Le suppresseur de tumeurs PALB2
: étude
fonctionnelle du domaine N-terminal de liaison à l'ADN
et établissement de modèles de létalité synthétique
Mémoire
Nadine Brahiti
Maîtrise en biologie cellulaire et moléculaire - avec mémoire
Maître ès sciences (M. Sc.)
Québec, Canada
Le suppresseur de tumeurs PALB2 :
Étude fonctionnelle du domaine N-terminal de
liaison à l’ADN et établissement de modèles de
létalité synthétique
Mémoire
Nadine Brahiti
Sous la direction de :
Jean-Yves Masson, directeur de recherche
©Nadine Brahiti, 2019
ii
Résumé
Les mécanismes de réparation de l’ADN sont cruciaux pour la survie cellulaire. Cependant,
ces mécanismes sont souvent altérés dans les processus cancéreux permettant l’accumulation
de mutations et d’instabilité génétique. Plusieurs voies de réparation sont utilisées par les
cellules pour réparer différents types de dommages à l’ADN. La réparation des cassures
double-brin (CDB) par la Recombinaison Homologue (RH) permet de réparer fidèlement
ces lésions. PALB2 est au cœur d’un réseau d’interactions protéiques comprenant BRCA1
et BRCA2. Tout comme ces dernières, PALB2 est un gène de prédisposition au cancer du
sein.
Ainsi, par ses fonctions dans la stabilité du génome et le cancer, l’étude fonctionnelle de
PALB2 et la compréhension du rôle de ses domaines sont essentiels. En effet, PALB2 se lie
à l’ADN, mais les mécanismes induisant sa liaison et la fonction de cette liaison sont toujours
mal compris. En ce sens, nos collaborateurs ont identifié quatre principaux acides aminés
dans ce domaine, comme étant importants car leurs mutations en alanine compromettent la
liaison de PALB2 à l’ADN. De ce fait, mon premier objectif porte sur l’étude fonctionnelle
de ce domaine de liaison à l’ADN dans la recombinaison homologue. En accord avec les
essais biochimiques conduits par nos collaborateurs, notre étude in vivo a pu démontrer que
la mutation de ces résidus en alanine réduit de 50% la formation de foyers RAD51 aux
dommages, réduisant ainsi l’efficacité de la RH dans ces cellules. Mon deuxième objectif,
vise l’établissement de modèles de létalité synthétique pour tuer les cellules déficientes en
réparation de l’ADN. Le but est d’identifier de nouveaux interacteurs de PALB2 par la
technique BioID et qui seraient synthétiquement létaux en combinaison avec d’autres
mutations génétiques. Enfin, il a été démontré que les inhibiteurs de PARP amènent à une
létalité synthétique dans les cellules déficientes en PALB2. Cependant, ces études ne
prennent pas en compte l’aspect tridimensionnel de la tumeur cancéreuse. C’est pourquoi,
dans mon projet j’ai initié le développement de modèles 3D qui pourraient possiblement
aider à l’évaluation des stratégies de létalité synthétique, se rapprochant ainsi du contexte
physiologique de la tumeur.
iii
Table des matières
Résumé ... ii
Table des matières... iii
Liste des figures ... v
Liste des tableaux ... vii
Liste des abréviations ... viii
Remerciements ... xii
Introduction ... 1
1. Maintenance de la stabilité génétique et le cancer ... 1
2. Les dommages à l’ADN et mécanismes de réparation... 2
2.1. Réparation des cassures simple-brin de l’ADN ... 5
2.2. Réparation des cassures double-brin CDB de l’ADN ... 5
2.2.1. Réparation par recombinaison homologue ... 8
2.2.2. PALB2 partenaire et localisateur de BRCA2 lors de la recombinaison homologue ... 11
A. Domaines protéiques de PALB2 et mécanismes de régulation ... 11
B. Domaines de liaison à l’ADN ... 14
C. La mutation de PALB2, gène suppresseur de tumeurs, dans le cancer du sein et l’Anémie de Fanconi ... 16
3. Ciblage des défauts de recombinaison homologue en thérapie contre le cancer... 17
3.1. Stratégie de létalité synthétique... 18
3.1.1. Les PARPi en stratégie de létalité synthétique dans les cellules BRCA1/2 déficientes ... 19
3.1.2. Stratégie de létalité synthétique dans les cellules PALB2 déficiente ... 22
4. La modélisation de tumeurs in vitro pour les essais thérapeutiques ... 23
4.1. Le modèle 3D multicellulaire de sphéroïdes ... 24
4.1.1. Caractéristiques du modèle MCS et mécanisme de formation ... 24 4.1.2. Utilisation des MCS hétérotypiques dans la compréhension du processus cancéreux 27
iv
4.1.3. Techniques de culture des sphéroïdes ... 27
4.2. Avantages de l’utilisation des sphéroïdes en thérapie contre le cancer... 29
4.3. Les sphéroïdes, un outil puissant dans le processus de découverte de traitements pour le cancer ... 32
A. Les sphéroides dans la modélisation des tumeurs ... 32
B. L’utilisation des sphéroïdes accélèrent l’étape d’identification de cibles thérapeutiques 33 C. L’utilisation des sphéroïdes est compatible avec le CHD des drogues ... 33
D. Les sphéroïdes dans l’étude du profil d’une drogue ... 34
Objectifs ... 35
1. Étude fonctionnelle du domaine N-terminal de liaison à l’ADN de PALB2 ... 35
2. Identification de nouveaux interacteurs de PALB2 pour l’établissement de modèles de létalité synthétique ... 35
3. Développement d’un modèle 3D de sphéroïdes déficient en RH pour le criblage des PARPi et d’autres stratégies de létalité synthétique ... 35
Matériels et méthodes ... 36 Résultats ... 52 Discussion ... 71 Conclusion ... 81 Bibliographie ... 83 Annexes ... 102
v
Liste des figures
Figure 1 : Origine et mécanismes de formation des cassures double-brin de l’ADN (modifiée) ... 7
Figure 2 : Les quatre mécanismes de réparation des CDB de l’ADN ... 8
Figure 3 : Les trois phases de la réparation par recombinaison homologue ... 11
Figure 4 : Domaines protéiques de PALB2 et leur fonction dans la régulation de sa fonction ... 16
Figure 5 : Mutations faux sens dans PALB2 retrouvées chez des patients atteints du cancer du sein ... 18
Figure 6 : Concept de la létalité synthétique ... 20
Figure 7 : L’implication de PARP dans la réparation de l’ADN et son inhibition en stratégie de létalité synthétique dans les cellules BRCA déficientes ... 23
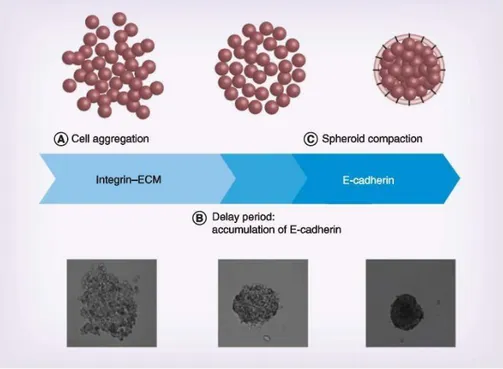
Figure 8 : Processus de formation d’un sphéroïde multicellulaire ... 28
Figure 9 : La diffusion par gradient dans les sphéroïdes ... 29
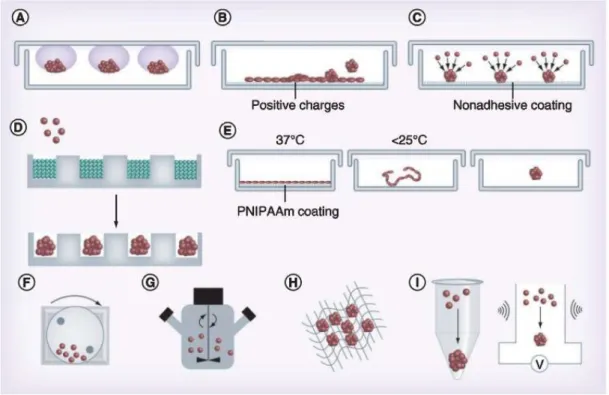
Figure 10 : Technique de culture in vitro des sphéroïdes ... 33
Figure 11 : Schéma explicatif de la démarche à suivre pour le test CRISPR-Cas9 mClover-LMNA1 dans les cellules U2OS ... 45
Figure 12 : Schéma des étapes suivies pour l’essai de la quantification des foyers RAD51 dans les cellules HeLa siPALB2 complémentées avec le PALB2 sauvage ou mutant (146AAAA) ... 47
Figure 13 : Schéma représentatif des étapes du système Flp-In dans les cellules Flp-In™ T-REx™ 293 pour la génération de lignées stables ... 49
