L'utilisation du système CRISPR-Cas9 pour l'étude des
protéines non structurales du bactériophage 2972
infectant
Streptococcus thermophilus
Mémoire
Ariane Renaud
Maîtrise en microbiologie - avec mémoire
Maître ès sciences (M. Sc.)
Québec, Canada
L’utilisation du système CRISPR-Cas9 pour
l’étude des protéines non structurales du
bactériophage 2972 infectant Streptococcus
thermophilus
Mémoire
Ariane Renaud
Sous la direction de :
iii
RÉSUMÉ
Les bactériophages sont des maîtres manipulateurs, prenant le contrôle complet d’une cellule bactérienne, contournant les systèmes de défense et détournant la machinerie de transcription et de traduction du génome bactérien pour la réplication virale. Les grandes étapes de la multiplication des phages sont connues, pourtant les mécanismes intrinsèques de cette prise de contrôle restent un mystère bien préservé. Malgré la petite taille et la simplicité relative des génomes de phages, seuls les gènes associés à la structure des virions sont amplement caractérisés. Toutefois, les protéines non structurales qui sont susceptibles d’être responsables de la prise de contrôle sont rarement étudiées. C’est effectivement le cas pour le phage modèle 2972, infectant la souche Streptococcus thermophilus DGCC7710 largement utilisée en industrie laitière. Son génome code pour 44 protéines putatives, dont 14 protéines sont classées comme étant non structurales et dont aucune fonction n’est encore associée. Lors de ce projet de maîtrise, le système CRISPR-Cas de type II-A, naturellement présent chez S. thermophilus, a été utilisé à des fins d’édition du génome de ce phage pour étudier le rôle des protéines non structurales. Ce système est idéal pour les manipulations génétiques des phages virulents généralement complexes à modifier. Ainsi, une connaissance approfondie des interactions entre le phage et son hôte sera un outil indispensable pour développer de meilleures méthodes de contrôle des phages en industrie laitière.
iv
ABSTRACT
Bacterial viruses are master manipulators of bacterial cells. They are able to take complete control of a bacterium, bypassing bacterial immune systems, hijacking core transcription and translation machinery, and typically resulting in lysis of the host. Although the major steps of phage replication are well understood, very little is known about the mechanisms of the host-cell takeover. Despite phages having relatively small and ‘simple’ genomes, generally only the structural proteins have been well characterized. In contrast, non-structural proteins, which include those involved in host cell takeover, tend to be completely uncharacterized.
This is certainly the case for the model of Streptococcus thermophilus phages, 2972, which infects the strain DGCC7710 widely used by the dairy industry. Its genome encodes for 44 putative proteins, 14 of which are non-structural and have no known function. In this master thesis, the type II-A CRISPR-Cas system naturally present in S. thermophilus was used for genome engineering purposes to investigate the role of non-structural proteins of phage 2972. This natural bacterial immune system provides an ideal means for genetic manipulation of virulent phages, which are otherwise intractable. This could lead to potentially valuable discoveries allowing us to further fine-tune the bacteria used in various biological processes.
v
TABLE DES MATIÈRES
RÉSUMÉ ... iii
ABSTRACT ... iv
LISTE DES FIGURES ... vii
LISTE DES TABLEAUX ... viii
REMERCIEMENTS ... xii
Introduction ... 1
1.1 Les bactériophages ... 1
1.1.1 Classification et morphologie ... 1
1.1.2 Mode d’infection ... 2
1.1.3 Les phages de Streptococcus thermophilus ... 7
1.1.4 Le phage 2972 ... 9
1.2 Les bactéries lactiques ... 11
1.2.1 Importance industrielle ... 12
1.2.2 Le genre Streptococcus ... 12
1.2.3 Sensibilité aux phages ... 14
1.2.4 Les mécanismes de défense ... 15
1.3 Les systèmes CRISPR ... 17
1.3.1 Composition des systèmes CRISPR-Cas ... 18
1.3.2 Classification ... 19
1.3.3 CRISPR-Cas chez Streptococcus thermophilus ... 21
1.3.4 Les mécanismes de fonctionnement ... 22
1.3.4.1 L’adaptation ... 23
1.3.4.2 La biogenèse des ARNcr ... 25
1.3.4.3 L’interférence ... 25
1.4 L’édition de génome... 27
1.5 Problématique et objectifs de recherche ... 30
CHAPITRE 2: Matériel et méthodes ... 32
2.1 Souches bactériennes, plasmides et phages utilisés ... 32
2.1.1 Conditions de culture des bactéries ... 34
2.1.2 Propagation, titration et conservation des lysats de phage ... 35
2.1.3 Transformation bactérienne et extraction de plasmides ... 36
2.2 Édition de génome du phage 2972 ... 36
2.2.1 Construction des BIM «sur demande» ... 38
2.2.2 Construction des gabarits de recombinaison ... 40
2.2.3 Génération et purification des phages mutants ... 45
2.3 Caractérisation des phages mutants ... 46
2.3.1 Courbes de croissance ... 46
2.3.2 Test d’adsorption ... 48
2.3.3 Microscopie électronique à transmission ... 48
2.3.4 Spectrométrie de masse ... 48
2.3.5 Production de BIM ... 49
2.4 Caractérisation des protéines sauvages ... 50
vi
2.4.2 Prédictions bio-informatiques ... 50
2.4.3 Dichroïsme circulaire ... 51
CHAPITRE 3: Résultats ... 53
3.1 Édition de génome du phage 2972 ... 53
3.1.1 Construction des BIM «sur demande» ... 53
3.1.2 Génération et purification des phages mutants ... 54
3.2 Caractérisation des phages mutants ... 56
3.2.1 Courbes de croissance ... 56
3.2.2 Test d’adsorption ... 58
3.2.3 Microscopie électronique à transmission ... 59
3.2.4 Spectrométrie de masse ... 60
3.2.5 Production de BIM ... 61
3.3 Caractérisation des protéines sauvages ... 65
3.3.1 Prédictions bio-informatiques ... 65
3.3.2 Dichroïsme circulaire ... 67
CHAPITRE 4: Discussion ... 72
4.1 Édition de génome du phage 2972 ... 72
4.2 Caractérisation des phages mutants ... 73
4.2.1 Le phage 2972 et 2972 RBP Courte ... 73 4.2.2 Le phage 2972Δ22... 75 4.2.3 Le phage 2972Δ24... 78 4.2.4 Les phages 2972Δ27 et 2972Δ28 ... 78 4.2.5 Le phage 2972Δ36... 79 4.2.6 Le phage 2972Δ41... 80 4.2.7 Le phage 2972Δ43... 82
4.3 Caractérisation des protéines sauvages ... 83
4.3.1 L’ORF32 ... 83
4.3.2 L’ORF40 ... 83
4.3.2 L’ORF 41 ... 84
Conclusion et perspectives ... 85
vii
LISTE DES FIGURES
Figure 1.1 : Morphologie des trois familles de l'ordre des Caudovirales. ... 2
Figure 1.2 : Le cycle lytique et lysogénique des bactériophages ... 3
Figure 1.3 : Modèle d’assemblage du bactériophage T4 ... 6
Figure 1.4 : Arbre phylogénétique montrant la divergence génétique entre le phage 5093 et cinq phages pour le groupe cos et le groupe pac... 8
Figure 1.5 : Représentation du génome du phage virulent 2972 infectant S. thermophilus ... 9
Figure 1.6 : Comparaison du génome des phages infectant Streptococcus thermophilus ... 10
Figure 1.7 : Arbre phylogénétique de 138 espèces du genre Streptococcus ... 14
Figure 1.8 : Représentation schématique des différents systèmes de défense bactériens ... 16
Figure 1.9 : Composition générale des systèmes CRISPR-Cas... 18
Figure 1.10 : Classification et organisation des types de systèmes CRISPR-Cas et domaines des protéines effectrices pour les classes 1 et 2 ... 21
Figure 1.11 : Les trois étapes de fonctionnement des systèmes CRISPR-Cas ... 23
