L’UNIVERSITE PAUL SABATIER TOULOUSE III Ecole doctorale
CHIMIE
En vue de l’obtention du titre de
DOCTEUR DE L’UNIVERSITE PAUL SABATIER Spécialité
CHIMIE-BIOLOGIE-SANTE par
Christine TALMARD
Interaction entre le zinc(II) et le peptide
amyloïde bêta lié à la maladie d’Alzheimer
Soutenue le 19 novembre 2007
Rapporteurs : P. DELANGLE Directeur de recherche CEA (DRFMC/LCIB, Grenoble) A.-M. ALBRECHT-GARY Directeur de recherche CNRS (LPCB, Strasbourg) Examinateurs : R. MELKI Directeur de recherche CNRS (LEBS,
Gif-sur-Yvette)
C. BLONSKI Directeur de recherche CNRS (LSPCMIB, Toulouse) B. FRANCES Professeur d’Université (UPS, Toulouse) Directeur de thèse : P. FALLER Professeur d’Université (UPS, Toulouse)
Laboratoire de Chimie de Coordination du CNRS 205, route de Narbonne, 31077 Toulouse
Liste des abréviations 5
Chapitre 1
9
Le peptide amyloïde bêta, les ions métalliques et la maladie
d’Alzheimer
I. La maladie d’Alzheimer, vue d’ensemble 11I.1. Définition clinique 11
I.2. Rappel historique 12
I.3. La maladie d’Alzheimer, priorité de santé publique 13 I.4. Le diagnostic 14
I.5. Facteurs génétiques, biologiques et environnementaux 16
I.5.a. Facteurs génétiques 16
I.5.b. Facteurs biologiques et environnementaux 18
II. Plaques amyloïdes et neurofibrilles 19
II.1. La protéine tau 20
II.2. Le peptide amyloïde bêta 23
II.2.a. Cas général des amyloïdoses 23
II.2.b. La protéolyse de l’APP 27
II.2.c. Le peptide Aβ 29
II.2.d. Structure des fibres amyloïdes 31
II.2.e. Relation entre Aβ et tau 33 II.2.f. Hypothèse de la cascade amyloïde –Toxicité liée à Aβ 35
II.3. Traitements 38
II.3.a. Traitements actuels 38
II.3.b. Voies de recherche 39
II.4. Influence des métaux 45
II.4.a. Introduction : les métaux et l’organisme 45
II.4.b. Modulation des métaux avec l’âge 50
II.4.c. Evolution du stress oxydant avec l’âge 50 II.4.d. Modulation des métaux dans la MA 51
II.4.e. Implication des métaux dans la MA 52
II.4.f. Approche thérapeutique en relation avec le cuivre et le zinc 61
III. Conclusion et contexte de l’étude 64
Etude de la liaison du zinc(II) au peptide amyloïde bêta
I. Résumé de l’étude 103
II. Publication 104
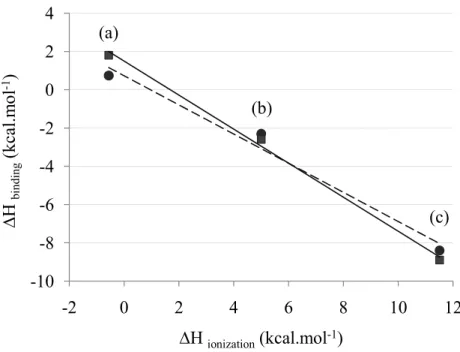
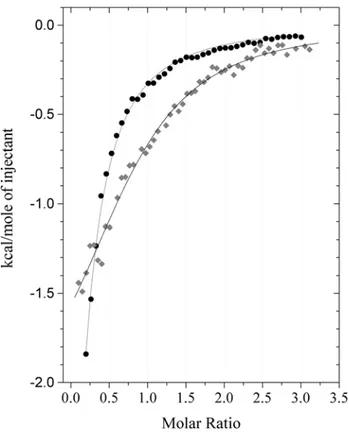
III. Complément : principe de la calorimétrie de titrage isotherme 128
Chapitre 3
133
Etude de l’oligomérisation du peptide amyloïde bêta durant
son agrégation en présence de zinc(II) ou de cuivre(II)
I. Contexte bibliographique et étude de l’influence des métaux sur le degré d’agrégation du peptide amyloïde bêta 135 II. Formation d’un complexe monomérique observable 137 III. Matériels et méthodes 142Références 148
Chapitre 4
151
Etude structurale du mécanisme de promotion de l’agrégation
du peptide amyloïde bêta par le zinc(II)
I. Introduction 153II. Résultats et discussion 155
III. Matériels et méthodes 162
Références 166
Chapitre 5
169
Etude de l’interaction entre la métallothionéine-3 et le peptide
amyloïde bêta, influence du stress oxydant
I. Introduction bibliographique 171III. Résultats et discussion 175
III.1. Hypothèse 175
III.2. Expériences avec le modèle Cd7-MT-3 176
III.2.a. Interaction entre Cd7-MT-3 et Aβ 176
III.2.b. Interaction entre Cd7-MT-3 et Aβ en présence de H2O2 177
III.2.c. Interaction entre Zn7-MT-3 et Aβ, en présence de H2O2 178
III.3. Agrégation de Aβ en présence de H2O2 et de Zn7-MT-3 180
IV. Matériels et méthodes 182
IV.1. Matériels 182
IV.1.a. Préparation de Cd7-MT-3 et Zn7-MT-3. 182
IV.1.b. Autres solutions 183
IV.2. Méthodes 183
IV.2.a. Analyse d’acides aminés 183
IV.2.b. Spectroscopie d’absorption atomique 184
IV.2.c. Chromatographie d’exclusion stérique 184
IV.2.d. Spectroscopie d’absorption UV-visible 184
Références 185
Chapitre 6
187
Etude de l’interaction du peptide amyloïde bêta avec les
membranes, influence du zinc
I. Bibliographie 189
II. Résultats et discussion 191
III. Méthode : la BLM 196 Références 201
Conclusion
et
perspectives
203
Annexes 207 Remerciements 235 Abstract 237Liste des abréviations
Aβ Amyloïde bêta
AD Alzheimer’s Disease
ADAM A Disintegrin And Metalloprotease
ADN Acide Désoxyribonucléique
ADRDA Alzheimer's Disease and Related Disorders Association AICD APP Intracellular Cytosolic Domain
AINS Anti-Inflammatoires Non Stéroïdiens
AMPA α-amino-3-hydroxy-5-méthylisoazol-4-propionate
apoE apolipoprotéine E
Aph-1 Anterior pharynx defective 1 APP Amyloid Precursor Protein
Arc Activity-regulated cytoskeleton-associated protein ARN(m) Acide Ribonucléique (messager)
BACE1 Beta site Cleaving Enzyme 1
BCA Acide Bicinchoninique
BLM Black Lipid Membrane
BPLED Bipolar Longitudinal Eddy current Delay
CDK Cyclin-Dependent Kinase CQ Clioquinol DC Dichroïsme Circulaire DFOA Desferrioxamine DMSO Diméthylsulfoxyde DOPC 1,2-dioleoyl-sn-glycero-3-phosphocoline EDTA Acide éthylène-diamine-tétraacétique
ESI-MS Electrospray Ionization Mass Spectrometry (Ionisation par electronébullisation – Spectrométrie de masse)
EXAFS Extended X-Ray Absorption Fine Structure GIF Growth Inhibitory Factor (MT-3)
GSK Glycogen Synthase Kinase
GTP Guanosine 5’-TriPhosphate
Hepes Acide [(hydroxy-2-éthyl)-4-pipérazinyl-1]-2éthanesulfonique HPLC High-Performance Liquid Chromatography
Hsp Heat shock protein
IDE Insuline Degrading Enzyme (ou insulinase) IRM Imagerie par Résonance Magnétique
ITC Calorimétrie de titrage isotherme (Isothermal Titration Calorimetry) IUPAC-IUB International Union of Pure and Applied Chemistry - International Union
of Biochemistry
JIP JNK Interacting Protein JNK c-Jun N-terminal Kinase kDa kiloDalton
Kdapp Constante de dissociation apparente
LA-ICPMS Analyse par couplage de l'ablation laser et de la spectrométrie de masse à source plasma
LCR Liquide Céphalorachidien
MA Maladie d’Alzheimer
Mint Munc18 interacting protein
MMP Matrices MétalloProtéinases
Mr Poids moléculaire
MT Métallothionéine
NADH Forme réduite du nicotinamide adénine dinucléotide NEP Néprilysine
NG Newport Green
NINCDS National Institute of Neurological and Communicative Diseases and Stroke (actuellement : National Institute of Neurological Disorders and Stroke) NMDA N-Méthyl-D-Aspartate
PAQUID Quid des personnes agées
PAR 4-(2-pyridylazo)-resorcinol Pen-2 Presenilin enhancer 2
PGSE NMR Pulse Gradient Spin-Echo Nuclear Magnetic Resonance PI-3K PhosphoInositol-3-Kinase
Pin1 Protein interacting with NIMA (never in mitosis A) 1 PPARγ Peroxisome Proliferator-Activated Receptor γ
PP2A Protéine Phosphatase 2A PS1 Préséniline-1 PS2 Préséniline-2 PSD Post Synaptic Density protein
Rh Rayon hydrodynamique
RMN Résonance Magnétique Nucléaire ROS Reactive Oxygen Species
SEC Size Exclusion Chromatography
TACE TNF-Alpha Converting Enzyme
TEP Tomographie par Emission de Positons TFE 2,2,2-trifluoroéthanol
ThT Thioflavine T
TNF Tumor Necrosis Factor
Tris Tris(Hydroxymethyl)aminométhane TSH Thyroid Stimulating Hormone
Zi Zincon
Chapitre 1
La maladie d’Alzheimer,
introduction bibliographique
I. La maladie d’Alzheimer, vue d’ensemble I.1. Définition clinique
La maladie d'Alzheimer (MA) est une maladie neurodégénérative du système nerveux central. Elle entraîne progressivement la perte des fonctions mentales, du fait de la détérioration du tissu cérébral (Figure 1.1), et conduit finalement à un syndrome démentiel. La nature neurodégénérative de la MA se traduit par des lésions histopathologiques bien précises qui sont les plaques séniles et les dégénérescences neurofibrillaires.
