THESE
THESE
En vue de l'obtention du
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
Délivré par L’université Toulouse III - Paul Sabatier Discipline : Biochimie et Biologie Moléculaire
Présentée et soutenue par Christophe PAPIN Le 19 novembre 2008
Mécanisme moléculaire de l’hélicase AAA+ TIP49B
JURY
Dr. Stefan DIMITROV, Directeur de Recherche, Institut Albert Bonniot, Grenoble, Rapporteur Dr. Emmanuelle DELAGOUTTE, Chargé de recherche, Institut Curie, Paris, Rapporteur Dr. Ali HAMICHE, Chargé de Recherche, IGBMC, Strasbourg, Rapporteur Pr. David CRIBBS, Professeur, CBD, Toulouse, Examinateur Dr. Solange MORERA, Chargé de Recherche, LEBS, Gif-sur-Yvette, Invitée Dr. Mikhail GRIGORIEV, Chargé de Recherche, LBME, Toulouse, Invité
École doctorale Biologie-Santé-Biotechnologie de Toulouse Laboratoire de Biologie Moléculaire des Eucaryotes – UMR 5099
Institut d’Exploration Fonctionnelle des Génomes 118, route de Narbonne, 31062 Toulouse Cedex
2
À mes parents,
à mes grands-parents.
3
« Ça me fait peur les gens prudents,
les gens précautionneux,
ils ont plus d’avenir que de présent,
ils sont assis mais ils se croient debout.
C’est effrayant, non ? »
4
Remerciements
Tout d’abord, je tiens à remercier l’ensemble des membres du jury Stefan Dimitrov, Ali Hamiche, Emmanuelle Delagoutte, Solange Morera et David Cribbs, pour avoir accepté de juger mon travail, ainsi que pour les commentaires pertinents et constructifs sur le manuscrit et pour la discussion enrichissante lors de la soutenance.
Je voudrais remercier également les collaborateurs Laszlo Tora, Patrick Schultz et Didier Trouche pour leur disponibilité et leur aide précieuse. Un grand merci à Marina Guerrin-Weber avec qui j’ai fait mes premiers pas dans un laboratoire de recherche. Ton encadrement et tes encouragements pendant mon stage de Master 1ère année auront été capitaux pour la suite de mon cursus universitaire.
Un très grand merci à Micha, qui m’a accueilli dans son équipe, d’abord pour un stage de Master 2ème année puis pour y effectuer une thèse. Merci au scientifique qui m’a toujours fait confiance tout au long de ces années, me laissant beaucoup de liberté sur l’avancement du projet, tout en sachant me recadrer quand il le fallait. Une très grande reconnaissance pour l’homme surtout, puisque tu as été présent du début à la fin pendant les moments les plus difficiles tant personnels que professionnels sans jamais baisser les bras. En quittant ton équipe, je ne quitte pas juste un directeur de thèse, mais une des personnes qui aura été parmi mes plus proches complices pendant ces 5 années.
Merci à Cynthia, qui m’a à peu près tout appris à la paillasse quand je suis arrivé dans l’équipe. Ce fût un vrai plaisir de travailler à tes côtés. Je suis particulièrement touché de ton déplacement de Londres jusqu’à Toulouse pour assister à ma soutenance. Merci aux membres actuels de l’équipe qui ont considérablement accéléré les choses en se greffant au projet TIP49. Anna, d’abord pour sa contribution précieuse sur la purification des protéines et les expériences de gel-retard. Bon courage et bonne chance pour la suite de ta thèse et sans rancune de m’avoir volé un ami ! François ensuite pour son expertise dans la manipulation et la modélisation des structures tridimensionnelles, et enfin Odile, qui a été un peu ma grande sœur ici. Nous nous ressemblons beaucoup toi et moi, si tous mes futurs collègues de bureau et/ou paillasse pouvaient te ressembler, alors mes journées de travail seront toujours joyeuses.
Merci à Martine qui m’a formé aux joies de l’enseignement, pour ta bonne humeur et ton implication dans l’enseignement en biologie moléculaire ici à l’Université Paul Sabatier.
Une pensée à tous les potes qui sont là et toujours là et depuis très longtemps pour certains, mille mercis à tous ces zigotos qui ont partagé beaucoup des mes nuits où nous décidions autour de trop de verres, de lâcher un peu la pression au cours de soirées
5
mémorables : Yann, Céd, Titi, Sandra, Adri, Anne, Tom, Guillaume, Élé, Yom, Sarah, Erwan, Ti’sylvain, Nico, Brice, Béa, Rami, Auré, Ananda, Céline, Isa, Lulu, Julie C, ZZJul, Olivier, Benjamin, Alex, Anja, Cat & Djul, Bob & Læt, Sandrine et Elodie.
Un remerciement tout particulier à la petite famille du 38 Grande-rue Saint-Michel, au rez-de-chaussée, au fond du couloir. Ces gens-là ont le cœur si large qu’on y entre sans frapper. J’ai eu l’immense privilège de partager deux ans de voisinage avec Paula, Salomé, Sabine et Thierry. Vous avez été ma famille ici à Toulouse pendant ces deux dernières années, mes confidents, mes épaules sur lesquels s’appuyer quand on a plus beaucoup de forces pour se relever. Ça va être difficile de quitter le 38, très très difficile !
