Magnétique) et les métastases à distance par scintigraphie osseuse;
- le grade histologique déterminé par le score de Gleason établi sur biopsies;
La tumeur est ensuite systématiquement "classée" à l’aide de la classification TNM (tableau 3). La lettre T, pour tumeur, précise son extension locale; la lettre N, pour node (adénopathie ou ganglion lymphatique) fait le point sur l’état des adénopathies régionales et la lettre M désigne les métastases (extension à distance).
T : Tumeur primitive
TX Tumeur primitive non évaluée
T0 Tumeur primitive non retrouvée
T1 Tumeur non palpable au toucher rectal et invisible en imagerie
T1a - découverte histologique : ≤ 5% du tissu réséqué et score de Gleason ≤ 7 T1b - découverte histologique : > 5% du tissu réséqué et/ou score de Gleason > 7 T1c - tumeur découverte sur une biopsie prostatique en raison d’un PSA élevé
T2 Tumeur palpable au toucher rectal, limitée à la prostate
T2a - atteinte de la moitié d’un lobe T2b - atteinte d’un seul lobe T2c - atteinte des deux lobes
T3 Tumeur dépassant la capsule prostatique
T3a - extension au-delà de la capsule
T3b - envahissement des vésicules séminales
T4 Tumeur envahissant les organes adjacents (col vésical, sphincter urétral,
rectum) ou tumeur fixée à la paroi pelvienne
N : Ganglions régionaux
NX Ganglions régionaux non évalués
N0 Absence de métastases ganglionnaires régionales
N1 Atteinte ganglionnaire régionale M : Métastases à distance
MX Métastases à distance non évaluées
M0 Absence de métastases à distance
M1 Métastases à distance M1a - ganglions non régionaux M1b - os
5. Les traitements
Le choix du traitement prend en compte le bilan d’extension (tableau 4) associé au taux de PSA (Prostate-Specific Antigen) dans le sang (Clin d'œil p.10) , le score de Gleason, l’âge, l’espérance de vie du patient et les effets secondaires des traitements.
- surveillance : ceci est justifié par le fait que si la tumeur évolue lentement et que le patient présente une espérance de vie limitée, celui-ci a plus de risque de décéder d’une autre cause que du cancer lui-même;
- prostatectomie radicale : il s’agit de l’ablation totale et en monobloc de la prostate et des vésicules séminales;
- radiothérapie : la radiothérapie est utilisée depuis les années 1950 pour le traitement du cancer de la prostate. Cette technique fait appel à des photons de haute énergie, un accélérateur linéaire et un repérage du volume cible et des organes critiques par un scanner préalable. La dose minimale est de 70 Gy en 35 fractions;
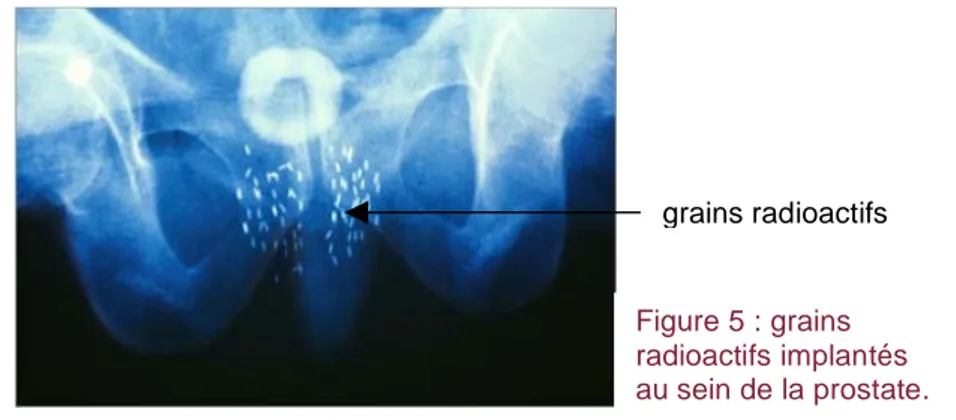
- curiethérapie : la curiethérapie consiste à implanter de façon définitive par voie périnéale et sous contrôle échographique des grains radioactifs directement au sein de la prostate ce qui permet une irradiation de la tumeur de l’intérieur (figure 5). Il s’agit en France le plus souvent d’iode (I125). Cette irradiation in situ permet d’administrer des doses supérieures à la radiothérapie externe;
Stade Recommandations thérapeutiques
Localisé T1-T2 N0-NX, M0 surveillance prostatectomie radicale radiothérapie curiethérapie Localement avancé T3-T4 N0-NX, M0 prostatectomie radicale radiothérapie + hormonothérapie hormonothérapie seule Métastatique T1-T4 N1, M0-M1 hormonothérapie
Échappement hormonal chimiothérapie
grains radioactifs
Figure 5 : grains radioactifs implantés au sein de la prostate.
- hormonothérapie : la privation androgénique reste le traitement le plus efficace des stades avancés de la maladie. Les différents traitements hormonaux utilisés seront détaillés ultérieurement;
- chimiothérapie : les travaux de Tannock et Petrylak ont montré l'intérêt des taxanes dans le traitement des formes de cancer de la prostate devenues réfractaires au traitement hormonal [Tannock IF., 2004; Petrylak DP., 2004].