Figure 1.1. Comparaison de cerveaux de personnes saines et d’individus atteints de la maladie d’Alzheimer (d’après (Mattson 2004)). (a) Volume global du cerveau (rétrécissement marqué du lobe temporal (partie basse) et des lobes frontaux (partie gauche)). (b) Imagerie TEP révélant le glucose : le patient atteint d’Alzheimer montre une forte diminution du métabolisme énergétique dans le cortex
frontal (haut du cerveau) et les lobes temporaux (sur les cotés).
Anatomiquement, elle est caractérisée par des lésions de l’hippocampe (impliqué dans le processus de mémorisation) et une atrophie corticale diffuse mais souvent à prédominance pariéto-occipitale, ce qui rend compte de la symptomatologie aphasie-apraxie-agnosie.
Ainsi, le premier symptôme frappant est la perte de la mémoire à court terme (amnésie) ; elle se manifeste initialement par des distractions mineures qui s'accentuent avec la progression de la maladie, tandis que les souvenirs plus anciens sont relativement préservés. En même temps que les désordres progressent, l'affaiblissement cognitif s'étend aux domaines de la langue (aphasie), de l'adresse des mouvements (apraxie), de la reconnaissance (agnosie), et aux
fonctions exécutives que sont la prise de décision et la planification, rattachées de près aux lobes frontaux, reflétant l'extension du processus pathologique.
I.2. Rappel historique
Le 4 novembre 1906, lors de la 37ème conférence des psychiatres allemands à Tübingen, Aloïs Alzheimer, médecin psychiatre à Francfort, décrit un cas de démence d’une femme de 51 ans Auguste D. (Alzheimer 1907; Alzheimer 1911).
Figure 1.2. Aloïs Alzheimer (1864-1915)
Cette patiente, admise à l’hôpital de Francfort le 25 novembre 1901, décède le 8 avril 1906. Elle présentait de très nombreux symptômes, allant d’une compréhension et d’une mémoire réduite jusqu’à l’aphasie, la perte du sens de l’orientation, des comportements imprévisibles, de la paranoïa, des hallucinations auditives et un délabrement psychosocial avancé.
L’autopsie, à Munich, du cerveau de la patiente révéla des « plaques » formées par une substance particulière dans le cortex (déjà observé sur un patient épileptique plus âgé par Blocq et Marinesco)(Blocq and Marinesco 1892) et, pour la première fois, des enchevêtrements de neurofibrilles (Figure 1.3) (Goedert and Spillantini 2006). Or, en ce temps-là, un état de démence du sujet âgé etait considéré par la grande majorité des psychiatres comme normal, lié à l'usure normale du temps, à la fameuse artériosclérose (maladie artérielle caractérisée par un dépôt de cholestérol et de calcium sur les parois internes des artères). Cependant, à la suite de cette première observation, Emil Kraepelin, un des rares psychiatres à croire en l'intérêt de l'étude histologique du cerveau dans les maladies mentales, parla pour la première fois, dans son traité de psychiatrie en 1912, de la " maladie d'Alzheimer " définie alors comme une démence du sujet jeune (Auguste D. est décédée à
l’àge de 56 ans seulement), rare et dégénérative, laissant au terme de "démence sénile", les démences vasculaires du sujet âgé (Kraepelin 1910). C’est pourquoi, jusque dans les années 1960, on supposait que la maladie était rare, mais plus tard on s'aperçut que dans beaucoup de cas, ce que l'on avait pris pour des aspects normaux de la sénescence relevait en fait de la MA.
Figure 1.3. Section du cortex d’Auguste D. révélée à l’argent de Bielschowsky (a) mettant en évidence la présence de neurofibrilles (b) et de plaques amyloïdes (c) (Goedert et al. 2006).
I.3. La maladie d’Alzheimer priorité de santé publique
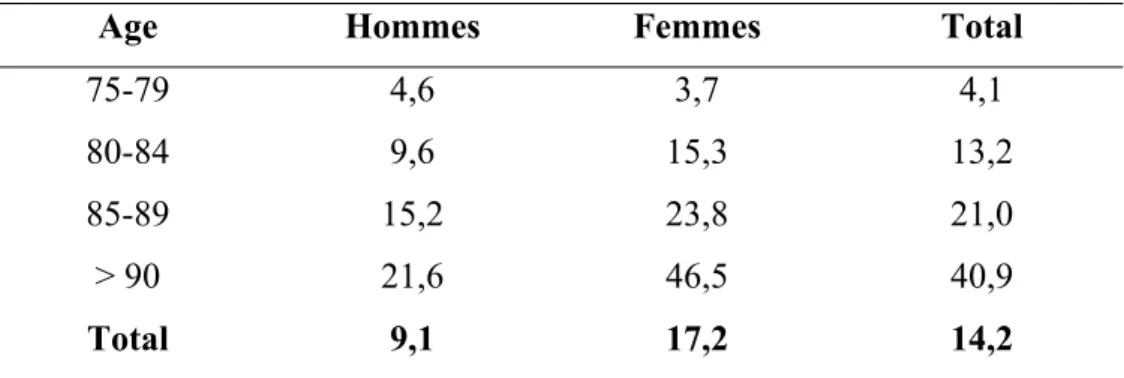
La MA est à présent la maladie neurodégénérative la plus répandue dans le monde avec environ 25 millions de malades. En France, on estime à 860 000 le nombre de personnes atteintes de la MA (Alzheimer 2006). Si les lésions se développent assez tôt dans la vie, la maladie ne s’exprime habituellement que tardivement, l’âge étant le premier facteur de risque (sauf dans le cas de mutations génétiques familiales, cf. section I.5.a). L’affection touche plus de femmes que d’hommes puisqu’au delà de 75 ans, les proportions sont de 13,2 % pour les hommes et de 20,5 % pour les femmes. Au-delà de 85 ans, la prévalence s’accroît de manière exponentielle avec une proportion de 25 % de sujets atteints. L’incidence s’élève à 225 000 nouveaux cas par an en France et l’espérance de vie après le début des symptômes est en moyenne de 8,5 ans (Tableau 1.1).
Actuellement 40 % des patients sont pris en charge dans une institution, ce qui signifie que 60% des sujets atteints sont à la charge des familles. La dépense moyenne pour la prise en charge d’un patient est de 22 000 euros par an, soit une charge annuelle de 10 milliards d’euros (moitié pour l’Etat et moitié pour les familles) qui se répartit en 75 % de dépenses médico-sociales et 25 % de dépenses médicales. Actuellement, ces dépenses s’élèvent à 0,6 % du PIB mais, compte tenu du vieillissement de la population, elles passeront à 0,8 % en 2020 (estimation à 1,3 millions de cas) et à 1,8 % en 2040 (2,1 millions). La prise en charge des
personnes atteintes de démence représentera alors, si la tendance actuelle continue, 7 % des dépenses de santé.
Age Hommes Femmes Total
75-79 4,6 3,7 4,1 80-84 9,6 15,3 13,2 85-89 15,2 23,8 21,0
> 90 21,6 46,5 40,9
Total 9,1 17,2 14,2
Tableau 1.1. Prévalence de la maladie d’Alzheimer en France pour les personnes âgées de 75 ans et plus (données issues de la cohorte PAQUID) (Ramaroson et al. 2003).
Il n’est donc pas étonnant que la MA ait été récemment désignée « grande cause nationale 2007 » par le gouvernement français et que le parlement européen la considère comme un « fléau » depuis déjà une dizaine d’années.
I.4. Le diagnostic
Aujourd’hui encore, le seul diagnostic définitif de la MA ne peut être établi que post mortem via l’autopsie du cerveau du malade. Ainsi, dans un premier temps, le diagnostic a reposé sur des tests cognitifs simples comme les échelles de Blessed (1968), de Pfeiffer (1975) et le célèbre Mini Mental Status Examination de Folstein (1975). Ces tests ont été conçus comme des outils de dépistage rapide et des indicateurs de sévérité de la démence, ils ne sont donc pas spécifiques de la MA.
En 1984, le NINCDS-ADRDA, groupe américain de spécialistes de la MA, propose les premiers critères diagnostic de la maladie. Peu à peu se dégage un consensus pour l'élaboration d'outils d'évaluation performants regroupés autour de 4 axes :
- tests cognitifs : Mattis Dementia Rating Scale ; Alzheimer Disease Assessment Scale-cognitive subscale (ADAS-cog) ; Syndrom Kurztest ;
- évaluation fonctionnelle (activité de la vie quotidienne) : Instrumental Activities of Daily Living (IADL) ; Physical Self-maintenance Scale ;
- évaluation comportementale (troubles comportementaux et de l'humeur) : ADAS-non cognitive subscale ; Behavioral pathology in Alzheimer Disease scale ; Neuropsychiatric
Inventory ;
- échelles de classification globale de sévérité : Global Deterioration Scale ; Clinical Dementia rating Scale.