Figure 14 : Vecteur d’expression pcDNA5/FRT des protéines recombinantes utilisé dans le système Flp-In. ... 50 Figure 15 : Schéma récapitulatif des étapes de la méthode BioID ... 52
Figure 16 : Technique de la gouttelette suspendue pour la formation de sphéroïdes. ... 55
Figure 17 : Les différents fragments de PALB2 testés pour l’affinité de liaison à l’ADN. ... 60 Figure 18 : Images représentatives et représentation schématique de la technique CRISPRCas9 / mClover-LMNA1. ... 62
Figure 19 : Effet de la mutation du domaine de liaison de PALB2 à l'ADN sur la recombinaison homologue ... 64
vi
Figure 20 : Principe de la stratégie de létalité synthétique impliquant PALB2 et RAD52. ... 66
Figure 21 : Induction de l’expression des contrôles (CTLs) et du gène d’intérêt (GOI) BirA*- PALB2 dans les lignées stables après traitement à la doxycycline. ... 67
Figure 22 : Immunofluorescence des protéines contrôles FLAG-BirA*-NLS/YFP et de FLAG- BirA*-PALB2 dans les cellules Flp-In T-REx HEK293 ... 68
Figure 23 : Coloration des protéines biotinylées sur gel par bleu de coomassie après purification par affinité sur billes de sépharose conjuguées à la streptavidine. ... 69
Figure 24 : Dot plot des protéines biotinylées et identifiées par spectrométrie de masse. ... 70
Figure 25 : Diagramme représentant la méthodologie suivie pour le développement du modèle 3D déficient en RH. ... 71
Figure 26 : Sphéroïdes de différentes lignées cellulaires formés par les deux techniques. ... 73
Figure 27 : Sphéroïdes DLD1 BRCA2 déficients. ... 74
Figure 28 : Sensibilité aux PARPi des DLD1 BRCA2-/- et +/- en 2D. ... 75
Figure 29 : Sphéroides DLD1 BRCA2+/- et -/- traités aux inhibiteurs de PARPi. ... 76 Figure 30 : Sphéroides DLD1 BRCA2+/- et -/- traités aux PARPi marqués aux Hoechst et EthD1. 78
vii
Liste des tableaux
Tableau 1 : Facteurs de dommage à l’ADN et type de lésion………...5 Tableau 2 : Amorces utilisées pour les constructions citées dans le texte... 58
viii
Liste des abréviations
% Pour cent
°C Degré Celcius
µg Microgramme
µm Micromètre
µM Micromolaire
53BP1 de l’anglais « p53-Binding Protein 1 »
AA Acide aminé
ADN Acide DésoxyriboNucléique
ADN DB ADN double-brin
ADN SB ADN simple-brin
ADP Adénosine DiPhosphate
Alt-EJ de l’anglais « Alternative-NHEJ » ANOVA de l’anglais « ANalysis Of Variance » ARN Acide RiboNucléique
ATM de l’anglais « Ataxia Telangiectasia Mutated » ATR de l’anglais « ATM and RAD3 related
BER Réparation par excision de bases (de l’anglais « Base Excision Repair ») BLM de l’anglais « BLoom syndrome Mutated protein »
BMN de l’anglais « BioMarin Pharmaceutical Inc., Novato, CA, USA » BRCA1/2 de l’anglais « BReast CAncer 1/2, early onset »
BSA de l’anglais « Bovine Serum Albumin » CDB Cassure Double-Brin
CDK de l’anglais « Cyclin-Dependent Kinase » CUL3 de l’anglais « Cullin protein family 3 »
CRISPR de l’anglais « Clustered Regularly Interspaced Short Palindromic Repeats » CSB Cassure simple-brin
CtIP de l’anglais « C-terminal binding protein Interacting Protein » CTL de l’anglais « Control »
DAPI 4', 6 - diamidino -2-phenylindole
DBD de l’anglais « DNA binding domain » DDR de l’anglais « DNA damage response » D-loop de l’anglais « Displacement loop »
DMEM de l’anglais « Dulbecco’s modified Eagle medium » DMSO DiMéthylSulfOxyde
DNA de l’anglais « Deoxyribonucleic acid »
DNA2 de l’anglais « DNA replication ATP-dependent helicase/nuclease DNA2 » EDTA de l’anglais « Ethylene Diamine Tetraacetic Acid »
EGTA de l’anglais « Ethylene Glycol Tetraacetic Acid » EMSA de l’anglais « Electrophoretic Mobility Shift Assay » EXO1 EXOnucléase 1
FACs Les fibroblastes associés aux carcinomes
ix
H2AX de l’anglais « H2A Histone Family, Member X »
HeLa cellules provenant d’un carcinome humain du col de l’utérus d’une patiente HEPES acide 4 - (2-hydroxyéthyl)-1 -pipérazine éthane sulfonique
HR de l’anglais «HomologousRecombination »
ICL de l’anglais « Interstrand crosslink » KCL de l’anglais « potassium chloride » kDa kiloDalton
KEAP1 de l’anglais « Kelch-like ECH- associated protein » LOH de l’anglais « Loss of heterozygosity »
MBD de l’anglais «MRG15 binding domain»
mg Milligramme
mL Millilitre
mM Millimolaire
mm Millimètre
MMR de l’anglais « Mismatch repair »
MRE11 de l’anglais « Meiotic REcombination protein 11 »
MRG15 de l’anglais « (Mortality factor)-related gene on chromosome 15 »
MRN MRE11-RAD50-NBS1
NBS1 de l’anglais « Nijmegen Breakage Syndrome protein 1 » NER de l’anglais « Nucleotide Excision Repair »
NHEJ de l’anglais « Non homologous end joining »
NLS de l’anglais « Nuclear Localization Signal »
nM nanomolaire
NRF2 de l’anglais « Nuclear factor (erythroid derived 2) like 2» PALB2 de l’anglais « Partner and Localizer of BRCA2 »
PARP de l’anglais « Poly (ADP-Ribose) Polymerase »
PBS de l’anglais « phosphate buffered saline » pH Potentiel Hydrogène
PI3K de l’anglais « Phosphoinositide 3-kinase » PMSF de l’anglais « PhenylMethylSulfonyl Fluoride » RAD50 de l’anglais « DNA repair protein RAD50 » RAD51 de l’anglais « DNA repair protein 51 homolog » RAD52 de l’anglais « DNA repair protein 52 homolog »
RH Recombinaison Homologue
RNA de l’anglais « Ribonucleotidic acid » RNF 168 de l’anglais « RiNg Finger protein 168» ROS de l’anglais « Reactive oxygene species » RPA de l’anglais « Replication protein A » SDS DodécylSulfate de Sodium
SEM de l’anglais « Standard Error of the Mean » siRNA de l’anglais « small interfering RNA » SS de l’anglais « Single-Strand »
x
TBS de l’anglais « Tris-Buffered Salin »
USP11 de l’anglais «Ubiquitin carboxyl-terminal hydrolase 11»
UV UltraViolet
V(D)J de l’anglais « Variable, Diversity and Joining » VUS de l’anglais « Variant of Unknown Significance»
WB de l’anglais « Western Blot »
WD40 Répétition d’environ 40 acides aminés souvent terminée par le dipeptide
tryptophane-aspartate
xi
À la plus courageuse des mamans, une battante
qui a lutté contre le cancer du sein. Celle qui a fait
de moi la femme que je suis aujourd’hui et qui m’a
toujours appris à ne jamais baisser les bras.
À mon époux qui a toujours cru en moi, qui m’a
comblé d’amour et aidé à surmonter toutes les
difficultés.
xii
Remerciements
Cette aventure n’aurait pas été possible sans Dr Jean-Yves Masson qui m’a accordé cette chance, qui m’a fait confiance et m’a accueilli dans son laboratoire. Il a toujours été à l’écoute, que ce soit pour l’évolution de mon projet, mes décisions professionnelles ou encore dans des situations difficiles que j’ai dû traverser. Je tiens à le remercier pour tout, les mots ne suffiraient pas pour lui exprimer ma gratitude. Intégrer un grand laboratoire, découvrir la recherche, pouvoir y contribuer était un de mes plus grands objectifs professionnels. Merci de m’avoir permis de réaliser ce rêve.