Spéciale dédicace à celui qui a été mon alter ego ici au LBME depuis le début, merci à Monsieur Simon. Nous avons, je crois, à peu près tout fait ensemble depuis qu’on se connaît, on a fait notre DEA ensemble, on a enseigné ensemble, on a vécu ensemble, on a aimé ensemble et nous aurons même (c’est pour bientôt !) co-signer un article ensemble… C’est dire si la vie a tout mis en œuvre pour souder entre nous une amitié indéfectible. Ce paragraphe est le moment opportun pour te dire toute l’amitié que je te porte mon salop ! comme le dit la chanson, lorsque je serais parti, il manquera un temps à ma vie, il me manquera toi, mon alter ego.
Je veux avoir une pensée plein de tendresse pour Rosie qui m’a porté (et supporté) pendant 4 longues années malgré la distance. Sans toi, je n’aurais tout simplement jamais réussi à tenir le coup ici. Merci infiniment pour tout ce que tu as fait pour moi. J’espère profondément que nous saurons gardé ce qui nous lie, ce qui nous a construit, c’est là mon souhait le plus cher. Je souhaite de tout cœur que ta future nouvelle vie en Belgique t’apportera tout ce que tu désires.
Merci à Audrey pour ton soutien dans cette dernière ligne droite difficile et non sans embûche que constitue une dernière année de thèse. Merci pour ton attitude plein d’amour, de sérénité et de tendresse pendant cette année. Merci pour ce regard dans lequel je me suis projeté pour mieux appréhender l’avenir. Il me tarde de te rejoindre à Strasbourg au plus vite et que l’on écrive ensemble la grande histoire que la vie nous offre.
Pour terminer, je veux dédier l’ensemble de ce travail à ma famille, à mes parents et à mes grands-parents. Des évidences biologiques font qu’effectivement sans eux, je ne serais pas là aujourd’hui. Mais plus que le lien de filiation, je veux vous dire tout l’amour que je vous porte. Merci à vous de m’avoir laissé suivre mon chemin, de m’avoir toujours fait confiance, même quand le chemin choisi était très chaotique. Je ne vous ai pas beaucoup épargné pendant toutes ces années, mais en me laissant libre de mes choix et en ne cessant jamais de me soutenir à chaque instant, j’ai réussi à me construire et à me trouver. Aujourd'hui, je suis un homme serein vous sachant toujours près de moi.
6
7 - SOMMAIRE - ____________________________________________________________ 6 AVANT-PROPOS__________________________________________________________ 10 - INTRODUCTION - _______________________________________________________ 12 I Les hélicases ___________________________________________________________ 13 A) Généralités _______________________________________________________________ 13 B) Variations autour d’un même thème__________________________________________ 13 1) Des motifs différents _____________________________________________________________ 14 2) Une structure conservée, les modules « RecA-like »/AAA+ _______________________________ 14 (a) Le module « RecA-like » _______________________________________________________ 14 (b) Le domaine AAA+, un module « RecA-like » allongé _________________________________ 15 3) Des propriétés biochimiques spécifiques ______________________________________________ 16 (a) Acide nucléique préférentiel_____________________________________________________ 16 (b) Etat oligomérique de la forme active ______________________________________________ 16 (c) Caractéristique de l’activité translocase ____________________________________________ 16 4) Des modules régulateurs___________________________________________________________ 17
II Hélicases et translocases non-hexamériques _________________________________ 17 A) La superfamille SF1 _______________________________________________________ 17
1) Les hélicases SF1A_______________________________________________________________ 17 (a) Données structurales ___________________________________________________________ 17
• L’hélicase Rep ______________________________________________________________ 18
• L’hélicase PcrA _____________________________________________________________ 18
• L’hélicase UvrD _____________________________________________________________ 19 (b) Le modèle « chenille » de déroulement de l’ADN ____________________________________ 19 (c) Implication sur l’état oligomérique de la forme active des hélicases SF1A _________________ 20 2) Les hélicases SF1B_______________________________________________________________ 21 (a) L’hélicase RecD au sein du complexe RecBCD. _____________________________________ 21 (b) La protéine Dda ______________________________________________________________ 22
B) La superfamille SF2 _______________________________________________________ 23 1) La famille SF2! _________________________________________________________________ 23 (a) Structure de la protéine virale NS3________________________________________________ 23 (b) Modèle par mouvement brownien de déroulement d’un acide nucléique double-brin _________ 24 (c) Implication sur le pas des hélicases SF2!___________________________________________ 25 2) La famille SF2" _________________________________________________________________ 26 (a) Structure de la protéine Rad54 ___________________________________________________ 26 (b) Modèle de translocation le long d’un ADN double-brin _______________________________ 27 (c) Implication dans les processus de remodelage _______________________________________ 28 3) Polarité des enzymes SF2 __________________________________________________________ 28 (a) La famille XPD, unique représentant d’hélicase SF2B_________________________________ 28 (b) La famille SF2" ______________________________________________________________ 29 (c) Cas particulier des ARN hélicases à boîte DEAD, modèle de déroulement d’un duplex par déstabilisation ____________________________________________________________________ 30 4) Domaines accessoires, ciblage des enzymes SF2________________________________________ 31 (a) Ciblage vers une structure d’acide nucléique spécifique _______________________________ 31
• La protéine RecG ____________________________________________________________ 31
• La protéine UvrB ____________________________________________________________ 32
• Les protéines recQ-like________________________________________________________ 32
• L’hélicase ARN à boîte DEAD DbpA ____________________________________________ 33 (b) Ciblage vers un complexe nucléoprotéique _________________________________________ 33 (c) Activités catalytiques nouvelles __________________________________________________ 34
III Hélicases et translocases hexamériques ____________________________________ 34 A) Généralités _______________________________________________________________ 34 B) Motifs et structure _________________________________________________________ 35
8
1) La famille SF3 __________________________________________________________________ 35 (a) Des hélicases réplicatives de virus.________________________________________________ 35 (b) Un module hélicase AAA+______________________________________________________ 35 (c) L’hélicase du virus SV40 _______________________________________________________ 36
• Structure de la protéine en absence de nucléotide ___________________________________ 36
• La liaison au nucléotide entraîne un mouvement en « iris »____________________________ 37