Actuellement, ces tests sont toujours d’actualité car il n’existe pas encore de marqueur spécifique de la MA, toutefois ils s’accompagnent de quelques examens complémentaires plus ou moins routiniers (Sarazin and Dubois 2005).
Le scanner cérébral ou l’IRM sont régulièrement pratiqués mais ces examens ne permettent pas de mettre en évidence de lésions spécifiques : l'amincissement du cortex (atrophie corticale ou sous-corticale) se voit dans d'autres maladies de la personne âgée ; ils servent essentiellement à éliminer d'autres causes : tumeurs, accident vasculaire cérébral, hématome intra-cérébral ou sous-dural... Des indices sont cependant en cours d'évaluation pour tenter de faire un diagnostic précoce (dont la diminution de la taille de l'hippocampe, Figure 1.4).
Figure 1.4. Progression de l’atrophie hippocampique chez un patient atteint de la MA, l’examen annuel par IRM montre l’augmentation de l’atrophie hippocampique et parahippocampique (> rouge) accompagnée d’un élargissement ventriculaire (; verte) et
des sillons corticaux (t grise) (service de neurologie, Hôpital la Salpêtrière).
Plus récemment, des techniques de neuroimagerie fonctionnelle telle l’imagerie par résonance magnétique fonctionnelle (IRMf) ou la tomographie par émission de positons (TEP) et la tomographie de simple photon (TEMP) qui utilisent des traceurs radioactifs, ont été développées. Elles permettent l’évaluation du métabolisme des cellules (TEP au 18 F-flurodéoxyglucose), l’analyse de la perfusion cérébrale et la réalisation d’études pharmacologiques. Des recherches visant à développer des marqueurs de plaques amyloïdes à l’aide de la TEP sont actuellement en cours.
Enfin, le dosage de la vitamine B12 et de la vitamine B9 (folates), ainsi qu'un bilan thyroïdien (TSH) sont systématiquement réalisés, car une carence en vitamine B12 ou B9 et une hypothyroïdie peuvent être causes de démence (démence curable).
présentant des signes de démence d'apparition progressive et pour lesquelles les autres causes ont été éliminées. Un des enjeux majeurs de la recherche sur la maladie d’Alzheimer concerne donc la découverte de marqueurs prédictifs fiables permettant de faire un diagnostic sûr et le plus précoce possible, avant l’apparition de la maladie, c’est-à-dire lors de l’étape dite asymptomatique qui peut durer plusieurs années. Ce dépistage précoce permettrait une meilleure prise en charge et un allongement de la durée de vie du patient. La mise au point de marqueurs spécifiques permettrait également un suivi et une évaluation plus efficaces des traitements pharmacologiques.
I.5. Facteurs génétiques, biologiques et environnementaux I.5.a. Facteurs génétiques
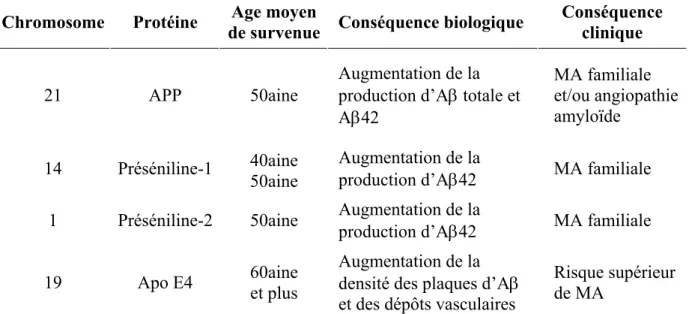
La MA est une affection polyfactorielle qui résulte de l’interaction entre un terrain génétique et des facteurs environnementaux. L’implication des gènes dans la maladie d’Alzheimer est double (Tableau 1.2) : d’une part, il existe des formes monogéniques exceptionnelles, caractérisées par un début précoce (inférieur à 60 ans) et par l’atteinte d’un sujet sur deux à chaque génération (formes autosomiques dominantes) ; d’autre part, dans les formes, de loin les plus courantes, dites sporadiques de la maladie, sont impliqués des facteurs de risque génétique (polymorphismes de l’ADN) qui constituent simplement un terrain génétique favorable.
Chromosome Protéine de survenueAge moyen Conséquence biologique Conséquence clinique
21 APP 50aine
Augmentation de la production d’Aβ totale et Aβ42 MA familiale et/ou angiopathie amyloïde 14 Préséniline-1 40aine 50aine Augmentation de la
production d’Aβ42 MA familiale 1 Préséniline-2 50aine Augmentation de la
production d’Aβ42 MA familiale 19 Apo E4 60aine et plus
Augmentation de la densité des plaques d’Aβ et des dépôts vasculaires
Risque supérieur de MA
Les formes familiales et héréditaires sont relativement rares (inférieure à 9%) et correspondent à plus de 150 mutations réparties sur 3 gènes : un gène du chromosome 21 codant pour la protéine APP (amyloid precursor protein), précurseur du peptide amyloïde bêta (Aβ) principal composant des plaques séniles ; un gène du chromosome 14 et du chromosome 1 codant respectivement pour le préséniline 1 (PS1) et 2 (PS2), les mutations sur ces derniers favorisent la libération de la forme longue Aβ42 (versus Aβ40), moins soluble.
Quant aux facteurs de susceptibilité génétique, la principale mutation concerne un gène situé sur le chromosome 19 qui code pour l’apolipoprotéine E (apoE) impliquée dans le transport du cholestérol (Corder et al. 1993; Strittmatter et al. 1993). En effet, les individus porteur d’un ou deux allèles ε4 (15 % de la population) ont plus de chance de développer la MA (40 à 65 % des cas sporadiques et familiales), contrairement aux allèles ε3, le plus courant, et ε2, protecteur (Farrer et al. 1997). L’allèle ε4 est également un facteur de risque pour d’autres maladies comme les démences vasculaires, la maladie de Creutzfeldt-Jacob et autres démences. Les mécanismes biologiques qui sous-tendent le rôle de l’apoE sont multiples et encore mal connus. Plusieurs hypothèses ont été émises : l’apoE E4 peut se lier au peptide Aβ et pourrait ainsi favoriser son agrégation (Stratman et al. 2005) ; elle augmenterait la production d’APP (Ye et al. 2005) ; l’apoE E4 est également la moins performante dans la restauration des membranes lésées via l’apport des lipides constitutifs nécessaires (Mahley et al. 2006) ; enfin elle pourrait, de manière indépendante au mécanisme amyloïde, avoir des effets toxiques sur les mitochondries via le clivage de sa partie C-terminale bioactive(Huang et al. 2001; Chang et al. 2005; Mahley and Huang 2006; Mahley et al. 2006).
Par ailleurs, deux gènes du chromosome 12 codant pour l’α2-macroglobuline et un récepteur des lipoprotéines de faible densité (tous deux impliqués dans la dégradation de Aβ) ont été identifiés comme facteurs de risque (Pericak-Vance et al. 1997; Myers and Goate 2001). Enfin la présence d’un ou plusieurs loci sur le chromosome 10 pourrait également être un facteur de risque pour la MA (Majores et al. 2000; Pericak-Vance et al. 2000). Cette région pourrait correspondre à l’enzyme de dégradation de l’insuline (qui jouerait un rôle dans la dégradation d’Aβ (Vekrellis et al. 2000) ; mais également dans l’augmentation du taux d’Aβ42 (Ertekin-Taner et al. 2000; Myers et al. 2001)).
I.5.b. Facteurs biologiques et environnementaux
Nous avons déjà vu que l’âge est un facteur prédominant pour l’apparition de la MA (cf. I.1.c). Le tableau 1.1 montre également que les femmes sont plus touchées que les hommes du fait de leur espérance de vie plus longue mais également à cause de la baisse en oestrogènes à la suite de la ménopause (Alberca et al. 2002; Candore et al. 2006). En effet les oestrogènes interviennent dans de nombreux processus au sein du cerveau (apoptose, stress oxydant, plasticité synaptique, effets sur les neurotransmetteurs, transport du glucose, réaction inflammatoire…) dont certains ont une influence dans le développement de la MA (Henderson 2000), cependant les mécanismes sous-jacents sont complexes et encore à élucider. En effet, paradoxalement, les femmes de plus de 65 ans ayant une supplémentation en œstrogène ont plus de chance de développer des démences (Henderson 2006).
De plus, un faible niveau d’éducation, ce qui est souvent le cas chez les femmes âgées, a également une incidence négative et favorise l’apparition de la MA.
De nombreuses études ont également démontré qu’un taux élevé de cholestérol est un facteur de risque pour la MA (Michikawa 2003). Des hypothèses, parfois contradictoires, ont également été avancées concernant l’influence des radeaux lipidiques lors de la coupure de l’APP (Riddell et al. 2001; Wahrle et al. 2002; Cordy et al. 2003; Ledesma et al. 2003; Chen et al. 2006). Mais il faut savoir que le taux de cholestérol dans le sang est largement indépendant de celui du système nerveux central (SNC), en effet ce dernier en synthétise la majeur partie de novo, le lien serait donc plutôt indirect. D’autant plus que les traitements à base de statines (visant à diminuer le taux cholestérol sanguin chez les patients atteint de problèmes cardiovasculaires) ont été supposés diminuer l’incidence de la MA (Jick et al. 2000; Wolozin et al. 2000), indépendamment de leur propriétés lipophiles et donc de leur capacité à passer la barrière hémato-méningée (Canevari and Clark 2007).