Figure 1.12 : Applications et outils reliés à l’utilisation de la protéine Cas9 en édition de génome.29 Figure 2.1 : Programmation du système CRISPR-Cas natif………..39
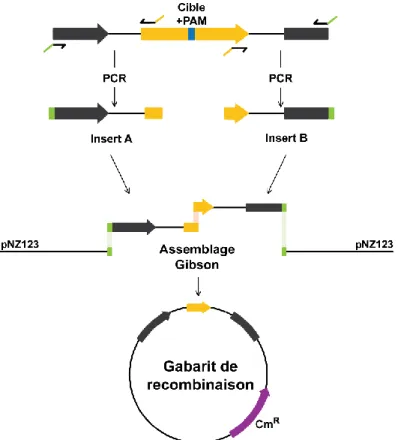
Figure 2.2 : Schématisation de la méthodologie permettant l’obtention des gabarits de recombinaison ... 41
Figure 2.3 : Exemple d’un graphique résultant d’une courbe de croissance de phage standard ... 47
Figure 2.4 : Signaux de dichroïsme circulaire émis par les différents types de structures secondaires en fonction de la longueur d’onde en nanomètre ... 52
Figure 3.1 : Migration des produits PCR des régions délétées et sauvages pour les sept gènes identifiés comme non essentiels au génome du phage 2972 ... 55
Figure 3.2 : Courbe de croissance à partir du titre relatif en fonction du temps pour cinq phages .. 57
Figure 3.3 : Photo en microscopie électronique à transmission des phages 2972 et 2972Δ22 ... 59
Figure 3.4 : Le ratio de la production de BIM avec deux phages mutants (2972Δ22 et CEM22) par rapport au phage 2972 sauvage en fonction de deux souches testées (DGCC7710 et BIM AR1) .... 62
Figure 3.5 : Le ratio de la production de BIM avec deux phages mutants (2972Δ41 et CEM41) par rapport au phage 2972 sauvage en fonction de deux souches testées (DGCC7710 et BIM A-35). .. 63
Figure 3.6 : Dispersion des proto-espaceurs visés sur le génome du phage 2972 du système CRISPR-Cas de S. thermophilus face aux phages ayant diverses mutations dans l’orf22 et l’orf41 ... 64
Figure 3.7 : Prédiction bio-informatique de la structure secondaire de l’ORF32 réalisée avec le logiciel I-TASSER ... 66
Figure 3.8 : Prédiction bio-informatique de la structure secondaire de l’ORF40 réalisée avec le logiciel QUARK (171). ... 66
Figure 3.9 : Prédiction bio-informatique de la structure secondaire de l’ORF41 réalisée avec le logiciel I-TASSER (171). ... 67
Figure 3.10 : Analyse par dichroïsme circulaire de la protéine ORF32 ... 68
Figure 3.11 : Courbe de dénaturation de la protéine ORF32. ... 69
Figure 3.12 : Analyse par dichroïsme circulaire de la protéine ORF40 ... 69
viii
LISTE DES TABLEAUX
Tableau 2.1 : Liste des souches bactériennes, des phages et des plasmides utilisés lors de l’étude et
leurs caractéristiques. ... 32
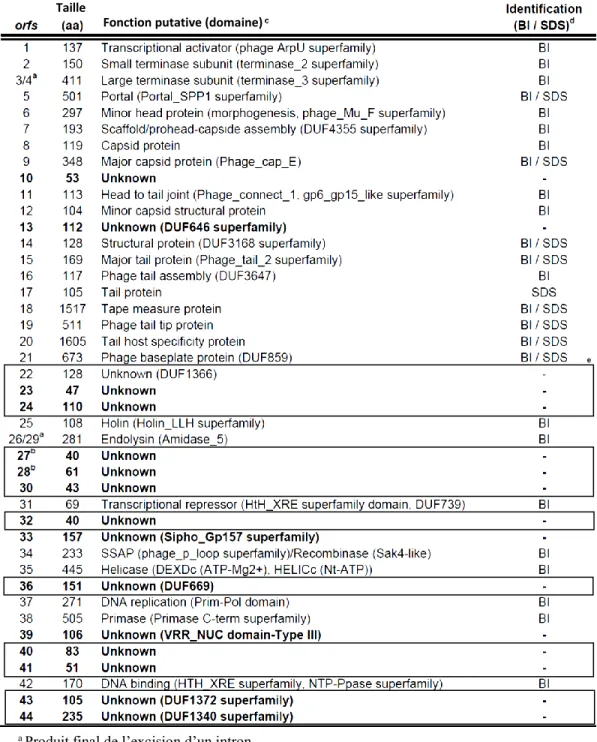
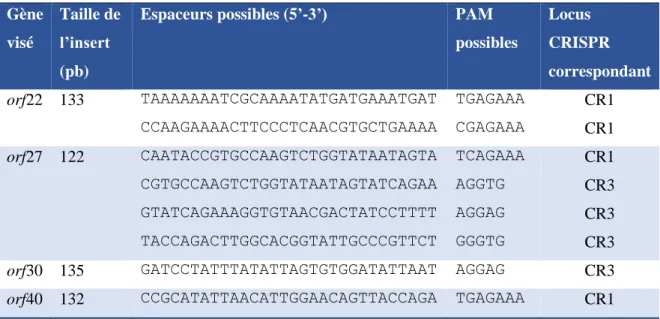
Tableau 2.2 : Les orfs prédits du phage 2972 ainsi que leurs fonctions putatives (48)... 37 Tableau 2.3 : Espaceurs possibles visés lors de la programmation du système CRISPR-Cas. ... 38 Tableau 2.4 : Informations sur les constructions génétiques pour chaque gène visé ainsi que sur les
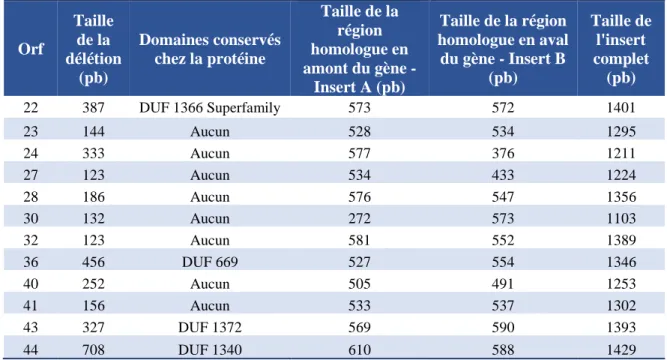
domaines conservés chez les protéines résultantes. ... 40
Tableau 2.5 : Oligonucléotides utilisés lors cette de recherche ainsi que leur utilisation. ... 42 Tableau 3.1 : Espaceurs intégrés dans les locus CRISPR1 ou CRISPR3 de la souche S. thermophilus
DGCC7710 suite à la programmation du système CRISPR-Cas natif………53
Tableau 3.2 : EOP du phage 2972 sur les BIM construits suite à la programmation du système
CRISPR-Cas. ... 53
Tableau 3.3 : Information génétique sur les gènes à l’étude du phage 2972. ... 54 Tableau 3.4 : Résultats du séquençage Illumina pour les phages 2972, 2972Δ22, 2972Δ28, 2972Δ41
et 2972Δ43. ... 56
Tableau 3.5 : Données correspondantes aux courbes de croissance des phages 2972, 2972 RBP
courte, 2972Δ22, 2972Δ41 et 2972Δ43. ... 58
Tableau 3.6 : Pourcentage d’adsorption après 10 minutes des phages 2972, 2972Δ22 et 2972Δ41 sur
la souche S. thermophilus DGCC7710. ... 58
Tableau 3.7 : Mesure des dimensions de la queue et de la capside du phage 2972 et du phage
2972Δ22. ... 60
Tableau 3.8 : Les protéines du phage 2972 détectées par spectrométrie de masse. ... 60 Tableau 3.9 : Capacité de production de BIM avec la souche DGCC7710 et le BIM AR1 contre les
phages 2972, 2972Δ22 et CEM22. ... 62
Tableau 3.10 : Capacité de production de BIM avec la souche DGCC7710 et le BIM A-35 contre les
phages 2972, 2972Δ41 et CEM41. ... 63
Tableau 3.11 : Résultats du calcul des structures secondaires en pourcentage de l’ORF32 à 25 °C
selon différentes bases de données et méthodes de calcul. ... 70
Tableau 3.12 : Résultats du calcul des structures secondaires en pourcentage de l’ORF40 à 25 °C
ix
LISTE DES ABRÉVIATIONS
a.a. Acide aminé
Abi Abortive infection mecanism (Mécanisme d’avortement de l’infection)
ADN Acide Désoxyribonucléique
ADNsb ADN simple brin
ARN Acide ribonucléique
ARNcr ARN CRISPR
ARNpré-cr ARN précusseur
ARNtracr ARN transactivateur des ARNcr
ATP Adénosine Triphosphate
BIM Bacteriophage Insensitive Mutant (Mutant résistant aux bactériophages)
Cas CRISPR associated (associé au système CRISPR)
CEM CRISPR Escaping Mutant (Mutant échappant au système CRISPR)
CHUL Centre Hospitalier de l’Université Laval
CRISPR Clustered Regularly Interspaced Short Palindromic Repeats (Agglomération de courtes répétitions palindromiques espacées par intervalle régulier)
dCas9 Dead Cas9 (Cas9 catalytiquement inactive)
DO600 Densité optique à une longueur d’onde de 600 nm DSB Double Stranded Break (Bris double brin)
DUF Domain of Unknown Function (Domaine de function inconnu)
EOP Efficiency of Plaquing (Efficacité d’infection)
EPS Exopolysaccharides
GRAS Generally Recognized as Safe (Reconnue comme sécuritaire)
HIF Host Intergration Factor (Facteur d’intégration de l’hôte)
BL Bactérie lactique
MOI Mutiplicity of Infection (Mutiplicité d’infection)
NAG N-acetylglucosamine
NAM Acide N-acetyl muramique
nCas9 Nickase Cas9 (Cas nickase)
NHEJ Non-Homologous End Joining
x
NmCas9 Cas9 de Neisseria meningitidis
nt. Nucléotide
Orf Open Reading Frame (Cadre de lecture ouvert)
PAM Protospacer Adjacent Motif (Motif adjacent au proto-espaceur)
pb Paire de bases
PCR Polymerase Chain Reaction (Réaction en chaine de la polymérase)
RBP Receptor Binding Protein (Protéine liant le récepteur)
RH Recombinaison homologue
rpm Rotation par minute
Sie Super infection exclusion (Exclusion de super-infection)
SpCas9 Cas9 de Streptococcus pyogenes
TALEN Transcription activator-like effectors nuclease (Nucléase semblable aux activateurs de la transcription)
TMP Tape Measure Protein (Protéine de mesure étalon)
UFP Unité formatrice de plage de lyse
xi
« Almost all aspects of life are engineered at a molecular level, and
without understanding molecules we can only have a very sketchy
understanding of life itself »
xii
REMERCIEMENTS
Je tiens à remercier mon directeur de recherche, le professeur Sylvain Moineau, de m’avoir donné l’occasion de travailler dans un laboratoire d’une grande expertise avec une équipe motivante et soudée. Mon amour pour les phages a commencé lorsque j’étais laborantine et j’ai pu ensuite découvrir ce monde merveilleux lors d’un stage de recherche et enfin, une maîtrise. Mes années passées dans le laboratoire du Prof. Moineau m’ont appris à repousser mes limites et à travailler pour ce qu’on aime. J’aimerais souligner l’appui des professionnelles de recherche Denise Tremblay et Geneviève Rousseau qui ont répondu à mes millions de questions chaque jour. Grâce aux membres de l’équipe, plus particulièrement Moïra, Marie-Laurence, Stéphanie, Rachel, Cécile, Alessandra, Alexander, Witold, Simon, Alice, Cas et Hany, j’ai trouvé ma deuxième famille et c’est avec cette famille que j’ai pu avancer dans la science !
