© Isabelle Guisle, 2020
Impact de la température corporelle sur la
phosphorylation de tau dans le contexte du sommeil
Thèse
Isabelle Guisle
Doctorat en neurobiologie
Philosophiæ doctor (Ph. D.)
Impact de la température corporelle sur la
phosphorylation de tau dans le contexte du sommeil
Thèse
Isabelle Guisle
Sous la direction de :
Emmanuel Planel, directeur de recherche
Sébastien Hébert, co-directeur de recherche
iii
Résumé :
La protéine tau est un marqueur pathologique important de la maladie d’Alzheimer car son niveau de phosphorylation et d’agrégation sont en corrélation avec la progression de la maladie. Les troubles du sommeil sont fréquents dans la maladie d'Alzheimer et inversement les études longitudinales montrent que les personnes présentant des troubles du sommeil sont plus à risque de développer la maladie d’Alzheimer. Cependant, on ne sait pas par quels mécanismes le manque de sommeil contribue au développement de la maladie. Mon laboratoire d’accueil à précédemment démontré que la température centrale peut affecter la phosphorylation de la protéine tau et que la température corporelle suit des oscillations circadiennes, nous avons émis l'hypothèse que la phosphorylation de la protéine tau pourrait être soumise à des oscillations circadiennes dues à la température corporelle. L’objectif de cette thèse était de déterminer si la phosphorylation de tau suit un rythme circadien et si la température corporelle et le sommeil sont impliqués dans le processus. Dans un premier temps, nous avons montré que la phosphorylation de tau suit un rythme circadien : lorsque les animaux dorment, leur température est plus basse et la protéine tau est plus phosphorylée, inversement, pendant l’activité, la température corporelle est plus haute, et tau est déphosphorylée. Pour déterminer si la température corporelle est directement impliquée dans le rythme circadien de la phosphorylation de tau, nous avons modifié les oscillations circadiennes de la température corporelle en exposant les animaux à 34ºC dans une étuve prévue à cet effet. L’exposition des animaux à cette température diminuait l’amplitude de la variation circadienne de la température corporelle et annulait les variations circadiennes de la phosphorylation de tau. Par la suite, nous avons déterminé si le sommeil pouvait avoir un impact sur la température corporelle et sur phosphorylation de tau en privant des souris de sommeil pendant 6 heures. La privation de sommeil augmentait la température corporelle et diminuait significativement la phosphorylation de tau. Pour vérifier que la température corporelle est impliquée dans le rythme circadien de la phosphorylation de tau nous avons exposé une lignée de cellules neuronales à la température rectale moyenne mesurée chez les souris pendant le sommeil (36.3ºC) et l’activité (37.4ºC). Une baisse de la température d’un degré était suffisante pour diminuer significativement la phosphorylation de tau. Globalement, nos résultats démontrent que la phosphorylation de la protéine tau suit un
iv
rythme circadien et qu’elle est influencée par le cycle veille/sommeil et par la température corporelle.
v
Abstract:
Tau protein is an important pathological marker of Alzheimer's disease because its level of phosphorylation and aggregation correlates with the progression of the disease. Sleep disorders are common in Alzheimer's disease and conversely longitudinal studies show that people with sleep disorders are at higher risk to develop Alzheimer's disease. However, the mechanisms by which poor sleep contributes to the development of the disease are unknown. As my host laboratory previously demonstrated that central body temperature can affect the phosphorylation of tau protein and that body temperature follows circadian oscillations, we hypothesized that phosphorylation of tau protein could be subjected to circadian oscillations due to body temperature. The objective of this thesis was to determine if phosphorylation of tau follows a circadian rhythm and if body temperature and sleep are involved in the process. First, we showed that the phosphorylation of tau follows a circadian rhythm: when the animals were sleeping, their temperature was lower and tau protein was more phosphorylated, conversely, during the activity, body temperature was higher, and tau was dephosphorylated. To determine whether body temperature was involved in the circadian rhythm of tau phosphorylation, we changed the circadian body temperature oscillations by exposing the animals to 34ºC in an incubator dedicated for animal housing. Exposure to 34ºC decreased the magnitude of circadian body temperature variation and abolished circadian changes in tau phosphorylation. Subsequently, we determined whether sleep had an impact on body temperature and tau phosphorylation by testing the effect of 6 hours sleep deprivation. Sleep deprivation increased body temperature and significantly decreased tau phosphorylation. To verify that body temperature is directly involved in circadian rhythm of phosphorylation of tau, we exposed a neuronal cell line at the mean rectal temperature measured during sleep (36.3ºC) and activity (37.4ºC). A decrease of one degree Celsius was sufficient to significantly decrease tau phosphorylation. Overall, our results demonstrate that phosphorylation of tau protein follows a circadian rhythm and is influenced by the sleep/wake cycle and body temperature.
vi
Table des matières :
Résumé : ... iii
Abstract: ... v
Table des matières : ... vi
Liste des figures et tableau: ... x
Liste des abréviations ... xi
Remerciements ... xiii
Avant-propos ... xiv
Introduction : ... 1
1. La maladie d’Alzheimer ... 1
1.1 Historique de la découverte de la maladie ... 1
1.2 Epidémiologie ... 2
1.2.1 Les troubles cognitifs légers ... 2
1.2.2 La Maladie d’Alzheimer ... 3
1.3 Etiologie ... 3
1.3.1 Maladie d’Alzheimer à développement précoce ... 3
1.3.2 Maladie d’Alzheimer à développement tardif ... 4
1.3.2.1 Les facteurs de risque reconnus... 4
1.3.2.2 Les facteurs de risque supposés... 4
1.4 Les symptômes ... 5
1.4.1 Troubles cognitifs légers ... 5
1.4.2 La démence de type Alzheimer ... 6
1.5 Le diagnostic ... 6
1.5.1 Les troubles cognitifs légers ... 6
1.5.2 La démence de type Alzheimer ... 7
1.6 Les manifestations neuropathologiques ... 9
1.6.1 Les plaques amyloïdes ... 9
1.6.1.1 Composition et structure ... 9
1.6.1.2 Distribution dans le cerveau ...10
vii
1.6.2.1 Composition et structure ...11
1.6.2.2 Distribution dans le cerveau ...11
1.6.3 La neuroinflammation ...13
1.6.4 Les pertes synaptiques ...13
1.6.5 Le métabolisme du glucose ...14
1.7 Les biomarqueurs ...15
1.7.1 Imagerie médicale ...15
1.7.2 Les marqueurs biochimiques ...17
1.7.2.1 Dans le liquide céphalo rachidien ...17
1.7.2.2 Dans le sang ...18
1.7.2.3 Autres fluides ...19
1.8 Les traitements ...19
1.8.1 Inhibiteurs de l’acétylcholinestérase ...19
1.8.2 Les antagonistes des récepteurs NMDA...21
1.8.3 Les essais cliniques en cours ...21
1.8.3.1 L’immunothérapie ...21
1.8.3.2 Les inhibiteurs d’enzyme ...22
1.8.3.3 La thérapie génique ...23
1.8.3.4 Les modulateurs de la réponse immunitaire ...23
1.8.3.5 L’administration de produits naturels ...24
2. Le peptide amyloïde ... 25
2.1 Généralités ...25
2.2 Rôle en conditions physiologiques...26
2.3 Rôle dans la pathologie ...26
3. La protéine tau ... 27
3.1 En conditions physiologiques ...27
3.2 Rôle dans la pathologie ...29
4. Maladie Alzheimer et troubles du sommeil ... 32
4.1 Les mécanismes du sommeil ...32
4.1.1 Le sommeil : définition et fonction ...32
4.1.2 Les rythmes circadiens ...35