• Modèle pour le déroulement de la double-hélice aux sites ori __________________________ 37 (d) L’hélicase du virus BPVE1 _____________________________________________________ 38 2) La famille SF4 __________________________________________________________________ 39
• Des hélicases réplicatives de type B______________________________________________ 39
• Cinq motifs conservés ________________________________________________________ 40
• Structure de la protéine codée par le gène 4 du bactériophage T7 _______________________ 40 3) La famille SF5 __________________________________________________________________ 41 (a) Structure du facteur Rho pleine taille, un hexamère ouvert _____________________________ 42 (b) Structure du facteur Rho déplété de 8 résidus, un hexamère fermé _______________________ 43 (c) La transition entre les formes ouvertes et fermées active une translocation 5’-3’ du facteur Rho 43 4) La superfamille SF6 ______________________________________________________________ 44 (a) Le complexe MCM____________________________________________________________ 44 (b) Les hélicases réplicatives _______________________________________________________ 45 (c) L’hélicase bactérienne RuvB ____________________________________________________ 46
• Structure du monomère de RuvB ________________________________________________ 46
• Reconstitution d’un hexamère de RuvB ___________________________________________ 47
• Assemblage du complexe RuvAB/jonction de holliday_______________________________ 48
• Structure atomique du complexe RuvAB/jonction de Holliday _________________________ 48 C) Mécanismes d’hydrolyse d’ATP _____________________________________________ 49 1) Modèle de l’ATPase F1 à trois sites séquentiels ________________________________________ 49 2) Modèle à six sites séquentiels_______________________________________________________ 50 3) Modèle concerté _________________________________________________________________ 51
D) Couplage entre les activités ATPase et translocases _____________________________ 51 E) Activité de déroulement d’ADN ______________________________________________ 52 IV Les protéines TIP49 ____________________________________________________ 52
A) Généralités _______________________________________________________________ 52 B) Fonction in vivo des protéines TIP49__________________________________________ 53 1) Remodelage de la chromatine_______________________________________________________ 54 (a) Les complexes TIP60 et NuA4 ___________________________________________________ 54
• TIP60 et NuA4 dans la réparation des CDB________________________________________ 54 - Fonction de la protéine Tip60 en amont d’ATM. ____________________________________ 55 - Cas particulier de la sous-unité TRRAP, rôle au sein ou à l’extérieur du complexe TIP60 ? ___ 57
• TIP60 et NuA4 dans la régulation transcriptionnelle _________________________________ 57 (b) Le complexe Ino80 ____________________________________________________________ 57
• Ino80 dans la réparation des CDB _______________________________________________ 58
• Ino80 dans la régulation transcriptionnelle_________________________________________ 58 (c) Le complexe SWR1 ___________________________________________________________ 59
• SWR1 dans la réparation des CDB_______________________________________________ 60
• SWR1 dans la régulation transcriptionnelle ________________________________________ 60 (d) Fonction des protéines TIP49 au sein de ces complexes _______________________________ 60 2) Régulation transcriptionnelle _______________________________________________________ 61 (a) Interaction avec c-Myc _________________________________________________________ 61 (b) Interaction avec "-caténine ______________________________________________________ 62
• Une activité antagoniste des protéines TIP49_______________________________________ 62
• Action des protéines TIP49 au sein de complexes différents ___________________________ 63 (c) Le complexe Uri1 _____________________________________________________________ 64 3) Biogenèse des snoARN ___________________________________________________________ 65 4) Division cellulaire________________________________________________________________ 66 5) Assemblage de la télomérase _______________________________________________________ 66
C) Propriétés enzymatiques des protéines TIP49 in vitro ____________________________ 67 1) Activités ATPase et hélicase des protéines TIP49 purifiées________________________________ 67 2) Structure tridimensionnelle de la protéine hTIP49a ______________________________________ 68
9
(a) Structure du monomère de hTIP49a _______________________________________________ 68 (b) Structure de l’hexamère de hTIP49a ______________________________________________ 68
V Présentation du projet ___________________________________________________ 69
- RÉSULTATS - ___________________________________________________________ 70
I Article 1 : Reptin is a slow-motion active 3'-5' DNA helicase ____________________ 71 A) Question posée et démarches expérimentales___________________________________ 71 B) Résultats et Discussions ____________________________________________________ 71 II hTIP49b est sensible aux modifications post-traductionnelles des histones_________ 93
A) Question posée et démarches expérimentales___________________________________ 93 B) Matériels et méthodes ______________________________________________________ 94 C) Résultats et discussions _____________________________________________________ 94 III Le complexe nucléoprotéique hTIP49b/ADN simple-brin agit comme une plateforme
de recrutement in vitro ______________________________________________________ 95 A) Question posée et démarches expérimentales___________________________________ 95 B) Matériels et méthodes ______________________________________________________ 96 C) Résultats et discussions _____________________________________________________ 96 IV Article 2 : A MRN/Tip60 complex involved in DNA double strand breaks repair____ 97
A) Question posée et démarches expérimentales___________________________________ 97 B) Résultats et Discussions ____________________________________________________ 97 - CONCLUSIONS & PERSPECTIVES - ______________________________________ 119 - RÉFÉRENCES - ________________________________________________________ 124
10
AVANT-PROPOS
Ce manuscrit présente le travail de caractérisation biochimique effectué sur les hélicases eucaryotes TIP49b. Les résultats obtenus mettent en évidence des propriétés enzymatiques singulières pour des protéines à activité hélicase. Aussi, l’introduction se focalisera tout particulièrement sur les caractéristiques biochimiques de cette vaste classe d’enzyme en présentant de manière détaillée à la fois les structures cristallographiques disponibles et les données apportées par des approches cinétiques. L’ensemble de ces travaux a permis de proposer pour ces enzymes plusieurs mécanistiques d’action à l’échelle moléculaire. Chacun des modèles est caractérisé par un certain nombre de propriétés enzymatiques. Dans le but de mieux comprendre les différents modèles présentés ici, il m’est apparu important de rappeler aux lecteurs, en guise d’avant-propos, quelques-unes de ces notions enzymologiques.