Je tiens aussi à remercier nos assistants de recherche, ou plutôt nos héros de laboratoire : Yan, Amélie et Marie-Christine, tant pour leur disponibilité, leur patience et leurs encouragements. Merci pour vos précieux conseils techniques qui transforment parfois notre désespoir en espoir. Un grand merci à Yan, notre MacGyver, qui m’a mentoré dès mes débuts pour mon apprentissage au laboratoire et m’a permis d’acquérir toutes les compétences techniques nécessaires au lancement de mon projet de recherche. Un immense merci à Amélie qui m’a aussi beaucoup assisté tout au long de mon projet. Merci Amélie pour ta disponibilité, ta patience et ton support et ton expertise qui étaient essentiels à l’évolution de mon projet et à mon évolution au laboratoire. Je ne peux ne pas inclure notre post-doc ou plutôt la meilleure des post-post-docs, Laure qui a toujours été là pour moi sur tous les plans, c’était la bonne fée du laboratoire. Ses conseils, ses idées, sa gentillesse, sa bonne humeur sont inégalables. Laure a toujours été là pour moi dans les moments difficiles et a su me transmettre son énergie positive quand il le fallait. Un grand merci à toi, continues à être la personne exceptionnelle que tu es et merci pour tous les moments exceptionnels qu’on a partagés.
Mon expérience au laboratoire m’a aussi permis de rencontrer de nouvelles personnes qui viennent des quatre coins du monde, je pense à Richa, Laura, Julia,Yuandi, Mandy, Aykut, Daryl, Thibaut, Franciele, Simone. Nos différences ont fait de cette expérience une fusion de cultures, de mentalités, d’expériences, très enrichissante où j’ai beaucoup appris tant sur le plan professionnel que personnel. Merci pour tous les bons moments qu’on a partagés.
J’aimerais aussi remercier nos collaborateurs Dr Korolev qui m’a accordé la chance de travailler sur le projet du domaine de liaison à l’ADN de PALB2 et qui a abouti en une publication scientifique pour ma maîtrise. Je remercie aussi Dr Jean Philippe Lambert pour son temps, son expertise et ses précieux conseils dans la réalisation de la BioID. Je remercie aussi Dr Samer Hussein, pour ses
xiii
conseils au lancement du projet sphéroïdes, ainsi que Dr Nicolas Bisson pour les outils fournis pour la réalisation de la BioID.
1
Introduction
1. Maintenance de la stabilité génétique et le cancer
L’ADN constitue l’unité de base dont est composé le matériel génétique et épigénétique d’une cellule. Il est considéré comme le support de toute l’information nécessaire aux différentes fonctions biologiques, mais aussi le pilier de l’hérédité. Ainsi, la protection de cette information est vitale pour la cellule qui est considérée comme l’unité fondamentale de tout être vivant. En effet, il existe des mécanismes sophistiqués qui assurent la protection de cette identité pour permettre la bonne transmission du code génétique d’une génération à une autre. Le maintien de cette intégrité génétique est donc crucial pour le développement normal d’un organisme mais aussi pour la protection contre le développement de diverses pathologies.
Au cours de sa vie la cellule va subir diverses attaques de nature endogène ou exogène qui menaceront l’intégrité de son matériel génétique. Pour contrer ces attaques la cellule a développé différents mécanismes qui assurent le maintien de la stabilité génétique. En détectant ces menaces cette dernière va enclencher des mécanismes de défenses pour réparer les dommages causés et prévenir la transmission d’erreurs.
À l’échelle du cycle cellulaire il existe des points de restrictions qui contrôlent cette intégrité permettant ou non le passage d’une phase à une autre. Ainsi, après la division cellulaire l’information génétique est transmise de façon stable à la cellule fille. Cependant, lorsque les processus de maintien deviennent défectueux, les dommages peuvent être tolérés et mener au développement de pathologies comme le cancer. Le bon fonctionnement des différents mécanismes de réparation de l’ADN est donc l’un des fondements sur lequel s’appuie la cellule pour maintenir sa stabilité génétique.
Il existe trois formes d’instabilité génétique : l’instabilité chromosomique CIN, l’instabilité microsatellitaire MIN et l’instabilité génétique caractérisée par une fréquence élevée des mutations de paires de bases. L’instabilité chromosomique est la plus répondue dans les cancers, elle est caractérisée par un nombre et/ou des structures chromosomiques anormales. Cependant, on ne sait toujours pas si ces évènements sont à l’origine du processus cancéreux et permettent son initiation ou une conséquence due à l’accumulation des mutations.1,2
2
En effet, l’accumulation de mutations dans les cellules cancéreuses est l’une des signatures du cancer. Ce phénomène conduit à un degré d’instabilité génétique qui favorise la transformation des cellules normales en cellules malignes. En fait, la cellule cancéreuse génétiquement instable devient non fidèle aux processus de réplication, de ségrégation des chromosomes et de la réparation du génome.3 Ces mutations ciblent principalement l’inactivation de gènes suppresseurs de tumeurs et la surexpression d’oncogènes qui permettent à la cellule transformée d’acquérir un avantage prolifératif et anti-apoptotique qui lui permettent d’échapper aux points de contrôle et assurer sa survie.4 Cette hypothèse appelée « Mutator hypothesis» est considérée comme l’une des forces majeures qui gouverne le processus tumoral. 5
Il est donc évident que le bon fonctionnement des mécanismes de réparation de l’ADN est étroitement lié au maintien de la stabilité génétique. Cette machinerie est considéré comme l’outil cellulaire majeur qui permet de conserver l’intégrité de l’ADN malgré toutes les menaces auxquelles il est exposé.
2. Les dommages à l’ADN et mécanismes de réparation
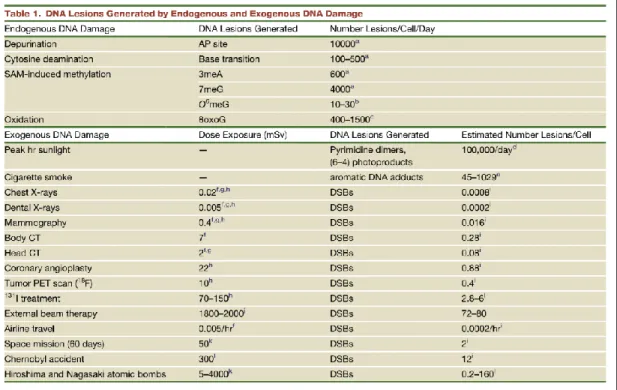
L’ADN est considéré comme l’unique macromolécule dans la cellule qui accumule des dommages mais qui doit être maintenue stable sans pour autant être reproduite entièrement. En effet, après exposition aux dommages, l’ADN comportant toute l’information génétique nécessaire au bon fonctionnement de la cellule, n’est jamais éliminée pour être resynthétisée de nouveau, mais en revanche elle est prise en charge par des mécanismes cellulaire qui rétablissent son intégrité. Il est donc clair que cela présente un défi pour la cellule dont plusieurs fonctions dépendent de l’intégrité de cette molécule. Le nombre de dommages que peut subir l’ADN d’une cellule s’élève à 105 lésions par jour, ces dommages peuvent provenir de différentes sources, certaines inévitables car endogènes et d’autres de nature exogène pouvant être évité jusqu’à un certain degré
.
6Ces
dommages sont classés en deux catégories (Tableau1).