6. L'hormonothérapie
Le traitement hormonal dans le cancer de la prostate est issu des travaux de Huggins publiés dans les années 1940, et honorés par le prix Nobel de Physiologie et Médecine en 1966. Huggins a mis en évidence, chez des patients atteints d’un cancer de la prostate métastatique, que l’élimination des androgènes par castration ou neutralisation par injection d’œstrogènes (Stilbestrol®) entraînait une diminution du taux de phosphatase acide qui était un marqueur sérique classique du cancer de la prostate [Huggins C., 1941]. Dans d'autres travaux [Huggins C., 1942], il démontre que la castration conduit à un réel bénéfice chez les patients avec notamment la régression des métastases.
a- la production des androgènes
Une cascade d'événements conduit à la synthèse et la sécrétion de la testostérone par les testicules. La première étape est la stimulation de la production de la LHRH (Luteinizing Hormone-Releasing Hormone) dans l'hypothalamus. L'interaction de la LHRH sécrétée avec des récepteurs se trouvant au niveau de l'hypophyse entraîne l'activation de la synthèse et la sécrétion de LH. Après fixation de la LH au niveau de ses récepteurs situés dans les cellules de Leydig des testicules, la testostérone est synthétisée à partir du cholestérol et libérée dans le sang (figure 6). 10% des androgènes sont également produits par les glandes surrénales. La testostérone circulant dans le sang pénètre dans les cellules prostatiques ce qui leur permet de proliférer et survivre. L'augmentation du niveau de testostérone peut entraîner une diminution de la production de LHRH et de LH via une boucle de rétrocontrôle négatif afin de maintenir la testostérone dans le sérum à des niveaux physiologiques.
b- les différents traitements hormonaux
Étant donné que les cellules prostatiques ont besoin d’androgènes pour proliférer et survivre, les différents traitements hormonaux visent à bloquer la synthèse ou l’action de l’hormone (figure 7).
i. castration chirurgicale
La castration chirurgicale peut être réalisée soit par exérèse des deux testicules (castration vraie ou orchidectomie) soit par exérèse de la pulpe testiculaire (pulpectomie). Cette intervention supprime de façon définitive et immédiate la source testiculaire des androgènes.
ii. castration chimique
Les traitements du cancer de la prostate par castration chimique ont été développés à la suite des travaux de Schally sur la LHRH menés au début des années 1970 et récompensés par le prix Nobel de Physiologie et Médecine en 1977.
- agonistes de la LHRH : il s’agit de décapeptides synthétiques présentant une structure analogue à celle de la LHRH. L’administration de ces agonistes entraîne une saturation des récepteurs de la LHRH au niveau de l’hypophyse. Après une hyperproduction initiale de testostérone appelée flare-up, cette stimulation permanente des récepteurs entraîne l’arrêt
de la sécrétion de la LH et secondairement, l’interruption de la production des androgènes d’origine testiculaire [Tolis, 1982];
- antagonistes de la LHRH : ils agissent en bloquant les récepteurs de la LHRH au niveau de l’hypophyse entraînant une castration rapide et réversible sans effet flare-up [Schally, 1983];
- anti-androgènes : ils bloquent la production de testostérone en inhibant de manière compétitive les récepteurs des androgènes au niveau hypothalamo-hypophysaire mais aussi au niveau des cellules prostatiques [Varenhorst E., 1982; Liao S., 1974];
- œstrogènes : ils agissent en inhibant la sécrétion de LH au niveau hypothalamo-hypophysaire et en inhibant le métabolisme de la testostérone dans les cellules prostatiques; - inhibiteurs de la 5α-réductase : ils bloquent la conversion de la testostérone en dihydrotestostérone (DHT) qui est une forme plus active de l'hormone [Kadohama N., 1984]; - blocage androgénique complet (BAC) : selon la théorie de Labrie, les cellules prostatiques seraient capables de s’adapter à un milieu pauvre en androgènes d’où l’idée de bloquer la production des androgènes d’origine surrénalienne. L’aminoglutéthimide [Sanford EJ., 1976] et le kétoconazole [Pont A., 1982] sont des composés qui suppriment la stéroïdogenèse dans les glandes surrénales. Actuellement, le BAC consiste à bloquer les sites de fixation des androgènes surrénaliens, via l'addition d'un analogue et d'un anti-androgène, au lieu de bloquer la synthèse de l'hormone.
L’efficacité du traitement hormonal n’est que transitoire jusqu’à la survenue inéluctable de l’androgéno-indépendance tumorale marquant l’entrée dans la phase terminale de la maladie.
Le PSA "Prostate-Specific Antigen" est une sérine protéase appartenant à la famille des kallikréines principalement sécrétée par les cellules épithéliales prostatiques. Dans les conditions physiologiques, cette protéine est présente en quantité importante dans le liquide séminal.
La fonction physiologique du PSA est de cliver des facteurs contenus dans le sperme pour faciliter sa liquéfaction ainsi que la fertilisation. Par ailleurs, le PSA pourrait être impliqué dans la carcinogenèse prostatique :
- le PSA clive la protéine IGFBP3 ce qui conduit à la libération d'IGF-1 qui est un facteur de croissance favorisant la prolifération des cellules prostatiques stromales et épithéliales;
- le PSA active le TGF-β qui peut stimuler le détachement cellulaire et faciliter la dissémination des cellules tumorales;
- le PSA clive des composants de la membrane basale ce qui facilite l'invasion des cellules tumorales et la formation de métastases.
Dans des conditions pathologiques telles que l'hypertrophie bénigne ou le cancer de la prostate une augmentation du taux de PSA dans de sang est observée. Il existe en effet une corrélation entre le taux sérique de PSA et le volume prostatique.
Les androgènes, connus pour induire la prolifération des cellules épithéliales prostatiques, stimulent l'expression du PSA dont le gène contient une séquence ARE "Androgen Responsive Element" dans son promoteur.
Depuis les années 1980, le PSA est utilisé comme un marqueur sérique pour le dépistage, le diagnostic et le suivi de l'évolution du cancer de la prostate.
Clin d'œil :
le PSALes fonctions du PSA [adapté de Bok
RA., 2002].
Abréviations : IGF-1, Insulin-like Growth Factor-1; IFGBP, IGF-Binding Protein; TGF-β, Transforming Growth Factor-β.
Chapitre II : De la cellule prostatique androgéno-dépendante au
cancer hormono-réfractaire.
1. Les modes d'action des androgènes
a- la voie génomique classique
i. description du mécanisme
La mise en évidence de la présence de DHT dans les cellules prostatiques et plus particulièrement dans les noyaux cellulaires date de la fin des années 1960. Il s'agissait d'administrer à des rats de la testostérone marquée au tritium, par intraveineuse, puis d'analyser la répartition de la radioactivité dans les cellules prostatiques par fractionnement subcellulaire [Bruchovsky N., 1968a; Bruchovsky N., 1968b].