Je voudrais aussi remercier les membres de mon comité aviseur. D’abord, Alexander Culley pour les conseils et le soutien lors de mon projet et Stéphane gagné pour son aide précieuse lorsqu’une microbiologiste s’aventure dans le monde de la détermination de structure des protéines.
Ensuite, j’aimerais remercier toutes les personnes présentes à mes côtés lors de mes études. Plus particulièrement mes parents et ma sœur, Gabrielle, pour le soutien moral et l’approvisionnement en café lors de mon écriture. Merci de m’avoir toujours encouragé dans mes projets.
Finalement, je désire remercier le regroupement PROTEO et le Conseil de recherche en sciences naturelles et en génie pour le soutien financier, ainsi que le Groupe de recherche en écologie buccale pour les locaux, les équipements et les évènements.
1
Introduction
1.1 Les bactériophages
Les bactériophages sont les entités biologiques les plus variées et nombreuses sur la planète (1). Il est estimé que le nombre de phages sur terre se situe entre 1030 et 1032, ce qui est dix fois plus que le
nombre de cellules bactériennes (2). Ils ont un impact écologique immense, notamment sur le recyclage des éléments nutritifs des cycles biogéochimiques et sur le maintien de la diversité microbienne (3, 4).
En 100 ans de recherche, les phages ont été impliqués dans de nombreuses de découvertes biologiques, contribuant à l’avancée significative de la génétique et de la biologie moléculaire (5). Les phages possèdent énormément de diversité morphologique et génétique, leur conférant une gamme de caractéristiques très diverse et répandue. L’étude des phages a notamment permis de découvrir que les attributs héréditaires sont transmis par l’ADN et non les protéines, que les phages sont responsables de conférer des facteurs de virulence à certaines bactéries pathogènes et que le code génétique est constitué de codons composés de trois nucléotides (5, 6). Les phages sont aussi au cœur de plusieurs technologies dont la thérapie par les phages et l’exposition sur phage (phage display) (7, 8). Les phages étant les entités biologiques les plus influentes sur Terre, la recherche qui en découle joue donc un rôle de haut niveau dans les avancées présentes et futures.
1.1.1 Classification et morphologie
Tous les virus sont construits selon le même principe, où une molécule génétique composée d’acide désoxyribonucléique (ADN) ou d’acide ribonucléique (ARN), sous forme double ou simple brin, est protégée par une enveloppe protéique. La forme, la taille et la composition de cette enveloppe varient grandement et elle est souvent constituée de sous-unités identiques qui respectent des règles géométriques très strictes (9). Les virus infectant les procaryotes peuvent avoir une morphologie polyédrique, filamenteuse, pléomorphique ou avec une queue (10). La classification de ces particules virales se fait principalement par la distinction morphologique de l’enveloppe protéique et par la différence du contenu génétique selon Bradley en 1967 (9). L’ordre le plus nombreux est celui des
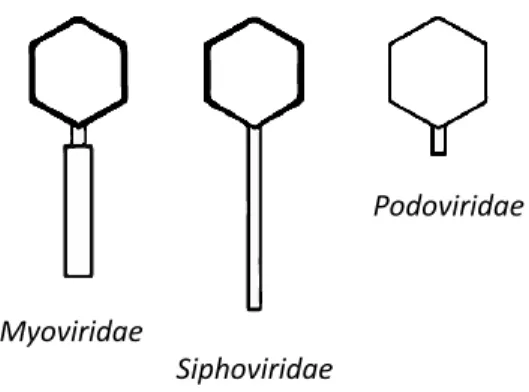
Caudovirales où 96,3 % des phages observés peuvent y être assignés, comprenant trois familles, soit
les Myoviridae, les Siphoviridae et les Podoviridae (figure 1.1), ayant une prévalence de respectivement 24,8%, 57,3% et 14,2% (10). Tous les phages appartenant à cet ordre possèdent une
2
tête icosaédrique, ou capside, formée de capsomères, une queue et un génome d’ADN double brin (11). Certains phages parmi ceux-ci peuvent être dotés d’appendices supplémentaires tels des fibres caudales ou des plaques basales. C’est la morphologie de la queue qui permet de distinguer les différentes familles. Les phages de la famille des Myoviridae ont une queue gainée qui a la capacité de se contracter et qui est assez large, contrairement à ceux de la famille des Podoviridae qui ont une queue très courte et non contractile. Pour ce qui est des phages de la famille des Siphoviridae, ils possèdent une queue plus longue, non contractile et moins large que celle des Myoviridae (11–13).
Figure 1.1 : Morphologiedes trois familles de l'ordre des Caudovirales. Adaptée de Ackermann, 2003 (12).
1.1.2 Mode d’infection
Les bactériophages ont plusieurs modes d’infection. En général, ils peuvent être virulents ou tempérés, et donc compléter respectivement un cycle lytique ou un cycle lysogénique (figure 1.2). Le cycle lytique comprend six grandes étapes. D’abord, il y a reconnaissance et adsorption du phage à la surface cellulaire. Ensuite, le phage injecte son ADN à l’intérieur du cytoplasme de la cellule, où celui-ci prend contrôle de la bactérie pour la réplication de son propre génome. Puis, il y a transcription et traduction des protéines du bactériophage. Les protéines structurales serviront à former les composantes nécessaires à l’assemblage des virions. Les éléments de la capside et de la queue s’associent indépendamment l’un de l’autre pour permettre à l’ADN d’entrer dans la capside. Le génome sera empaqueté dans la capside vide, puis les virions complets seront assemblés et libérés par lyse cellulaire (4). Pour ce qui est des bactériophages tempérés, ils vont s’intégrer au génome bactérien suite à l’entrée de l’ADN dans la cellule pour entamer un cycle lysogénique. Ils pourront être transmis aux cellules filles sous forme de prophage, jusqu’à ce qu’un stress provoque son excision et que le cycle lytique soit engagé (3, 4).
Podoviridae
Siphoviridae Myoviridae
3
L’étape initiale de l’adsorption implique les protéines de la queue du phage (fibres, plaque basale, etc.) qui reconnaîtront un récepteur à la surface bactérienne. Les récepteurs sont variables en fonction du type de bactérie (Gram positif ou Gram négatif) et sont généralement des protéines membranaires, des polysaccharides ou des composantes du peptidoglycane, tels les acides téichoïques et lipotéichoiques (14, 15). Le type de récepteur reconnu est spécifique à chaque phage, ce qui détermine son spectre lytique. Certains phages vont aussi posséder des enzymes associées à leur queue pour dégrader les constituants de la paroi cellulaire et permettre une meilleure adsorption (16). L’attachement initial à la cellule hôte est réversible jusqu’à ce qu’un changement de conformation des protéines de reconnaissance rendent le lien irréversible, assurant le début du cycle lytique (14).
Figure 1.2 : Le cycle lytique (à gauche) et lysogénique (embranchement à droite) des bactériophages. Adaptée de Labrie et al., 2010 (17).
Puis, lorsque cet attachement devient irréversible, le cycle lytique peut alors commencer. La queue du bactériophage va subir un changement de conformation majeur, lui permettant d’injecter son génome à travers la membrane bactérienne. Les phages appartenant à des familles différentes n’auront pas les mêmes mécanismes d’injection de l’ADN dû aux composantes de la queue qui sont distinctes entre ces familles. Les Myoviridae ont deux composantes majeures qui jouent un rôle important lors de cette étape, soit le tube interne rigide qui connecte la capside du phage au cytoplasme de la bactérie
4
et la gaine contractile qui entoure le tube interne (18). Lors de l’attachement du phage par la plaque basale ou les fibres caudales, celle-ci va entamer un changement de conformation qui provoque la contraction de la gaine de la queue. Ensuite, le tube interne, grâce à sa nature rigide, va percer l’enveloppe cellulaire pour permettre la translocation de l’ADN et des protéines contenues dans la capside à l’intérieur de la bactérie (19). Les phages appartenant à la famille des Siphoviridae ont une stratégie plutôt différente. Les détails du mécanisme de translocation de l’ADN n’ont pas encore été élucidés, en revanche il a été établi que la protéine majeure étalon (TMP) joue un rôle très important. Suite à la reconnaissance de la surface bactérienne par les fibres de la queue ou les protéines se liant aux récepteurs (RBP), un changement de conformation de ce complexe protéique force le contact du tube interne de la queue sur la paroi cellulaire. La liaison de ces deux structures engendre l’éjection de l’ADN, qui force la sortie de la TMP, contenue à l’intérieur du tube rigide et crée ainsi un tunnel pour l’ADN jusqu’au cytoplasme (20). Pour ce qui est des Podoviridae, comme cette famille est définie par une queue très courte, beaucoup de phages ont des RBP qui possèdent une activité enzymatique permettant au phage de percer un canal jusqu’à la surface bactérienne (21). Il y a souvent une digestion du récepteur primaire, fréquemment des polysaccharides, pour générer l’accès au récepteur secondaire. Suite à la liaison irréversible du phage, les protéines internes de la queue qui forme la machinerie d’éjection complexe vont former un passage à travers la paroi. L’ADN encapsidé se trouve dans un état énergétique non favorable, créant une extrême pression qui va donc forcer son éjection dans le cytoplasme (22, 23).