•
Translocases et hélicases :
Les translocases sont des enzymes qui couplent l’hydrolyse d’ATP en un mouvement directionnel le long d’un acide nucléique simple-brin ou double-brin. Les hélicases sont une sous-classe de la famille des translocases, qui en plus de transloquer, sont capable de dérouler un duplex d’acide nucléique en composants simple-brins.
•
Processivité :
Pour une hélicase (ou translocase), la processivité mesure le nombre de paires de bases (et/ou nucléotides) déroulées (ou transloqués) avant la dissociation de l’acide nucléique.
•
Polarité :
L’ADN est une molécule bipolaire composée de deux brins parallèles en orientation opposée. La séquence à l’intérieur d’un acide nucléique est donc le plus souvent assymétrique. Les moteurs protéiques se déplacent donc le long d’un acide nucléique dans une direction précise. Pour les translocases simple-brins, cela ne pose pas de problème. Une
11
fois chargée sur son substrat, la polarité de l’enzyme est définie par la direction du déplacement de celle-ci le long de sa matrice monocaténaire. Pour les translocases double-brins, le squelette phosphodiester antiparallèle confère au duplex une asymétrie intrinsèque. Pour ces enzymes, la polarité est déterminée par le brin utilisé comme « rail » et ceci bien que la protéine se déplace le long d’un ADN bicaténaire. Par conséquent, le brin choisi lors du chargement de la protéine sur son substrat double-brin, est une étape critique car elle déterminera la direction vers laquelle l’enzyme va transloquer.
•
Pas de l’enzyme :
Le pas d’une hélicase (ou translocase) correspond au nombre de paires de bases (et/ou nucléotides) déroulées (ou transloqués) pour chaque cycle d’hydrolyse d’ATP.
•
Stoechiométrie de couplage à l’ATP:
La stoechiométrie de couplage à l’ATP définit le nombre d’ATP hydrolysé par paire de base déroulée (et/ou par nucléotide transloqué). Cette notion est parfois assimilée au pas de l’enzyme, mais ces deux notions ne sont pas toujours équivalentes. En effet, l’hydrolyse de l’ATP n’est pas toujours accompagnée d’un déplacement unidirectionnel de l’enzyme. Dans ce cas, le nombre de paires de bases déroulées par molécule d’ATP hydrolysé sera une sous-estimation du pas réel de l’enzyme.
•
Mécanisme passif versus mécanisme passif :
Dans le cas des hélicases, on décrit deux mécanismes de déroulement qualifiés de passif et actif. Au sens strict, les hélicases sont toutes des protéines actives dans la mesure où elles utilisent l’énergie de l’hydrolyse de l’ATP pour catalyser leur réaction. Cette nomenclature a été utilisée pour distinguer les enzymes qui participent activement au déroulement d’un duplex, des enzymes qui le déroulent de manière passive. Pour les hélicases actives, il y a une déstabilisation du duplex ADN à la jonction simple-brin/double-brin par la protéine, alors que pour les hélicases passives, le déroulement n’est qu’une conséquence de l’activité de translocation sur la partie simple-brin.
12
13
La double hélice d’ADN constitue une structure stable in vivo bien adaptée à la transmission et à la sauvegarde de l’information génétique. Cependant, de nombreux aspects du métabolisme de l’ADN demandent l’accès à une forme simple-brin dans laquelle les bases nucléotidiques sont accessibles. C’est pourquoi le déroulement des molécules d’acide nucléique est un mécanisme nécessaire à tous les processus génétiques fondamentaux comme la réplication, la réparation, la recombinaison, la transcription, la synthèse des ribosomes, le remodelage de la chromatine, la maturation des ARN ou encore les processus d’exports nucléaires. Dans chacun de ces processus, le déroulement des duplex d’ADN et/ou ARN est catalysé par des enzymes connues sous le nom d’hélicase.
I
Les hélicases
A)
Généralités
Les hélicases sont essentielles dans tous les aspects du métabolisme des acides nucléiques. Pour cette raison, ce sont des protéines ubiquitaires identifiées chez les procaryotes, les eucaryotes, les bactériophages et les virus. Ces enzymes fonctionnent en déstabilisant les liaisons hydrogènes entre les paires de bases nucléotidiques complémentaires dans une réaction couplée à la liaison et l’hydrolyse de nucléoside 5’-triphosphate (NTP). Elles sont ainsi considérées comme des « protéines motrices » car elles transduisent une énergie chimique en une force mécanique (Caruthers and McKay, 2002; Lohman et al., 2008; Singleton et al., 2007; Tuteja and Tuteja, 2004a, b).