4.1.3 L’homéostasie du sommeil ...37
4.1.4 La température corporelle et le rythme veille/sommeil ...37
4.1.4.1 Impact de la température corporelle sur le sommeil ...38
4.1.4.2 Qualité du sommeil et anomalies du rythme circadien de la température corporelle ...38
viii
4.1.4.4 Mécanismes de l’induction du sommeil ...40
4.1.4.5. Les structures anatomiques du lien entre température corporelle et sommeil ...41
4.2 Le risque de déclarer la maladie d’Alzheimer est plus important chez les personnes âgées présentant des troubles du sommeil. ...42
4.3 Sommeil et marqueurs de la MA chez les individus sains ...43
4.4 Les troubles du sommeil sont fréquents chez les personnes atteintes de troubles cognitifs légers ou de la MA ...44
4.5 Les troubles du sommeil dans les modèles murins de la MA ...45
Hypothèses et objectifs : ... 46
Chapitre 1 : Les variations circadiennes de la phosphorylation de tau sont guidées par la température et par le cycle veille/sommeil ... 47
1.1 Résumé ...49
1.2 Abstract ...51
1.3 Introduction ...53
1.4 Material and methods ...55
1.4.1 Animals ...55
1.4.2 Core body temperature ...55
1.4.3 Circadian rhythmicity and temperature exposure ...55
1.4.4 Sleep deprivation ...56
1.4.5 Protein extraction ...57
1.4.6 Cell culture ...57
1.4.7 SDS-PAGE and Western blot ...58
1.4.8 PP2A activity ...58
1.4.9 Statistical analyses ...59
1.5 Results ...60
1.5.1 Circadian variations in endogenous tau phosphorylation ...60
1.5.2 Circadian variations in tau phosphorylation are correlated with body temperature ...60
1.5.3 Increased tau phosphorylation is driven by mild temperature drop in neuron-like cells ...61
1.5.4 Mechanisms of tau hyper phosphorylation during sleep ...61
1.5.5 Sleep deprivation prevents sleep-induced hyperphosphorylation of tau ...62
1.6 Discussion ...63 1.7 Acknowledgments: ...67 1.8 Author Contributions: ...67 1.9 Funding: ...67 1.10 Competing Interest: ...67 Discussion et perspectives: ... 78
ix
1. Retour sur les résultats ... 78
2. La phosphorylation de tau est corrélée avec la température corporelle ... 78
3. Rôle de tau pendant le sommeil ... 79
4. PP2A est un régulateur potentiel de la phosphorylation de tau pendant l’activité ... 79
5. Le rythme circadien de la phosphorylation de tau ... 80
6. Translocation de tau dans le noyau ... 81
7. Ablation du noyau suprachiasmatique et rythme circadien de la température ... 81
Conclusion générale : ... 82
Annexe : Article non inclus dans la thèse ... 83
x
Liste des figures et tableau:
Figure 1 : Extrait des notes d’Aloïs Alzheimer le 26 Novembre 1901 ... 1
Figure 2 : Extrait des écrits de Auguste Deter ... 2
Figure 3 : Progression de la pathologie amyloïde selon les travaux de Thal ... 10
Figure 4 : Progression de la pathologie tau selon les travaux de Braak ... 12
Figure 5 : Cycle de l’acétylcholine ... 20
Figure 6 : Le clivage de la protéine APP ... 25
Figure 7 : Oligomérisation du peptide A ... 27
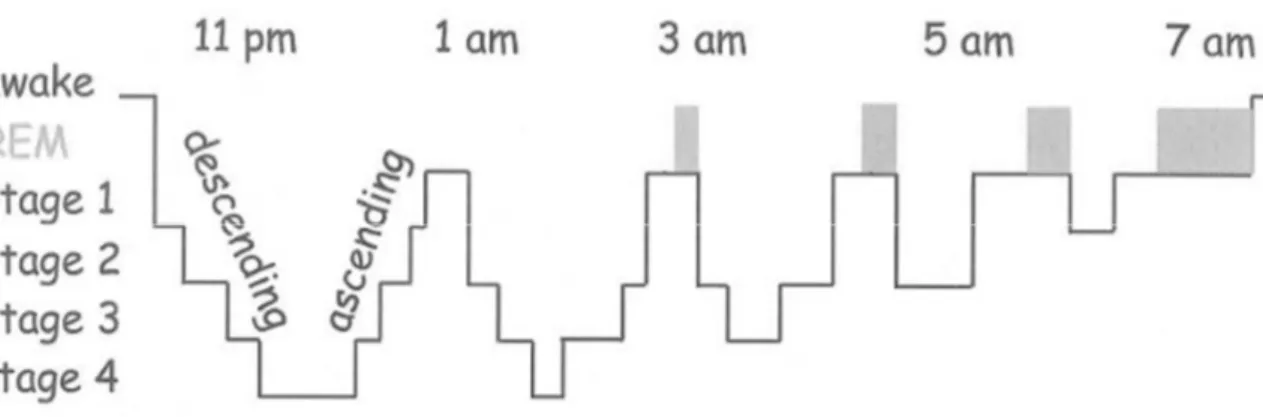
Figure 8 : Activité électrique du cerveau enregistrée par EEG pendant l’activité et le sommeil. ... 33
Figure 9 : Le cycle du sommeil ... 34
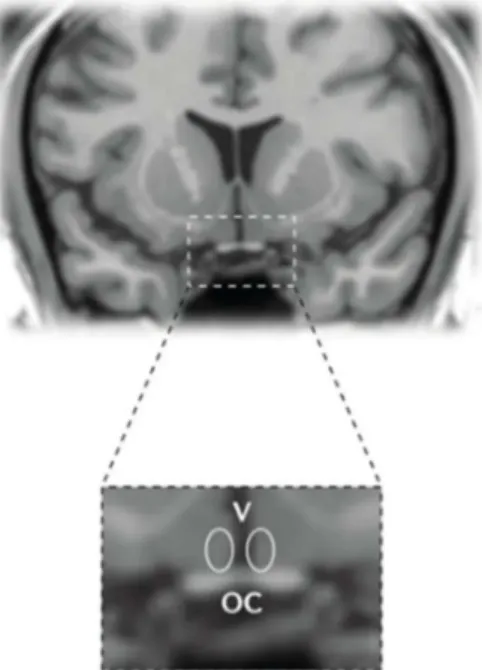
Figure 10 : Localisation du noyau suprachiasmatique dans une section coronale de cerveau chez l’homme ... 36
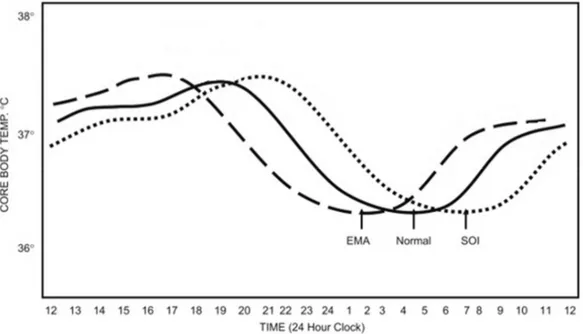
Figure 11 : Diagramme représentant les courbes de températures ... 39
Figure 1.1: Phosphorylation of tau observed by Western blotting during subjective day and subjective night in the cerebral cortex of mice housed at room temperature or at 34°C: ... 68
Figure 1.2: Body temperature recordings of mice during the entire experiment (A) and at the time of death (B): ... 70
Figure 1.3: Phosphorylation of tau observed by Western blotting in neuron-like cells (SH3R cells): ... 72
Figure 1.4: Acute sleep deprivation is associated with dephosphorylation of tau: ... 73
Figure 1.5: Acute sleep restriction drives hyperthermia: ... 74
Table 1: antibodies used in the study. ... 75
Figure S1: Phosphorylation of tau observed by Western blotting during subjective day and subjective night in the cerebral cortex of mice housed at room temperature or at 34°C in the first cohort of mice: ... 76
xi
Liste des abréviations
MA Maladie d’Alzheimer
ENF Enchevêtrements Neurofibrillaires TEP Tomographie à Emission de Positrons LCR Liquide Céphalo Rachidien
Amyloïde Beta
A42 Amyloïde Beta 42
OMS Organisation Mondiale de la Santé APP Amyloid Precursor Protein
PSEN1 Presenilin 1
PSEN2 Presenilin 2
NMDA N-Methyl-D-Aspartate
ApoE4 Apolipoprotéine E4
ARN Acide RiboNucleique
ADN Acide DésoxyRibonucléique
GSK3 Glycogen Synthase Kinase 3 PP1 Protéine Phosphatase 1 PP2A Protéine Phosphatase 2A PP2B Protéine Phosphatase 2B PP5 Protéine Phosphatase 5
CaMKII Calcium Calmoduline Kinase II PKA cAMP dependent Protein Kinase A
JNK cJun N terminal Kinase
AKT PKB, Protéine Kinase B
AMPK AMP activated protein Kinase CDK5 Cyclin Dependent Kinase 5
Dyrk1 dual-specificity tyrosine-phosphorylated and -regulated kinase 1A EEG Electroencéphalogramme
xii
xiii
Remerciements
Je tiens tout d’abord à remercier mon directeur de recherche Emmanuel Planel, pour m’avoir accueillie dans son laboratoire malgré mon parcours atypique. Vous m’avez fait confiance, guidée et vous m’avez donnée la possibilité de tester mes idées. Vous dirigez votre équipe avec tact, gentillesse et bonne humeur et cela a été un réel plaisir de travailler à vos côtés. Je tiens également à remercier Sébastien Hébert mon co-directeur de recherche pour son soutien et ses précieux conseils donnés tout au long de mon doctorat. J’ai eu aussi la chance de bénéficier du soutien et de l’expertise de Valérie Mongrain au cours de mon doctorat. Je tiens à remercier les membres du jury pour le temps que vous avez accordé à mon travail de thèse.
Je tiens aussi à remercier Serena Petry pour son aide précieuse tout au long de mon doctorat. Elle m’a montré comment faire des Western blot au début de mon doctorat, et elle a été d’une aide précieuse pour l’article sur le sommeil quand il a fallu refaire des expérimentations. Merci à Françoise pour son aide technique. Merci à Maud pour ses conseils au début de mon doctorat.
Je tiens aussi à remercier Sara, Emmanuelle, Léa, Baya, Rémi, Andréanne, Claudia et Chloé pour leur gentillesse et leur bonne humeur. Ils ont été pour moi un vrai rayon de soleil dans le laboratoire et ils étaient aussi présents dans les moments difficiles. De plus ils étaient toujours là pour aider, conseiller quand j’ai eu besoin d’aide.
Le personnel de l’animalerie a joué un rôle très important pendant mon doctorat. Depuis les préposés jusqu’aux membres de la direction, j’ai toujours eu une aide substantielle et une écoute de mes besoins. Je tiens particulièrement à remercier Anne-Marie pour son aide technique et sa bonne humeur, Geneviève pour sa disponibilité et sa gentillesse, Mélanie et Emilie pour leur disponibilité et leur aide très précieuse.
Ma famille a joué un rôle très important également. Mes parents et mes grands-parents maternels ont été un soutien très important. Merci à ma maman pour la correction des fautes d’orthographe dans ce manuscrit. Mon mari et mes enfants ont traversé l’Atlantique pour me suivre et se sont bien adaptés à notre nouvelle vie. Je les remercie pour leur soutien car ce parcours n’aurait pas pu être possible s’ils ne m’avaient pas suivi.
xiv
Avant-propos
La première partie de cette thèse est une revue de la littérature permettant d’introduire mes résultats. Le chapitre 1 est présenté sous forme d’un article publié. Une autre publication issue de mon doctorat non inclue dans cette thèse est mentionnée en annexe.