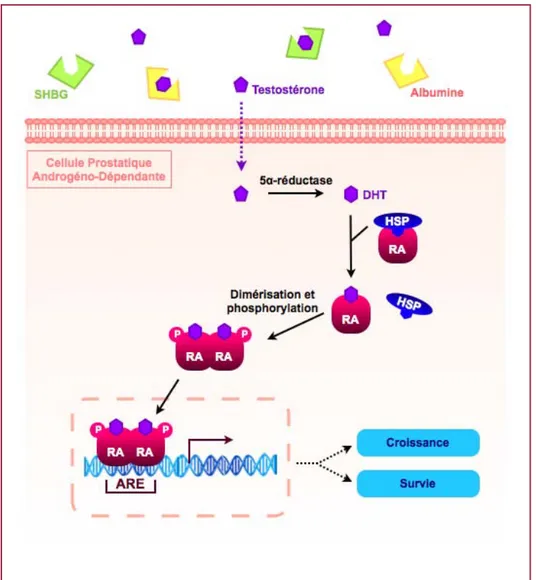
La testostérone circulant dans le sang est majoritairement liée à des protéines de transport telles que l'albumine ou la SHBG (Sex-Hormone Binding Globulin). La testostérone libre est capable de pénétrer dans les cellules prostatiques soit par diffusion passive à travers la membrane plasmique soit de manière assistée par un transporteur de protéines. Une fois dans le cytosol, la testostérone est convertie en DHT par une enzyme appelée 5α-réductase. Le récepteur des androgènes (RA) qui est un membre de la superfamille des récepteurs nucléaires [Mangelsdorf DJ., 1995] a simultanément été découvert par les équipes de Liao, Bruchovsky et Mainwaring [Anderson KM., 1968; Fang S., 1971; Mainwaring WI., 1969]. Comme tous les récepteurs nucléaires, le RA comprend :
- un domaine N-terminal impliqué dans l'activation de la transcription;
- un domaine central de liaison à l'ADN et qui participe également à la dimérisation du récepteur;
- un domaine charnière abritant une séquence de localisation nucléaire;
- un domaine C-terminal responsable de la liaison de l'hormone mais possédant aussi une surface d'interaction avec les protéines de choc thermique HSP (Heat-Shock Protein), une fonction de transactivation dépendante de la liaison du ligand, un site impliqué dans la dimérisation ainsi qu'une autre séquence de localisation nucléaire [Poujol N., 2000].
A l'état basal, le RA est lié à des protéines HSP (HSP 90, HSP 70, HSP 56) ainsi que d'autres protéines chaperonnes telle que FKBP52 (FK506-Binding Protein 52) [Tai PK., 1986] dans une conformation qui empêche l'interaction entre le récepteur et l'ADN [Veldscholte J., 1992]. La liaison de la DHT au niveau des RA présents dans le cytoplasme provoque un changement de conformation du récepteur et la dissociation des protéines qui lui sont associées [Kuil CW, 1995] ce qui démasque le site de liaison du ligand, les sites de
dimérisation ainsi que la séquence de localisation nucléaire [Wong C., 1993]. Il a été mis en évidence que la liaison des androgènes conduit à une hyperphosphorylation [Kuiper GG., 1995] et dimérisation du RA nécessaires pour la liaison du récepteur au niveau de l'ADN [Wong C., 1993]. Le complexe RA/DHT transloque dans le noyau en passant par les pores nucléaires et se lie au niveau de séquences spécifiques de l'ADN appelées ARE (Androgen Response Element) et situées dans les régions promotrices de gènes cibles. Cette interaction stimule ou inhibe la transcription de gènes impliqués dans la régulation de la prolifération et de la survie des cellules épithéliales prostatiques. Le recrutement de protéines co-régulatrices telle que ARA70 (AR-Associated protein 70) [Gao T., 1999; McKenna NJ., 1999] est nécessaire pour permettre l'interaction du RA avec l'appareillage de la transcription.
Les mécanismes impliqués dans la régulation de la prolifération et de la survie des cellules épithéliales prostatiques induite par les androgènes ne sont pas clairement identifiés.
ii. régulation de la prolifération : cycle cellulaire et facteurs de
croissance
Un des mécanismes responsables de la prolifération des cellules épithéliales prostatiques est la régulation par les androgènes des gènes codant pour des protéines contrôlant le cycle cellulaire. Il a été mis en évidence que les androgènes stimulent l'expression des cyclines et CDK (Cyclin-Dependent Kinase) permettant la progression en G1 et la transition G1/S du cycle cellulaire conduisant ainsi à une augmentation de la prolifération. Dans les cellules tumorales prostatiques LNCaP, l'addition de DHT stimule en quelques heures l'expression des cyclines A,B, D1 et des CDKs 1, 2 et 4 associé à une augmentation de l'activité kinase de CDK-2. Par ailleurs, l'addition de DHT entraîne une diminution de l'expression de p16 qui est une protéine inhibitrice des CDKs (CDKi). Ceci est également associé à l'augmentation de la phosphorylation de la protéine Rb (Ser780) qui est nécessaire pour effectuer la transition de la phase G1 vers la phase S [Lu S., 1997; Chen Y., 1998; Taneja SS., 2002]. La phosphorylation de Rb précède l'accumulation de la cycline A dans les cellules [Fribourg AF., 2000].
Cifuentes et al. ont démontré, dans le même modèle cellulaire, que le RA est nécessaire pour réaliser la transition G1/S [Cifuentes E., 2003]. En effet, le traitement des cellules avec un anti-androgène (bicalutamide) conduit à une diminution de l'expression de gènes régulant le cycle cellulaire et empêche les cellules d'entrer en phase S [Bai VU., 2005].