Pour catalyser le déroulement de duplex ADN et/ou ARN, une hélicase passe à travers une série d’états conformationnels et énergétiques, dirigée par la liaison au NTP, son hydrolyse et le relargage subséquent des produits de la réaction (nucléoside 5’-diphosphate + phosphate inorganique = NDP + PO42-). Par conséquent, comprendre la mécanistique
moléculaire de déroulement des acides nucléiques par les hélicases, requiert des informations sur le couplage de l’hydrolyse du nucléotide à la translocation de l’enzyme le long de son substrat, mais également sur les modifications conformationnelles acide nucléique/protéine qui ont lieu pendant le déroulement. Une telle connaissance nécessite des études quantitatives sur la thermodynamique et la cinétique des réactions enzymatiques associées à des approches structurales.
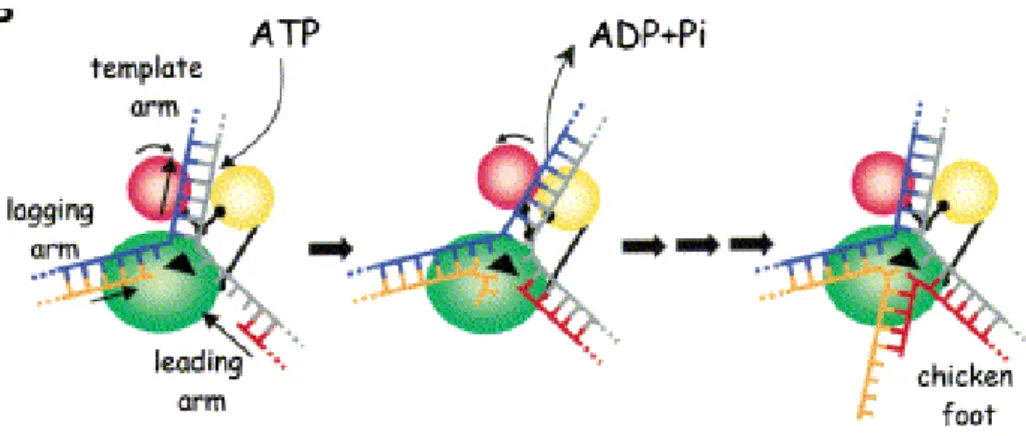
Figure 1 : Classification des hélicases et des translocases. (a) Le nom d'un membre de chacune des six familles est donnée à titre d’exemple entre parenthèses. Les deux domaines structuralement conservés « RecA-like » (en bleu et rouge) ainsi que la position des séquences signatures à l’intérieur de ce noyau protéique sont indiqués pour chaque classe d’hélicase. La position précise de ces motifs est représentative de la famille tout entière. Les motifs colorés en jaune représentent les séquences signatures conservées présentes chez toutes les hélicases. La position et la fonction des domaines accessoires sont spécifiques de la protéine donnée en exemple. Ils existent une grande variété pour ces domaines accessoires à l’intérieur d’une même famille. (b) et (c) Structure représentative du noyau protéique. Les éléments structuraux universels impliqués dans la liaison et l’hydrolyse du NTP, et le couplage de ces activités au changement conformationnel de la protéine sont indiqués en jaune. (b) Les enzymes de la famille SF1 et SF2 possèdent un noyau catalytique monomérique formé d’une répétition en tandem de deux modules « RecA-like » (notés N-core et C-core). Le NTP (en noir) est lié à l’interface des deux modules « RecA-like ». Les motifs 1 (Walker A) et 2 (Walker B) sont localisés sur le domaine N-core. Le motif 6, contenant le résidu R « arginine finger », est situéé sur le domaine C-core. La structure représentative donnée est celle de l’hélicase PcrA de la famille SF1. Ces deux modules répétés constituent une structure minimale pour former un moteur protéique. (c) Les membres de la famille SF3, 4, 5 et 6 contiennent un noyau protéique composé de six modules individuels RecA-like (en rouge) assemblés en anneau. De la même façon que les enzymes SF1 et SF2, les motifs conservés Walker A et Walker B sont localisés à l’opposé du résidu « arginine finger » R. La structure représentative donnée est celle de la protéine T7 du gène 4 de la famille SF4. (d) La nomenclature pour les sous-familles est basée sur la direction de la translocation (3’-5’, de type A ou 5’-3’, de type B) et si l’acide nucléique est simple-brin (!) ou double brin ("). Le brin le long duquel la translocation a lieu est indiquée en mauve.
14
1)
Des motifs différents
En 1993, Gorbalenya & Koonin proposent pour la première fois une classification des hélicases et décrivent cinq superfamilles (SF1, SF2, SF3, SF4 et SF5) d’hélicases « putatives » sur la base de l’analyse de leur séquence, et plus précisément sur la présence de séquences signatures appelées motifs hélicases (Gorbalenya and Koonin, 1993). On sait aujourd’hui que ces motifs sont en réalité caractéristiques de la superfamille d’enzymes appelées translocases, protéines capables de se déplacer le long d’un acide nucléique. Or, la translocation de ces moteurs protéiques le long d’un acide nucléique n’est pas toujours accompagnée d’une activité hélicase (séparation physique d’un duplex en deux composants simple-brins). En d’autres termes, toutes les hélicases sont des translocases, mais l’inverse n’est pas vrai.
Par ailleurs, de nombreuses protéines de la famille AAA+ (ATPase Associées à diverses Activités cellulaires) ont été décrites comme des moteurs protéiques spécifiques des acides nucléiques. Dans la classification présentée ici, ces dernières ont été regroupées dans une sixième superfamille (SF6). Les six superfamilles et leurs motifs associés sont représentés Figure 1a.