Chapitre 1 : Les variations circadiennes de la phosphorylation de tau sont
guidées par la température et par le cycle veille/sommeil.
Ce chapitre est constitué à partir de l’insertion de l’article suivant :
Guisle, I., Gratuze, M., Petry, S., Morin, F., Keraudren, R., Whittington, R. A., Hebert, S. S., Mongrain, V., & Planel, E. (2019). Circadian and sleep/wake-dependent variations in tau phosphorylation are driven by temperature. Sleep. doi:10.1093/sleep/zsz266.
Cet article a été accepté pour publication en octobre 2019 dans le journal « Sleep ». J’ai effectué les expériences et les analyses à l'exception du Western blot concernant la privation de sommeil, j’ai contribué à la conception de l'étude et rédigé le manuscrit. Maud Gratuze a effectué le Western blot et l'analyse concernant la privation de sommeil. Françoise Morin a aidé à la décapitation des animaux, à l'échantillonnage des tissus et aux chirurgies. Serena Pétry et Rémi Kéraudren ont effectué des Western blot sur la deuxième cohorte d'animaux. Robert Wittington a participé à l’écriture du manuscrit et a apporté son expertise. Sébastien Hébert a participé à l’écriture du manuscrit et a apporté son expertise. Valérie Morin a fourni son assistance pour la conception de l'étude, effectué l'expérience de privation de sommeil, aidé pour les analyses statistiques et participé à la rédaction du manuscrit. Emmanuel Planel a conçu l'étude et participé à l’écriture du manuscrit.
xv
Annexe 1 : Sauna-like conditions induce tau dephosphorylation through
hyperthermia.
Isabelle Guisle; Serena Pétry; Françoise Morin; Robert A. Whittington; Frédéric Calon ; Sébastien S. Hébert; Emmanuel Planel.Cet article est en préparation pour soumission prochaine. Le titre de cet article a changé, le titre précédent était : « Targetting Alzheimer disease by sauna like conditions ». J’ai effectué les expériences et les analyses. J’ai contribué à la conception de l'étude et rédigé le manuscrit. Françoise Morin a aidé à la décapitation des animaux et à l'échantillonnage des tissus. Robert Wittington a participé à l’écriture du manuscrit et a apporté son expertise. Fréderic Calon a participé à l’écriture du manuscrit et a apporté son expertise.Sébastien Hébert a participé à l’écriture du manuscrit et a apporté son expertise. Emmanuel Planel a conçu l'étude et participé à l’écriture du manuscrit.
1
Introduction :
1. La maladie d’Alzheimer
1.1 Historique de la découverte de la maladie
La patiente Auguste Deter fut examinée pour la première fois par le clinicien Aloïs Alzheimer le 25 novembre 1901 (Maurer et al., 1997). Elle souffrait d’une forme de démence incluant des troubles de la mémoire, la perte partielle de sa propre identité (Fig.1) et la perte de la capacité à écrire (Fig.2). Les autres symptômes associés à sa maladie étaient « l’aphasie, la confusion, les changements d’humeur non prédictibles, la paranoïa, les hallucinations auditives et les inaptitudes psychosociales » (Maurer et al., 1997). Plus tard, au décès de sa patiente, Aloïs Alzheimer caractérisa le cerveau de sa patiente et divulgua ses premiers résultats dès novembre 1906 (Alzheimer, 1906, 1907; Maurer et al., 1997). Le docteur Alzheimer décrivait déjà à l’époque la présence de plaques et d’enchevêtrements de neurofibrilles (Alzheimer, 1911; Maurer et al., 1997).
Figure 1 : Extrait des notes d’Aloïs Alzheimer le 26 Novembre 1901. Aloïs Alzheimer rapporte les troubles de l’identité et de la mémoire à court therme de sa patiente Auguste Deter (Maurer et al., 1997).
2
Figure 2 : Extrait des écrits de Auguste Deter. La patiente Auguste Deter n’est plus capable d’écrire une phrase correctement (Maurer et al., 1997)
1.2 Epidémiologie
1.2.1 Les troubles cognitifs légers
L’incidence et la prévalence des troubles cognitifs légers est très variable (Ganguli et al., 2004; Ward et al., 2012). Selon les études, l’incidence peut varier entre 21 et 71 pour 1000 par an pour les troubles cognitifs légers non amnésiques et entre 8.5 et 25.9 pour 1000 par an pour les troubles cognitifs amnésiques (Ward et al., 2012). La prévalence elle, peut varier entre 0.5 et 42% selon les études et selon les critères donnés pour les troubles cognitifs légers (Ward et al., 2012). Il est important de noter que les troubles cognitifs légers sur un suivi de 2 à 5 ans en général, peuvent, soit progresser vers la démence (4 à 40% des cas), soit rester stable (37 à 67%) soit reverser vers une cognition normale (30 à 55% des cas) (Pandya et al., 2016). Sur des suivis plus longs de 10 à 17ans, la progression vers la démence peut varier de 17 à 55% (Ganguli et al., 2004; Pandya et al., 2016), la réversion vers une cognition normale peut varier de 10 à 53.5% (Lopez et al., 2012; Pandya et al., 2016) et le diagnostic MCI peut rester stable dans 35% des cas (Lopez et al., 2012; Pandya et al., 2016).
3 1.2.2 La Maladie d’Alzheimer
Selon un rapport de Alzheimer’s Disease International, on estime que environs 50 millions de personnes souffrent de démence à travers le monde (Prina, 2013) et selon l’Organisation Mondiale de la Santé (OMS), 60 à 70% de ces cas de démence relèvent de la maladie d’Alzheimer (World Health Organization, 2019). Une méta-analyse de 2016 évalue que la prévalence de la MA est aux alentours de 30.4 pour 1000 et que l’incidence est de 15.8 pour 1000 par an (Fiest et al., 2016). L’OMS estime que d’ici 2030, environs 75 millions de personnes souffriront de démence et 135 millions d’ici 2050 (World Health Organization, 2019) : la progression de la démence suit une courbe exponentielle (World Health Organization, 2012).
1.3 Etiologie
Le principal facteur de risque de la MA est l’âge (Carr et al., 1997). En effet, 90% des formes de la maladie d’Alzheimer se développent tardivement (après 65 ans) (Alzheimer’s Disease International report, 2015) et sont d’origine sporadique (Carr et al., 1997; Guerreiro et al., 2012). Dans 10% des cas, la maladie se développe précocement (avant 65 ans) (Alzheimer’s Disease International report, 2015).
1.3.1 Maladie d’Alzheimer à développement précoce
On considère que la plupart les formes précoces de la maladie d’Alzheimer sont d’origine génétique (Wingo et al., 2012). Principalement, trois gènes sont identifiés dans les formes génétiques de la MA : le gène codant pour l’APP (Amyloid Precursor Protein) et ceux codant pour les présinilines 1 et 2 (PSEN1 et PSEN2)(Guerreiro et al., 2012), deux protéines faisant partie du complexe sécrétase. Ces 3 gènes sont impliqués dans 71% des cas de forme précoce (Campion et al., 1999).
4
1.3.2 Maladie d’Alzheimer à développement tardif
1.3.2.1 Les facteurs de risque reconnus
Un certain nombre de facteurs de risque sont maintenant bien acceptés par la communauté scientifique. Pour ce qui est de la forme tardive de la maladie d’Alzheimer, après l’âge, le deuxième facteur de risque pour la MA est l’héritabilité. En effet, on considère que un certain nombre de formes tardives de la MA ont une composante génétique transmissible, la plupart du temps non identifiée (Gatz et al., 2006; Pedersen et al., 2001; Wingo et al., 2012). Seuls certains facteurs de risque génétiques tels que le port de l’allèle 4 de l’apolipoprotéine E (Guerreiro et al., 2012), une protéine chargée entre autre, de redistribuer le cholestérol dans l’organisme (Mahley, 1988), augmentent le risque de développer la MA. Les individus porteurs d’un seul allèle 4 ont un risque augmenté de 3 fois de déclarer la MA et ceux porteurs des 2 allèles ont un risque augmenté de 15 fois de déclarer la maladie d’Alzheimer (Ashford, 2004). Les variants hétérozygotes de TREM2 (Triggering Receptor found on myeloid cells 2), augmentent aussi le risque de développer la MA (Jonsson et al., 2013).
Également, les femmes, les fumeurs (Launer et al., 1999), les personnes obèses (Obisesan et al., 2012), diabétiques (Arvanitakis et al., 2004; Whittington et al., 2010) ou ayant un faible niveau d’éducation (Launer et al., 1999; Letenneur et al., 1999) ont un risque plus élevé de développer la MA. Pour finir les personnes atteintes de maladie cardiovasculaire, ayant souffert de traumatisme crânien, ou ayant une consommation d’alcool trop importante présentent un risque plus élevé de déclarer la MA (Edwards Iii et al., 2019).
Avec la modernisation récente de notre société, nous sommes exposés à la présence de polluants dans l’environnement. De nombreuses études montrent que l’exposition aux métaux lourds, aux pesticides, à l’aluminium peuvent être des facteurs faisant progresser la pathologie amyloïde et la pathologie tau (Chin-Chan et al., 2015).