Dans la prostate de rat, les androgènes régulent également l'expression des cyclines de la phase G1, leurs CDKs et inhibiteurs : dans des rats castrés, l'augmentation de la prolifération des cellules prostatiques après injection de testostérone est associée à une augmentation de l'expression des messagers de la cycline C, cyclines D1, D2, D3, cycline E, cycline A et CDKs 2, 4 et 6, ceci étant associé à une augmentation du nombre de cellules en phase S [Furuya Y., 1995; Chen Y., 1996]. L'augmentation de la prolifération est maximale à 48h puis diminue grâce à la stimulation de l'expression de p21 et p27 qui sont des CDKi. La coopération entre ces régulateurs du cycle cellulaire conduit à une régénération de la prostate très bien contrôlée.
Les facteurs de croissance dont IGF-1 (Insulin Growth Factor-1), EGF (Epidermal Growth factor) et TGF-β (Tumor Growth Factor-β) sont produits par les cellules stromales prostatiques en réponse aux androgènes et agissent de manière paracrine en stimulant les cellules épithéliales prostatiques. Les androgènes favorisent l'action de ces facteurs en augmentant le nombre de récepteurs à la surface des cellules épithéliales. Il a été mis en évidence dans les cellules LNCaP et PC3-RA (exprimant de manière stable le RA) que l'addition d'androgènes stimule l'expression du récepteur de EGF (EGFR) à la surface des cellules et augmente la liaison EGF/EGFR [Schuurmans AL., 1988; Brass AL., 1995]. A l'inverse, les androgènes diminuent l'expression du récepteur de TGF-β, un facteur de
croissance négatif, qui agit de manière paracrine en inhibant la prolifération et induisant l'apoptose des cellules épithéliales prostatiques [Wikstrom, 1999; Zhu B., 2005].
Il a également été démontré dans des cellules LNCaP ainsi que dans des cellules prostatiques immortalisées PNT1 que les androgènes stimulent la synthèse et la sécrétion du facteur de croissance VEGF (Vascular Endothelial Growth Factor) [Joseph I., 1997b; Sordello S.,1998].
iii. régulation de la survie : inactivation de l'apoptose
Les androgènes protègent les cellules de cancer de la prostate de l'apoptose induite par divers stimuli en bloquant l'activation des caspases dans les voies intrinsèques et extrinsèques de la mort cellulaire programmée.
Berchem a montré que les androgènes bloquent l'apoptose induite par l'addition d'étoposide +/- irradiations [Berchem GJ., 1995] ou de TNF-α (ou anti-Fas) +/- irradiations [Kimura K., 2001]. Des études menées par Kimura et Coffey ont permis de mettre en évidence une inhibition des caspases 8, 7, 2, 9, 3 et Rokhlin a montré que le promoteur du gène codant pour la caspase 2 contient deux séquences ARE [Rokhlin CW., 2005]. Par ailleurs, l'inactivation des caspases est associée à une diminution de (1) l'expression des membres pro-apoptotiques de la famille Bcl-2 comme Bik, Bak et Bax, (2) clivage de Bid, (3) la dépolarisation mitochondriale et (4) la libération du cytochrome c [Kimura K., 2001; Coffey RNT., 2002].
A l'inverse, les androgènes régulent positivement l'expression de gènes codant pour des protéines anti-apoptotiques. Dans les cellules LNCaP, les androgènes entraînent une augmentation de l'expression des protéines anti-apoptotiques Bcl-2 et Bcl-xL[Berchem GJ.,
1995; Coffey RNT., 2002].
b- les effets rapides des androgènes : la voie non classique
Dans les années 1950, Hans Selye a découvert que les hormones stéroïdes pouvaient induire des effets seulement quelques minutes après leur application. Ce phénomène n'est pas conforme au mode d'action classique des hormones qui peut prendre plusieurs heures entre la pénétration de l'hormone dans la cellule et l'accumulation de protéines nouvellement synthétisées. Il s'agit d'un mode d'action non-génomique des hormones stéroïdes. Il existe plusieurs critères pour définir ce mode d'action :
- action rapide, effets observés entre quelques secondes et quelques minutes : preuve la plus évidente d'une action non génomique;
- insensibilité à l'action d'inhibiteurs de la transcription comme l'actinomicyne D ou d'inhibiteurs de la traduction tel que le cycloheximide;
- la transmission d'un effet rapide via un récepteur membranaire différent de RA doit être mise en évidence par l'utilisation d'analogues aux stéroïdes non-perméants ou par démonstration de la liaison du stéroïde à la surface cellulaire ou sur membranes isolées [Lösel R., 2003].
Les effets non-génomiques de la testostérone ont été mis en évidence au début des années 1980 notamment grâce aux travaux que Koenig. Il a montré que des concentrations physiologiques de testostérone (1-10 nM) induisaient une stimulation rapide (< 1min) de l'endocytose, du transport des acides aminés et hexose dans le cortex rénal de la souris [Koenig H., 1982].
L'action non-génomique des androgènes est impliquée dans de nombreuses fonctions physiologiques comme la reproduction, le système cardiovasculaire, le système immunitaire ou au niveau des muscles squelettiques [Rahman F., 2007]. Dans les cellules prostatiques, différents modes d'action rapide des androgènes ont été décrits.
i. implication du récepteur des androgènes "classique"
De nombreuses études impliquent le RA dans un mode d'action rapide des androgènes. Steinsapir et al. ont montré que le traitement à la DHT de cellules LNCaP entraîne une augmentation rapide et dose-dépendante du calcium intracellulaire (figure 9). Cette mobilisation est bloquée par l'addition de vérapamil ce qui indique qu'il s'agit d'un influx calcique passant à travers les canaux membranaires dépendants du voltage de type-L [Steinsapir J., 1991; Sun YH., 2006].
Figure 9 : l'effet de la DHT sur la concentration intracellulaire en Ca2+ dans les cellules LNCaP pré-incubées avec la sonde calcique Fura-2/AM [Sun YH., 2006].