L’ADN contenu dans la capside se trouve sous forme linéaire, toutefois la majorité des génomes de phages vont prendre une forme circulaire dès l’entrée dans le cytoplasme. La circularisation assure la transcription des extrémités du génome, cependant plusieurs stratégies ont été développées par les phages se répliquant de manière linéaire pour contrer ce problème. Par exemple, le phage T7 possède des extrémités répétées qui seront régénérées lors de l’empaquetage de l’ADN, ce qui empêche la perte de matériel génétique (24). Quant à eux, les génomes circulaires ont un type de réplication thêta (θ) ou en cercle roulant.
Ensuite, les différents modules de l’architecture des génomes sont transcrits et traduits selon leur expression temporelle, soit les gènes précoces, médians et tardifs, en fonction des étapes du cycle infectieux (figure 1.2). Les gènes précoces et médians codent normalement pour des protéines ayant un rôle dans la réplication et la transcription virale, ainsi que la régulation des gènes. Comme l’assemblage des virions est une des dernières étapes, les gènes tardifs, exprimés tard suite à l’infection formeront les composantes structurales du virion (25, 26).
5
Puis, les protéines de la capside vont s’assembler pour permettre l’empaquetage de l’ADN, où un minimum de 60 copies de la protéine majeure de la capside est nécessaire pour former une procapside vide (figure 1.3). Chaque génome est transloqué dans la capside par une ouverture dans celle-ci, comme une bobine de fil. Le complexe protéique d’empaquetage, composé de plusieurs protéines structurales, utilise l’hydrolyse de l’ATP pour accomplir cette tâche (27, 28). Enfin, la terminase, faisant partie de ce complexe et attachée à l’ouverture de la capside, coupe le génome des phages de différente façon selon le type de phage. Par exemple, le génome des phages de type cos sera coupé au site portant le même nom, afin d’obtenir une copie du génome par capside et donc une population de phages identiques. Pour les phages de type pac, la terminase coupe le génome lorsque la capside est pleine et donc celle-ci contient généralement 102% à 110% du génome total (29). Lorsque l’ADN est transloqué, le connecteur et les protéines du col vont venir bloquer l’ouverture de la capside et permettre l’attachement de la queue préalablement assemblée. Finalement, les protéines accessoires de la queue, s’il-y-a lieu, vont venir se joindre à l’extrémité de la queue pour former un virion mature (4).
Finalement, les virions matures sont prêts à être libérés dans l’environnement par lyse cellulaire. Les phages à ADN double brin ont une lyse précisément programmée qui est possible grâce à deux principales protéines, la holine et l’endolysine. La holine forme des agrégats résultant en un trou dans la membrane interne, permettant le passage de l’endolysine et l’accès au peptidoglycane. La lyse est donc dépendante de l’expression de la holine pour contrôler son temps de latence, soit le temps nécessaire pour compléter un cycle lytique (14). Trois classes de holines sont connues chez les phages, mais chaque phage ne possède qu’une holine. Les holines de classe I ont trois domaines transmembranaires et une courte séquence en C-terminal chargée positivement. Les holines de classe II n’ont que deux domaines transmembranaires dont les extrémités de la protéine sont cytoplasmiques. Puis, les holines de classe III possèdent un domaine transmembranaire et un large segment hydrophile en C-terminal. Quant à elle, l’endolysine est responsable de la dégradation du peptidoglycane et s’accumule dans le cytoplasme généralement durant la transcription et la traduction tardive du génome (14, 30)
Pour les endolysines, il existe quatre principales classes qui attaquent différents liens de la couche du peptidoglycane. Encore une fois, un phage ne peut posséder qu’une seule endolysine. Deux types de lysozymes, les endo-β-N-acetylglucosaminidases et les N-acetylmuraminidases agissent sur deux liaisons majeures entre le squelette glucosé du N-acetylglucosamine (NAG) et de l’acide N-acetyl
6
muramique (NAM). Les endopeptidases dégradent les liens interpeptidiques et les amidases, la classe d’endolysine la plus fréquente, hydrolysent les liaisons amides des oligopeptides (14).
Figure 1.3 : Modèle d’assemblage du bactériophage T4. Tirée de Wiley, 2013 (4).
Les phages tempérés sont particuliers du fait qu’ils peuvent prendre la décision d’entrer soit dans le cycle lytique ou lysogénique. Il y a l’établissement d’une communication, dirigée entre autres par de petites protéines ou des déterminants génétiques, entre l’hôte et le phage pour coordonner l’un ou l’autre des cycles (31). Le cycle lysogénique comporte trois grandes étapes, dont les mécanismes moléculaires varient beaucoup en fonction de la nature du phage. D’abord, il y a l’intégration du génome viral, où celui-ci va s’intégrer dans le chromosome bactérien au hasard ou par recombinaison via des sites spécifiques (32, 33). Certains phages vont rester sous forme circulaire ou linéaire sans intégration au génome bactérien. Puis, il y a l’étape de persistance où le génome viral, alors nommé prophage, est répliqué avec celui de la bactérie et donc transmis aux cellules filles. Les prophages extra-chromosomaux codent alors pour des gènes d’origine plasmidique nécessaires à leur transfert et à leur maintien. Finalement, il y a l’induction du prophage, où il va s’exciser du génome bactérien
7
et entreprendre le cycle lytique. Cet évènement se fait de façon spontanée à très basse fréquence ou survient suite à un stress externe chez la bactérie (34).
1.1.3 Les phages de Streptococcus thermophilus
Les phages infectant l’espèce Streptococcus thermophilus appartiennent tous à la famille des
Siphoviridae par la présence de leur longue queue fine et non contractile. La plupart de ces phages
ont été isolés à partir d’échantillons de lait ou de fromage, car ceux-ci sont responsables de certains problèmes dans l’industrie laitière. En effet, comme les phages attaquent les bactéries présentes dans les ferments, ils peuvent causer un ralentissement de la fermentation laitière (voir section 1.2.1) (35– 38). La classification de ces phages se fait maintenant en quatre groupes en fonction du nombre de protéines majeures structurales, de leur mécanisme d’empaquetage de l’ADN et de la séquence génomique (39). Les phages appartenant au groupe cos ont des extrémités génomiques cohésives (site
cos) qui sont reconnues et clivées lors de l’empaquetage de l’ADN dans la procapside, résultant en
une seule copie du génome par virion (29, 40). Les phages de ce type possèdent deux protéines majeures structurales tandis que les phages de type pac possèdent trois protéines majeures structurales. Les phages de type pac ne possèdent pas de sites cos et utilisent un mode d’empaquetage dit «tête pleine» (headful packaging), où l’ADN est transloqué dans la capside et clivé seulement lorsque celle-ci est pleine (27, 40).
Un autre groupe de phages, nommé 5093, moins prévalent, infecte aussi l’espèce thermophilus (41). Il affiche certaines similitudes de séquence avec des phages infectant le genre Streptococcus, mais appartenant à des espèces non utilisées dans l’industrie laitière (S. pyogenes, S. pneumoniae et S.
gordonii), en plus de son incapacité à être détecté par un PCR Multiplex utilisé pour grouper les
phages appartenant aux groupes précédents (37, 42). Ce groupe est distinct par un gène codant pour la protéine de liaison au récepteur complètement différent, généralement très conservé parmi les autres groupes et par une plaque basale contenant 6 fibres d’apparence globulaire suggérant que son mode d’attachement à la cellule bactérienne est différent (41, 42). D’un point de vue évolutif, ce groupe est divergent des autres et il a emprunté un chemin phylogénétique tout à fait unique des phages des groupes cos et pac (figure 4).
8
Figure 1.4 : Arbre phylogénétique montrant la divergence génétique entre le phage 5093 et cinq phages pour le groupe cos et le groupe pac (41).
Récemment, un nouveau groupe de phages infectant S. thermophilus a été découvert, le groupe 987 (43). Ce groupe, détecté en 2016, est le résultat probable d’un évènement d’échange génétique, puisqu’il présente plusieurs caractéristiques des phages du groupe P335 infectant Lactococcus lactis. La morphologie de la queue présente de grandes ressemblances avec ces phages, étant significativement plus petite, soit environ 135 nm contrairement aux phages typiques de S.
thermophilus qui varie entre 220 et 330 nm (35). De plus, la plaque basale est visible en microscopie
électronique et s’apparente à celles observées chez les phages du groupe P335. Ces traits sont le produit d’un génome hybride, où les modules codant pour les protéines structurales ont un haut pourcentage d’identité en acides aminés avec les phages du groupe P335 (plus précisément le phage TP901-1) et où les modules de réplication du groupe 987 présentent de fortes similarités avec les autres phages de S. thermophilus. De plus, certains des phages provenant de ce groupe ont la capacité de s’adsorber à la surface des souches de Streptococcus thermophilus et de Lactococcus lactis (43).