1.3.2.2 Les facteurs de risque supposés
Il existe d’autres facteurs qui pourraient expliquer le développement de la maladie d’Alzheimer tels que la dépression et les crises d’épilepsie (Edwards Iii et al., 2019).
5
Les troubles du sommeil, également, sont un autre paramètre potentiellement capable d’augmenter le risque de développer la maladie d’Alzheimer. En effet un certain nombre d’études montrent que la restriction de sommeil augmente la pathologie amyloïde et la pathologie tau chez l’homme comme chez l’animal (Macedo et al., 2017; Van Egroo et al., 2019; Wu et al., 2019a). Inversement, entre 25 et 40% des personnes souffrant de la MA ont des troubles du sommeil (Macedo et al., 2017). (Revu en détail dans la partie 4. Maladie Alzheimer et troubles du sommeil)
Certaines études montrent aussi que des altérations de la capacité à thermoréguler contribuent aussi probablement au développement de la MA. L’hypothermie associée au diabète, à l’âge, à l’altération du métabolisme du glucose ou à l’administration d’une drogue telle qu’un anesthésique augmente probablement le risque de développer la MA (Whittington et al., 2010). Dans des modèles animaux de la MA, l’hypothermie augmente la pathologie tau et la pathologie A(Planel et al., 2004; Tournissac et al., 2019; Tournissac et al., 2017; Vandal et al., 2016).
1.4 Les symptômes
1.4.1 Troubles cognitifs légers
La maladie d’Alzheimer (MA) est une maladie neurodégénérative qui apparait progressivement avec l’âge. Les premiers symptômes répertoriés chez les patients sont les troubles cognitifs légers (Albert et al., 2011; Diagnostic and Statistical Manual of Mental Disorders. 5., 2013). Ils sont caractérisés par un déclin des capacités cognitives ne relevant ni de la vieillesse, ni de la démence et n’affectant pas la vie quotidienne (Albert et al., 2011; Diagnostic and Statistical Manual of Mental Disorders. 5., 2013; Petersen, 2004; Petersen et al., 2014; Winblad et al., 2004). La manifestation la plus fréquente est l’apparition de pertes de la mémoire antérograde épisodique (Galton et al., 2000) c’est-à-dire une perte de la capacité à fabriquer de nouveaux souvenirs personnels (Matthews, 2015).
6 1.4.2 La démence de type Alzheimer
La maladie d’Alzheimer est une forme de démence c’est-à-dire « un syndrome de déclin cognitif suffisamment sévère pour interférer avec la vie sociale ou professionnelle » (Chertkow et al., 2013). Le symptôme le plus fréquemment répertorié chez les patients atteints de la maladie d’Alzheimer est la perte progressive de la mémoire épisodique (capacité à se rappeler des évènements personnels (Matthews, 2015)), puis l’attention et les fonctions exécutrices, la mémoire sémantique (capacité à sa rappeler des connaissances générales fondamentales (Matthews, 2015)) sont affectées (Chertkow et al., 2013; Galton et al., 2000; Grady et al., 1988; Greene et al., 1995; Hodges et al., 1995; Locascio et al., 1995; McKhann et al., 1984; Morris, 1993; Perry et al., 1999). Également, la capacité à initier des mouvements sur commande ou « praxis » est affectée ainsi que les perceptions visuelles et spatiales (Binetti et al., 1998; Galton et al., 2000; Grady et al., 1988; Locascio et al., 1995; Mendez et al., 1990). Les biopsies étant beaucoup trop invasives, le diagnostic de la maladie est donné avec plusieurs degrés de probabilités par les cliniciens. Le diagnostic est posé de manière définitive post mortem sur la base d’analyses histopathologiques réalisées sur les patients cliniquement classés « maladie d’Alzheimer probable » (Montine et al., 2012). Il existe aussi des formes atypiques de la maladie d’Alzheimer. Elles sont classées ainsi car le symptôme principal n’est pas l’amnésie mais plutôt des troubles cognitifs tels que l’aphasie progressive, une atteinte des compétences visuo-spatiales et visuo-perceptuelles, une atteinte des capacités à lire ou à écrire tel que répertorié dans la démence de type « atrophie postérieure du cortex » (Crutch et al., 2012; Dubois et al., 2010).
1.5 Le diagnostic
1.5.1 Les troubles cognitifs légers
Les troubles cognitifs légers sont les premiers symptômes cliniques pouvant être diagnostiqués chez les patients. Ce n’est que récemment en 2011, que les troubles cognitifs légers ont été inclus dans le manuel « Diagnostic and Statistical Manual for mental disorders V» (DSMV) (Albert et al., 2011; Diagnostic and Statistical Manual of Mental
7
Disorders. 5., 2013; Langa et al., 2014). Le diagnostic clinique est posé sur la base de tests fonctionnels visant à évaluer les capacités du patient dans les tâches de la vie quotidienne et sur la base de l’histoire du patient (Langa et al., 2014; Marshall et al., 2012; Petersen, 2011; Pfeffer et al., 1982). La plupart du temps, l’entourage du patient va également être interrogé de manière à compléter l’évaluation du patient (Marshall et al., 2012). Il existe une multitude de tests fonctionnels permettant d’évaluer le stade du patient (Marshall et al., 2012). Parmi ces tests, le « Functionnal Activities Questionnaire » est utilisé en clinique pour évaluer les troubles cognitifs légers (Langa et al., 2014; Marshall et al., 2012).
Les performances cognitives du patient peuvent également être évaluées sous la forme d’un questionnaire soumis à un proche : IQCODE (Informant Questionnaire on Cognitive Decline in Elderly), le Dementia Severity Rating Scale (DSRS) ou le AD8 (Langa et al., 2014). D’autres tests peuvent être utilisés en recherche clinique pour évaluer directement les performances cognitives du patient mais ils ne font pas partie des recommandations du DSMV pour le diagnostic (Langa et al., 2014). Le seul test développé pour tester spécifiquement les troubles cognitifs légers est le MoCA test (Montreal Cognitive Assessment) (Nasreddine et al., 2005). Les autres tests existants (plus d’une quarantaine) permettent de tester l’état de démence du patient (Tsoi et al., 2015). Ces tests sont aussi utilisés pour évaluer les troubles cognitifs légers cependant leur sensibilité et leur spécificité sont moins bonnes (Tsoi et al., 2015; Tsoi et al., 2017). Une méta-analyse récente réalisée sur 108 études regroupant 23546 participants a mis en évidence que les meilleurs tests permettant de détecter les troubles cognitifs légers sont ceux qui testent la mémoire à long ou à court terme (Tsoi et al., 2017).
Pour finir, il est important de noter que le diagnostic de troubles cognitifs légers n’est pas définitivement acquis : il peut s’inverser dans certains cas, rester stable ou progresser vers la MA (voir section 1.7 Epidémiologie) (Pandya et al., 2016).
1.5.2 La démence de type Alzheimer
Le diagnostic de la maladie est donné avec plusieurs degrés de probabilités par les cliniciens : « Alzheimer probable », « Alzheimer possible » sur la base de critères récemment révisés en
8
2011 (McKhann et al., 2011). Les méthodes de diagnostic actuelles sont basées sur l’histoire du patient (très souvent racontée par un proche), ses performances fonctionnelles et cognitives (Knopman et al., 2001). Elles ont une sensibilité de 81% (capacité à diagnostiquer pour la MA) et une spécificité de 70% (capacité à identifier quelqu’un sans la MA) (Knopman et al., 2001). Tout comme pour les troubles cognitifs légers, les performances cognitives peuvent être évaluées sous la forme de tests tels que le MoCA, le Mini Mental State Examination (MMSE), ou le MiniCog (Mini Cognitive Test). Le MMSE est un test très fréquemment retrouvé dans la littérature. Il permet d’évaluer en 10 minutes les performances cognitives du patient dans plusieurs domaines : l’orientation spatio-temporelle, l’attention, la mémoire de travail, la mémoire à court terme, le langage, ainsi que les compétences visuo-spatiales (Folstein et al., 1975). Moins d’une vingtaine de questions permettent d’évaluer sur 30 points le statut cognitif des individus testés. Un score entre 24-25 et 30 correspond à un individu normal et un score inférieur à 24 correspond à un individu malade. Une méta-analyse récente réalisée dans des populations communautaires indique que le test MMSE à une sensibilité pour la démence est entre 87 et 97% et une spécificité entre 70 et 82% selon les seuils de détection choisis (score de 24 ou 25)(Creavin et al., 2016). Cette méta-analyse ne précise pas si les études sont longitudinales ou transversales.
Le diagnostic post mortem de la maladie d’Alzheimer se fait selon trois critères définis par le « National Institute on Aging-Alzheimer’s Association » (NIA-AA) (Montine et al., 2012): la présence de plaques amyloïdes, d’enchevêtrements de neurofibrilles et de plaques neuritiques. Dans 93% des cas, le diagnostic neuropathologique est en accord avec le diagnostic clinique (Berg et al., 1998), c’est-à-dire que les tissus présentent les 3 critères désignés ci-dessus. Dans 40% des cas, les tissus peuvent porter d’autres marqueurs que ceux de la maladie d’Alzheimer tels que par exemple l’alpha-synucléine (maladie de parkinson), des lésions vasculaires (démence vasculaire), la protéine TDP-43 (Transactive response DNA binding Protein 43kD, marqueur de la sclérose latérale amyotrophique et de la démence fronto temporale lobaire) (Berg et al., 1998; Rahimi et al., 2014; Robinson et al., 2018). Il arrive aussi que des individus cognitivement sains soient porteurs des marqueurs neuropathologiques de la maladie d’Alzheimer (Bennett et al., 2012; Knopman et al., 2003; Rahimi et al., 2014).