[Ca2+]i élevée
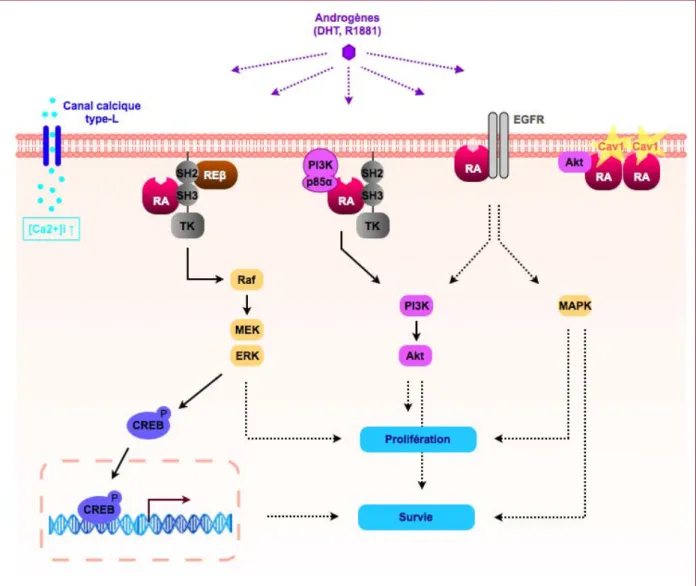
Sous l'action des androgènes, le RA interagit avec de nombreux partenaires entraînant l'activation de voies de signalisations impliquées dans la prolifération et la survie des cellules épithéliales prostatiques (figure 10) :
- la voie MAPK (Mitogen Activated Protein Kinase)
Les travaux de Migliaccio ont montré que le traitement de cellules LNCaP avec un androgène synthétique appelé R1881 conduisait à la formation d'un complexe entre le RA, la protéine tyrosine kinase Src et le récepteur des œstrogènes de type bêta (REβ). Src occupe une position centrale dans la formation de ce complexe puisque son domaine SH2 (Src Homology 2 domain) interagit avec la tyrosine 537 phosphorylée de RE et que son domaine SH3 (Src Homology 3 domain) se lie avec un domaine riche en proline du RA [Migliaccio A., 2000]. La formation d'un autre complexe a été mise en évidence par Unni E. et al. après stimulation des cellules LNCaP avec de la DHT. Il s'agit de l'interaction entre le RA, Src et la protéine MNAR (Modulator of Nongenomic Activity of estrogen Receptor) [Unni E., 2004]. La formation de ces complexes conduit à la stimulation de l'activité kinase de Src suivi de l'activation de Raf-1 et de la phosphorylation consécutive des protéines de la voie des MAPK : MEK1/2 (Mitogen-activeted protein kinase kinase) et ERK1/2 (Extracellular signal-Regulated protein Kinase 1/2). Il a ainsi été montré que l'activation de ERK1/2 entraîne la stimulation de la prolifération des cellules LNCaP [Migliaccio A., 2000; Unni E., 2004]. D'autre part, la phosphorylation de ERK1/2 conduit à l'activation du facteur de transcription CREB (c-AMP Response Element Binding protein) qui est impliqué dans l'induction d'effets anti-apoptotiques [Unni E., 2004].
- la voie PI3K/Akt (PhosphoInositide 3-kinase/Akt)
Dans les cellules LNCaP, les androgènes activent la voie PI3K/Akt via la formation d'un complexe entre le RA, Src et la PI3K. Il a été mis en évidence que le domaine N-terminal du RA interagit avec le domaine SH2 C-terminal de p85α qui est la sous-unité régulatrice de la PI3K. L'affinité de cette interaction est augmentée par une stimulation androgénique. L'activation de la voie PI3K/Akt ne fait pas intervenir une interaction directe entre RA et son ligand. En effet, RA présentant une délétion de la région C-terminale incluant le domaine de liaison du ligand est malgré tout toujours capable d'activer la voie PI3K/Akt après une stimulation androgénique.
Il a été démontré que EGFR joue un rôle en amont de PI3K/Akt car l'addition d'un inhibiteur de ce récepteur (AG1478) entraîne une diminution de l'activation de cette voie de signalisation [Sun M., 2003]. De plus, il a été montré qu'un pool de RA relocalise au niveau de la membrane plasmique et interagit avec EGFR après stimulation avec R1881 [Bonaccorsi L., 2004]. Des expériences réalisées par Sun ont démontré que que la voie
PI3K/Akt est impliquée dans la croissance cellulaire ainsi que dans la survie des cellules tumorales prostatiques induites par les androgènes [Sun M., 2003].
- la localisation membranaire
Le recrutement du RA au niveau de la membrane plasmique lui permet d'être au contact de ses partenaires.
L'utilisation d'un anticorps dirigé contre le RA a permis de mettre en évidence, dans les cellules PC3-RA [Bonaccorsi L., 2004] et LNCaP [Pedram AN., 2007], que le récepteur relocalise au niveau de la membrane plasmique après stimulation androgénique. L'équipe de Pedram a identifié un motif très conservé de neuf acides aminés dans le domaine de liaison du ligand du RA murin et humain qui est essentiel à la localisation membranaire du récepteur. Des mutations dans cette séquence induisent une diminution significative de la localisation membranaire du RA, de l'activation de MAPK et de la PI3K ainsi que de la croissance et de la viabilité stimulées par les androgènes [Pedram AN., 2007]. Par ailleurs, il a été mis en évidence que le RA interagit avec la cavéoline-1 de manière androgéno-indépendante au niveau de la membrane plasmique. En effet, la région N-terminale de la cavéoline-1 interagit avec le domaine N-terminal ainsi que le domaine de liaison du ligand du RA [Lu ML., 2001]. La cavéoline-1 est une protéine membranaire qui est présente de manière abondante dans les cavéoles, ces structures étant des invaginations de la membrane plasmique formées à partir de radeaux lipidiques par polymérisation des cavéolines [Simons K., 2000]. Une sous population de RA a été mise en évidence au niveau de ces radeaux dans les cellules LNCaP [Cinar B., 2007]. Ces cellules ne contiennent pas de cavéoline cependant elles possèdent des radeaux lipidiques qui sont impliqués dans le survie cellulaire [Zhuang L., 2002]. Dans une étude récente, Cinar a montré la formation androgéno-dépendante d'un complexe RA/Akt-1 au niveau des radeaux lipidiques. L'interaction entre ces deux protéines entraînerait l'augmentation, d'une manière indépendante de PI3K, de l'activité d'Akt-1 après 10 minutes de traitement [Cinar B., 2007].