A. Les dommages endogènes
a) Dommages liés aux réactions spontanées d’hydrolyse de l’ADN :
L’hydrolyse spontanée de l’ADN peut créer des sites abasiques ou une déamination de certaines bases. Cette réaction est due à l’instabilité chimique de l’ADN en réponse à différentes conditions de pH, température, force ionique ou encore due à la structure secondaire des acides nucléiques en
3
solution. Plus spécifiquement, cette instabilité est un résultat de la labilité de liaisons glycosidiques dans un environnement aqueux causant l’hydrolyse des purines et des pyrimidines (avec un taux moins élevé). D’autre part, la déamination hydrolytique des bases peut aussi se produire spontanément en réponse aux différentes conditions physiologiques, comme la conversion de la cytosine en uracile ou thymine. Ces deux réactions intrinsèques bien que différentes se produisent à des taux semblables dans la cellule. 6,7
b) Dommages liés au métabolisme :
Les dérivés réactifs d’oxygène DRO et de nitrogène sont continuellement synthétisés par notre
métabolisme et peuvent créer , par oxydation, des dommages à l’ADN. Les DRO peuvent exister sous différentes formes : du peroxyde d’hydrogène (H2O2), des radicaux d’hydroxyle (OH), superoxide (O2-) et de l’oxygène singulet (1O2). Ces DRO provoquent l’oxydation des bases de l’ADN qui peuvent causer des cassures simple et double-brin de l’ADN. Un bon exemple d’oxydation est la production continuelle du superoxide (O2) durant la respiration mitochondriale. 8 Ces dérives créent un stress oxydatif qui est pris en charge par une réponse biologique qui limite les niveaux de ces réactifs et leurs dommages. Cependant, ces réactifs peuvent engendrer un stress oxydatif qui est défini par un niveau plus élevé de DRO comparé à la réponse anti-oxydative ce qui génère des dommages dans l’ADN et dans d’autres molécules. 9 Similairement, les dérivés de nitrogènes produits par le métabolisme ou par la réponse inflammatoire comme le nitro-deoxyguanosine ONO2 conduit à la dépurination des bases, mais aussi à la déamination ce qui résulte en la formation de cassures des brins de l’ADN.10 Il existe d’autres dommages de nature endogène autres que les DRO qui causent des dommages à l’ADN. Deux bons exemples de ces dommages sont : la mauvaise incorporation de dNTPs par les polymérases lors de la réplication ou encore la perte de bases due à l’alkylation (Tableau 1). 7
B. Dommages liés aux facteurs exogènes :
Cette catégorie inclue des facteurs environnementaux de source physique ou chimique auxquels est
exposée la cellule, et qui menacent l’ADN. L’exposition aux rayons ultra-violets induit la formation de dimers de pyrimidines et de photo-produits, dus à une liaison covalente entre deux bases consécutives sur le même brin de l’ADN, qui altère la liaison normale des bases au brin complémentaire. On estime qu’une exposition au soleil toute une journée induit 105 lésions dans l’ADN de chaque kératinocyte exposé (Tableau1). 6,11 L’autre source physique importante qui crée des dommages à l’ADN est l’irradiation qui peut provenir d’une simple radiographie médicale aux
4
rayons X ou encore lors d’un vol aérien. Ou encore, la radiothérapie et la chimiothérapie qui sont employés dans le traitement de plusieurs cancers. De nombreux agents chimio thérapeutiques comme les agents alkylants tel que le temozolomide ou les agents créant des pontages inter/intra-brins comme le cisplatin, la mitomycine C et autres sont utilisés pour créer des dommages à l’ADN dans les cellules cancéreuses. Il existe d’autres facteurs de nature exogène qui peuvent altérer l’ADN et qui sont résumés dans le Tableau 1. 11
L’ensemble de ces dommages sont pris en charge par des mécanismes de réparation qui participent en grande partie au maintien de l’intégrité de l’ADN. Le choix de la voie de réparation dépend de plusieurs facteurs qui permettent un contrôle strict de ces mécanismes, mais aussi oriente le choix de la voie selon le type de dommage au bon endroit et au bon moment. En effet, il existe une multitude de protéines intervenant dans ces processus, certaines permettent la détection du dommage, d’autres se chargent de la préparation de l’ADN pour accueillir la machinerie et enfin des protéines effectrices qui permettent la réparation. 11
Tableau 1 : Facteurs de dommage à l’ADN et type de lésion. Tiré de Ciccia et Elledge.
(2010).11
Tableau récapitulatif des facteurs de nature endogène ou exogène qui causent des dommages à l’ADN, ainsi que du type et de la fréquence des lésions générées par jour dans une cellule.
5
2.1. Réparation des cassures simple-brin de l’ADN
Il existe au minimum six voies de réparation distinctes pour les CSB. La réparation par excision de base BER est connue pour prendre en charge les dommages mineurs altérants les bases de l’ADN, telle que l’oxydation ou l’alkylation. Comme son nom l’indique ce mécanisme excise la base altérée à l’aide d’une endonucléase APE1, générant un site abasique AP pour apurinique/apyrimidinique. Ce qui résulte en une cassure simple-brin qui est prise en charge par ce mécanisme. La brèche est re-synthétisée par une polymérase suivie par une ligation par une ligase qui résulte en une restauration du brin intact. 7 Les mésappariements de bases sont tant qu’à eux pris en charge par le MMR. Cette voie prend en charge les mésappariements engendrés par les polymérases non fidèles mais aussi les loupes de délétions/insertions IDLs, qui sont le résultat de la synthèse de régions de l’ADN microsatellitaires. Ce mécanisme possède deux étapes cruciales à son bon fonctionnement, la première étant la reconnaissance du mésappariement ou de l’IDL et la deuxième la distinction du brin nouvellement synthétisé ayant la mauvaise base. Le MMR fait intervenir différents facteurs qui assurent la fidélité de ce mécanisme. 12
L’autre voie prenant en charge les cassures simple-brin est la réparation par excision de nucléotides NER. Cette voie répare les dommages causant des distorsions majeures à l’un des deux brins de l’ADN, comme les dimères de pyrimidines ou les pontages intra-brins. Le NER permet la suppression d’une section de l’ADN d’approximativement 30 nucléotides puis son remplacement et restauration d’un ADN intact. Ce mécanisme ainsi que le BER sont connus pour être couplés à la transcription. Le NER est le plus communément associé et qui permet de réparer les dommages engendrés lors de cette fonction biologique et permettre son bon déroulement. 13
2.2. Réparation des cassures double-brin CDB de l’ADN
Les cassures double-brin sont des lésions hautement toxiques qui peuvent induire de grands réarrangements génomiques. Leur prise en charge et réparation sont indispensables à la survie d’une cellule. Un défaut dans leur réparation peut mener au développement de plusieurs pathologies dont le cancer. Ce type de lésions est parfois programmé par la cellule et leur réparation participe dans certains cas à une fonction biologique (Figure 1). En effet, l’induction de cassures double-brin et leur réparation dans les gènes d’immunoglobulines participent à leur diversité immunologique, ce qui permet de générer une multitude de combinaisons pour les récepteurs d’antigènes. 14 La première division méiotique est aussi un processus biologique où les CDB sont créés par la cellule et jouent
6
un rôle important dans la bonne ségrégation des chromosomes et à la combinaison des allèles parentaux dans les gamètes. 15.
Figure 1 : Origine et mécanismes de formation des cassures double-brin de l’ADN. Tirée de
Chapman et al. (2012).16
Les dommages de l’ADN peuvent être d’origine exogène ou endogène. Les cassures double-brin peuvent posséder une (bulle verte) ou deux extrémités (bulle jaune). Ces dernières peuvent être formées à la suite d’une exposition à un facteur exogène, comme l’irradiation, ou être le résultat d’un processus endogène programmé comme la recombinaison V(D)J des immunoglobulines, la méiose, ou encore à l’origine d’un évènement spontané comme l’effondrement des fourches de réplication au niveau d’un ICL ou d’une CSB de l’ADN. Les CDB sont ensuite réparées par NHEJ ou par RH.
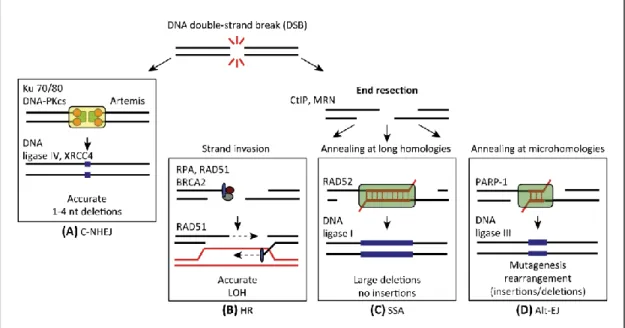
Il existe au moins quatre mécanismes de réparation des CDB, dont deux voies majeures, la réparation par jonction d’extrémités non homologues NHEJ et la réparation par recombinaison homologue RH (Figure 2). Le choix entre ces deux mécanismes dépend majoritairement de la phase du cycle cellulaire, car la RH utilise la chromatide sœur comme substrat et qui n’est disponible qu’en phase S et G2 du cycle cellulaire. Alors que la NHEJ qui ne nécessite pas de substrat, peut se produire à travers tout le cycle cellulaire en G0/G1 et G2 et se fait par jointure des deux bouts de la CDB. 17 La
7
RH est le mécanisme de réparation le plus fidèle qui permet la restauration de la séquence originale, alors que la NHEJ est hautement mutagène et peut créer de grands réarrangements, d’où sa dominance dans les processus biologiques qui nécessitent des réarrangements comme pour la diversité des immunoglobulines. 18 Cependant, le choix entre la RH et les deux voies de réparation restantes, la voie de réparation alternative par jonction d’extrémités Alt-EJ et la réparation par hybridation simple-brin SSA, ne dépend pas seulement de la phase du cycle cellulaire mais aussi d’un autre facteur important qui est le niveau de résection. Ces voies de réparation se produisent aussi en phase S et G2 du cycle cellulaire et dépendent des niveaux de digestion des extrémités 3’ de la CDB, mais qui sont contrairement à la RH hautement mutagènes (Figure 2). 19
Figure 2 : Les quatre mécanismes de réparation des CDB de l’ADN. Tirée de Ceccaldi et al.