9
Désormais, il est aussi possible de détecter certains biomarqueurs par des méthodes d’imagerie médicale ou par analyse biochimique du liquide céphalo-rachidien par exemple (voir section « 1.5 Les biomarqueurs »). Ces méthodes ne sont pas recommandées en routine clinique en Amérique du nord pour la détection des troubles cognitifs légers ou pour la détection de la maladie d’Alzheimer (Jack et al., 2011; McKhann et al., 2011; National Institute of Health, 2019) mais peuvent être utilisées en recherche clinique.
1.6 Les manifestations neuropathologiques
Même si ce n’est pas une caractéristique spécifique de la MA, une des manifestations neuropathologiques observable dans un cerveau atteint de la maladie d’Alzheimer est l’atrophie cérébrale (Pini et al., 2016; Pirttila et al., 1993). Les autres caractéristiques neuropathologiques spécifiques de la MA sont la présence de plaques amyloïdes et d’enchevêtrements de neurofibrilles.
1.6.1 Les plaques amyloïdes
1.6.1.1 Composition et structure
Les plaques amyloïdes sont des dépôts extracellulaires insolubles composées principalement du peptide amyloïde beta, sous sa forme agrégée. Le peptide amyloïde beta a été isolée pour la première fois des plaques amyloïdes par l’équipe de Glenner et Wong en 1984 (Glenner et al., 2012). Avec l’arrivée des technologies d’analyse haut débit au début des années 2000, il a été possible de caractériser l’ensemble des protéines présentes dans les plaques amyloïdes par analyse protéomique (Liao et al., 2004). Les plaques amyloïdes sont enrichies en 26 protéines dont la protéine amyloïde beta, le constituant majeur, 14-3-3, des protéines de l’adhésion cellulaire (collagène, fibrinogène), du cytosquelette (tau), de l’inflammation (Glial Fibrillary Acidic Protein (GFAP), Vimentine), du trafic membranaire, du métabolisme et de la protéolyse.
Les plaques amyloïdes sont organisées en feuillets . Avec le développement de la pathologie, le peptide amyloïde beta passe progressivement d’une structure en hélice
10
soluble et monomérique à une structure feuillet insoluble et oligomérique (Serpell, 2000a; Shao et al., 1999; Zagorski et al., 1992).Sous l’effet de l’augmentation de leur concentration (Lansbury, 1999; Serpell, 2000a), les oligomères du peptide amyloïde s’organisent en micelles (Lomakin et al., 1996; Serpell, 2000a), en protofibrilles (Serpell, 2000a; Stine et al., 1996), puis en fibrilles (Inouye et al., 1993; Serpell, 2000a).
1.6.1.2 Distribution dans le cerveau
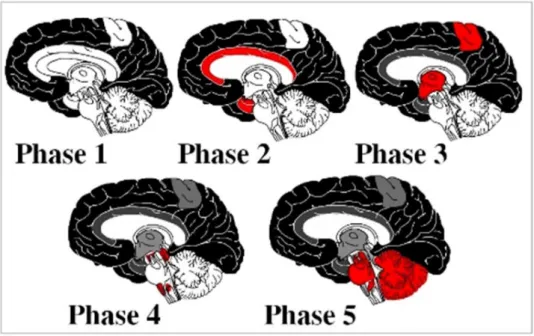
La topographie des plaques amyloïdes est généralement très peu prédictive de la progression de la maladie (Braak et al., 1991). Braak et Thal ont néanmoins tenté de retrouver des grandes tendances et ont chacun édité leur classification. Braak décrit trois stades : (A) les plaques amyloïdes sont présentes dans la partie basale de l’isocortex, (B) les plaques amyloïdes sont présentes dans les aires d’association de l’isocortex et un peu dans l’hippocampe, (C) toutes les zones de l’isocortex sont atteintes (Braak et al., 1991). Thal lui a décrit 5 stades (Hyman et al., 2012; Thal, 2013; Thal et al., 2002) (Fig.3) et une progression descendante allant de l’isocortex jusqu’aux zones sous corticales (Serrano-Pozo et al., 2011; Thal et al., 2002).
Figure 3 : Progression de la pathologie amyloïde selon les travaux de Thal. Selon Thal, la pathologie amyloïde progresse du néocortex (noir, phase 1), vers les zones allocorticales
11
(rouge) puis vers les noyaux du tronc cérébral et du striatum (phase3-4, rouge), jusqu’à des structures telles que le cervelet (phase 5, rouge). Abbréviations : Cg : Cingulate cortex, Die : Diencephalon, Mid : Midbrain, Med :Medulla Oblongata, Cblm : Cerebellum. (Serrano-Pozo et al., 2011; Thal et al., 2002).
1.6.2 Les enchevêtrements de neurofibrilles
1.6.2.1 Composition et structure
L’étude de J.P. Brion a démontré en 1985 que les enchevêtrements de neurofibrilles (ENF) sont constitués majoritairement de la protéine tau, le deuxième marqueur de la maladie d’Alzheimer (Brion, 1985). Dans les années 2000, les analyses biochimiques des enchevêtrements de neurofibrilles ont montré que ceux-ci sont effectivement composés de la protéine tau (Wang et al., 2005) et d’autres protéines telles que l’Apolipoprotéine E (Richey et al., 1995; Wang et al., 2005), et l’-synucléin (Marui et al., 2000; Wang et al., 2005). Également, les enchevêtrements de neurofibrilles sont composés de 18% de protéines du cytosquelette/de la structure, 29% de protéines du métabolisme et de l’énergie, 10% de protéines de la matrice extracellulaire, 12% de protéines du trafic intracellulaire ou de la synapse, 4% de protéines nucléaires, 10% de protéines du signalement cellulaire, 10% de protéines du stress et de protéines chaperonnes (Wang et al., 2005).
Les enchevêtrements de neurofibrilles sont des dépôts insolubles intracellulaires dans lesquelles la protéine tau s’agrège progressivement sous la forme de filament en double hélice (Kidd, 1963). D’abord soluble, tau s’agrège ensuite en oligomères, puis en protomères pour terminer en enchevêtrements de neurofibrilles (Martin et al., 2011).
1.6.2.2 Distribution dans le cerveau
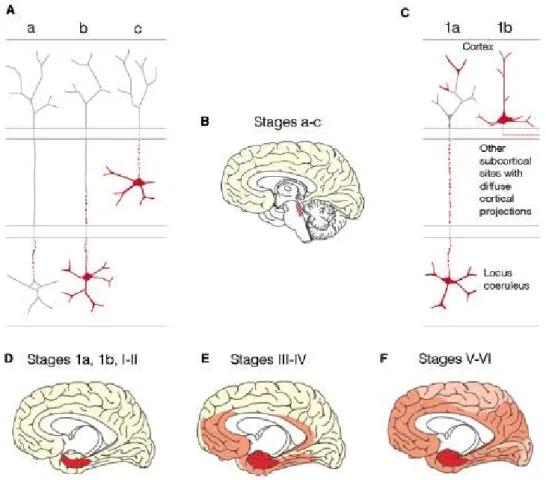
La protéine tau, à l’inverse de la protéine amyloïde beta se propage avec une topographie représentative de l’avancement de la MA (Braak et al., 2016) (Fig. 4). Les travaux de Braak ont permis de mettre en évidence que les premiers dépôts de tau (pré-enchevêtrements de
12
neurofibrilles) s’accumulent dans la partie somatodendritique des neurones du locus coeruleus très tôt (dès l’âge 6 ans) (Fig.4. A-C) (Braak et al., 2011). Etant donné que ces lésions sont asymptomatiques, on ne sait pas si elles sont caractéristiques de la « normalité » ou si elles sont les prémisses d’un développement ultérieur de la maladie d’Alzheimer. Avec le vieillissement, les enchevêtrements de neurofibrilles s’accumulent progressivement dans le cortex transentorhinal et l’hippocampe (Fig.4.D), dans le système limbique (Fig.4.E) puis dans tout le cortex (Fig.4.F). Les premiers troubles cognitifs appelés troubles cognitifs légers peuvent apparaître lorsque les dépôts d’enchevêtrement de neurofibrilles sont dans le système limbique (Braak et al., 2012).
Figure 4 : Progression de la pathologie tau selon les travaux de Braak. Les pré-enchevêtrements de neurofibrilles s’accumulent d’abord dans la partie somatodendritique des neurones du locus coeruleus (rouge) (A-C), puis dans le cortex transentorhinal (D), les zones
13
incluant système limbique (rose foncé) (E) et tout le cortex (rose foncé et rose clair). (Braak et al., 2016).
1.6.3 La neuroinflammation
De nombreux mécanismes inflammatoires sont actifs dans les cerveaux de patients atteints de la maladie d’Alzheimer (Heneka et al., 2015; Kinney et al., 2018). Par exemple, les cellules microgliales, quiescentes en condition physiologique (Glenn et al., 1989; Glenn et al., 1992), sont activées dans le cerveau de patients atteints de la MA (Heneka et al., 2015; Kinney et al., 2018). Les études montrent que la microglie phagocyte autant le peptide A que la protéine tau en condition pathologique (Perea et al., 2018). Avec le développement de la pathologie, la microglie perd ses capacités à phagocyter le peptide A et tau (Perea et al., 2018). Cette perte de la capacité de la microglie à nettoyer les débris serait, selon certains à l’origine de la neurodégénérescence (Kinney et al., 2018; Perea et al., 2018). Les astrocytes sont également activés dans la MA (Gonzalez-Reyes et al., 2017). Ils sont responsables du nettoyage des plaques amyloïdes et des enchevêtrements de neurofibrilles (Perez-Nievas et al., 2018) et forment une cicatrice autour des plaques amyloïdes (Fakhoury, 2018). Une fois activés, la microglie et les astrocytes sécrètent des cytokines pro-inflammatoires telles que l’interleukine 1 et 6 et le facteur de nécrose tumorale (TNF) qui ont pour propriétés d’augmenter le stress oxydatif, l’apoptose et la démyélinisation (Fakhoury, 2018; Gonzalez-Reyes et al., 2017) et ceci contribue probablement à la neuro-dégénescence.