Pour les deux groupes SF1 et SF2, sept séquences signatures ont été originalement décrites (motifs notés 1, 1a, 2, 3, 4, 5, et 6, Figure 1a) (Gorbalenya and Koonin, 1993). Ces sept motifs originaux ne sont pas tous comparables entre les membres de SF1 et SF2. Par exemple, les motifs 3 des enzymes SF1 et SF2 ne partagent pas de séquences similaires et sont situés à différents endroits de la protéine. Pour ces deux premières familles, trois nouveaux motifs ont été caractérisés depuis la première classification (Figure 1a), le motif TxGx (Schmid and Linder, 1992), le motif 4a (Korolev et al., 1998) et le motif Q (non-représenté Figure 1a, situé à l’extrémité N-terminale des protéines qui le possèdent en amont du motif 1 (Tanner et al., 2003)). Notez qu’il existe aujourd’hui dans la littérature une grande confusion sur la fonction de ces différents motifs et leurs équivalents potentiels dans les quatre autres familles. Ces notions seront discutées de manière détaillée plus bas pour chaque famille.
2)
Une structure conservée, les modules « RecA-like »/AAA
+(a)
Le module « RecA-like »Figure 2 : Représentation en diagramme topologique du module « RecA-like » des protéines RecA et SV40 (SF3). Les hélices ! sont représentées par des rectangles et les feuillets " par des flèches pleines. (RecA) Le module « RecA-like » est composé des 5 feuillets " colorés en jaune et des 6 hélices ! colorées en bleu. La flèche verte indique l’extrémité N-Terminal du module RecA-like. Les feuillets " représentés en gris sont spécifiques de la protéine RecA. (SFIII) L’hélice ! représentée en bleu foncé et les 5 hélices ! situées à l’extrémité C-terminal sont spécifiques du module AAA+
. Les feuillets " représentés en mauve sont spécifiques d’une branche de la superfamille AAA+ appelée PS1BH (« pre-sensor 1 "-hairpin » )de la famille SFIII. Ils forment une structure en épingle à cheveux responsable du couplage de la liaison et l’hydrolyse de l’ATP à la translocation et au déroulement de l’enzyme (voir section III-A-1).
15
et/ou la position de ces différents motifs, il existe pour l’ensemble de ces protéines une structure tridimensionnelle conservée et par conséquent une mécanistique moléculaire partagée. En effet, les motifs cités au-dessus se retrouvent à l’intérieur d’un module catalytique dont la topologie est conservée pour l’ensemble des enzymes des six familles (Figure 1b). Le domaine ainsi formé est appelé module « RecA-like » en référence à la protéine bactérienne RecA, qui fut la première enzyme à activité ATPasique dont la structure tridimensionnelle a été determinée (Story and Steitz, 1992; Story et al., 1992). Chez la bactérie, RecA est impliquée dans les étapes précoces de la recombinaison homologue (RH). Pendant ce mécanisme, RecA est capable (1) de s’oligomériser en forme de filament hélicoïdal le long d’un ADN simple-brin, (2) d’induire la recherche d’une matrice bicaténaire possédant une séquence similaire, et (3) d’échanger les brins entre les deux molécules d’ADN de manière ATP-dépendante.
Chez les hélicases, les modules « RecA-like », composés de 5 feuillets " et de 5 hélices !, sont situés en tandem, soit sur une même chaîne polypeptidique (familles SF1 et SF2, Figures 1a et b), soit sur des sous-unités différentes arrangées en anneau hexamérique (familles SF3, SF4, SF5 et SF6, Figures 1a-c). Ils convertissent une énergie chimique en une force mécanique en couplant la liaison et l’hydrolyse du NTP aux changements conformationnels de la protéine. En effet, le module catalytique « RecA-like » contient les résidus impliqués dans la liaison et l’hydrolyse du NTP équivalents aux boîtes de Walker A et B (Walker et al., 1982) de toutes les protéines à activité ATPase (notés motifs 1 et 2 respectivement pour les familles SF1, SF2 et SF5 ; A et B pour les familles SF3 et SF6 ; H1 et H2 pour la famille SF4 ; Figure 1a) et un « doigt arginine » qui joue un rôle clés dans le couplage énergétique (Scheffzek et al., 1997), inclus dans le domaine 6 pour les familles SF1 et SF2, notés R pour les familles SF3, SF4, SF5 et SF6 (Figure 1a).
(b)
Le domaine AAA+, un module « RecA-like » allongéPour les hélicases hexamériques de la famille SF3 et SF6, le module « RecA-like » est allongé par la présence de 4 hélices ! supplémentaires situées à l’extrémité C-terminale du module « RecA-like » classique (Figure 2). Ce module « RecA-like » allongé est caractérisé par ailleurs par une orientation spécifique de la première hélice !, adjacente à l’extrémité N-terminale de la boîte de Walker A. Cette structure particulière du module « RecA-like » est spécifique des protéines AAA+ (ATPases Associées à différentes Activités cellulaires), une vaste classe d’enzyme comprenant des membres aux fonctions aussi variées que des protéases, des activateurs de transcription, des régulateurs d’enzymes du métabolisme et des
16
hélicases (Ammelburg et al., 2006; Frickey and Lupas, 2004; Iyer et al., 2004).
Le module AAA+ se caractérise par ailleurs par la présence de deux motifs très conservés appelés senseur 1 et 2. Ces deux motifs lient le phosphate ! de l’ATP et sont requis pour l’activité ATPase des protéines AAA+. En référence à ce module « RecA-like » particulier appelé module AAA+, les hélicases hexamériques SF3 et SF6 sont appelées hélicases AAA+.