(2016).20
Le choix de la voie de réparation de la CDB par NHEJ ou la RH et les autres mécanismes restants est premièrement influencé par les niveaux de résection. En absence de résection la CDB est réparée par NHEJ (A). Lorsque la résection est bloquée, la réparation par C-NHEJ est privilégiée. Cependant, lorsque la résection n’est pas bloquée les trois voies impliquées RH (B), Alt-EJ (D) et SSA (C) peuvent entrer en compétition pour la réparation de la CDB. Les quatre voies possèdent différentes conséquences génétiques, la RH est considérée comme la voie la plus efficace et fidèle des quatre. Les deux voies restantes SSA et Alt-EJ entrainent de larges délétions ou des insertions/délétions respectivement. Abréviations : nt, nucléotides ; LOH, perte d'hétérozygotie.
8
2.2.1. Réparation par recombinaison homologue
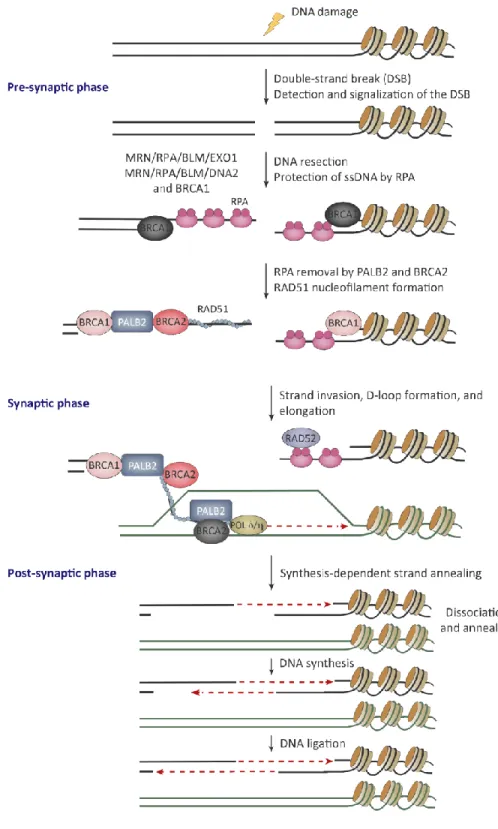
La réparation par recombinaison homologue des CDB peut être divisée en trois phases, la phase présynaptique, la phase synaptique et la phase post-synaptique (Figure 2). 21
A- La phase pré-synaptique
La première phase appelée pré-synaptique est marquée par la digestion de l’ADN au niveau de la cassure double-brin, qui est une étape importante pour initier la réparation par recombinaison homologue. Cette digestion des bouts de l’ADN en 3’ est accomplie par les nucléases MRE11-RAD50-NBS1 (MRN) et CtIP. Environs 20 pb sont digérées au début, mais ce n’est qu’après la résection extensive induite par BRCA1 et exercée par des exonucléases et des hélicases (BLM, WRN, DNA2, CtIP et EXO1) et qui génèrent de l’ADN simple-brin, que le choix de la voie de réparation pourrait être orienté vers la RH. 21–23 Cet ADN simple-brin généré est protégé par la protéine de liaison à l’ADN simple-brin RPA, qui est par la suite retirée pour permettre le recrutement et la formation du filament RAD51 qui marque le début de la phase synaptique. La fin de l’étape pré-synaptique est marquée par l’intervention de quatre protéines cruciales pour la RH, BRCA1, PALB2, BRCA2 et RAD51. BRCA1 permet le recrutement de PALB2 qui à son tour recrute BRCA2 pour décharger la protéine RPA de l’ADN et laisser place à la recombinase RAD51 qui va former le filament synaptique.
B- La phase synaptique
Durant cette phase le filament nucléoprotéique de RAD51 formé va permettre la recherche d’homologie sur le chromatide sœur et induire l’invasion du brin. L’hybridation du brin ayant la lésion avec le brin contenant l’homologie déplace le brin complémentaire de la chromatide sœur générant ainsi une structure appelée D-loop (pour loupe de déplacement). Par la suite, une polymérase appelée delta ou êta va initier la synthétise l’ADN manquant sur le brin de l’ADN.
9
C- La phase post-synaptique
Durant cette phase la D-loop va être résolue pour permettre à l’ADN simple-brin réparé de rejoindre sa chromatide originale et servir de substrat pour la synthèse du brin complémentaire. La résolution de la D-loop dans les cellules mitotiques est prise en charge par la recombinase RAD52. Après la complémentation de la brèche les brins de l’ADN sont ligués et un ADN intact est rétabli.
10
Figure 3 : Les trois phases de la réparation par recombinaison homologue. Tirée de Ducy et al.
(2018).21
La réparation des CDB par recombinaison homologue se déroule en trois étapes (présynaptique, synaptique et postsynaptique (voir texte pour plus d’information).
11
2.2.2. PALB2 partenaire et localisateur de BRCA2 lors de la recombinaison homologue
L’identification des protéines impliquées dans les mécanismes de réparation et la compréhension de leur rôle est cruciale pour l’interprétation des processus de maintien de l’intégrité génomique, mais aussi pour le développement de thérapies. Dans ce contexte, BRCA2 est un gène suppresseur de tumeurs codant pour une protéine majeure dans le maintien de la stabilité génétique, et ce de par son rôle central dans la recombinaison homologue. L’altération de ce gène est considérée comme une des sources d’apparition du cancer du sein familial, d’où son nom Breast Cancer 2. De ce fait, l’intérêt de comprendre les étapes lui permettant de remplir sa fonction dans la RH, entre autres ses interactions protéiques a fait l’objet de plusieurs études. 24,25 Ainsi, c’est en étudiant BRCA2 que
PALB2 a été découvert par Xia et al en 2006, un gène est localisé sur le chromosome 16p2.2 qui
comporte 13exons et code pour une protéine de 1186 acides aminés interagissant avec BRCA2.26 Comme son nom l’indique, PALB2 a été identifiée comme partenaire mais aussi localisateur de BRCA2, qui à son tour induit la localisation de RAD51 sur l’ADN simple-brin de la cassure pour permettre la formation du filament RAD51. Cette dernière est considérée comme une étape critique pour le bon fonctionnement de la RH et assurer une réparation fidèle de la CDB. Ainsi, la découverte de PALB2 a permis de mieux comprendre les étapes qui précèdent le recrutement de BRCA2 à la cassure double-brin lors de la RH. En effet il a été prouvé que l’accumulation et la localisation intranucléaire de BRCA2 dépendait entièrement de sa liaison à PALB2, interconnectant ainsi PALB2 directement à BRCA2. Des travaux qui ont permis d’identifier PALB2 ont démontré la formation de foyers nucléaires de PALB2 qui co-localisaient avec BRCA2 et BRCA1, mais aussi avec γ-H2AX,, un variant de l’histone H2A qui est phosphorylé lors de la création d’une CDB. Dans la même étude l’équipe de Xia et al. ont pu mettre en évidence l’importance de PALB2 pour la localisation et la stabilité de BRCA2, soulignant ainsi son rôle dans l’ancrage de BRCA2 aux dommages et sa stabilisation. 26
De plus, la purification de PALB2 et sa reconstitution in vitro a permis de caractériser son implication dans l’étape d’invasion et la formation de la structure D-loop (Figure 3). Ce qui a été démontré par son affinité de liaison à cette structure et son interaction directe avec RAD51 pour stimuler l’étape d’invasion. 27
A. Domaines protéiques de PALB2 et mécanismes de régulation
Comme pour toutes les protéines de la RH, l’activité de PALB2 doit être restreinte aux phases S/G2 du cycle cellulaire où la chromatide sœur est rendue disponible. Cette protéine est contrôlée à trois niveaux par des modifications post-traductionnelles d’oligomérisation, de phosphorylation et
12
d’ubiquitination. La protéine PALB2 comporte plusieurs domaines protéiques et des sites de modification post-traductionnelle qui permettent un contrôle strict de son recrutement, son expression, son transport, mais aussi de ses interactions et sa spécificité (Figure 4).