De manière générale, une carence alimentaire en vitamines antioxydantes (notamment C et E), fréquentes chez les personnes âgées alors que le stress oxydant est important, est un facteur aggravant de la MA (Morris et al. 2006).
L’hypertension artérielle, qui est connue pour être un facteur aggravant de la MA, pourrait également être un facteur de risque. De plus, des études ont montré que des personnes traitées
contre l’hypertension artérielle avaient un risque moindre de développer la MA, reste à savoir si ce résultat est lié à l'effet direct des molécules ou uniquement au contrôle de la tension (Birkenhager and Staessen 2004). Paradoxalement, il a été rapporté qu’une faible pression diastolique (<65 mmHg) était également un facteur de risque pour la MA (Qiu et al. 2003).
La MA s’accompagne généralement de mécanismes inflammatoires (probablement dus au stress oxydant), ainsi on note un rôle protecteur des anti-inflammatoires non stéroïdiens (AINS) comme l’aspirine et l’ibuprofène. Mais les effets secondaires de ces médicaments ne permettent pas d’envisager une prévention systématique. Cependant, de récentes et prometteuses recherches ont démontré que certains de ces AINS, dont l’ibuprofène, ont la propriété étonnante de diminuer le taux de peptides Aβ42 (forme longue plus agrégeante), via peut-être une modulation de l’activité des γ-secrétases (Czirr and Weggen 2006).
Enfin on peut citer comme derniers facteurs de risque : les traumatismes crâniens (Fleminger et al. 2003), l’exposition aux pesticides (Baldi et al. 2003), les dépressions nerveuses (Jorm et al. 1991; Alexopoulos et al. 1993; Devanand et al. 1996). Il semblerait finalement qu’une consommation modérée de vin ou d’alcool diminue le risque de démence et de MA (Letenneur 2004).
II. Plaques amyloïdes et neurofibrilles
La maladie d'Alzheimer est caractérisée par la rencontre de deux processus dégénératifs différents, qui semblent se conjuguer pour provoquer la dégénérescence des cellules nerveuses : l'agrégation de la protéine tau sous forme de filaments dans les cellules nerveuses (processus de dégénérescence neurofibrillaire ou pathologie tau) et l’agrégation du peptide amyloïde bêta (Aβ) sous forme de plaques amyloïdes à l’extérieur des neurones. L’état actuel des connaissances sur l’agrégation de la protéine tau sera présenté succinctement, puis nous nous intéresserons plus longuement au peptide Aβ sur lequel porte cette thèse.
II.1. La protéine tau
45 à 62 kilodaltons. Elles sont codées par un seul gène contenant 16 exons et situé sur le chromosome 17. L'épissage alternatif de ce gène génère la formation de six isoformes (de 48 à 67 kDa), co-existant dans les neurones dans certains rapports (Wischik et al. 1988; Goedert et al. 1989; Goedert et al. 1989).
Ces protéines comportent des régions répétitives du côté C-terminal (en noir sur la figure 1.5) qui permettent l'accrochage aux microtubules1 (Lee et al. 1989). En effet, le rôle des protéines tau est de réguler l’assemblage des sous-unités de tubuline en microtubules : les protéines phosphorylées induisent la dépolymérisation des microtubules alors que les protéines déphosphorylées les stabilisent.
Figure 1.5. Représentation schématique des six isoformes de la protéine tau exprimées dans le cerveau humain adulte. Les régions communes à tous les isoformes sont représentées en
bleu, en noir les régions s’associant aux microtubules.
Dans la MA, la dégénérescence neurofibrillaire (Figure 1.6) résulte de l’agrégation intraneuronale de protéines tau anormalement phosphorylées sous la forme de paires de filaments appariés en hélice (Kidd 1963; Grundke-Iqbal et al. 1986; Ihara et al. 1986; Goedert et al. 1988; Wischik et al. 1988; Wischik et al. 1988; Goedert et al. 1992). Ces filaments ont un diamètre de 10 nm et un pas d'hélice de 80 nm, ils s'accumulent dans les corps cellulaires des neurones ainsi que dans leurs prolongements neuritiques.
1 Les microtubules, fibres constitutives du cytosquelette, sont des tubes dont la paroi est constituée de plusieurs
protofilaments de tubuline. Très nombreux dans les neurones, en particulier dans les dendrites et les axones, ils permettent d'acheminer divers composants vers leurs extrémités.
Figure 1.6. exemples de dégénérescences neurofibrillaires ou neurofibrilles (a : localisation himmunohistochimique avec anticorps spécifiques de l’état de phosphorylation de tau (Goedert et al. 2006) ; b : section du cortex cérébral d’Auguste D. marqué à l’argent de
Bielscowsky (Graeber et al. 1998)).
Plusieurs observations et hypothèses ont été faites pour tenter d’expliquer la formation de ces neurofibrilles à partir de protéines tau non mutées et donc a priori fonctionnelles.
• Les protéines tau hyperphosphorylées ont également été décrites comme étant anormalement glycosylées, ce qui induirait une stabilisation l’hélicité des paires de filaments (Wang et al. 1996). Cette glycosylation précède et favoriserait l’hyperphosphorylation et ainsi la formation des filaments (à noter que la glycosylation seule n’a pas d’effet sur la capacité d’assemblage des microtubules) (Liu et al. 2002; Liu et al. 2004).
• L’état de phosphorylation d’une phosphoprotéine est fonction d’un équilibre entre l’activité de protéines kinases et de protéines phosphatase. Or il se trouve que l’activité d’une de ces dernières, la protéine phosphatase 2A (PP2A), est diminuée d’environ 20 % dans le cerveau de personnes atteintes de la MA (Gong et al. 1993). Cette diminution influe sur l’hyperphosphorylation de le protéine tau (Gong et al. 1995) et ainsi sur sa capacité d’assemblage des microtubules (Gong et al. 1994; Gong et al. 2000; Bennecib et al. 2001). La diminution de PP2A est elle-même probablement due à l’augmentation (d’environ 20 %) d’une de ces deux protéines inhibitrices IPP2A 1 (protéine cytosolique de 30 kDa) (Li et al. 1995).
• La protéine tau est également plus facilement phosphorylée quand elle est libre que liée aux microtubules.
• La séquence peptidique joue aussi un rôle, la vitesse et le taux de phosphorylation
variant avec le nombre de domaines de liaison aux microtubules (3 ou 4, en noir sur la figure 1.5) et d’inserts N-terminaux (0, 1 ou 2, en rose et vert) (Singh et al. 1997).
Mais quelles sont les conséquences de cette hyperphosphorylation en terme de toxicité ? La quantité globale en protéine tau est équivalente dans un cerveau sain et dans celui atteint de la MA. Cependant la concentration en protéine tau hyperphosphorylée est environ 8 fois supérieure dans ce dernier (Khatoon et al. 1992; Khatoon et al. 1994). Or l’hyperphosphorylation diminue la capacité de la protéine à s’apparier avec les microtubules (Lindwall and Cole 1984; Bramblett et al. 1993; Yoshida and Ihara 1993; Tanaka et al. 1995). Pire encore, ces protéines tau anormalement phosphorylées désassemblent les MT déjà formés en séquestrant les protéines tau normales (Alonso et al. 1994; Santacruz et al. 2005) La perte en microtubules induit vraisemblablement une inhibition du transport axoplasmique suivie d’une dégénérescence synaptique et neuronale.
Tauopathies Maladie d’Alzheimer
Sclérose latérale amyotrophique ou maladie de Charcot Maladie des grains argyrophiles
Maladie de Parkinson précoce autosomique dominante Parkinsonisme autosomique dominant
Maladie de Parkinson juvénile autosomique récessive Dégénérescence cortico-basale
Dementia pugilistica (démence des boxeurs)
Démence avec dégénerescence neurofibrillaire diffuse et calcifications intracrâniennes
Syndrome de Down (trisomie 21) Démence britannique familiale
Démence frontotemporale avec parkinsonisme liée au chromosome 17 Syndrome de Gerstmann-Sträussler-Scheinker
Parkinsonisme atypique de Guadeloupe Maladie de Hallervorden-Spatz
Dystrophie myotonique (maladie de Steinert) Maladie de niemann-pick de type C
Maladie de Pick
Parkinsonisme post-encéphalitique
Angiopathie amyloïde cérébrale à protéine prion Gliose sous-corticale progressive
Paralysie supranucléaire progressive Panencéphalite sclérosante subaiguë
Démence à dégénérescence neurofibrillaire seule
La formation de dépôts de protéines tau n’est pas spécifique à la MA. Ce processus dégénératif, qui n’est pas forcément doublée de la présence de plaques amyloïdes (Spillantini et al. 1996; Spillantini et al. 1998; Crowther and Goedert 2000), est en effet le plus répandu parmi les pathologies démentielles et neurodégénératives. Une grande partie de ces maladies, nommées tauopathies, est listée dans le tableau 1.3. Les sites de phosphorylation sont similaires, avec des différences mineures entre les maladies. Cependant la distribution des différents isoformes dans la composition des filaments varie. Ainsi la MA et le complexe ‘‘sclérose latérale amyotrophique / syndrome parkinsonien’’ de l’île de Guam sont caractérisés par l’hyperphosphorylation des isoformes de 60, 64 et 68 kDa (présence également, mais minoritaire, de l’isoforme de 72 kDa). En comparaison, l’isoforme de 60 kDa n’apparaît pas dans les filaments correspondant à la paralysie supranucléaire progressive et la dégénération corticobasale (Flament et al. 1991; Ksiezak-Reding et al. 1994), mais est majoritaire, avec l’isoforme à 64 kDa, dans la maladie de Pick.