3)
Des propriétés biochimiques spécifiques
Les hélicases et les translocases sont classées en sous-famille sur la base de leurs propriétés biochimiques.
(a)
Acide nucléique préférentielOn distingue des enzymes spécifiques de l’ADN, de l’ARN ou encore d’hybrides ADN/ARN. Il n’y a aucune règle concernant le substrat acide nucléique préféré et la famille d’appartenance de la protéine.
(b)
Etat oligomérique de la forme activeL’état oligomérique des protéines des familles SF1 et SF2 a fait l’objet d’un débat vigoureux et elles sont considérées comme agissant principalement à l’état monomérique ou dimérique. Plusieurs études ont cependant montré que l’état oligomérique « minimum » requis pour fournir une activité motrice à la protéine était la forme monomérique (Brendza et al., 2005; Nanduri et al., 2002). Pour les hélicases SF1 et SF2, le noyau catalytique n’est constitué que d’une seule chaîne polypeptidique contenant deux modules « RecA-like » (Figure 1b). Les autres superfamilles SF3, SF4, SF5 et SF6 sont des protéines qui s’assemblent en anneau hexamérique (ou dodécamérique) composés de six (ou douze) modules « RecA-like » individuels.
(c)
Caractéristique de l’activité translocaseLa translocation peut se faire de 3’ vers 5’ (noté 3’-5’ dans ce manuscrit, appelé de type A, Figure 1d) ou de 5’ vers 3’ (noté 5’-3’, appelé de type B, Figure 1d). Les membres des familles SF1, SF2 et SF6 peuvent être de type A ou B, alors que pour la famille SF3, il
17
n’a été caractérisé jusqu’à maintenant que des hélicases de type A, et pour les familles SF4 et SF5, uniquement des hélicases de type B.
Enfin, on distingue parmi ces enzymes, celles qui transloquent le long d’un acide nucléique simple-brin (appelé de type ", Figure 1d) de celles qui se déplacent le long d’un duplex double-brin (appelé de type #, Figure 1d). Mais pour la plupart d’entre elles, cette dernière distinction n’est pas claire. Les enzymes caractérisées de type # sont souvent référencées comme des translocases dans la littérature récente, dans le but de les distinguer des enzymes qui possèdent une « vraie » activité hélicase (séparation physique d’un duplex en deux composants simple-brin). À ce jour, toutes les enzymes SF1 sont de type " alors que la famille SF2 contient les deux classes de protéines. Les hélicases en anneau peuvent être de type " ou#.
4)
Des modules régulateurs
Un dernier élément dans la complexité de ces protéines motrices est la présence de domaines accessoires spécifiques de chaque membre d’une famille (Figure 1a). Leur architecture est très diverse et ils peuvent être situés aussi bien en partie N-terminale, à l’intérieur du module « RecA-like » ou à l’extrémité C-terminale de celui-ci (Figure 1a). Ces domaines modulent l’activité enzymatique en ciblant la protéine sur un substrat spécifique, ou en modifiant les fonctions catalytiques de celle-ci. Ainsi en associant des domaines accessoires modulateurs spécifiques à un noyau catalytique structuralement très conservé, la nature a créé une gamme de moteurs protéiques très diversifiée pour les acides nucléiques.
II
Hélicases et translocases non-hexamériques
A)
La superfamille SF1
La famille SF1 est probablement la classe la mieux caractérisée au niveau structural. Tous les membres caractérisés à ce jour sont des hélicases qui se déplacent spécifiquement sur l’ADN simple-brin (type "), mais peuvent transloquer dans les deux directions (type A ou B).
1)
Les hélicases SF1A
(a)
Données structuralesFigure 3 : Structure et motifs de la famille SF1. (a) Représentation des domaines structuraux d’une hélicase typique de la famille SF1, la protéine PcrA. Les deux modules RecA-like (notés N-core et C-core) sont colorés en rouge et bleu, respectivement, et les insertions à l’intérieur de ces domaines (1B et 2B) sont colorés en bleu pâle et vert. (b) Structure cristallographique d’un complexe ternaire composé de la protéine PcrA lié à un substrat ADN comprenant une partie duplex et une partie 3’ sortante, et de l’ADP. Les domaines protéiques sont colorés comme en (a). (c) Structure identique à celle présentée en (b) mais cette fois-ci sont colorés les motifs caractéristiques de la famille SF1. (d) Représentation en encombrement stérique utilisant les mêmes couleurs qu’en (c). Notez que la queue d’ADN simple-brin passe à travers un large sillon à l’intérieur de la protéine. Les surfaces sont représentées par transparence afin de révéler le chemin emprunté par l’ADN simple-brin.
18
UvrD et PcrA. Des études cristallographiques ont révélé une conformation monomérique commune pour ces trois protéines (Korolev et al., 1997; Lee and Yang, 2006; Subramanya et al., 1996) dans laquelle chaque monomère est composé de deux modules « RecA-like », eux-mêmes découpés en deux sous-domaines (Figures 3a-b). Dans cette structure, le site de liaison à l’ATP se fait via des résidus appartenant aux motifs 1 et 4, situés à l’interface des deux modules « RecA-like » (plus précisément entre les sous-domaines 1A et 2A, Figure 3a).