ii. implication du récepteur de SHBG
SHBG est une glycoprotéine plasmatique qui lie certains androgènes et œstrogènes et régule la concentration en stéroïdes libres dans le sang. Le récepteur de SHBG (SHBGR) est un récepteur couplé aux protéines G [Nakhla AM., 1999] qui a été solubilisé et caractérisé dans la prostate humaine [Hryb DJ., 1989] et également mis en évidence à la surface des cellules tumorales prostatiques LNCaP [Nakhla AM., 1990] et ALVA-41 [Nakhla AM., 1996]. Nakhla a montré que l'addition de DHT, en présence de SHBG, entraînait une augmentation dose-dépendante de la concentration d'AMPc (Adénosine Mono-Phosphate cyclique) intracellulaire. La DHT ou SHBG seules ne produisent pas cet effet indiquant que le
Il a été mis en évidence que la voie
stéroïde/SHBG/SMBGR conduit in fine à l'activation de la voie classique d'action des androgènes et à la stimulation de la prolifération cellulaire. En effet, une étude menée par Ding a montré que les androgènes entraînaient non seulement l'augmentation de l'AMPc mais aussi de l'activité de la protéine arginine
estérase, enzyme dont l'expression est sous la dépendance d'un ARE, dans du tissu prostatique de chien pré-incubé avec SHBG. De plus, cet effet est bloqué par l'addition d'hydroxyflutamide qui est un anti-androgène [Ding VDH., 1998].
Par ailleurs, l'augmentation de la sécrétion du PSA à partir de tissu prostatique humain induit par le duo œstradiol/SMBG est bloqué par l'utilisation d'un inhibiteur de la PKA (Protéine Kinase A) et par des anti-androgènes impliquant l'intervention de la PKA et du RA classique [Nakhla AM., 1997].
L'ensemble de ces données nous a permis d'établir la cascade de signalisation représentée dans la figure 11.
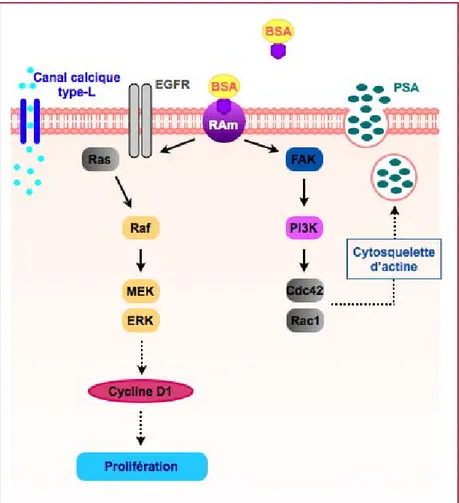
iii. implication d'un récepteur des androgènes "non conventionnel"
Un récepteur des androgènes "non conventionnel" (RAm) a été mis en évidence au niveau de la membrane plasmique. Des sites membranaires de liaison de la testostérone ont été identifiés dans les cellules LNCaP grâce à l'utilisation d'un analogue fluorescent et non-internalisable : la testostérone-BSA-FITC (figure 12). Ces sites ne sont pas reconnus par un anticorps dirigé contre le RA classique et les expériences ont été réalisées en privation de sérum donc en l'absence de SHBG. Aussi ce travail est une preuve supplémentaire de l'existence de récepteurs membranaires des androgènes différents des RA classiques et indépendants des SHBGR [Kampa M., 2002].
Figure 11 : implication du récepteur de SHBG dans les effets rapides des androgènes.
Figure 12 : microscopie confocale de cellules LNCaP marquées avec la testostérone-BSA-FITC [Kampa M., 2002].
Des études ont mis en évidence que la présence de RAm est corrélée au développement du cancer de la prostate : le récepteur membranaire est préférentiellement exprimé dans les cellules de carcinome prostatique. Les cellules épithéliales saines présentent peu ou pas de sites de liaison [Stathopoulos EN, 2003]. De plus, l'analyse de 109 carcinomes prostatiques et 103 hyperplasies bénignes de la prostate a mis en évidence que le niveau d'expression était corrélé à l'agressivité du cancer (score de Gleason) [Dambaki C., 2005].
Paradoxalement, il a été montré que l'activation de RAm avec la T-BSA conduit à l'inhibition de la croissance et induction de l'apoptose dans les cellules LNCaP et la diminution de la migration, adhésion, invasion et activation de l'apoptose dans les cellules DU-145 (négatives pour RA). De plus, le traitement à la T-BSA de souris nude xénotransplantées avec des cellules LNCaP entraîne 60% de réduction du volume tumoral en comparaison du groupe contrôle [Hatzoglou A., 2005].
Il a été mis en évidence que RAm est capable de médier les effets rapides des androgènes. En effet, la concentration en calcium est augmentée de manière rapide et transitoire par stimulation de RAm avec la T-BSA dans les cellules LNCaP [Sun YH., 2006] ainsi que par stimulation androgénique de cellules PC3 qui ne contiennent pas de RA classique [Lyng FM., 2000]. L'utilisation d'inhibiteurs tels que la nifédipine, le vérapamil ou le diltiazem a permis de mettre en évidence que cette augmentation de la concentration intracellulaire en calcium était due aux canaux calciques membranaires de type-L.
Par ailleurs, Kampa M. et al. ont montré que la stimulation des cellules LNCaP avec de la DHT ou de la T-BSA entraînait une sécrétion rapide (à partir de 10 min) de PSA dans le milieu de culture et que ceci était dû à un remodelage du cytosquelette d'actine [Kampa M., 2002]. En effet, la cytochalasine B, un inhibiteur de la polymérisation de l’actine, inhibe cette sécrétion de PSA. Papakonstanti et al. ont mis en évidence que la réorganisation du cytosquelette d'actine initiée au niveau de la membrane plasmique était médiée par la phosphorylation de la protéine kinase FAK (Focal Adhesion Kinase), l'interaction entre FAK et PI3K suivie de l'activation successive de PI3K et des petites protéines G Cdc-42 et Rac-1 [Papakonstanti EA., 2003].