- Recrutement de PALB2 aux dommages par son interaction avec BRCA1 en phase S/G2 du cycle cellulaire
PALB2 possède un domaine coiled-coil (CC) en N-terminal qui permet son oligomérisation pour inhiber son interaction avec BRCA1 en absence de dommages. 28 Ce changement entre l’état d’oligomère et monomère est régulé par les kinases ATM/ATR. De telle sorte qu’après avoir détecté un stress génotoxique ces kinases traductrices du signal phosphorylent les serines (S59, S157 et S376) de PALB2, cette modification post-traductionnelle va permettre d’inhiber son oligomérisation le rendant prêt à interagir avec BRCA1 pour son recrutement au site de la cassure. D’autre part, il existe un modèle bi-phasique qui contrôle son recrutement et qui dépend du choix de phosphorylation de la serine S64 par une CDK ou de la serine S59 par ATR. Dans ce modèle l’activation d’ATR dépend de la CDK et l’inhibition de cette dernière dépend d’ATR. Dans une première phase, CDK phosphoryle PALB2 sur la S64 activant ainsi la voie ATR-Chk1 qui stimule la résection de l’ADN au site du dommage. Dans une deuxième phase, ATR inhibe la CDK et phosphoryle PALB2 sur la S59 permettant son recrutement au site de la CDB à travers son interaction avec BRCA1 (Figure 4B). 29,30 Une autre interaction protéique avec une ubiquitine ligase RNF168, impliquant le domaine WD40 de PALB2, est connue pour stimuler le recrutement de PALB2 au site du dommage. Cette ubiquitine ligase agit sur les lysines K13/K15 de l’histone H2A, une marque qui est lue par PALB2 grâce à son interaction avec la région C-terminal de RNF168 pour promouvoir le recrutement de la machinerie de réparation par recombinaison homologue aux dommages en phase S/G2 du cycle cellulaire (Figure 4A). 31
- L’interaction de PALB2 et KEAP dans la réponse au stress oxydatif et sa dégradation
en phase G1 du cycle cellulaire
En 2015, une étude conduite par l’équipe d’Orthwein et al. a démontré que le complexe KEAP1 CUL3 induisait l’ubiquitination des lysines K20/25/30 du domaine coiled-coil de PALB2.32 Cette modification est nécessaire pour empêcher son interaction avec BRCA1 et promouvoir sa dégradation en phase G1 du cycle cellulaire.32 Cette ubiquitination est contrecarrée en S/G2 par la dé-ubiquitylase USP11 rendant PALB2 disponible pour interagir avec BRCA1 lors d’un dommage
13
durant ces phases du cycle cellulaire où la chromatide sœur est disponible pour une réparation par RH (Figure 4C). 32
L’interaction de PALB2 avec KEAP1 est aussi importante pour le maintien de l’homéostasie redox cellulaire. Dans des conditions de stress oxydatif, PALB2 se lie à KEAP1 à travers son motif de type ETGE pour empêcher sa liaison avec NRF2 (facteur de transcription clé dans la réponse anti-oxydative) et donc empêcher sa dégradation (Figure 4D). Le relargage de NRF2 va permettre son accumulation nucléaire et l’expression de plusieurs éléments anti-oxydants. PALB2 participe ainsi indirectement au maintien de la balance redox cellulaire et la régulation de la réponse au stress oxydatif. 33
- Le recrutement de PALB2 dans un contexte chromatinien
Dans un contexte cellulaire, l’ADN n’est pas nu mais présent sous forme de chromatine qui est constituée de nucléosomes en mouvement. PALB2, étant une protéine de réparation de l’ADN, doit s’ancrer à la chromatine pour rejoindre le site de la cassure lors d’un dommage. La chromatine est hautement dynamique et est constituée de l’ADN enroulé autour des histones formant les nucléosomes. Ces changements d’états peuvent être dus à l’ADN qui peut subir une méthylation, des modifications post-traductionnelles des histones ou encore une incorporation de variants d’histones. Ces changements permettent l’ouverture ou la fermeture de la chromatine afin d’autoriser ou limiter l’accès entre autres aux protéines de réparation. 34,35 PALB2 contient un domaine d’association directe à la chromatine (ChAM) et deux autres domaines MBD de liaison à MRG15, une protéine du complexe NuA4/TIP60, qui permettent son attachement à la chromatine. 36,37 En fait, il existerait une association entre le ChAM et le MBD pour l’ancrage de PALB2 à la chromatine, ce qui a été démontré par une délétion du domaine ChAM qui n’affectait pas totalement la liaison de PALB2 à la chromatine en présence d’un domaine MBD intact. 26,38
L’association à MRG15 aurait aussi une fonction préventive en réponse au stress réplicatif. Il a été suggéré que MRG15 permettrait l’ancrage de PALB2 à la chromatine en absence de dommages dans les régions hautement actives. 39 Cette localisation serait possible grâce à la lecture d’une marque épigénétique K36me3 sur l’histone H3 par le chromodomaine de MRG15, rendant PALB2 toujours disponible en cas de dommages dans ces régions hautement transcrites (Figure 4F). 40, 41
14
- Domaine WD40 de PALB2, une plateforme d’interactions protéiques
Ce domaine situé en C-terminal replié en sept hélices beta constitue un site d’interaction avec plusieurs protéines : BRCA2, RAD51, RAD51C, RNF168 et la polymérase êta (Figure 4G). 42, 43,44 Cette structure lui confère des caractéristiques biochimiques spécifiques qui sont hautement conservées. En effet, leur perturbation peut provoquer une dégradation de l’ARNm de PALB2 par le NMD nonsense mediated decay, un processus de contrôle de qualité des ARNm. 43,45 D’autre part, le WD40 contient un signal d’export nucléaire qui peut devenir exposé si la structure de ce domaine est altérée par des mutations, résultant en l’expulsion de PALB2 au cytoplasme (Figure 4H). 46
B. Domaines de liaison à l’ADN
Les études biochimiques ont pu démontrer l’affinité de liaison de PALB2 à l’ADN et identifier deux domaines de liaison. Pour cela, les chercheurs ont effectué des tests d’affinité en utilisant des versions tronquées de PALB2 avec différentes structures de l’ADN et ont pu mettre en évidence deux fragments ayant une forte affinité pour les structures D-loop et l’ADN simple-brin qui s’étendaient de l’aa (1-200) pour le premier, et de l’aa (372-561) pour le deuxième. Ces résultats d’affinité avec les D-loop ont permis aux chercheurs d’appuyer la possible implication de PALB2 dans la stimulation de l’étape d’invasion. En effet, cette étude suggérerait qu’en plus de la localisation de BRCA2 aux cassures double-brin, PALB2 stimulerait l’invasion par RAD51 et stabiliserait la formation du filament nucléoprotéique lors de l’étape synaptique de la RH.27
15
Figure 4 : Domaines protéiques de PALB2 et leur fonction dans la régulation de sa fonction.
Tirée de Ducy et al. (2018). 21 (A-G) Voir texte pour la régulation de PALB2 par le cycle cellulaire et ces domaines protéiques.
16
C. La mutation de PALB2, gène suppresseur de tumeurs, dans le cancer du sein et
l’Anémie de Fanconi
Après l’identification de BRCA1/2 comme deux gènes de prédisposition au cancer du sein dont l’expression était dans un même complexe mais dont on ignorait le lien, PALB2 a été identifiée comme troisième candidat de prédisposition à cette pathologie permettant d’établir le lien entre ces deux dernières. 24,47–49En effet, les individus porteurs de mutations dans PALB2 présentent un risque élevé de développer le cancer du sein. Ces dernières exposent les personnes âgées de moins de 40 ans à un risque relatif de 8-9 et de 6-8 fois plus élevé pour ceux âgés entre 40 et 60 ans et à un risque 5 fois plus élevé pour les individus de plus de 60 ans.50 Le risque de développer le cancer du sein s’étend aussi aux individus de sexe masculin porteurs de mutations dans PALB2 qui sont exposés à un risque 8.3 fois plus élevé relativement à la population masculine générale.50
Additionnement, il a été démontré que les familles porteuses d’une mutation germinale dans PALB2 présentaient un risque 9.5 fois plus élevé de développer le cancer du sein. Alors que PALB2 était considéré comme un gène à risque modéré, aujourd’hui les individus porteurs de mutation dans PALB2 sont exposés à un risque élevé de développer le cancer du sein similairement aux porteurs de mutations dans BRCA1/2. 50
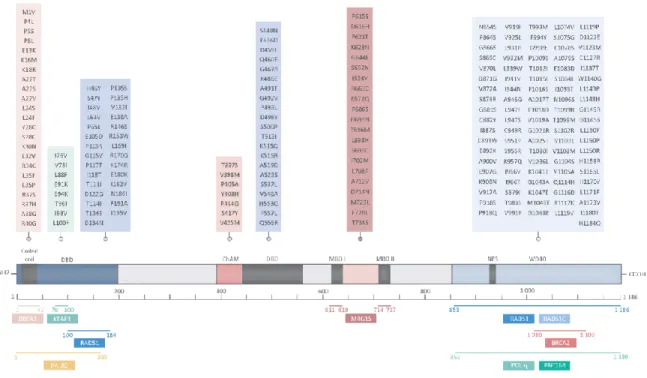
L’étude de l’implication de PALB2 dans le cancer du sein et l’évaluation de la pathogénicité des variants génétique de signification inconnue (VUS) représente un vrai défi. En effet, la plupart des études d’association de PALB2 au cancer du sein se sont concentrées sur les mutations délétères alors que plusieurs VUS présents dans tous les domaines fonctionnels de PALB2 (Figure 5) ont été retrouvés chez des patients ayant le cancer du sein. Il serait donc important qu’en plus de leur répertoriage dans des bases de données de développer des tests fonctionnels pour permettre l’évaluation de leur effet sur la fonction de PALB2 et mieux comprendre leur implication dans le cancer du sein.