Pour ce qui est de la ressemblance génétique entre les phages infectant S. thermophilus, peu de cadres de lecture ouverts (orf) peuvent être inclus dans le génome de cœur, soit la portion du génome partagée entre tous les phages. Selon douze séquences complètes des phages de S. thermophilus des groupes cos ou pac, il est possible d’identifier 26 orfs conservés parmi les phages de type cos et 24
orfs conservés parmi les phages de type pac, pour seulement 5 orfs tous types confondus (44). Les
gènes généralement conservés dans le génome de cœur font partie du module de la lyse cellulaire (holine et endolysine) et du module d’empaquetage de l’ADN (35). De plus, les phages de S.
9
thermophilus possèdent un gène codant pour la protéine de liaison au récepteur bien conservé (sauf
pour le groupe 5093 décrit ci-dessus). Il est intéressant de noter qu’une région hautement variable dans ce gène, nommée VR2, est spécifique à chaque phage et que cette région est le principal déterminant du spectre lytique (45). Alternativement, il est possible de grouper les phages en fonction de cette région, cependant cette méthode ne permet pas un classement exhaustif, car il a été suggéré que VR2 n’est probablement pas le seul déterminant de la reconnaissance bactérienne (42, 45, 46). Bref, il est clair qu’une grande diversité génétique est présente chez les phages infectant S.
thermophilus. Jusqu’à ce jour, quatre groupes de phages ont été découverts et caractérisés. Cette
disparité est due à beaucoup d’évènements de réarrangement génétique et d’échange de modules, résultant en une gamme variée de phages pouvant infecter les souches utilisées dans l’industrie laitière (42, 47).
1.1.4 Le phage 2972
Le phage 2972 est un phage virulent, appartenant au groupe de type pac, qui infecte la souche S.
thermophilus DGCC7710. Ce phage de la famille des Siphoviridae possède une capside de 55 nm et
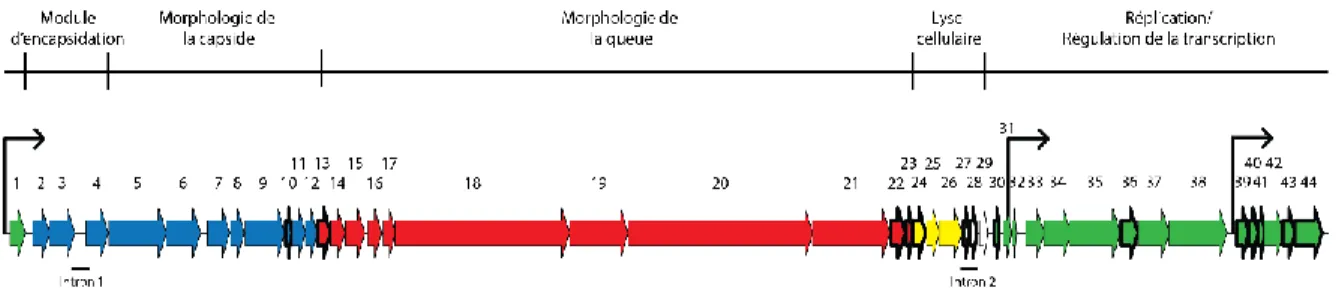
une queue d’une longueur de 260 nm. Il possède un génome d’ADN double brin de 34 704 paires de base (pb), un des plus petits chez les phages infectant l’espèce thermophilus et code pour 44 orfs putatifs de 40 codons et plus ayant tous la même orientation (figure 5) (26).
Figure 1.5 : Représentation du génome du phage virulent 2972 infectant S. thermophilus. Les flèches représentent des orfs. Les orfs en vert indiquent des gènes exprimés tôt suite à l’infection (0-7 minutes post-infection), les orfs en bleu indiquent des gènes exprimés au milieu de l’infection (7-22 minutes post-infection) et les orfs en rouge ou jaune indiquent des gènes exprimés tard suite à l’infection (12-27 minutes post-infection). Les orfs ayant un contour gras codent pour des protéines aux fonctions inconnues. Les flèches noires représentent les promoteurs putatifs, inspirée de Martel et Moineau, 2014, Duplessis et al., 2005 et Lévesque et al., 2005 (26, 48, 49).
10
Son génome a un pourcentage G+C de 40,15%, ce qui est semblable au pourcentage présent dans le génome de son hôte DGCC7710, qui est de 39% (50, 51). Il est divisé en trois groupes de gènes déterminés selon le profil d’expression temporelle. Les gènes précoces (de l’orf30 jusqu’à l’orf44 et l’orf1), en vert, sont exprimés au niveau maximal de 0 à 7 minutes suite à l’infection. Les gènes médians (de l’orf2 à l’orf12), en bleu, sont exprimés au plus haut niveau de 7 à 22 minutes après le début du cycle lytique. Puis les gènes tardifs (de l’orf13 à l’orf26), en rouge, sont exprimés majoritairement de 12 à 27 minutes. De plus, les gènes peuvent aussi être divisés en module de fonction dans le génome : le module d’encapsidation, la morphogénèse de la capside et de la queue, le module de lyse et le module de réplication de l’ADN et de régulation de la transcription (48, 49).
Figure 1.6 : Comparaison du génome des phages infectant Streptococcus thermophilus. Les orfs sont représentés par les flèches. Les flèches de la même couleur et reliées en gris si possible ont 70% ou plus d’identité en acides aminés entre les protéines déduites. Les couleurs de fond représentent les différents modules des génomes. Le rouge représente le module de morphologie, le jaune est pour le module de lyse cellulaire et le bleu regroupe les orfs du module de lysogénie. Finalement, le vert représente le module de réplication du génome et activation de la transcription. Tirée de Martel et Moineau, 2014 (44).
Le génome du phage 2972possède aussi deux introns. Le premier se situe entre les orfs 3 et 4, a une longueur de 307 pb et interrompt la grande sous-unité de la terminase. Le deuxième se trouve entre les orfs 26 et 29, a une longueur de 442 pb et coupe le gène de l’endolysine. Ces introns font partie
11
des introns du groupe I. Ils sont auto-épissable et s’excisent de l’ARNm, donc ils n’empêchent pas l’expression des gènes dans lesquels ils se trouvent (52). Trois promoteurs putatifs ont été déterminés selon la séquence consensus en position -35 et -10. Le promoteur P1 se trouve à 358 pb en amont de l’orf2, le promoteur P2 se situe 30 pb en amont de l’orf31 et le promoteur P3 a été localisé à 166 pb en amont de l’orf39 (26).
Plusieurs régions du génome du phage 2972 sont conservées parmi les phages infectant S.
thermophilus, dont le module de réplication de l’ADN et les modules codant pour les protéines
structurales (figure 6). Toutefois, l’orf25 codant pour la holine possède un pourcentage d’identité >50% avec des prophages infectant d’autres espèces appartenant au genre Streptococcus. La prédiction de la topologie a identifié que cette protéine possèderait trois domaines transmembranaires, ce qui est caractéristique des holines de classe I. L’orf20 a été identifiée comme codant pour la protéine structurale la plus divergente parmi celles du protéome. Cette protéine possède plusieurs motifs collagen-like, ce qui est typique des RBP chez les phages infectant des hôtes ayant un faible pourcentage G+C. Elle a donc été désignée comme étant la RBP, dû aux caractéristiques énumérées précédemment et sa position dans le génome (26). La plupart des gènes structuraux ont été identifiés pour ce phage, principalement par analyses bio-informatiques et SDS-PAGE (tableau 2.2). Toutefois, une grande partie des protéines codées par les gènes précoces dans le module de réplication du génome et de régulation de la transcription ont encore une fonction inconnue (48).
1.2 Les bactéries lactiques
Les bactéries lactiques (BL) sont des bactéries à Gram positif et catalase négative. Elles sont majoritairement retrouvées sous forme de coques ou de bâtonnets, ayant un mode de fermentation anaérobie facultative (53). Le produit final de la fermentation des sucres se trouve à être en grande partie l’acide lactique. Les BL incluent parmi leur rang les genres Lactobacillus, Lactococcus,
Leuconostoc, Streptococcus, Oenococcus, Pediococcus et Denococcus (54). Les BL ont un
métabolisme homofermentaire ou hétérofermentaire. Les espèces homofermentaires convertissent les sucres en acide lactique et les espèces hétérofermentaires ont une production d’acide lactique accompagnée d’acétate, de CO2 et d’éthanol (55).
Les bactéries lactiques sont des organismes commensaux et leurs habitats sont très diversifiés. Elles peuvent se retrouver dans la cavité orale, vaginale et la voie gastro-intestinale des humains et des animaux, ainsi que sur les plantes et les produits de fermentation. Certains de ces genres bactériens
12
sont producteurs d’exopolysaccharides (EPS), un polymère de grande masse moléculaire attaché ou non à la paroi ou la capsule de la bactérie (56, 57).