La voie des MAPK est impliquée dans l'action rapide des androgènes passant par RAm. Dans des cellules DU-145 (négatives pour RA) les androgènes entraînent l'activation de la cascade de signalisation Ras/Raf/MEK1/2conduisant à une phosphorylation de ERK1/2en 15 minutes puis à l'augmentation de l'expression de la cycline D1 [Lee YF., 2002]. La phosphorylation de ERK1/2 est inhibée par AG1478 ce qui implique l'intervention du EGFR.
L'action des androgènes au niveau des cellules épithéliales prostatiques normales et tumorales est médiée par la modulation simultanée de multiples voies de signalisation. Dans l'épithélium prostatique normal, il existe un équilibre entre la prolifération et la mort cellulaire de façon à maintenir l'intégrité de la glande. Par contre, dans le cancer de la prostate, cette balance est déséquilibrée en faveur de la prolifération ce qui conduit à une croissance exagérée.
Figure 13 : implication d'un récepteur des androgènes "non conventionnel" (RAm) dans les effets rapides des androgènes.
2. Les effets de la privation androgénique
La privation androgénique qui est le traitement de référence des cancers de la prostate avancés et métastatiques agit au niveau de plusieurs processus biologiques dans les cellules épithéliales sécrétrices de la prostate.
a- la prolifération et le cycle cellulaire
La privation androgénique conduit à une inhibition de la prolifération cellulaire in vitro [Sonnenschein C., 1989] et une diminution de la croissance tumorale dans les modèles animaux de cancer de la prostate. Dans des souris SCID (Severe Combined ImmunoDeficiency) xénotransplantées avec des cellules LNCaP et après trois semaines de castration, Joseph I. et al ont montré que l'inhibition de la croissance conduisait à 75% de diminution de la masse tumorale en comparaison des souris non castrées [Joseph I., 1997b]. Il a également été mis en évidence un ralentissement de la croissance tumorale dans les modèles Dunning G et H de cancer de la prostate chez le rat [Joseph I., 1997b] ainsi que dans le modèle CWR22 de tumeur prostatique humaine implanté dans des souris nude [Gregory CW., 2001c]. Il a largement été démontré que cette diminution de la prolifération était associée à un arrêt du cycle cellulaire en G0/G1. Dans la prostate ventrale de rat, la castration entraîne une diminution de l'expression des cyclines D1 et C, responsables de la transition G0/G1, mais aussi de la cycline E, cycline A et de la CDK-2 qui permettent la transition vers la phase S ainsi que sa progression [Furuya Y., 1995]. Dans les cellules LNCaP, la privation androgénique conduit également à un arrêt du cycle cellulaire qui semble être dû à une diminution de l'expression des cyclines D1, D3 et A associée à des activités CDK-4 et CDK-2 réduites [Knudsen KE., 1998; Fribourg AF., 2000]. Par ailleurs, la privation n'a pas d'effet au niveau du cycle cellulaire dans les cellules DU-145 et PC-3 qui sont des lignées tumorales prostatiques humaines androgéno-insensibles [Eto M., 2003]. L'inhibition des cyclines et CDKs responsables de la progression du cycle cellulaire pendant les phases G1 et S est également présente dans les tumeurs CWR22, implantées dans des souris nude, après castration [Gregory CW., 2001c]. La diminution de ces protéines motrices du cycle cellulaire est associée à une déphosphorylation de la protéine Rb [Knudsen KE., 1998; Fribourg AF., 2000; Gregory CW., 2001c]. Un autre événement intervenant dans l'arrêt du cycle cellulaire est l'augmentation de l'expression de la protéine inhibitrice p27Kip1 qui a été mise en évidence dans les cellules LNCaP après castration [Knudsen KE., 1998].
b- l'apoptose
De nombreuses études ont été menées pour mettre en évidence les effets de la privation androgénique que ce soit dans la prostate ventrale de rat, dans des lignées humaines de
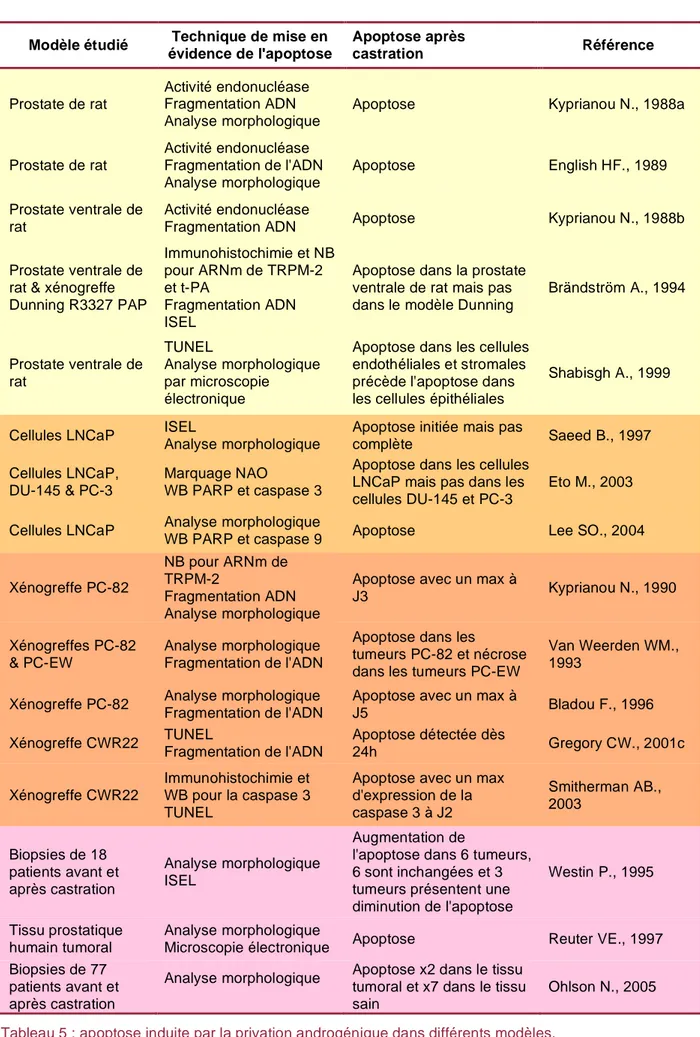
cancer de la prostate, dans des modèles de xénogreffe tumorale mais aussi dans des biopsies de patients. Les résultats de ces études sont controversés en ce qui concerne l'activation de la mort cellulaire programmée (tableau 5).