L’identification dans les années 90, de mutations dans le gène de l’APP et de la préséniline dans les cas héréditaires de la MA (environ 1% des personnes atteintes) (Goate et al. 1991; Murrell et al. 1991; Levy-Lahad et al. 1995; Rogaev et al. 1995; Sherrington et al. 1995) et le fait que les dépôts de filaments de protéines tau soient présents dans différentes conditions sans relations apparentes, a conduit une partie de la communauté scientifique à considérer cette agrégation comme un épiphénomène de peu d’importance. A l’heure actuelle, le débat reste ouvert.
II.2. Le peptide amyloïde bêta
II.2.a. Cas général des amyloïdoses
Lors de la synthèse de protéines au sein d’une cellule, il y a toujours un risque qu’elles s’agrègent. Mais la formation d’agrégats est normalement inhibée par les molécules chaperons et les processus de dégradation, de plus ils sont défavorisés par les séquences d’acides aminés choisies par l’évolution. Il arrive pourtant que certaines protéines, du fait de mutations ou de facteurs environnementaux défavorables, s’agrègent ; dans le cas où les agrégats sont de type fibrillaire (à distinguer des agrégats amorphes), il s’agit d’une amyloïdose (ou amylose). Il existe ainsi de nombreuses maladies humaines associées à la formation de fibres amyloïdes extracellulaires ou d’inclusions intracellulaires de type
amyloïde, à partir de protéines normalement solubles. Quelques exemples d’amyloses sont donnés dans le tableau 1.4 où elles sont classées en trois groupes distincts : les maladies neurodégénératives, comme la MA, dans lesquelles l’agrégation a lieu dans le cerveau ; les amyloses non neuropathiques localisées pour lesquelles l’agrégation a lieu dans un type de tissu précis autre que le cerveau et enfin les amyloses non neuropathiques systémiques dont les dépôts sont retrouvés dans différents tissus de l’organisme (Chiti and Dobson 2006; Gillmore and Hawkins 2006).
Maladie Protéine / peptide impliqué Nombre
de résidus
Structure native Maladies neurodégénératives
Maladie d’Alzheimer a Peptide amyloïde bêta 40-42 Non structuré Encephalopathie
spongiforme a, c Prion 253 Résidus non structuré 1-120
Résidus 120-230 hélice alpha
Maladie de Parkinson a Α-synucléine 140 Non structuré
Maladie d’Huntington Huntingtine – poly Q 3144 Principalement non structuré Démence britannique
familiale b
Abri 23 Non structuré
Démence familiale danoise b
ADan 23 Non structuré
Amyloses non neuropathiques localisées
Diabète du type II a Amyline 37 Non structuré
Dystrophie héréditaire de la cornée b
kérato-épithéline (fragment C-terminal principalement)
50-200 Inconnu
cataracte a Cristalline γ variable Inconnu
myosite à inclusions (myopathie) a
Peptide bêta amyloïde 40-42 Non structuré
Amyloses non neuropathiques systémiques Amylose AL
(primaire) a
Immunoglobuline (chaîne légère) ~90 Feuillet bêta Amylose AA
Amylose ApoAI a, II a
ou IV b Fragment N-terminal de l’apolipoprotéine AI, II ou IV 98, ~70 ou 71 Non structurée l’amylose de la
lysozyme b
Lysozyme muté 130 Hélice alpha +
feuillet bêta l’amylose du
fibrinogène b
Fibrinogène (chaîne α) 27-81 Inconnu
Amylose systémique sénile a
Transthyrétine normale 127 Feuillet bêta Polyneuropathie
amyloïde familiale b
Transthyrétine mutée 127 Feuillet bêta
a majoriatirement sporadique b majoritairement héréditaire c 5% de cas par transmission
Figure 1.4. Exemples et classification des amyloses (ou amyloïdoses) (Chiti et al. 2006; Gillmore et al. 2006).
Certaines de ces amyloses sont majoritairement sporadiques, c’est le cas de la MA et de la maladie de Parkinson, bien que des formes héréditaires soient également présentes (Baltasar-Rodriguez et al. 2006). D’autres, comme l’amylose de la lysozyme ou celle du fibrinogène, sont dues à des mutations spécifiques et sont héréditaires. A cela faut-il ajouter l’encéphalopathie spongiforme qui peut être transmissible (5 % des cas contre 85 % de formes sporadiques et 10 % héréditaires).
De façon générale, les dépôts amyloïdes extracellulaires sont majoritairement constitués de la protéine incriminée, qui forme le cœur du dépôt, associée à diverses molécules (glycosaminoglycanes, apolipoprotéine E, collagène, ions métalliques,… et bien d’autre). Les fibres qui composent en partie ces dépôts, sont généralement constituées de 2 à 6 protofilaments de 2 à 5 nm de diamètre chacun (Serpell et al. 2000) (à ne pas confondre avec les protofibrilles, agrégats sphériques de diamètre similaire constitués d’une vingtaine de protéines en feuillets bêta) (Harper et al. 1997; Harper et al. 1997; Walsh et al. 1997; Walsh et al. 1999). Une description plus précise de la structure des fibres amyloïdes sera donnée pour le cas de la MA dans la section II.2.d.
Ces différentes protéines susceptibles de former des dépôts amyloïdes n’ont paradoxalement pas de séquence peptidique commune ou d’homologie structurale évidente. En effet, autant du point de vue des structures secondaires que des longueurs / compositions des chaînes
peptidiques, l’hétérogénéité est de mise. Cependant, un nombre élevé de résidus hydrophobes, l’absence de charge nette locale ou globale élevée, et une propension à former des structures en feuillets bêta, semblent être les trois dénominateurs communs de ces protéines (Broome and Hecht 2000; Otzen et al. 2000; Schwartz et al. 2001; Chiti et al. 2002; Uversky 2002; Wurth et al. 2002).
Enfin, il faut noter que, dans quelques cas relativement rares, la formation de fibres amyloïdes a été conservée par l’évolution pour générer des nanostructures naturelles exploitées par l’organisme. La plupart de ces amyloses naturelles connues sont développées par des bactéries, champignons ou levures (Coustou et al. 1997; Eaglestone et al. 1999; True and Lindquist 2000; Mackay et al. 2001; Chapman et al. 2002; Claessen et al. 2003; Chien et al. 2004; True et al. 2004). Mais il en existe aussi par exemple chez l’araignée : les fibres de spidroïne forment la soie de la toile de la Nephila Edulis (Kenney et al. 2002). Chez l’être humain, un fragment de la glycoprotéine Pmel17 polymérise en fibres amyloïdes afin de séquestrer la mélanine, durant la biogenèse des mélanosomes. Cette fibrillation est régulée via la coupure de Pmel17 par la furine, une convertase de proprotéine, ce qui n’est pas sans rappeler la coupure de l’APP (Amyloid Precurseur Protein) par les γ et β-sécrétases impliquées dans la MA (cf. section II.2.b). Le mécanisme de ces amyloses naturelles contrôlées pourrait ainsi inspirer l’élaboration de traitement permettant de réguler les amyloses pathologiques.
II.2.b. La protéolyse de l’APP Description et fonction de l’APP
L’APP (amyloid precursor protein) est une protéine transmembranaire de type 1 du système nerveux central, d’environ 700 acides aminés. Elle est constituée d’un large domaine extracellulaire (environ 88% du poids total, pour l’isoforme majoritaire), d’une unique région transmembranaire et d’une petite partie cytosolique. Son rôle exact n’est pas encore élucidé, mais il est clair qu’elle est impliquée dans la synaptogénèse et la plasticité synaptique (Gralle and Ferreira 2007). Il a été montré que l’APP interagissait avec de nombreuses protéines : la protéine Go (protéine liant le GTP et intervenant donc dans la signalisation intracellulaire) (Nishimoto et al. 1993), des protéines adaptatrices comme, par exemple, Fe65 (impliquée dans la mobilité cellulaire, elle stimulerait la production d’APP/Aβ et interagirait également
avec la protéine tau) (Sabo et al. 2001; Sabo et al. 2003; Barbato et al. 2005; Yoon et al. 2005), X11/Mint (impliquée, entre autres, dans la formation des synapses, diminuerait la production d’APP/Aβ) (Lau et al. 2000; Mueller et al. 2000) et JIP (dont l’interaction avec APP déboucherait sur une dégradation du signal axonal via la protéine kinésine-1) (Matsuda et al. 2003).
Génération du peptide Aβ
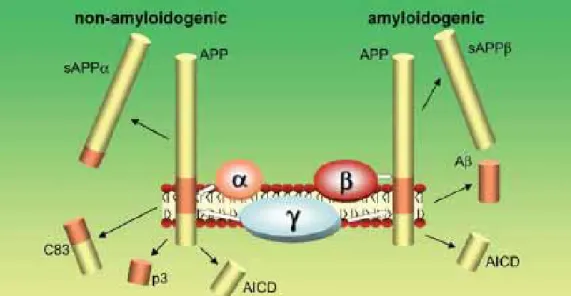
La région Aβ se situe au niveau de la partie membranaire de la protéine APP. Elle est composée des 28 acides aminés jouxtant le domaine transmembranaire et des 12 à 14 résidus voisins faisant partie du domaine transmembranaire. L’Aβ résulte d’un clivage séquentiel de l’APP par deux enzymes protéolytiques (Figure 1.7) : la β- et la γ-sécrétase (Wolfe 2006). Dans un premier temps, la β-sécrétase, une protéase aspartique aussi nommée BACE1 (pour Beta site APP Cleaving Enzyme 1, également appelée memapsin 2) et dont l’activité enzymatique n’est possible qu’en milieu subcellulaire (à pH acide), clive l’APP au niveau du site bêta, libérant dans le milieu extracellulaire un large ectodomaine soluble (sAPPβ) (Vassar et al. 1999). Puis le fragment transmembranaire C-terminal restant, C99, est clivé au niveau de la bicouche lipidique près de la frontière cytosolique par la γ-sécrétase, générant ainsi les peptide Aβ40 ou Aβ42 et le fragment AICD (APP Intracellular Cytosolic Domain) libéré dans le cytosol.