1.6.4 Les pertes synaptiques
La perte de synapses est une autre caractéristique bien connue de la maladie d’Alzheimer (Hamos et al., 1989; Jackson et al., 2019). Dès le stade des troubles cognitifs légers, la perte synaptique est présente chez l’homme dans l’hippocampe et dans le gyrus temporal inférieur par comparaison avec des individus cognitivement normaux du même âge (Scheff et al., 2006; Scheff et al., 2011). Le peptide A et Tau sous leur forme soluble ou oligomérique sont probablement impliqués dans la perte synaptique alors que les plaques amyloïdes et les ENF seraient inertes (Guerrero-Munoz et al., 2015; Jackson et al., 2019). En effet, la
14
présence de plaques A et la destruction synaptique sont corrélées dans les modèles animaux de pathologie amyloïde (Koffie et al., 2009; Mucke et al., 2000) mais il semblerait que ceci ne soit pas retrouvé chez l’homme (Perez-Nievas et al., 2013). Également, la présence de multimères de tau hyper-phosphorylée à la synapse corrèle inversement avec la perte synaptique chez l’homme (Guerrero-Munoz et al., 2015; Perez-Nievas et al., 2013).
1.6.5 Le métabolisme du glucose
Le métabolisme du glucose est altéré dans la maladie d’Alzheimer (Calsolaro et al., 2016; Fukuyama et al., 1994). L’altération du métabolisme du glucose est retrouvée dans de nombreuses autres maladies neurodégénératives telles la démence fronto-temporale, la maladie de Parkinson avec ou sans démence, la maladie de Huntington, la dégénération cortico-basale, la maladie de Pick (Diehl-Schmid et al., 2007; Ishii et al., 1998; Jeong et al., 2005; Martin et al., 1992; Nagahama et al., 1997; Otsuka et al., 1989; Peppard et al., 1992). Le métabolisme du glucose est donc présent dans d’autres maladies neurodégénératives que la maladie d’Alzheimer.
Le métabolisme du glucose est altéré chez l’homme dès les troubles cognitifs légers (Croteau et al., 2018; Weise et al., 2018). Également, l’altération du métabolisme du glucose est présente chez les personnes non symptomatiques porteuses des marqueurs génétiques de la MA précoce (Croteau et al., 2018; Hoyer et al., 1988) ou de la maladie de Huntington (Mazziotta et al., 1987). Les troubles du métabolisme du glucose sont retrouvés chez les personnes nouvellement diagnostiquées pour la maladie de Parkinson (Firbank et al., 2017). Il semble donc que les troubles du métabolisme soient un marqueur précoce des maladies neurodégénératives dont la MA.
La diminution du métabolisme du glucose semble être une caractéristique commune retrouvée chez la plupart des personnes à risque pour la maladie d’Alzheimer. En effet, le métabolisme du glucose est diminué chez les femmes (Daulatzai, 2017), chez les personnes âgées, chez les personnes obèses, hypertendues, diabétiques, dépressives, atteintes de l’apnée du sommeil ou encore ayant souffert d’un traumatisme crânien (Varlamov et al., 2014). Également, le métabolisme du glucose est fonction de l’activité cérébrale (Gobel et al., 2013),
15
un paramètre augmenté chez les personnes à plus haut niveau d’éducation. Les fumeurs, une autre population à risque pour la MA, ont des concentrations post prandiales en glucose plasmatique faibles en comparaison à des non-fumeurs (Grondahl et al., 2018). L’alcoolisme est aussi associé à un métabolisme du glucose altéré si le statut nutritionnel des individus en question est mauvais ou si leur fonction hépatique est atteinte (Steiner et al., 2015).
1.7 Les biomarqueurs
Les biomarqueurs les plus utilisés sont des marqueurs des dépôts d’amyloïde beta et de la protéine tau.
1.7.1 Imagerie médicale
Il est possible de détecter les plaques amyloïdes et les ENF dans le cerveau in vivo grâce à l'utilisation de traceurs marqués avec des composés radioactifs à demi-vie courte combinés à une imagerie par tomographie TEP (tomographie par émission de positrons)(Ichise et al., 2008; Klunk et al., 2004; Shoghi-Jadid et al., 2002). Trois composés sont les plus
couramment utilisés. L’un d’eux, le composé B de Pittsburg (PIB), un composé radiomarqué au carbone 11, marque préférentiellement les plaques amyloïdes mais se fixe aussi sur les ENF (Klunk et al., 2004). Les 2 autres sont des composés marqués au fluor 18 (Ichise et al., 2008; Shoghi-Jadid et al., 2002). Ces molécules peuvent se lier de manière non spécifique sur les fibrilles de tau (Harada et al., 2013). Les dépôts amyloïdes beta sont significativement augmentés dans le cerveau des individus atteints de la MA, mais pas dans le cerveau de personnes atteintes de troubles cognitifs légers (Rodrigue et al., 2009). Néanmoins, avec le vieillissement, il existe une grande variabilité dans les dépôts d'amyloïde beta : des individus cognitivement normaux peuvent avoir des niveaux élevés ou faibles d'amyloïde beta (Jack et al., 2008). En conséquence le marquage des dépôts amyloïdes in vivo ne peut pas être utilisé seul comme outil de diagnostic.
Les fibrilles de tau peuvent être détectés in vivo chez l’homme dans les enchevêtrements de neurofibrilles et les plaques neuritiques grâce à des traceurs (Leuzy et al., 2019; Okamura et al., 2018). Etant donné que la progression spatio-temporelle de tau dans le cerveau est
16
corrélée avec la progression de la maladie, cette approche est très intéressante. Néanmoins, les premières générations de traceurs de tau pouvaient se fixer sur d’autres structures/molécules telles que les plaques amyloïdes, la mélanine, la lipofuscine, la mono-amine oxydase (Leuzy et al., 2019; Okamura et al., 2018). Les deuxièmes générations de marqueurs semblent très prometteurs car ils ne se fixent pas sur la mono-amine oxidase mais leur spécificité complète reste encore à être évaluée (Leuzy et al., 2019).
Le métabolisme du glucose cérébral ainsi que la consommation d’oxygène sont réduits dans le cerveau des personnes présentant des troubles cognitifs légers ou la maladie d’Alzheimer (Ishii et al., 1996; Lajoie et al., 2017; Ma et al., 2018). Il est assez facile de détecter par tomographie à émission de positrons (TEP) la consommation de glucose ou la consommation d’oxygène dans le cerveau humain. Il est ainsi possible de tracer le glucose en utilisant le 18-Fluoro-Deoxyglucose (18FDG-TEP)(Ma et al., 2018), ou l’oxygène en utilisant l’oxygène 15
radioactif (15O-PET)(Ishii et al., 1996). L’imagerie fonctionnelle par résonance magnétique
peut aussi être utilisée pour détecter la consommation d’oxygène du cerveau ou signal BOLD (Blood Oxygen Level Dependent) (Lajoie et al., 2017; Logothetis, 2002; Ogawa et al., 1990a; Ogawa et al., 1990b). Cette méthode est basée sur le principe selon lequel la désoxyhémoglobine, grâce à son ion ferreux (Fe2+) localisé au centre du noyau hémique,
possède des propriétés paramagnétiques différentes quand elle fixe l’oxygène (Logothetis, 2002). Globalement, le suivi du métabolisme du glucose et de l’oxygène présente l’avantage d’avoir une bonne sensibilité pour le dépistage des troubles cognitifs légers et/ou de la MA (Ishii et al., 1996; Lajoie et al., 2017; Ma et al., 2018).
Pour finir, il est maintenant possible de détecter les niveaux de neuromélanine directement dans le locus coeruleus par imagerie par résonance magnétique (Betts et al., 2019b; Liu et al., 2019). Tout comme pour l’oxygène, la neuromélanine possède des propriétés paramagnétiques exploitables en imagerie. Néanmoins, même si une étude très récente montre que la détection de la neuromélanine permet de discriminer les individus normaux des patients Alzheimer (Betts et al., 2019a), il reste encore un certain nombre de limitations. Par exemple, la neuromélanine peut fixer des ions tels que le cuivre et le fer qui eux aussi possèdent des propriétés paramagnétiques pouvant induire des biais de détection (Enochs et al., 1989; Enochs et al., 1997; Trujillo et al., 2017). Également, les variabilités
17
interindividuelles importantes de la neuromélanine, les confirmations histologiques post mortem peu nombreuses, et les divergences entre les études sur le profil d’accumulation de la neuromélanine avec l’âge font que des études supplémentaires doivent être réalisées (Betts et al., 2019b).
1.7.2 Les marqueurs biochimiques
Les deux marqueurs biochimiques les plus étudiés dans les troubles cognitifs légers et la maladie d’Alzheimer sont le peptide amyloïde beta et la protéine tau. Ils peuvent être détectés principalement dans le liquide céphalo rachidien (LCR) ou dans le sang/plasma. D’autres fluides corporels tels que la salive et l’urine sont aussi à l’étude pour le développement de biomarqueurs.