L'induction de l'apoptose dans la prostate ventrale de rat après castration a été mise en évidence par une augmentation de l'activité endonucléase Ca2+ et Mg2+ dépendante, fragmentation de l'ADN nucléaire [Kyprianou N., 1988a; English HF., 1989; Kyprianou N., 1988b] et expression de protéines marqueurs de l'apoptose comme TRPM-2 (Transient Receptor Potential cation channel M2) et t-PA (tissue Plasminogen Activator) [Brändström A., 1994]. Dans une étude menée par Shabisgh, l'apoptose est mise en évidence dans des sections de prostate ventrale de rat par la technique TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling). L'apoptose apparaît dans les cellules endothéliales et stromales dès 12 heures après la castration. Dans les cellules épithéliales, l'apoptose n'est pas significative et n'apparaît qu'à partir de 24 heures après la castration [Shabisgh A., 1999].
Les résultats des études réalisées dans la lignée LNCaP sont plus contrastés. Après privation androgénique, ces cellules ne présentent pas de signes évidents d'apoptose. Elles se trouvent dans un état cytostatique qui peut être réversé par l'addition de DHT [Saeed B., 1997; Sonnenschein C., 1989]. Par ailleurs, il a été mis en évidence que la privation androgénique conduit à une augmentation de l'activité PI3K et de la phosphorylation de la protéine Akt sur la sérine 473. Ceci est observé dès J1 après le début de la privation et augmente progressivement jusqu'à J4 [Murillo H., 2001]. La stimulation de la voie PI3K/Akt pourrait être un élément permettant d'expliquer la survie des cellules LNCaP en absence d'androgènes. A contrario, il a été montré que la privation pouvait conduire à l'apoptose des cellules LNCaP caractérisée par l'accumulation de C16-céramide qui est un second messager lipidique suivi de l'activation de la caspase 3 et du clivage de PARP (Poly(ADP-Ribose)Polymerase) [Eto M., 2003]. Une autre étude a montré une condensation et une fragmentation nucléaire ainsi que le clivage de la caspase 9 [Lee SO., 2004]. La privation androgénique n'induit pas l'apoptose dans les cellules androgéno-insensibles DU-145 et PC-3 [Eto M., 2003].
Dans des modèles de xénogreffes d'adénocarcinomes prostatiques androgéno-sensibles implantés dans des souris nude (PC-82, PC-EW, LuCaP-23.1, CWR22) ou dans des rats (Dunning R3327 PAP) la majorité des études mettent en évidence l'activation du processus apoptotique. La castration des animaux xénotransplantés avec le modèle tumoral PC-82, LuCaP ou CWR22 induit une régression des tumeurs qui est due à l'activation de l'apoptose [Kyprianou N., 1990; Van Weerden WM., 1993; Bladou F., 1996; Gregory CW., 2001b; Smitherman AB., 2003]. Kyprianou et al. ont mis en évidence une augmentation de l'expression de TRPM-2, la fragmentation de l'ADN ainsi que la formation de corps
montré que la régression des tumeurs après castration est due à une mort qui est majoritairement de type nécrotique [Van Weerden WM., 1993]. Enfin, Brändström et al. n'ont pas trouvé de signes évidents d'apoptose après castration dans le modèle Dunning R3327 PAP [Brändström A., 1994].
Dans des biopsies de patients ayant suivi une hormonothérapie, la mise en évidence de l'induction de l'apoptose est également controversée. Reuter et al. ont montré une activation de l'apoptose dans les adénocarcinomes prostatiques notamment via l'apparition de noyaux picnotiques [Reuter VE., 1997]. Cependant, Ohlson démontre que l'apoptose dans le tissu tumoral est plus faible que dans le tissu normal [Ohlson N., 2005]. Enfin, les travaux de Westin montrent une activation de la mort cellulaire programmée dans seulement 33% des tumeurs étudiées [Westin P., 1995].
![Figure 1 : l'appareil génital masculin (a) [ adapté de National Cancer Institute ] et la prostate (b)](https://thumb-eu.123doks.com/thumbv2/123doknet/2257265.19252/17.892.110.780.382.714/figure-appareil-genital-masculin-adapte-national-institute-prostate.webp)
![Figure 6 : la production des androgènes [ adapté de Denmeade SR., 2002 ] .](https://thumb-eu.123doks.com/thumbv2/123doknet/2257265.19252/24.892.157.739.114.561/figure-la-production-des-androgenes-adapte-denmeade-sr.webp)
![Figure 9 : l'effet de la DHT sur la concentration intracellulaire en Ca 2+ dans les cellules LNCaP pré-incubées avec la sonde calcique Fura-2/AM [ Sun YH., 2006 ]](https://thumb-eu.123doks.com/thumbv2/123doknet/2257265.19252/31.892.112.770.743.1052/figure-effet-concentration-intracellulaire-cellules-lncap-incubees-calcique.webp)
![Figure 12 : microscopie confocale de cellules LNCaP marquées avec la testostérone-BSA-FITC [ Kampa M., 2002 ]](https://thumb-eu.123doks.com/thumbv2/123doknet/2257265.19252/35.892.249.604.999.1159/figure-microscopie-confocale-cellules-lncap-marquees-testosterone-kampa.webp)