L’APP peut également, et plus couramment, être clivé par l’α-sécrétase plutôt que la β-sécrétase, c’est la voie non amyloïdogénique. L’α-sécrétase est une protéine de la famille ADAM (A Disintegrin And Metalloprotease, protéines intervenant dans l’adhésion, la migration cellulaire et le clivage protéolytique de cytokines et facteurs de croissance), la littérature propose 3 candidates : ADAM-10, ADAM-17 (TACE) et ADAM-9 (Kojro and Fahrenholz 2005). Le site alpha se trouvant au milieu de la région Aβ, le clivage correspondant libère un large ectodomaine soluble (sAPPα), puis l’action de la γ-sécrétase sur le fragment C-terminal restant C83 génère le peptide p3 non pathogène.
Figure 1.7. Représentation schématique du clivage de l’APP libérant le peptide Aβ (Pearson and Peers 2006).
BACE1 est une protéine membranaire de type 1 caractérisée par un large domaine extracellulaire comprenant deux résidus aspartiques impliqués dans l’activité β-sécrétase (Hussain et al. 1999). Elle semble être particulièrement active dans les endosomes (son activité requérant un pH acide), ce qui pourrait indiquer que le clivage de l’APP se fait préférentiellement durant son internalisation (Koo and Squazzo 1994; Koo et al. 1996; Perez et al. 1999). Au niveau de la membrane, elle est localisée dans les radeaux lipidiques (microdomaines de la membrane biologique enrichis notamment en cholestérol) (Riddell et al. 2001; Cordy et al. 2003; Ehehalt et al. 2003; Abad-Rodriguez et al. 2004), ces derniers renforçant son activité (Kalvodova et al. 2005). De récentes études ont également avancé que BACE1 pourrait se dimériser, acquérant ainsi une spécificité catalytique envers l’APP, en particulier si celui-ci est sous forme d’homodimères ou tétramères (Multhaup 2006).
La γ-sécrétase, qui ne coupe que les liaisons peptidiques situées à l’intérieur de la bicouche lipidique (Weihofen and Martoglio 2003; Wolfe and Kopan 2004), est un complexe composé de 4 protéines : la préséniline (PS1 et PS2), la nicastrine, Aph-1 et Pen-2 (De Strooper et al. 1998; Yu et al. 2000; Francis et al. 2002; Goutte et al. 2002). La stoechiométrie et la fonction de chacune de ces sous-unités au sein du complexe restent à déterminer, on sait cependant que l’activité catalytique du complexe est très probablement portée par PS1 (Herreman et al. 2000; Zhang et al. 2000). Il est à noter que la préséniline joue également un rôle essentiel dans la voie de signalisation Notch (mis-en jeu dans la différenciation cellulaire) (Artavanis-Tsakonas et al. 1995; De Strooper et al. 1999; Radtke et al. 2004), limitant ainsi les applications
thérapeutiques la ciblant. La majorité des cas héréditaires de la MA proviennent de mutations au niveau des gènes codant pour PS1 ou PS2.
La phosphorylation de APP sur certains résidus sérine et thréonine joue également un rôle important dans la production de Aβ (Lee et al. 2003; Lu et al. 2003). Plus précisément, il a été montré que la phosphorylation du motif Thr668-Pro induit un changement conformationnel (trans → cis, passage de ~0 à ~10% de formes cis) favorisant la voie amyloïdogénique (clivage par la β-sécrétase au lieu de l’α-sécrétase) (Ramelot and Nicholson 2001). Pour rétablir l’équilibre initial, la peptidyl-prolyl isomérase Pin1 catalyse le changement cis → trans, favorisant ainsi à nouveau la voie non-amyloïdogénique (Figure 1.8).
Figure 1.8. Diminution de la production de Aβ sous l’effet de la protéine Pin1, catalyseur de l’isomérisation du motif pThr 668-Pro de l’APP (Pastorino et al. 2006).
Une dérégulation de la protéine Pin1 et/ou son inhibition par oxydation pourrait donc favoriser le développement de la MA (Pastorino et al. 2006).
Pin1 pourrait également avoir un effet protecteur dans les tauopathies (cf. section II.1) et le cancer (Lu 2003; Lu et al. 2006).
II.2.c. Le peptide Aβ Description
Le clivage de l’APP par la β- et la γ-sécrétase conduit principalement à un peptide Aβ de 40 acides aminés noté Aβ40. Cependant, d’autres formes plus courtes ou plus longues sont également produites (entre 39 et 43 acides aminés), en particulier la forme Aβ42, dont la séquence peptidique est indiquée sur la figure 1.9, représente environ 10 % des espèces d’Aβ. Les deux peptides Aβ40 et Aβ42 sont normalement présents en tant qu’espèces solubles dans les fluides biologiques de tous les individus. Cependant l’Aβ42, plus hydrophobe et principal constituant des plaques séniles, est supposé jouer un rôle plus important qu’Aβ40 dans la pathologie.
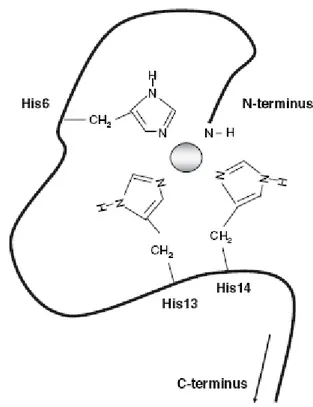
Asp1-Ala-Glu-Phe-Arg-His6-Asp-Ser-Gly-Tyr10-Glu11-Val-His13-His14
-Gln- Lys-Leu-Val-Phe-Phe-Ala-Glu-Asp-Val-Gly-Ser-Asn-Lys-Gly-Ala-Ile-Ile-Gly-Leu-Met35-Val-Gly-Gly-Val-Val40-Ile41-Ala42
Figure 1.9. Séquence peptidique du peptide Aβ42, écrite du N- vers le C-terminal. La partie en gras correspond au domaine intramembranaire. En bleu sont indiqués les acides aminés correspondant uniquement à Aβ42, en rouge ceux ayant été évoqués comme participant à la
chélation des ions métalliques (dans le site de plus forte affinité) (Clippingdale et al. 2001).
Dans le cadre des études portant sur l’interaction du peptide avec les métaux, les formes tronquées Aβ16 et Aβ28 (c’est-à-dire comprenant les acides aminés respectivement 1 à 16 et 1 à 28) sont couramment utilisées. En effet, elles contiennent les acides aminés correspondant au principal site de fixation des métaux et ont l’avantage d’être entièrement (Aβ16) ou partiellement (Aβ28) solubles, ce qui peut dans certains cas faciliter l’expérimentation mais ne permet généralement pas l’étude à l’état agrégé.
Rôle du peptide Aβ chez le sujet sain
Le fait que le peptide Aβ soit généré tout au long de la vie sans forcément induire une pathologie, permet de penser qu’il a peut-être une fonction physiologique. Cependant cette dernière est encore largement méconnue : une activité neurotrophique et un effet stimulant sur la viabilité neuronale ont été évoqués (Whitson et al. 1990; Yankner et al. 1990), notamment en contrôlant un relargage excessif de glutamate dans la synapse (Lesne and Kotilinek 2005; Pearson et al. 2006), l’hypothèse d’une régulation au niveau de canaux ioniques et dans l’homéostasie du calcium a également été avancée (Abramov et al. 2004; Plant et al. 2006). Il
a également été montré qu’Aβ42 pouvait réguler négativement le taux de sphingomyéline et Aβ40 la synthèse de cholestérol (Grimm et al. 2005). Il faut cependant être prudent dans l’assignation d’un rôle physiologique à Aβ, sa séquence peptidique étant très peu conservée chez les humains et rongeurs (bien que ce ne soit pas le cas chez d’autres mammifères), il est possible que ce peptide n’ait pas de fonction physiologique essentielle.
II.2.d. Structure des fibres amyloïdes
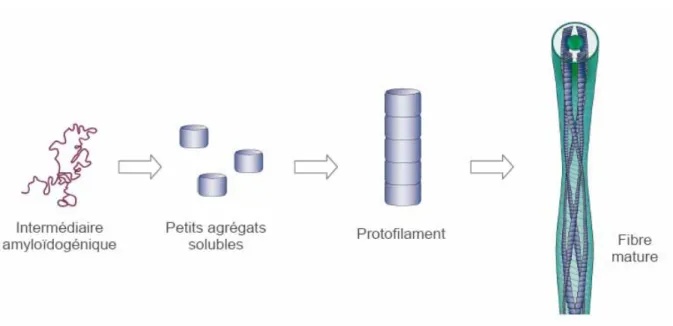
Le processus de formation de fibres et de plaques amyloïdes à partir du peptide monomérique soluble, sujet d’intenses recherches, n’a pas encore été élucidé. Mais il est admis que ce processus passe par l'association de petits agrégats ordonnés ou unités de nucléation, dont la croissance va déboucher sur de longs protofilaments qui s'enroulent pour former des fibres amyloïdes matures (figure 1.10).
Figure 1.10. Représentation schématique du processus de formation des fibres amyloïdes.