En plus de son implication dans le cancer du sein, PALB2 fait partie d’un groupe de protéines appelées FANC (protéine du Groupe N d’Anémie de Fanconi) dont les variants pathogènes en homozygotie causent le syndrome de l’Anémie de Fanconi. Cette maladie est caractérisée par une insuffisance médullaire héréditaire, des malformations congénitales et un risque élevé de développer le cancer.51,52 En fait, il existe plusieurs groupes de protéines FANC faisant partie de la voie de réparation de l’Anémie de Fanconi, dont la fonction est la réparation des pontages inter-brins ICL. Conséquemment, les patients ayant une mutation bi-allélique dans PALB2 ont un défaut de réparation de ce type de dommages et développent cette maladie. 53,54
17
Figure 5 : Mutations faux sens dans PALB2 retrouvées chez des patients atteints du cancer du sein. Tirée de Ducy et al. (2018).21
3. Ciblage des défauts de recombinaison homologue en thérapie contre le
cancer
Les traitements conventionnels de chimiothérapie utilisés en clinique pour différents types de cancers génèrent à court et long terme plusieurs effets secondaires. En fait, malgré l’efficacité de cette thérapie, ces effets secondaires ajoutent aux patients souffrants de l’inconfort, parfois des intolérances, ainsi que des séquelles qui inquiètent les patients mais aussi les cliniciens. 55 C’est la question sur laquelle plusieurs recherches se sont penchées afin de venir à bout de ces limites, mais surtout pour résoudre la majeure problématique de ces traitements qui est sa non sélectivité. En effet, la chimiothérapie dont le principe est le ciblage des cellules qui se divisent rapidement, affectent aussi les cellules non cancéreuses du corps ayant aussi cette caractéristique, ce qui représente une grosse limite de ces thérapies conventionnelles.
18
3.1. Stratégie de létalité synthétique
La létalité synthétique est une nouvelle stratégie très prometteuse qui peut répondre aux limites que présentent les thérapies conventionnelles et plus précisément la non-sélectivité des drogues en chimiothérapie. Cette stratégie spécifique a été initialement décrite par le généticien Calvin Bridges chez la drosophile, où l’inactivation d’un seul gène n’avait peu ou pas d’effet, alors que l’inactivation de deux de ces gènes se traduisait par un défaut dans la cellule ou l’organisme.194 D’autre part, les études de criblage à grande échelle des interactions génétiques chez la levure Saccharomyces
Cerevisiae ont non seulement permis de mieux comprendre la structure globale des réseaux
biologiques, mais aussi de cartographier plusieurs interactions synthétiques léthales impliquant des gènes de la réparation de l’ADN ou encore des gènes codants pour des protéines de ségrégation chromosomique lors de la division cellulaire.195-196 Ce concept a ensuite été convoité dans différents domaines, dont la recherche thérapeutique contre le cancer, où l’on teste si l’inactivation d’un gène ou l’inhibition d’une molécule serait létale en combinaison avec un défaut manifesté par la cellule cancéreuse. En d’autres termes, il s’agit d’une inactivation combinée de deux éléments qui provoquerait une instabilité génétique et conduirait la cellule cancéreuse à la mort, alors que l’inactivation individuelle de ces deux éléments pourrait lui être avantageuse permettant sa survie (Figure 6). 56,57Ce concept a déclenché une course dans le domaine de la recherche sur le cancer afin d’identifier des interactions synthétiques létales (SL) qui pourraient être de nouveaux traitements plus adaptés et spécifiques pour différents types de cancers. Les gènes des voies de réparation de l’ADN représentent de bonnes cibles dans ces stratégies, et ce dû au fait qu’elle présente différentes voies de contournement dans la prise en charge des défauts de l’ADN. 58,59Dans une cellule normale il existe plusieurs voies de réparation qui peuvent servir de voie de soutien si l’une est défectueuse, ce critère est exploité par les cellules cancéreuses qui doivent se répliquer malgré leur déficience dans une voie de réparation donnée. 60En fait, les cellules cancéreuses peuvent accumuler des défauts dans une voie de réparation pour favoriser leur prolifération, mais aussi pour échapper à la mort en devenant dépendantes d’une autre voie de soutien, et c’est ce trait de dépendance qui est ciblé par les stratégies de létalité synthétique. 61,62
19
Figure 6 : Concept de la létalité synthétique. Tirée de Neil et al. (2017).63
(a) Cette partie démontre qu’en présence d’une copie intact d’un des deux gènes A ou B, en combinaison avec la mutation ou la surexpression du deuxième, cela résulte en un état viable. Tandis que la mutation ou l’inhibition du produit protéique (partie b et c, respectivement) du gène B dans les cellules présentant une mutation (parties b, c) ou une surexpression (partie d) du gène A, entraîne une létalité synthétique. La flèche plus épaisse indique une surexpression accrue. L'étoile représente une mutation. Les croix rouges indiquent l'inhibition du produit du gène B.
3.1.1. Les PARPi en stratégie de létalité synthétique dans les cellules BRCA1/2 déficientes
L’utilisation des inhibiteurs de PARP pour les patients ayant une mutation germinale dans BRCA1/BRCA2, est la première interaction synthétique létale à entrer en clinique.61 Cette dernière consiste en l’inhibition pharmacologique des PARPs (Poly(ADP-ribose) polymérases, essentiellement PARP1) dont les cellules cancéreuses déficientes en BRCA1/BRCA2 sont dépendantes. La Poly(ADP-ribose) polymérase 1/ 2 PARP 1 et 2 sont des transducteurs de signal dans la réponse aux dommages de l’ADN. En fait, après la détection d’une CSB ou d’autres dommages, les PARPs vont synthétiser des chaines poly (ADP-ribose) PAR, cette parylation sert à
20
modifier post-traductionnellement plusieurs protéines de la réponse aux dommages de l’ADN.67–69 Il existe maintenant plusieurs inhibiteurs de PARP, dont trois (olaparib, rucaparib et niraparib) qui ont été approuvés en clinique pour le cancer de l’ovaire et l’un d’eux (l’olaparib) pour le cancer du sein déficient en BRCA.64–66 Ainsi, l’inhibition de PARP dans des cellules ayant des protéines BRCA non fonctionnelles est supposée conduire à l’accumulation de cassures DBs, l’effondrement des fourches de réplication et donc engendrer une instabilité génétique qui peut être létale pour la cellule cancéreuse sans pour autant affecter les cellules normales ayant une RH fonctionnelle qui peut prendre en charge les CDBs (Figure 7).70–72
Le modèle initial proposé pour le mécanisme d’action des PARPi, propose que l’inhibition de PARP empêche la réparation des CSBs causant leur conversion en des CDBs, celles-ci résulteraient en l’effondrement de la fourche de réplication qui est très toxique pour la cellule, conduisant à des catastrophes mitotiques et à l’apoptose, si celles-ci ne sont pas réparées. 61, 62,73 Des études ont ensuite mis à jour ce modèle en démontrant que certains inhibiteurs de PARP, appelés trapping PARPi, piégeaient PARP1 dans l’ADN empêchant ainsi son autoPARylation et sa libération du site du dommage provoquant des lésions cytotoxique. 74 Le trapping des PARPi peut différer d’un agent à un autre, par exemple le talazoparib est connu pour avoir une capacité de trapping qui est 100 fois plus efficace que le niraparib, et qui à son tour est plus efficace que l’olaparib et le rucaparib dans le
trapping de PARP1. Ces derniers sont considérés plus efficaces que les PARPi inhibant la
PARylation tel que le veliparib dans les cellules mutées dans BRCA.75,76
Additionnellement aux dommages et la cytotoxicité causés par les PARPi, PARP1 et 2 sont impliqués dans la transcription, l’apoptose et la fonction immunitaire ce qui pourrait aussi participer dans leur potentiel anti-tumoral. 77–80
Le traitement aux PARPi génère dans certains cas, comme la plupart des thérapies ciblées, des formes de résistances. 81 Parmi ces résistances certaines ont été identifiées in vitro chez des patients ayant des cancers à stage avancé. Ces mécanismes de résistance incluent la restauration partielle de la RH par l’inactivation des protéines de réparation 53BP1 ou REV7, ou encore la perte d’expression de PARP1 qui résulte en une résistance aux inhibiteurs.82 La restauration de l’expression de BRCA1/2 par des mutations qui rétablissent le cadre de lecture dans ces gènes est la seule forme de résistance qui a été validée cliniquement chez certains patients. 83Cette forme de résistance génère des protéines de nouveau fonctionnelles dans les cellules auparavant BRCA déficientes et parviendrait à rétablir la RH, ce qui se traduit en une résistance aux inhibiteurs de PARP.