Ces bactéries sont connues comme étant un groupe hétérogène possédant une diversité énorme de plasmides (58). Les plasmides sont des modules autoréplicatifs indépendants du chromosome bactérien, qui confère souvent un avantage évolutif dans un environnement particulier (4). Plusieurs fonctions importantes pour les bactéries lactiques sont codées par des gènes retrouvés sur ces éléments génétiques. Par exemple, ils peuvent porter des gènes codant pour l’hydrolyse de protéines, la production d’exopolysaccharides, la résistance aux antibiotiques et le métabolisme de certaines molécules. Les souches de BL qui possèdent un plasmide augmentant l’hydrolyse de la caséine, permettant l’utilisation du lactose, du galactose, ou de tout autre métabolite qui modifie le goût et la texture des produits de consommation, ont donc des caractéristiques industrielles intéressantes (58).
1.2.1 Importance industrielle
L’industrie laitière est le principal exploitant des ferments composés de ces bactéries lactiques, productrices ou non d’exopolysaccharides, pour la production de yogourt, fromage, kéfir et autres produits d’origine laitière. Toutefois, les bactéries lactiques sont aussi utilisées lors du processus de production de la choucroute, du pain de seigle, de certains types de saucissons et de beaucoup d’autres produits de fermentation (53). Les ferments de départ utilisés vont contribuer à la saveur du produit grâce à la diversité des métabolites secondaires produits.
Les bactéries lactiques sont aussi utilisées sous forme de probiotiques. Les probiotiques sont définis comme une préparation de microorganismes vivants qui sont destinés à la consommation et qui lorsqu’ils sont consommés en quantité adéquate procurent des bienfaits sur la santé (59). Plusieurs études ont montré que le microbiote, soit la communauté de bactéries qui colonise le corps humain, influence la prévalence de plusieurs maladies tels le diabète, l’obésité et quelques problèmes inflammatoires des intestins. L’administration de probiotiques promeut donc un microbiote équilibré pour de meilleurs bénéfices sur la santé globale (59).
1.2.2 Le genre Streptococcus
Les bactéries appartenant au genre Streptococcus ont une morphologie en coques sphériques ou allongées d’une longueur variant entre 0,8 µm et 1,2 µm, en pairs ou chaînes et non motiles (60).
13
Elles ont une respiration anaérobie facultative et un métabolisme homofermentaire, où l’acide lactique est le seul produit de la fermentation des sucres. Les besoins nutritifs de ce genre sont assez complexes et variables selon l’espèce et les souches. De plus, la grande production d’acide lactique chez ce genre bactérien cause une baisse du pH rapide qui inhibe la croissance bactérienne et c’est pour cette raison qu’il est important de tamponner le milieu de culture. Beaucoup d’espèces présentes dans ce genre, telles S. pneumoniae et S. pyogenes, sont connues comme des organismes pathogènes ayant une grande virulence. Leur température optimale de croissance varie de 37 °C à 42°C (55, 61).
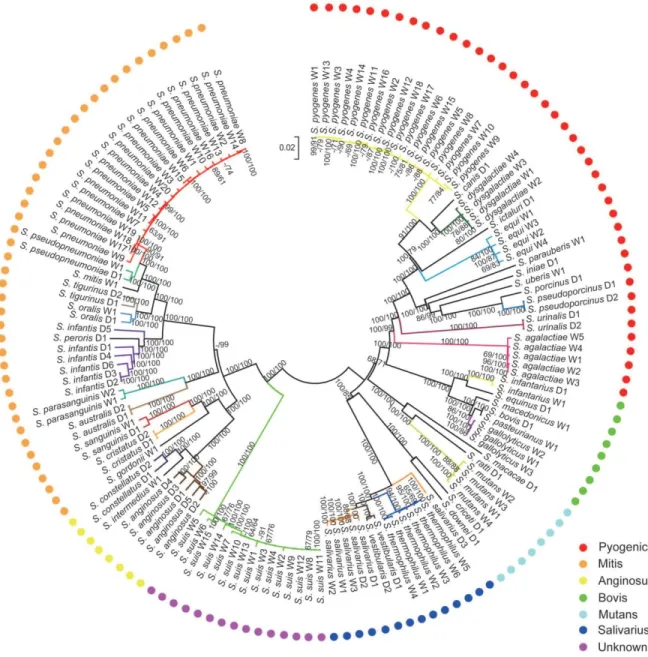
Une classification du genre Streptococcus regroupe les espèces en sept distincts groupes nommés par l’espèce modèle de chacun (figure 1.7). La classification phylogénétique s’est faite selon l’ARN16S.
S. thermophilus, contenu dans le groupe Salivarius, est une espèce ayant une distance phylogénétique
proche des espèces pathogènes, pourtant elle est la seule à être définie comme un organisme sécuritaire Generally Recognized As Safe (GRAS), ce qui lui permet d’être utilisée dans l’industrie alimentaire (62).
Effectivement, les gènes codant pour des déterminants génétiques de la virulence sont absents, inactifs ou sous forme de pseudogènes chez S. thermophilus. Ceci témoigne de l’évolution de cette espèce à s’adapter à sa niche écologique, le lait. L’acclimatation de Streptococcus thermophilus s’est fort probablement faite par une pression sélective du milieu et par échange de gènes horizontaux (61). Plusieurs séquences d’insertions dans certains génomes de cette espèce ont plus de 90% d’identité avec d’autres bactéries lactiques, telles Lactobacillus bulgaricus, Lactococcus lactis sous-espèce
cremoris et lactis. Ces microorganismes, dont le métabolisme est optimisé pour la croissance dans le
lait, sont fréquemment utilisés en co-culture lors de la fermentation de produits laitiers et donc cela suggère que leur proximité physique a favorisé l’échange de gènes (62).
C’est grâce à cette évolution que l’espèce S. thermophilus est la plus utilisée pour les ferments de yogourt, puisqu’elle procure une texture remarquable ayant de grandes propriétés organoleptiques (56, 63–65). Ceci est dû en partie à son métabolisme homofermentaire et à sa production unique d’exopolysaccharides. En effet, cette espèce bactérienne à Gram positif et à faible pourcentage G+C, permet l’obtention d’un ferment avec une plus grande stabilité et viscosité que les autres genres bactériens, grâce à la capacité de ses EPS à lier les molécules d’eau (1, 54, 66). Les bactéries lactiques productrices d’EPS font partie de la plus importante application industrielle et sont utilisées partout à travers le monde (56). L’utilisation des ferments producteurs d’exopolysaccharides est destinée à augmenter la qualité et la texture des produits de fermentation sans ajout d’additifs alimentaires.
14
Figure 1.7 : Arbre phylogénétique de 138 espèces du genre Streptococcus. Les espèces sont
regroupées en sept groupes en fonction de l’espèce modèle, Pyogenic, Mitis, Anginosus, Bovis,
Mutans, Salivarius et Inconnu (Unknown) selon les points de couleurs différentes (67).
1.2.3 Sensibilité aux phages
Cette espèce bactérienne, comme tant d’autres, est sensible aux bactériophages qui peuvent affecter les productions (68). Ceci peut mener à un ralentissement de la fermentation et une perte de la qualité des produits (69). Les phages se retrouvent naturellement dans le lait et résistent à la pasteurisation, ils sont donc ubiquitaires dans les usines laitières. De plus, ils ont un temps de latence relativement court et une taille de la progéniture très grande ce qui leur permet de se reproduire rapidement lors
15
d’une production. Les industries peuvent être impactées par plusieurs facteurs, cependant les phages sont la source majeure de problèmes d’origine biologique. L’observation d’un haut pH, d’un haut niveau de lactose résiduel et d’un contenu en acide lactique pauvre lors d’une fermentation sont les indicateurs d’une infection par les phages. Ils proviennent principalement du lait, mais ils peuvent être dispersés sous forme d’aérosols par le mouvement du personnel ou des équipements, causant la contamination des zones à risques (36). Une autre source de contamination importante est le recyclage des sous-produits de la fermentation, telles les protéines de lactosérum. Les producteurs les utilisent pour augmenter le rendement fromagé, cependant ces protéines ont un effet protecteur sur les phages lors du traitement thermique. Le début d’une production est donc contaminé par les sous-produits de la précédente (70).
Plusieurs mesures pour tenter de contrôler les phages et leur dissémination doivent être mises en place dans une industrie laitière. Les stratégies de contrôle concernent souvent la configuration de l’usine, l’utilisation de désinfectants appropriés et la gestion des ferments. D’abord, la conception de l’usine doit séparer les zones à risques et permettre une réception du lait isolée de la production. Une filtration de l’air appropriée, ainsi qu’une pression atmosphérique positive sont des facteurs pouvant contrôler le déplacement des phages (36). Puis, l’utilisation d’agents assainissant efficace est très importante. Toutefois, certains aspects sont à prendre en compte. L’assainissant ne doit pas avoir d’impacts négatifs sur la production, il doit avoir un mécanisme d’action rapide et stable dans le temps, il ne doit pas posséder d’effets toxiques et doit pouvoir agir sur les phages et les microorganismes. Les agents oxydant tels l’acide peracétique, l’acide acétique et le peroxyde d’hydrogène, et les composés d’ammonium quaternaire sont parmi les agents assainissant les plus efficaces pour le contrôle des phages infectant les BL (71). Finalement, une bonne gestion des ferments permet de réduire la fréquence des infections. Il doit y avoir une rotation des cultures pour éviter les infections récurrentes par le même phage qui devient de plus en plus nombreux. De plus, l’utilisation de ferments naturellement résistants aux phages est employée par plusieurs compagnies, car les bactéries possèdent de nombreux mécanismes de résistance (36, 69).