Cependant, la structure de fibres formées in vitro est, elle, bien décrite dans la littérature. Cette description a été élaborée à partir de données expérimentales obtenues principalement par RMN du solide (en accord avec d’autres techniques RMN, diffraction des rayons X et microscopie électronique) (Malinchik et al. 1998; Antzutkin et al. 2000; Goldsbury et al. 2000; Antzutkin et al. 2002; Balbach et al. 2002; Petkova et al. 2002; Antzutkin et al. 2003).
Figure 1.11. Modèle structural des protofilaments d’Aβ40 (Tycko 2003),(a) représentation en ruban, (b) représentation atomique (en vert les résidus hydrophobes, en magenta les résidus
polaires, en bleu ceux chargés positivement, en rouge ceux chargés négativement).
Dans le modèle structural des protofilaments obtenu par RMN du solide, figure 1.11, les peptides sont agencés en couches de deux feuillets bêta, perpendiculairement à l’axe de la fibre (structure dite « cross bêta »). Les résidus 12-24 et 30-40 sont impliqués dans les feuillets bêta, 25-29 dans le coude, les 10 premiers acides aminés ne sont pas structurés. Les liaisons C=O et N-H du squelette sont approximativement perpendiculaires à la page. Le cœur du protofilament est majoritairement hydrophobe (interactions hydrophobes entre brins), à l’exception des résidus chargés D23 et K28 qui forment un pont salin. La diversité de morphologie des fibres observées en microscopie électronique reflète les différences d’association latérale des protofilaments (Goldsbury et al. 1997; Jimenez et al. 2002).
Les protofilaments formés par le peptide Aβ42 ont vraisemblablement une structure très similaire, leur plus grande vitesse de formation serait d’avantage due à la moindre solubilité d’Aβ42 par rapport Aβ40 plutôt qu’à une différence structurale (Antzutkin et al. 2002; Torok et al. 2002; Tycko 2003).
Les caractéristiques fondamentales des fibres amyloïdes sont une coloration au rouge Congo (associée à une biréfringence en lumière polarisée), une augmentation/déplacement de la fluorescence de la Thioflavine T et leur résistance aux protéases.
II.2.e. Relation entre Aβ et tau
Bien que les deux caractéristiques de la MA, les plaques amyloïdes et les neurofibrilles, aient été identifiées depuis maintenant plus de cent ans, les relations existant entre Aβ et tau sont encore mal définies. Actuellement, les dernières recherches tendent à montrer qu’Aβ pourrait directement ou indirectement interagir avec tau pour accélérer la formation de neurofibrilles (Gotz et al. 2001; Lewis et al. 2001; Oddo et al. 2003), confirmant ainsi l’hypothèse de la cascade amyloïde. A titre d’exemple, l’accumulation de plaques amyloïdes précède de quelques mois le dévelopement de neurofibrilles chez des souris triplement mutées (sur l’APP, tau et PS1) (Oddo et al. 2003). Il semble donc qu’Aβ, via des mécanismes cellulaires et moléculaires distincts, facilite la phosphorylation, l’agrégation, une mauvaise localisation et l’accumulation de tau, aboutissant sur l’apparition de neurofibrilles (Blurton-Jones and Laferla 2006).
Il a été montré qu’un grand nombre de kinases, en particulier GSK3β et CDK5 (impliquées dans la phosphorylation de tau et donc indirectement dans la formation de neurofibrilles) (Cruz et al. 2003; Necula and Kuret 2004; Necula and Kuret 2005), voient leur expression augmentée dans la MA (Pei et al. 2001; Ferrer et al. 2002; Swatton et al. 2004; Derkinderen et al. 2005; Griffin et al. 2005; Ho et al. 2005; Stoothoff and Johnson 2005). Or des expériences in vitro ont montré que la présence d’Aβ dans les cultures cellulaires entraîne une activation de GSK3β et une augmentation de la phosphorylation de tau (Takashima et al. 1993; Busciglio et al. 1995; Alvarez et al. 1999). De plus, certaines études semblent indiquer que la toxicité induite par Aβ est dépendante de tau. Par exemple, le blocage in vitro de GSK3β et CDK5 protège les neurones contre Aβ (Michaelis et al. 1998; Rapoport et al. 2002). De même, in vivo, des souris transgéniques porteuses du gène muté de l’APP exhibent une augmentation de l’activation de kinases de tau (Hwang et al. 2004; Puig et al. 2004). Plus récemment, une réduction in vivo de la quantité d’oligomères solubles d’Aβ (mais pas des insolubles) a été corrélée avec une réduction de l’activité de GSK3β et de la phosphorylation de tau (Ma et al. 2006). Il faut cependant rappeler que les souris transgéniques porteuses du gène muté de
l’APP ne développent pas la pathologie correspondante à l’hyperphosphorylation de tau et à la formation de neurofibrilles (excepté une très faible quantité au niveau des plaques neuritiques extracellulaires). En dehors de l’espérance de vie limitée des rongeurs, il est possible que la différence d’espèce implique l’absence d’une kinase nécessaire au lien entre Aβ et tau.
Il est établi que les cerveaux atteints de la MA sont particulièrement sujets à des réactions inflammatoires (cellesci étant à la fois bénéfiques – phagocytose d’Aβ et préjudiciables -activation des cytokines proinflammatoires) (Duffy et al. 1980; Sheng et al. 1995; Akiyama et al. 2000; Wyss-Coray and Mucke 2002; Koenigsknecht-Talboo and Landreth 2005). De plus, des études in vitro ont également montré qu’Aβ était capable d’induire directement une réponse inflammatoire (Galimberti et al. 1999; Tan et al. 2000; Combs et al. 2001). Or certaines cytokines proinflammatoires induisent, in vitro et parfois in vivo, une accélération de l’hyper phosphorylation de tau et de la formation de neurofibrilles (Sheng et al. 2000; Li et al. 2003; Quintanilla et al. 2004). Ce dernier mécanisme semble impliquer CDK5 (Cruz et al. 2003; Kitazawa et al. 2005).
Plusieurs études ont également montré que la voie ubiquitine-protéasome, mécanisme principal de dégradation des protéines, était déficient chez les patients atteints de la MA (réduction de 48 % de l’activité du protéasome dans l’hippocampe) (Keller et al. 2000) et que cette déficience pourrait contribuer à la formation d’agrégats de tau. En effet de récentes études semblent indiquer qu’Aβ est impliqué directement ou indirectement dans le dysfonctionnement du protéasome (Gregori et al. 1997; Lopez Salon et al. 2003; Oh et al. 2005), tandis qu’il existe de nombreux indices attestant d’un lien fort entre la déficience du protéasome et l’accumulation de tau, même s’il n’est pas encore bien établi si le phénomène est en aval ou en amont dans la cascade moléculaire et cellulaire (Mori et al. 1987; Perry et al. 1987; Keller et al. 2000; Shimura et al. 2004; Sahara et al. 2005; Qian et al. 2006).
Enfin, une déficience dans le transport neuronal pourrait aussi jouer un rôle important dans la MA, ainsi que d’autres maladies neurodégénératives (Roy et al. 2005). En effet, des études montrent une interaction entre l’APP, Aβ et le transport axonal, mais ces interactions sont complexes et il n’est pas encore clair à partir duquel, du dysfonctionnement du transport axonal ou de l’augmentation de la production d’Aβ, le mécanisme débute (Koo et al. 1990; Kamal et al. 2000; Kamal et al. 2001; Pigino et al. 2003; Lazarov et al. 2005; Stokin et al.
2005). Parallèlement, certaines données indiquent que l’augmentation de la production ou la diminution de la dégradation de tau conduirait à une déficience du transport axonal (Ebneth et al. 1998; Stamer et al. 2002; Mandelkow et al. 2004). De plus, la protéine tau et son ARNm sont eux-mêmes transportés le long des axones, ainsi un déficit du transport axonal (du fait par exemple d’une phosphorylation des kinésines, protéines motrices des microtubules, induit par Aβ) pourrait conduire à une localisation erronée de tau et de son ARNm (cf. revue (Blurton-Jones et al. 2006)).
L’ensemble de ces données bibliographiques met clairement en évidence une interaction plus ou moins directe, via des mécanismes distincts, entre Aβ et tau. Il est à noter que la majorité des ces données pointe plutôt en faveur de l’hypothèse de la cascade amyloïde : la pathologie amyloïde induirait la formation de neurofibrilles et serait ainsi la cause première de la MA.
II.2.f. Hypothèse de la cascade amyloïde –Toxicité liée à Aβ
L’hypothèse de la cascade amyloïde (Figure 1.12) considère l’agrégation d’Aβ comme l’initiateur de la MA, plaçant les pathologies associées à tau, ainsi que d’autres modifications dégénératives, en aval (Hardy and Higgins 1992; Hardy and Selkoe 2002). Cette hypothèse provient de l’observation des plaques amyloïdes dans le cerveau des patients atteints de la MA qui est caractéristique de la pathologie, ce qui n’est pas le cas des dégénérescences neurofibrillaires (cf le tableau 1.3 des tauopathies) ; mais elle a surtout été renforcée par la découverte des mutations génétiques responsables de la forme familiale autosomale dominante de la MA sur les gènes codant pour l’APP, PS1 ou PS2 (Hardy et al. 2002). Ainsi, d’après cette hypothèse, l’agrégation anormale d’Aβ entraînerait une série de déséquilibres et de processus neurotoxiques débouchant sur la mort neuronale et la démence.