21
Les inhibiteurs de PARPs ont aussi démontré leur efficacité pour les tumeurs n’ayant pas de mutations germinales dans les gènes BRCA, mais partageant certains phénotypes avec ces dernières. Ces tumeurs appelées BRCAness, sont aussi caractérisées par des défauts dans la RH qui peuvent être dus à des mutations somatiques dans BRCA, telle que la mutation somatique causant une hyperméthylation du promoteur de BRCA1, mais aussi des mutations dans d’autres gènes de la RH qui causeraient des défauts dans la RH et qui phéno-copieraient l’effet des mutations germinales dans BRCA. Ce qui pourrait élargir le spectre d’utilisation des inhibiteurs de PARP pour les tumeurs dites BRCAness. 84,85
Figure 7 : L’implication de PARP dans la réparation de l’ADN et son inhibition en stratégie de létalité synthétique dans les cellules BRCA déficientes. Tirée de
Dziadkowiec et al. (2016).
71 A) Représentation simplifiée des étapes de réparation d’une CSB faisant intervenir une protéine PARP fonctionnelle pour le recrutement des protéines de réparation dans des cellules normales. B) Inhibition de PARP en présence d’une CSB et sa conversion en une CDB. Cette conséquence peut être prise en charge dans des cellules exprimant des protéines BRCA fonctionnelles, alors que dans des cellules déficientes, celle si peut entrainer une accumulation de dommages qui peuvent conduire à la mort.22
3.1.2. Stratégie de létalité synthétique dans les cellules PALB2 déficiente
Les efforts fournis dans le criblage de nouvelles interactions SL autres que PARP et BRCA, pourraient être justifiés par deux raisons. La première étant le nombre important de cancers déficients en RH ayant des protéines BRCA fonctionnelles et qui partagent les mêmes caractéristiques moléculaires que les cancers BRCA déficients. Telle que l’implication de PALB2 dans le cancer du sein qui est un bon exemple de tumeur qui phénocopie le cancer du sein déficient en BRCA. 85La deuxième raison est la résistance aux PARPi qui se manifeste chez des patients diagnostiqués pour bien répondre à cette thérapie mais dont les cellules cancéreuses ciblées sont transformées par l’un des mécanismes cité plus haut.82,83 Dans cette optique, les PARPi ont aussi démontré leur efficacité
in vitro dans les tumeurs BRCAness incluant les cellules PALB2 déficientes. En effet, des études in vitro ont démontré que l’Olaparib et le Talazoparib seuls ou en combinaison avec le temolozomide
étaient synthétiques létaux dans un contexte cellulaire déficient en PALB2.27,86,87 Ces résultats étaient très prometteurs pour la prise en charge des tumeurs PALB2 déficientes et ont poussé la recherche d’autres interactions synthétiques létales qui pourraient présenter une alternative dans les cas de résistance. Cependant, les études mettant en évidence la sensibilité des cellules PALB2 déficientes aux PARPi sont des études in vitro préliminaires qui devraient être validées en clinique avant que les patients puissent en bénéficier. Ce type de tumeurs BRCAness qui phéno-copient les cellules BRCA déficientes restent quand même différentes de ces dernières. Un exemple de cette différence est le niveau d’expression des PARPs dans ces cellules qui peut être différent d’une tumeur à une autre, et qui peut conséquemment se traduire en des niveaux sensibilité aux PARPi qui seraient de même différents.88,89 Dans ce contexte, des essais de diagnostic moléculaire ont été développés pour pouvoir prédire l’efficacité des PARPi dans les tumeurs déficientes en PALB2 ou d’autres gènes de RH.90 Comme précédemment expliqué la létalité synthétique des PARPi dans les tumeurs BRCA ou BRCAness est essentiellement due à la déficience en RH. De ce fait, une étude a établi un test qui permettrait de prédire l’efficacité des PARPi, et ce en attribuant des scores à la quantification de foyers RAD51 par immunofluorescence dans les tumeurs.90 Ces études ont démontré une corrélation entre le score de foyers RAD51 de cellules dérivées de xénogreffes PDX déficientes en PALB2 et leur degré de déficience en RH. Proposant ainsi ce test en routine clinique pour prédire la sensibilité ou la résistance des cancers du sein déficients en PALB2.
L’une de ces nouvelles interactions synthétiques létales dans les cellules BRCA déficientes, est celle résultante de l’inactivation de la protéine RAD52.91 Cette dernière intervient dans la voie de soutien de la RH qui est la « SSA », décrite plus haut, mais a aussi été prouvé être le médiateur de la voie alternative pour le recrutement de RAD51 et l’initiation de l’invasion de brin dans les cellules
23
BRCA2 déficientes.20,91,92 Ainsi, plusieurs études ont démontré l’interaction synthétique létale existante après
l’inactivation de RAD52 dans les cellules BRCA et PALB2 déficientes. 91,93,94 Ces résultats ont poussé les chercheurs à effectuer des criblages de drogues à haut débit pour l’identification d’inhibiteurs de RAD52.95,96 Ces drogues identifiées étaient efficaces dans les essais impliquant des cellules BRCA déficientes. Etant donné la relation étroite que partage la déficience en BRCA et PALB2 il serait intéressant d’investiguer cette interaction SL dans les cellules PALB2 déficientes et de rechercher des inhibiteurs adaptés pour ce type de tumeurs.
Ainsi, le développement et le choix de bons modèles in vitro qui faciliteraient l’étude de ces stratégies thérapeutiques et qui prennent en considération l’environnement tumoral retrouvé dans les cancers, représente une étape critique dans la recherche thérapeutique contre le cancer.
4. La modélisation de tumeurs in vitro pour les essais thérapeutiques
Malgré les efforts fournis par l’industrie pharmacologique pour le développement des traitements existants ou la production de nouvelles drogues pour le cancer, peu ont aboutis. En effet, le nombre important de drogues prometteuses testées dans les cellules tumorales in vitro mais qui sont par la suite rejetées en phase II ou III de validation clinique représente une grande limite.97 Cette dernière est souvent due à plusieurs facteurs incluant la stabilité et la toxicité des molécules, mais l’utilisation des cellules en monocouche lors des évaluations in vitro représente parfois une limite importante pour la traduction de ces traitements en clinique. En fait, les essais conduits sur les cellules cultivées en monocouche fournissent parfois des résultats qui sont difficiles à reproduire en clinique, car les cellules en 2D ne sont souvent pas représentatives de la structure, de l’hétérogénéité, ni de la complexité des tumeurs qui sont présentes dans un environnement tridimensionnel in vivo. 98,99 Ainsi, les modèles cellulaires 3D représente une bonne alternative qui pourrait mettre fin à ce type de limites.
Le modèle de culture in vitro 3D n’est pas un nouveau concept mais une idée développée par Alexi Carrel en 1912 et qui a pu cultiver un fragment d’embryon de poulet et l’a maintenu pendant 3 mois en culture.100 Ce qui a été suivi par plusieurs autres études qui ont optimisé cette culture 3D en utilisant différents supports, ce qui a permis son utilisation dans la compréhension de certains processus biologiques, comme le mécanisme gouvernant la gastrulation.101 L’intérêt pour ce type de culture a augmenté au cours des années pour aboutir à son adoption dans la recherche sur le cancer. La majeure raison de cet intérêt dans la recherche sur le cancer est leur bonne représentation des tumeurs. En fait, ces modèles récapitulent d’une meilleure façon les caractéristiques tumorales