1.2.4 Les mécanismes de défense
Il y a plusieurs mécanismes de résistance aux bactériophages qui visent les différentes étapes du cycle d’infection (figure 1.8). D’abord, il est possible de bloquer l’adsorption des phages à la surface cellulaire et ainsi empêcher le cycle lytique de commencer. Les bactéries peuvent accomplir cette action par une variété de stratégies. Elles peuvent muter le récepteur cellulaire et donc il n’y a plus
16
de reconnaissance moléculaire par la protéine de liaison au récepteur du phage ou alors elles peuvent bloquer l’accès de ce récepteur par la production d’une matrice extracellulaire (EPS) qui devient alors une barrière physique contre les phages (17, 72).
Figure 1.8 : Représentation schématique des différents systèmes de défense bactériens. Plusieurs mécanismes sont présents chez les procaryotes pour lutter contre les éléments d’ADN étrangers, comme les génomes de bactériophages et les plasmides. Ces systèmes incluent le blocage de l’adsorption en masquant ou en mutant les récepteurs cellulaires, le blocage de la translocation de l’ADN dans le cytoplasme, les systèmes Abi où la cellule provoque sa propre mort suite à une infection, les systèmes de restriction/modification et les systèmes CRISPR-Cas qui clivent l’ADN de façon différente avec ou sans reconnaissance spécifique de l’ADN envahisseur. M : méthyltransférase, R : endonucléase de restriction, C : Protéine Cas (73).
Ensuite, il est possible de bloquer l’entrée de l’ADN dans la cellule. C’est un phénomène souvent associé à la présence d’un prophage qui se nomme l’exclusion de surinfection (Sie) et qui va contrecarrer la réinfection par le même phage ou un phage similaire (74). Ceci va empêcher la compétition pour les ressources de la bactérie entre plusieurs phages et assurer la survie du prophage déjà en place (17). Les prophages codent souvent pour des protéines qui vont s’associer à la
17
membrane, avec les éléments qui la composent ou qui interagissent avec le récepteur de la queue des phages. Celles-ci peuvent bloquer physiquement l’éjection de l’ADN ou bloquer le processus (75). Il est important de noter que l’exclusion de surinfection ne bloque pas l’adsorption des phages.
Puis, il y a les systèmes d’avortement de l’infection (Abi) qui cause la mort prématurée de la cellule suite au début de la réplication du phage. Le sacrifice d’une cellule infectée résulte en moins de phages relâchés pour préserver la population bactérienne (17, 76). Lorsqu’infectés par un phage, les procaryotes possédant un système Abi ciblent des étapes importantes de la réplication de la cellule ou clivent les composantes cellulaires majeures. Par exemple, un système Abi peut cliver des composantes de la membrane et ainsi provoquer une baisse du potentiel membranaire pour arrêter la production d’ATP, ou alors un autre genre de système Abi va inhiber la synthèse protéique (73, 77).
La cellule peut aussi couper l’ADN étranger qui entre dans le cytoplasme pour inhiber la réplication de celui-ci. Les systèmes de restriction/modification sont des systèmes très répandus qui coupent l’ADN non méthylé lors de son entrée dans la cellule (78). Ces systèmes fonctionnent en trois grandes étapes. D’abord, une séquence nucléotidique présente dans le génome de l’hôte est reconnue par la méthylase. Puis, un groupement méthyle est ajouté au site de reconnaissance sur l’ADN par cette enzyme, ce qui protège le chromosome bactérien. Finalement, l’ADN non méthylé qui se retrouve dans le cytoplasme de la cellule bactérienne sera digéré par une endonucléase de restriction pour arrêter la propagation de l’infection. (79).
Finalement, les systèmes CRISPR-Cas (CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats, Cas: CRISPR associated), présents dans un grand nombre de bactéries et d’archées, font partie des systèmes capables de couper et digérer l’ADN étranger. Ces systèmes fonctionnent par reconnaissance et clivage spécifique d’une courte séquence d’ADN ou d’ARN (80–83).
1.3 Les systèmes CRISPR
Les structures génétiques CRISPR et leurs gènes Cas sont des systèmes d’immunité adaptative bactériens observés pour la première fois en 1987 chez Escherichia coli (84). Ces systèmes sont présents chez près de 90% des archées et près de 50% des procaryotes, sans aucun homologue chez les eucaryotes (85, 86). Ils jouent un rôle de défense contre les éléments d’ADN envahisseurs parmi lesquels se trouvent les phages et les plasmides, tout comme le système immunitaire humain joue un rôle de défense contre les infections (1, 87). Les systèmes CRISPR-Cas sont décrits comme étant
18
adaptatif, car ils ont la capacité d’acquérir leur immunité et de la transmettre aux cellules filles suite à la division cellulaire.
1.3.1 Composition des systèmes CRISPR-Cas
Les systèmes sont composés de loci d’un nombre variable de courtes séquences nucléotidiques identiques, nommées répétitions, entrecoupées par des espaceurs uniques de longueurs variables selon l’espèce (figure 1.9). Les répétitions ont une longueur entre 23 pb et 47 pb et une séquence généralement partiellement palindromique, ce qui leur confère une structure secondaire nécessaire à la maturation des ARNcr (85, 86). Les répétitions sont habituellement bien conservées parmi les espèces proches, toutefois le nombre de répétitions et la séquence des espaceurs varient grandement en fonction des souches (86). Ces espaceurs, de 21 pb à 72 pb, sont des séquences hypervariables (85). Les espaceurs ont longtemps été considérés comme de l’ADN insignifiant, jusqu’à la réalisation qu’ils correspondent à des séquences d’ADN identiques aux génomes des bactériophages ou à certains plasmides (1, 80). En effet, un espaceur peut souvent être associé à une séquence complémentaire dans le génome correspondant d’un phage, ou à la séquence d’un plasmide, d’où il provient (80, 81, 88). Cette séquence correspondante, nommée proto-espaceur, est un élément clé lors de la reconnaissance de l’ADN envahisseur. Pour certains types, il est obligatoirement adjacent à un court motif nommé PAM (Protospacer Adjacent Motif) et il est nécessairement absent du chromosome bactérien pour éviter que le système CRISPR cible son propre génome (1, 80).
Figure 1.9 : Composition générale des systèmes CRISPR-Cas. Les flèches en rouge représentent les gènes cas, la ligne rose représente la séquence leader suivie du locus CRISPR composé des répétitions (losanges noirs) et des espaceurs (rectangles de couleurs individuelles). Adaptée de Hille et al. (87).
Les loci CRISPR sont généralement tous adjacents à une séquence leader, riche en adénine et en thymine qui ne possède pas de cadre de lecture ouvert, donc elle ne code pas pour une protéine. Cette séquence se situe directement en amont ou en aval du locus CRISPR et elle s’oriente toujours dans le
19
même sens que le locus (86). Il a été montré qu’elle est le point de départ de la transcription du locus CRISPR et qu’elle joue donc un rôle de promoteur lors de la biogenèse (87).
Puis, les loci CRISPR fonctionnels sont accompagnés de gènes cas qui varient en nombre et en composition selon le type de système. Ils peuvent se situer en amont ou en aval du locus et ils sont couramment éloignés de celui-ci par environ une centaine de paires de base. L’orientation de l’opéron des gènes cas n’est pas obligatoirement identique à celle du locus et il possède habituellement le même contenu en guanine et en cytosine que le chromosome bactérien (86). Les protéines codées par les gènes cas ont plusieurs domaines fonctionnels caractéristiques des nucléases, des hélicases et des protéines liants les polynucléotides. Ces gènes accomplissent quatre rôles distincts au sein des systèmes CRISPR-Cas en fonction de ces domaines : le module d’adaptation pour l’acquisition de nouveaux espaceurs, le module d’expression pour la maturation des ARNcr, le module d’interférence pour le clivage de l’ADN envahisseur et le module auxiliaire pour la régulation ou toute autre fonction (89).
1.3.2 Classification
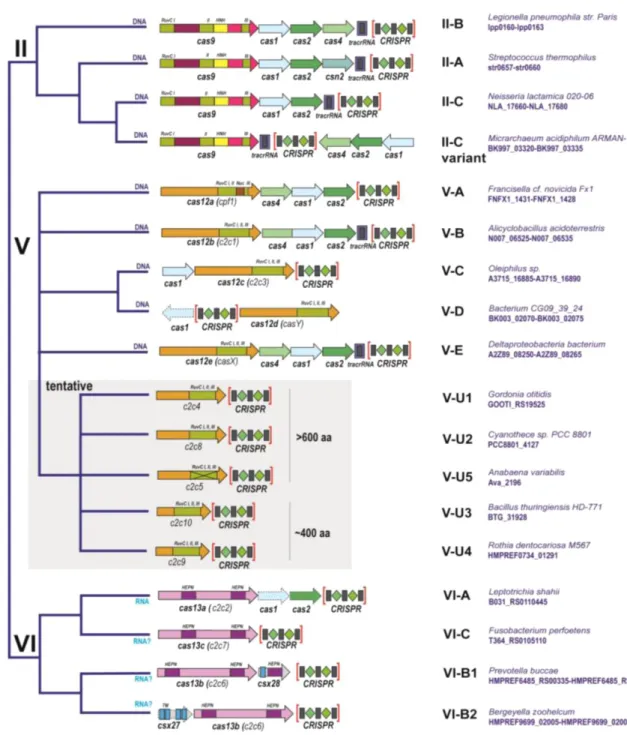
La classification des systèmes CRISPR-Cas est une tâche ardue et complexe. Plusieurs classifications ont été nécessaires avant d’arriver à celle établie à ce jour. En 2011, une première classification des systèmes a été proposée qui les divisait en trois types en fonction des gènes cas signatures associés au locus, où tous les systèmes fonctionnels possèdent les gènes cas1 et cas2. Le type I est caractérisé par la présence de cas3, le type II est caractérisé par la présence de cas9 et le type III possède cas10 (90). L’architecture génomique et la disposition des gènes cas est responsable des différents sous-types.