Cependant, l’hypothèse de la cascade amyloïde, même si elle est considérée par une grande partie de la communauté scientifique comme une base pour la compréhension du processus de développement de la MA, est en constante évolution. En effet, la corrélation entre plaques séniles et pertes cognitives n’était pas satisfaisante (« [plaques appear] at the wrong time and in the wrong places » (Naslund et al. 2000)). Depuis, un grand nombre d’articles ont attesté d’une meilleure corrélation et d’une toxicité comparativement bien plus importante pour les oligomères de faible poids moléculaire (protofilaments, petits oligomères, monomères, agrégats sphériques…mais la difficulté d’isoler ces différents types d’agrégats n’a pas permis
d’apporter une réponse définitive à ce jour) (Lambert et al. 1998; Haass and Steiner 2001; Walsh et al. 2002; Kayed et al. 2003; Stefani and Dobson 2003; Klein et al. 2004).
Figure 1.12. Hypothèse classique de la cascade amyloïde pour les formes familiales de la maladie d’Alzheimer (d’après (Hardy et al. 2002)). Pour les formes sporadiques, les causes
de l’augmentation de la production et de l’accumulation d’Aβ sont moins bien définies.
D’un point de vue de la toxicité, ces espèces de faibles poids moléculaire auraient la capacité d’induire une dépolarisation et une perméabilisation des membranes (celles-ci facilitant probablement l’étape de nucléation), altérant ainsi l’activité neuronale et débouchant finalement sur la mort cellulaire (Hartley et al. 1999; Stefani 2006). Plus précisément, un déséquilibre au niveau du calcium intracellulaire a été plusieurs fois observé (voir revue (Stefani et al. 2003)), ce déséquilibre pourrait affecter le bon fonctionnement des mitochondries (perméabilité, libération de cytochrome C) et/ou directement activer les caspases, conduisant ainsi à l’apoptose (Orrenius et al. 2003). D’autres voies de toxicité ont également été avancées : déséquilibre dans l’homéostasie des lipides (Janciauskiene and Ahren 2000), interactions avec des récepteurs membranaires en quantité croissante (du fait de
l’accumulation de produits terminaux de glycation avancée) (Mruthinti et al. 2003; Mruthinti et al. 2006), liaisons à des intégrines (protéines d’adhésion) activant des voies de signalisation intracellulaires (Caltagarone et al. 2007), formation de pores au niveau des membranes (Arispe et al. 1993; Yip and McLaurin 2001; Ji et al. 2002; Quist et al. 2005) (cf. chapitre 5), accumulation d’Aβ dans les mitochondries couplée à des interactions enzymatiques perturbant son fonctionnement (Chen and Yan 2006).
Les premiers symptômes de la maladie, comme les défauts de stockage mnésique, pourraient s’expliquer par une localisation préférentiellement synaptique des oligomères d’Aβ (en particulier au niveau des clusters de protéines PSD-95) (Allison et al. 2000), associée à la surexpression de protéines Arc (activity-regulated cytoskeletal-associated protein) inhibitrices du processus de mémorisation à long terme (Guzowski 2002).
La formation d’un complexe entre Aβ et l’hème pourrait également être source de production d’espèces oxydantes par les mitochondries, le complexe se comportant comme une peroxydase (Atamna 2006).
La toxicité liée à l’interaction avec les métaux sera abordée dans la section II.4. de ce chapitre.
Cependant, en marge des courants tauistes (qui place les neurofibrilles à l’origine du développement de la MA) et βAPPtistes (soutenant l’hypothèse de la cascade amyloïde), une partie de la communauté scientifique s’interroge encore sur le bien-fondé de ces deux axes de recherche, argumentant que les deux types de lésions observés chez les patients atteints de la MA sont pathognomoniques (caractéristiques de la maladie) mais pas forcément pathologiques, pointant le manque de corrélation entre neurofibrilles / dépôts amyloïdes et pertes cognitives / mort neuronale (Knowles et al. 1998; Andorfer et al. 2005; Santacruz et al. 2005; Nunomura et al. 2006). En se basant sur (1) l’observation d’une diminution des lésions représentatives de stress oxydant au voisinage des dépôts amyloïdes (Nunomura et al. 1999; Nunomura et al. 2000; Nunomura et al. 2004), (2) sur le fait que l’apparition de stress oxydant semble précéder les dépôts amyloïdes (Misonou et al. 2000; Nunomura et al. 2000; Paola et al. 2000; Pratico et al. 2001; Bayer et al. 2003; Drake et al. 2003; Li et al. 2004; Sung et al. 2004), (3) un effet protecteur d’Aβ40 et 42 sur le stress oxydant induit par le fer et le cuivre (Zou et al. 2002; Bishop and Robinson 2003) (probablement par chélation des ions métalliques), ils suggèrent que la production d’Aβ serait une voie de défense contre le stress oxydant, faisant ainsi de la conséquence (dans l’hypothèse de la cascade amyloïde), la cause. De même, en attribuant à tau des propriétés anti-oxydantes, la formation de neurofibrilles peut
être vu comme une conséquence du stress oxydant (Nunomura et al. 2001; Wataya et al. 2002; Gomez-Ramos et al. 2003; Nakashima et al. 2004). Dans le cas où cette théorie s’avèrerait exacte, il faudra alors rechercher les causes de ce stress oxydant, décrit comme relativement faible (insuffisant pour entraîner une mort cellulaire rapide) mais chronique (Keyse and Tyrrell 1989; Rushmore et al. 1990; Davies et al. 1995; Wiese et al. 1995) et débouchant sur des modifications compensatoires, réversibles dans un premier temps, puis permanentes (incluant entre autre la formation de neurofilaments, de plaques séniles et, en parallèle, une dérégulation mitotique) (Zhu et al. 2007).
Ainsi, malgré d’intenses recherches menées en ce sens, l’étiologie de la MA est encore loin d’être élucidée, retardant d’autant la découverte de traitements curatifs, faute d’axes de stratégie thérapeutique clairs.
II.3. Traitements
Actuellement, il n’existe aucun traitement thérapeutique curatif et/ou étiologique pour la MA, seuls sont disponibles des traitements symptomatiques (et, en dernier lieu, palliatifs), qui ont pour but de retarder le développement de la maladie et d’améliorer les conditions de vie des malades.
II.3.a.Traitements actuels
Différents traitements sont actuellement utilisés pour améliorer les conditions de vie des patients atteints de la MA, mais aucun d’entre eux n’est curatifs.
Concernant les troubles cognitifs, les inhibiteurs de cholinestérase (donepezil (Shintani and Uchida 1997; Rogers et al. 1998; Rogers et al. 1998; Burns et al. 1999), tacrine (Davis et al. 1992; Knapp et al. 1994; Samuels and Davis 1997; Gracon et al. 1998), galantamine (Bickel et al. 1991; Bores et al. 1996), rivastigmine (Sramek et al. 1996; Rosler et al. 1999)) sont les plus couramment utilisés, aux différents stades de la maladie. Ils inhibent les enzymes responsables de l’hydrolyse de l’acétylcholine, compensant ainsi la déficience de ce neurotransmetteur.
A un stade avancé de la MA, la mémantine, un antagoniste non compétitif des récepteurs glutamatergiques NMDA (N-methyl-D-aspartate), permettrait de contrebalancer le taux excessif de glutamate en partie responsable de l’augmentation des troubles mnésiques
(Fleischhacker et al. 1986; Robinson and Keating 2006; Cosman et al. 2007; Schmitt et al. 2007).
D’autres traitements thérapeutiques sont évoqués dans la littérature, mais leur efficacité reste encore à confirmer, les résultats des précédentes études réalisées étant parfois contradictoires. Les traitements à base d’œstrogène pour les femmes en fin de ménopause semblent retarder de quelques années l’apparition de la maladie, mais aucun résultat positif n’a été vérifié avec les personnes plus âgées atteintes de la MA (Henderson et al. 1994; Paganini-Hill and Henderson 1996; Schmidt et al. 1996; Asthana et al. 1999).
Des études rétrospectives ont montré que les anti-inflammatoires non stéroïdiens pouvaient ralentir la progression de la maladie (via l’inhibition des cyclooxygénases, enzymes de la biosynthèse des dérivés de type prostaglandine intervenant dans les processus inflammatoires) (Breitner et al. 1994; Stewart et al. 1997), mais ces résultats n’ont malheureusement pas pu être reproduits dans des études plus récentes (Aisen et al. 2000; Reines et al. 2004). Certains d’entre eux, cependant, agiraient selon un mécanisme différent et prometteur, ils seront évoqués dans la section suivante.
La prise d’anti-oxydant (vitamine E, sélégiline qui stimule la production de superoxyde dismutase) devrait permettre de compenser la baisse d’enzyme anti-oxydante associée à l’augmentation d’espèces radicalaires, mais là encore les différences observées entre le placebo et la molécule active ne sont pas significatives (Pratico and Delanty 2000; Ames and Ritchie 2007).
Quant aux troubles comportementaux (dépression, psychose), ils sont généralement traités à l’aide d’anti-dépresseur et d’anti-psychotique jugés compatibles avec la maladie (Schachter et al. 2000).
En dehors des pharmacopées, l’environnement psychosocial joue un rôle primordial. Une alimentation saine, des exercices physiques et intellectuels réguliers et variés, une bonne hygiène permettent de retarder significativement l’avancé de la maladie. Un environnement social apaisant (soutien familial, maintien au domicile quand c’est possible) est également nécessaire pour ces personnes fragilisées, plus enclins aux insomnies, syndromes d’anxiété et dépressions (Mittelman et al. 1996; Stern et al. 1997).
II.3.b.Voies de recherche