1.7.2.1 Dans le liquide céphalo rachidien
Un grand nombre d’études ont exploré les quantités de tau et d’amyloïde beta dans le contexte de la maladie d’Alzheimer (Fagan et al., 2007; Ritchie et al., 2017). Avec le développement de la maladie d’Alzheimer, la quantité du peptide amyloïde beta de 42 acides aminés (A42) dans le liquide céphalo rachidien diminue et celle de tau total (Fagan et al., 2007; Galasko et al., 1998; Motter et al., 1995; Sunderland et al., 2003) et tau phosphorylée en position thréonine 181 augmentent (Thomann et al., 2009). Les études montrent que c’est le ratio tau/amyloïde beta 42 qui est le meilleur prédicteur de la maladie d’Alzheimer (Galasko et al., 1998; Ganguli et al., 2004; Janelidze et al., 2018; Ward et al., 2012). Cependant cet outil n’est utilisé qu’en recherche clinique. Le ratio tau/A42 ne permet pas non plus de diagnostiquer les troubles cognitifs légers (Ritchie et al., 2017). Quelques études longitudinales prometteuses montrent en revanche que le ratio phospho-tau 181/A42 pourrait être un bon prédicteur du déclenchement de la MA chez les personnes présentant des troubles cognitifs légers, mais les cohortes sont de petites tailles (140 individus présentant des troubles cognitifs légers et 293 individus sains) (Hansson et al., 2006; Koivunen et al., 2008; Monge-Argiles et al., 2011; Parnetti et al., 2012; Ritchie et al., 2017; Visser et al., 2009).
18
Chez les individus sains, la quantité de tau dans le LCR augmente avec l’âge alors que pour le peptide amyloïde beta 42, il n’y a pas de corrélation entre la quantité de A42 et le vieillissement (Sjogren et al., 2001).
D’autres marqueurs pourraient aussi être tracés dans le LCR pour suivre le développement de la MA. Une étude longitudinale récente réalisée sur une cohorte de 821 personnes suivies pendant 1 à 6 ans (durée moyenne de 3ans) montre que 5 marqueurs de l’inflammation (Intercellular Adhesion Molecule 1, Vascular Cell Adhesion Molecule 1, Chitinase 3 Like Protein 1 (YKL40), interleukine 15, Fms Related Tyrosine Kinase1) pourraient être de bons prédicteurs du développement de la maladie d’Alzheimer et également de bons marqueurs des troubles cognitifs légers (Janelidze et al., 2018). Cependant cette étude ne révèle rien sur la spécificité de ces marqueurs de l’inflammation pour la MA.
1.7.2.2 Dans le sang
La protéine tau et le peptide A sont aussi sécrétés dans le sang en très faible quantité(Lue et al., 2017). Il fut très longtemps difficile de détecter de manière fiable ces 2 marqueurs dans le sang en raison de limitations technologiques. Des progrès technologiques récents ont néanmoins permis de détecter de très faibles quantités de tau et de A dans le sang (Randall et al., 2013). Ces études révèlent que le niveau de la protéine tau plasmatique augmente de manière significative dans le plasma des patients atteints de la MA (Olsson et al., 2016; Pase et al., 2019; Zetterberg et al., 2013). Également, il est possible de détecter la protéine tau phosphorylée en position thréonine 181 mais les études sont contradictoires et de petite taille (Mielke et al., 2018; Park et al., 2019; Tatebe et al., 2017). En ce qui concerne les quantités de A aussi les études sont contradictoires : certaines études montrent que le niveau de Adans le sang diminue (Janelidze et al., 2016; Park et al., 2019) alors que d’autres montrent qu’il ne change pas (Olsson et al., 2016).
D’autres marqueurs tels que ApoJ ou les sphingolipides sont aussi à l’étude pour le diagnostic ou le développement de la MA mais des études complémentaires sont requises (O'Bryant et al., 2017). Également, les marqueurs épigénétiques circulants représentent une piste
19
intéressante pour le suivi de la MA mais il semblerait qu’il y a de grandes disparités entre les études (Fransquet et al., 2019; Kumar et al., 2016; Wu et al., 2016).
1.7.2.3 Autres fluides
Il est intéressant de noter que certaines équipes de recherche s’intéressent à la détection de tau et de A42 dans la salive (Gleerup et al., 2019) et les urines (Takata et al., 2008) mais ces études sont très peu nombreuses, concernent de petites cohortes et nécessitent par conséquent d’être répliquées.
1.8 Les traitements
Cent années environs après la découverte de la MA par Aloïs Alzheimer, il n’existe toujours aucun traitement (Cummings et al., 2014). Les seuls médicaments existants sont des drogues qui diminuent les symptômes temporairement : les inhibiteurs de l’acétylcholinestérase (Howard et al., 2012; Hyde et al., 2013), les antagonistes des récepteurs NMDA (Howard et al., 2012; Hyde et al., 2013) (N-methyl-D-aspartate) ou encore la thérapie combinée des deux (Food Drug Administration, 2014; Greig, 2015; Matsunaga et al., 2014). Également, de nombreux essais cliniques prometteurs sont actuellement en cours mais à ce jour aucune thérapie ou prophylaxie n’est sur le marché (Long et al., 2019).
1.8.1 Inhibiteurs de l’acétylcholinestérase
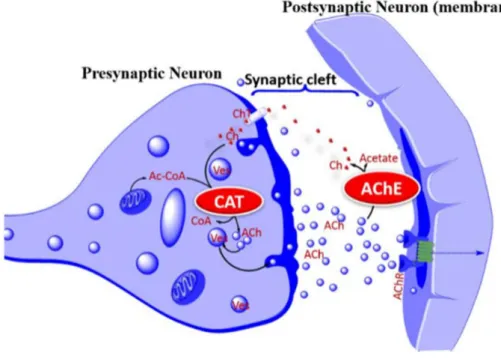
Les inhibiteurs de l’acétylcholinestérase permettent d’augmenter la quantité d’acétylcholine présente dans la fente synaptique (Fig.5). Ils ont été développés sur la base des découvertes montrant des déficits cholinergiques observés dans les noyaux de Meynert et le cortex d’individus atteints de la maladie d’Alzheimer (Coyle et al., 1983; Davies et al., 1976; Whitehouse et al., 1982). L’acétylcholine est un neuromodulateur important du système nerveux central. Elle joue un rôle dans de grandes fonctions telles que le sommeil (Vazquez et al., 2001), les circuits de l’addiction et de la récompense, l’attention et le stress (Picciotto et al., 2012), la thermorégulation (Takahashi et al., 2001), la mémoire (Hasselmo, 2006). On
20
pourrait donc s’attendre à restaurer tout ou partie de ces fonctions en administrant des inhibiteurs de l’acétylcholinestérase. En fait, les patients sous inhibiteurs de l’acétylcholinestérase ont une légère amélioration de leur fonction cognitive (mémoire en particulier (Schredl et al., 2001)) mais pas de changement dans leur qualité de vie (Birks et al., 2018). Les inhibiteurs de l’acétylcholinestérase améliorent aussi la qualité du sommeil (dos Santos Moraes et al., 2006; Moraes et al., 2008; Schredl et al., 2001) et l’attention (Ancoli-Israel et al., 2005; Vila-Castelar et al., 2018) mais ne ralentissent pas la progression de la maladie d’Alzheimer.
Figure 5 : Cycle de l’acétylcholine. Dans les structures présynaptiques, la choline acétyle transférase (CAT) transforme la choline (ch) et l’acétyle Coenzyme A (AcCoA) en acétylcholine (Ach). L’acétylcholine est ensuite relâchée dans la synapse ou elle active les récepteurs cholinergiques puis est ensuite dégradée par l’acétylcholine estérase (AchE) en choline et en acétate. La choline est alors réabsorbée dans le bouton présynaptique pour refaire de l’acétylcholine (Habtemariam, 2019; Purves D, 2001).
21 1.8.2 Les antagonistes des récepteurs NMDA
La dégénérescence du système glutamatergique est une autre caractéristique observée dans le cerveau de patients atteints de la maladie d’Alzheimer (Greenamyre et al., 1987). Très vite l’hypothèse selon laquelle l’excitotoxicité du glutamate jouait un rôle important dans la neurodégénérescence a été émise (Arias et al., 1998) et a conduit à l’utilisation de la mémantine, un antagoniste des récepteurs NMDA (Kuns B, 2019). Ce médicament fut d’abord mis au point chez l’homme pour soigner le diabète, mais une fois son inefficacité prouvée, la compagnie pharmaceutique décida de rediriger le traitement vers les maladies neurodégénératives (Folch et al., 2018). Les études chez les patients atteints de la MA ont ensuite pu démontrer que la mémantine réduit principalement l’agitation et l’agressivité (Gauthier et al., 2005; Kishi et al., 2017), diminue les illusions, la désinhibition et les hallucinations (Kishi et al., 2017). Elle diminue aussi les perturbations nocturnes et diurnes liées à la MA (Kishi et al., 2017).
1.8.3 Les essais cliniques en cours
Différentes stratégies sont actuellement en test clinique dont l’immunothérapie active ou passive, les inhibiteurs d’enzymes, la thérapie génique, les modulateurs de la réponse immune ou encore l’administration de produits naturels (Habtemariam, 2019; Long et al., 2019).