Puis, la recherche accrue sur les systèmes CRISPR-Cas, ainsi qu’une augmentation des données génomiques disponibles, a permis la découverte de nouveaux systèmes ne correspondant pas à la classification établie. En 2015, la catégorisation a été révisée pour donner le jour à deux classes, cinq types et seize sous-types, classés en fonction des modules d’interférence (89). Finalement, une dernière mise à jour a été faite en 2017 pour ajouter un nouveau type, le type VI (figure 1.10). La classe 1 regroupe les types I, III qui sont les plus fréquents parmi les archaea et le type IV, qui est très rare et ne possède pas de module d’adaptation. Les types appartenant à cette classe possèdent beaucoup de petits gènes cas pour former le complexe d’interférence, tandis que les types rattachés à la classe 2 possèdent une seule grosse protéine, ayant de multiples domaines, capables d’effectuer
20
l’interférence. Le type II, ayant la protéine Cas9, le type V, ayant l’effecteur Cfp-1 et le nouveau type VI, possédant le gène cas13 font partie de la classe 2 (91). Finalement, l’évolution rapide des systèmes CRISPR-Cas, son énorme variabilité et l’absence de gènes cas universels démontrent qu’une classification robuste doit constamment être mise à jour pour permettre la caractérisation précise de ces systèmes (89, 92).
21
Figure 1.10 : Classification et organisation des types de systèmes CRISPR-Cas et domaines des protéines effectrices pour les classes 1 (A) et 2 (B). Tirée de Koonin, 2017 (93).
1.3.3 CRISPR-Cas chez Streptococcus thermophilus
Chez l’espèce Streptococcus thermophilus, il y a quatre loci CRISPR possibles (1 à 4) identifiés dans plusieurs génomes. Chez la souche DGCC7710, les loci CR1 et CR3 sont de type II-A, le locus CR2
22
est de type III-A et le locus CR4 est de type I-E, où chaque séquence des répétions est propre à chaque locus (88, 94–96). Il a été démontré que tous les loci CRISPR, ainsi que leurs gènes cas associés, présents dans cette souche sont transcrits indépendamment lors d’une infection phagique (95, 97). En effet, une séquence leader a été identifiée pour les loci CR1, CR2 et CR3, toutefois la transcription du locus CR4 serait probablement une continuation de la transcription des gènes cas (95). Cependant, seuls les locus CR1 et CR3 sont actifs et confèrent une résistance contre les bactériophages lorsqu’il y a acquisition de nouveaux espaceurs (96, 98). Ces locus possèdent les protéines Cas1, Cas2 et Csn2 en amont de la séquence répétition-espaceur en plus du gène cas9, qui est le gène signature pour le type II.
Les répétitions des loci de type II-A, chez la souche DGCC7710, ont une longueur de 36 pb. Les répétitions et les gènes cas sont spécifiques à chaque locus et ils ne reconnaissent pas le même motif PAM. Le locus CR1 est associé au motif NNAGAAW et le locus CR3 est associé au motif NGGNG, où N représente n’importe quel nucléotique de l’ADN et W représente une adénine ou une thymine (88, 98). Toutes les souches de S. thermophilus possèdent le locus CR1, qui possède en moyenne 23 espaceurs par locus d’une longueur entre 28 pb et 32 pb. Environ 91 % des souches de S. thermophilus possèdent un locus CR2, avec un maximum de 7 espaceurs, d’une longueur qui varie entre 35 pb et 40 pb. Puis, 80% des souches possèdent le locus CR3 avec environ 13 espaceurs de longueur entre 29 pb et 32 pb. Puisqu’ils sont rares, la fréquence des loci CR4 n’a pas été déterminée. En général, la grande majorité des espaceurs, tous loci confondus, proviennent de séquences virales (77%), puis de plasmides (16%) et finalement 7% proviennent du chromosome bactérien (98).
1.3.4 Les mécanismes de fonctionnement
Les systèmes CRISPR-Cas ont tous un mécanisme de fonctionnement propre à leur type et parfois à leur sous-type, toutefois il y a trois étapes communes à tous (99, 100). Il y a d’abord l’étape d’acquisition d’un espaceur dans le locus CRISPR, suivi de la biogenèse, ou la maturation, des ARNcr et finalement l’étape d’interférence (figure 1.11). Ici, les étapes générales seront décrites en soulignant plus en détail le système de type II-A, présent dans la souche S. thermophilus DGCC7710.
23
Figure 1.11 : Les trois étapes de fonctionnement des systèmes CRISPR-Cas. Lors de l’adaptation, le complexe Cas1-Cas2 est responsable de la sélection et de l’intégration d’un nouvel espaceur dans le locus. Puis, le locus est transcrit sous forme d’ARN précurseur et sera traité en ARNcr mature lors de la biogenèse. Finalement, il y a reconnaissance de l’ADN envahisseur par le module d’interférence et clivage de cet ADN (87).
1.3.4.1 L’adaptation
L’adaptation est le processus par lequel le système CRISPR-Cas acquiert de nouveaux espaceurs dans son locus, formant ainsi une mémoire moléculaire des infections précédentes. (87). Les protéines Cas1 et Cas2 sont communes à presque tous les types et semblent jouer un rôle clé lors de l’adaptation (91). Le processus d’acquisition est accompli en plusieurs étapes complexes, soit la détection de l’infection, la sélection du proto-espaceur et son intégration dans le locus (87). D’abord, il y a la détection d’un élément d’ADN étranger qui stimule la machinerie d’adaptation. Ici, il est nécessaire
24
que la machinerie puisse différencier et préférer l’ADN envahisseur de son propre ADN pour éviter une auto-immunité. Chez E. coli, il a été démontré qu’une source importante de proto-espaceurs provenait de la dégradation des fragments d’ADN lors de la réparation des bris double brins par le sentier de réparation RecBCD. Le système de réparation RecBCD déroule et dégrade l’ADN jusqu’à ce qu’il atteigne un site Chi. Le nombre de sites Chi dans les ADN étrangers étant beaucoup plus faibles que dans le génome d’E. coli, une plus grande portion de l’ADN étranger est dégradée et intégrée dans le locus CRISPR, induisant ainsi un biais envers l’ADN envahisseur (101). De plus, un mécanisme similaire a été décrit chez Streptococcus pyogenes qui possède un CRISPR de type II-A, utilisant la machinerie AddAB semblable à la voie de réparation RecBCD (102).
Toutefois, la sélection des proto-espaceurs intégrés dans le locus CRISPR n’est pas un processus laissé au hasard, d’où l’existence des PAM. Les protéines Cas des systèmes de types I et II sélectionnent des proto-espaceurs adjacents aux PAM compatibles avec la machinerie d’interférence (1, 87, 103). Dans certains cas, seul le complexe formé de Cas1 et Cas2 est nécessaire à la reconnaissance du PAM, tel que retrouvé dans le type I-E (104). Cependant, d’autres types nécessitent des protéines additionnelles à la machinerie d’acquisition, tel le type II-A. Ce type nécessite aussi l’assistance de Cas9 qui aide à la reconnaissance du PAM, de Csn2 et de l’ARN transactivateur (ARNtracr) (105). Ainsi, différents mécanismes servent à sélectionner les proto-espaceurs qui seront intégrés dans le locus. Cette intégration se fait de préférence à l’extrémité 5’ de la séquence répétition-espaceurs du locus CRISPR et permet d’obtenir un ordre chronologique des infections passées (80). L’intégration des nouveaux espaceurs peut se faire de cette façon, car la machinerie d’adaptation sera en mesure de reconnaître la séquence riche en A-T de la région leader. Encore une fois, les mécanismes d’intégration varient en fonction du type et du sous-type. Par exemple, le type I-E, parmi les plus étudiés, a besoin entres-autres d’un facteur d’intégration provenant de l’hôte (HIF, Host Integration factor), tandis que le type II-A nécessite un court site d’encrage nommé LAS présent dans la séquence leader permettant d’incorporer l’espaceur (106, 107). Cette séquence de 5 pb à l’extrémité adjacente aux répétitions est directement reconnue par le complexe Cas1-Cas2, jouant le rôle d’intégrase. Puis, l’espaceur est intégré par deux réactions de «demi-site», où une extrémité 3’OH du futur espaceur fait une attaque nucléophile à l’extrémité 5’ à proximité de la séquence leader de la répétition et l’autre extrémité 3’ de l’espaceur ira se lier à l’extrémité 5’ distale de la séquence leader du brin complémentaire de la répétition. Le clivage de l’ADN ainsi que la ligature des extrémités est effectuée par transestérification à l’aide de Cas1. Donc l’intégration d’un espaceur duplique une répétition pour chaque espaceur intégré (108–110).