1.8.3.1 L’immunothérapie
L’immunothérapie active consiste à immuniser l’organisme contre un des marqueurs de la MA comme A ou tau par exemple, de manière à diminuer la concentration du marqueur en question. L’immunisation par le peptide amyloïde a déjà été testée chez des patients diagnostiqués maladie d’Alzheimer probable, mais les premières études cliniques ont été arrêtées car quelques patients ont déclaré des méningo-encéphalites aseptiques (Gilman et al., 2005). L’immunisation active, chez les patients n’ayant pas souffert de méningite, a mis en évidence une diminution de la charge amyloïde, mais n’a pas amélioré la cognition des patients traités (Gilman et al., 2005) et n’a pas freiné le déclin cognitif 14 ans après
22
l’immunisation (Long et al., 2019; Nicoll et al., 2019). Néanmoins, deux autres essais cliniques sont toujours en cours avec d’autres versions moins immunogènes du peptide (Lacosta et al., 2018; Long et al., 2019; Vandenberghe et al., 2017). Pour la protéine tau, des essais d’immunisation sont également en cours avec des fragments de la protéine tau (Novak et al., 2019; Zhejiang Hisun Pharmaceuticals, 2019).
L’immunothérapie passive consiste à injecter des immunoglobulines dirigées contre un des marqueurs de la MA. Plusieurs essais cliniques utilisant des anticorps dirigés contre Acontre tau ou encore contre l’apolipoprotéine E4 (ApoE4) sont en cours (Long et al., 2019). De nombreux essais d’immunisation passive réalisés contre le peptide Aont échoué soit parce qu’ils avaient des effets secondaires de type œdème ou microhémorragie, soit parce qu’ils n’amélioraient pas la cognition malgré parfois une diminution de la charge amyloïde (Long et al., 2019). Les essais d’immunisation contre ApoE4 ont des résultats prometteurs mais les études cliniques n’ont pas encore été commencées. Les résultats chez la souris montrent une réduction de la charge amyloïde (Liao et al., 2018) mais ils n’ont pas testé si l’immunisation passive contre ApoE4 améliorait la cognition. Seul un essai d’immunisation contre la protéine tau a été arrêté les autres essais cliniques sont toujours en cours (Long et al., 2019; Yanamandra et al., 2017).
1.8.3.2 Les inhibiteurs d’enzyme
De nombreux inhibiteurs d’enzyme ont été testés que ce soient des enzymes de la voie amylogénique ou des enzymes impliquées dans la progression de la pathologie tau. Les essais cliniques incluant les inhibiteurs de la sécrétase et de la sécrétase, 2 enzymes responsables du clivage du peptide Aont été arrêtés soit parce qu’ils provoquaient un plus grand déclin cognitif soit parce qu’ils avaient des effets secondaires très importants tels que le développement de cancer, la perte de poids ou des nausées (Coric et al., 2015; Doody et al., 2013; Egan et al., 2018; Egan et al., 2019; Long et al., 2019; Panza et al., 2018). Pour ce qui concerne les inhibiteurs d’enzymes impliquées dans la progression des tauopathies, des inhibiteurs de GSK3 (Glycogen Synthase Kinase 3) et de Fyn, deux kinases de tau, ont été testés en clinique sans succès (Tolosa et al., 2014; van Dyck et al., 2019). Il est intéressant de voir que toutes ces enzymes ont en commun de nombreux effets pléiotropes (Beurel et al.,
23
2015; Nygaard, 2018; Strooper et al., 2010; Vassar et al., 2014) qui pourraient expliquer les échecs cliniques observés.
1.8.3.3 La thérapie génique
La thérapie génique est une autre piste explorée en recherche clinique. Cette méthode consiste à introduire du matériel génétique ciblant un gène d’intérêt dans des tissus. Les outils de thérapie génique utilisés en essai clinique pour la maladie d’Alzheimer sont principalement des oligonucléotides antisens (Long et al., 2019). Ce sont de courtes séquences d’ADN ou d’ARN (13 à 25 bases) qui peuvent s’hybrider par complémentarité sur une séquence cible (Dias et al., 2002). Il en résulte une extinction ou une diminution transitoire de l’expression d’un gène ou un blocage transitoire de l’épissage d’un gène. Un essai clinique avec un oligonucléotide antisens anti tau injecté par voie intrathécale est en cours pour tenter de diminuer la quantité de tau dans la MA (Ionis Pharmaceuticals, 2019; Mignon et al., 2018). Il est important de considérer que les effets non spécifiques (effets « OFF target » en anglais) des oligonucléotides antisens utilisés sont à évaluer. En effet, bien que dessinés pour atteindre spécifiquement leur cible sur la base de leur complémentarité parfaite avec celle-ci, il est envisageable que des hybridations non spécifiques aillent cibler d’autres gènes. Également, il est important de prendre en compte le coût très élevé de ces méthodes une fois transposées chez l’homme (plusieurs centaines de milliers de dollars américains)(Wurster et al., 2018). Un tel coût ne pourra pas être supporté par les systèmes de santé public actuels et pourra être supporté avec beaucoup de restrictions uniquement par certains assureurs privés (Wurster et al., 2018).
1.8.3.4 Les modulateurs de la réponse immunitaire
La réponse inflammatoire est une autre manifestation de la maladie d’Alzheimer (McGeer et al., 2015) et on ne sait pas si cette réponse immunitaire est une cause ou une conséquence de la MA (Armstrong, 2013). Ainsi certaines stratégies de recherche visent à employer des modulateurs de la réponse immunitaire à des fins thérapeutiques (Long et al., 2019). Plusieurs stratégies telles que l’utilisation d’anti-inflammatoires, les injections
24
d’immunoglobulines ont étés testées chez l’homme en vain (Long et al., 2019). Une stratégie visant à stimuler la microglie et la phagocytose est également en phase clinique (Long et al., 2019). Elle consiste à utiliser des anticorps activateurs ou inhibiteurs de certaines voies de signalisation (Angata et al., 2015; Cheng et al., 2018).
1.8.3.5 L’administration de produits naturels
Avec les nombreux échecs de la médecine moderne, de plus en plus d’études se tournent vers le test de produits naturels pour contrer la progression de la maladie d’Alzheimer (Habtemariam, 2019). Deux inhibiteurs de l’acétyle choline estérase connus pour leurs effets sur les symptômes de la MA sont issus de produits naturels : la galantamine est issue du bulbe du perce neige d’où elle tient son nom (Galanthus nivalis) et la physostigmine est issue de la fève de Calabar (Physostigma venenosum) une plante tropicale originaire de l’Afrique tropicale (Habtemariam, 2019; Mucke, 2015; Scheindlin, 2010). Deux études menées chez l’homme rapportent que le cumin par exemple (Nigella sativa), améliore la cognition (Tavakkoli et al., 2017) particulièrement chez des volontaires en santé (Bin Sayeed et al., 2013; Bin Sayeed et al., 2014) mais les mécanismes d’action méritent d’être investigués (Cascella et al., 2018). Également, la curcumine (le composant actif du curcuma) pourrait avoir des effets bénéfiques sur la MA mais pour l’instant les résultats sont négatifs en raison d’une faible biodisponibilité de la curcumine dans le sang et du stade avancé des patients traités (Goozee et al., 2016; Mishra et al., 2008). En revanche, une étude récente réalisée en double aveugle sur des personnes âgées en santé pendant un mois, a mis en évidence que, l’administration d’une formulation de curcumine augmentant sa biodisponibilité plasmatique et cérébrale (Begum et al., 2008; Gota et al., 2010), améliore significativement l’attention, la mémoire de travail et l’humeur et ce dès le début du traitement (Cox et al., 2015). Également, plusieurs études menées chez l’homme montrent une corrélation négative entre la consommation du piment rouge et le déclin cognitif (Liu et al., 2016; Shi et al., 2019).
25
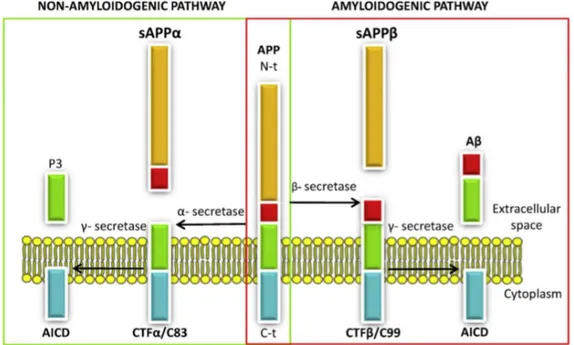
2. Le peptide amyloïde
2.1 Généralités
Le peptide Aest issu du clivage de la protéine transmembranaire Amyloid Precursor Protein (APP) par la sécrétase et la sécrétase (Fig.6). La taille du peptide amyloïde peut varier entre 39 et 43 acides aminés (Serpell, 2000b). Les peptides majoritairement présents dans le cerveau sain sont A40 (~90%) et A42 (~10%) (Duff et al., 1996; Suzuki et al., 1994).
Figure 6 : Le clivage de la protéine APP . Le clivage de la protéine APP par la sécrétase et la sécrétase permet la production du peptide A dans l’espace extracellulaire (rectangle vert et rouge), d’un fragment N-terminal sécrété (secreted APP sAPPrectangle orange) et d’un fragment C-terminal membranaire (AICD : Amyloid Intracellular Domain, rectangle bleu). Cette voie de synthèse est appelée la voie amyloïdogénique. L’APP peut aussi être clivé par l’ sécrétase et la sécrétase. Dans ce cas, le peptide P3 (rectangle vert), l’ectodomaine N terminal (secreted APP : sAPP rectangle orange et rouge) et l’AICD sont produits. Cette voie de synthèse est la voie non-amyloïdogénique (Montoliu-Gaya et al., 2015).