• L’hélicase Rep
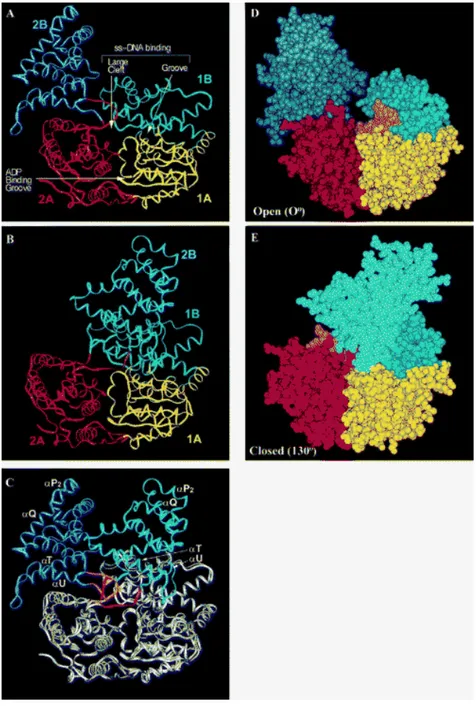
Concernant la protéine Rep, la structure a été obtenue en présence d’ADP et d’un ADN simple-brin de 16 bases (Korolev et al., 1997). Dans ces conditions, le cristal est composé d’une molécule d’ADN simple-brin liée à un dimère de Rep. Cette étude, en plus de confirmer le rôle des motifs hélicases 1 et 4 dans la liaison au nucléotide, montre que les motifs 1a, 3 et 5 sont impliqués dans la liaison à l’acide nucléique, alors que les motifs 2 et 6 fonctionnent en couplant les effets allostériques des nucléotides sur la liaison à l’ADN. Les auteurs ont par ailleurs révélé une asymétrie dans la structure des deux monomères de Rep particulièrement intéressante. En effet, alors qu’un monomère de Rep est lié à l’extrémité 5’ de la molécule d’ADN simple-brin dans une conformation ouverte parfaitement symétrique à la structure du monomère de PcrA présentée Figure 3, le second monomère de Rep lié à l’extrémité 3’ de l’oligonucléotide est dans une structure fermée (Figure 4). La transition entre les deux conformations se fait par une rotation du domaine 2B de 130° qui dans la structure fermée vient refermer le sillon situé entre les domaines 1A et 1B, sillon qui correspond au site de liaison à l’ADN simple-brin.
• L’hélicase PcrA
Plus tard, l’équipe de Wigley a caractérisé la structure de PcrA complexée à un substrat ADN contenant une région duplex de 10 paires de bases et une extrémité simple-brin 3’ sortante de 7 bases. La structure a été obtenue en présence, soit d’un ion sulfate, soit d’un analogue non-hydrolysable de l’ATP (Velankar et al., 1999). Dans le premier complexe, l’ion sulfate est situé à la position normalement occupée par l’ion phosphate après l’hydrolyse de l’ATP, représentant ainsi un « complexe produit » alors que le second mime un « complexe substrat » (l’analogue de l’ATP n’étant pas hydrolysable, il est maintenu dans la poche catalytique). La comparaison de ces deux structures révèle un changement conformationnel induit par le ligand. Le nucléotide entraîne une fermeture du site situé entre les domaines 1A et 2A autour du nucléotide. En parallèle, on observe un mouvement des domaines 1B et 2B
Figure 4 : Les deux formes ouvertes et fermées du monomère Rep. Les sous-domaines 1A, 1B, 2A et 2B sont représentés en jaune, vert, rouge et bleu respectivement. Le domaine 2B est coloré en bleu clair foncé pour la forme ouverte et en bleu ciel pour la forme fermée. Dans les représentations en encombrement stérique (d) et (e), l’ADN est représenté en rose. (a) Structure du conformère ouvert de la protéine Rep. (b) Structure du conformère fermé. (c) Superposition des domaines 1A, 1B et 2A de chaque monomère. Les codes couleurs pour le domaine 2B sont identiques à ceux en (a) et (b), alors que les domaines superposés sont en gris. (d) La représentation en encombrement stérique du conformère ouvert est dans la même orientation qu’en (a). Dans cette conformation, une large poche est clairement visible entre les domaines 1B et 2B (notés « large cleft » en (a)). (e) La représentation en encombrement stérique du conformère fermé est dans la même orientation qu’en (b). Dans cette conformation, le domaine 2B effectue une rotation de 130° (c) et referme la poche en venant se caler entre les domaines 1A et 1B, ce qui obstrue le site de liaison à l’ADN simple-brin situé entre ces deux domaines (notés « groove) » en (a)).
19
qui se repositionnent pour former une surface compatible avec la forme et la charge de l’ADN double-brin. Cette surface n’est pas correctement structurée en absence de liaison au nucléotide, résultant en une faible affinité de cette surface pour l’ADN duplex. Par conséquent, les changements conformationnels appropriés pour les domaines 1B et 2B ne peuvent pas prendre place avant la liaison au nucléotide. Ce travail met ainsi en évidence un couplage entre la liaison et l’hydrolyse du nucléotide, et l’affinité de l’enzyme pour son substrat.
Les différences entre les deux complexes « substrats » et « produits » ne sont pas confinés aux changements conformationnels de la protéine, puisqu’on observe une modification importante dans la structure de la queue d’ADN simple-brin. Pour le conformère « substrat », le fragment d’ADN simple-brin qui s’étend à l’intérieur du sillon de liaison à l’ADN situé entre les domaines 1A et 1B (sillon identique à celui observé pour la protéine Rep), est long de 4 bases. Pour le complexe « produit », la région d’ADN simple-brin à l’intérieur de ce sillon s’étend sur 5 bases. La base supplémentaire occupe une poche du domaine 1A qui n’est pas accessible dans le complexe substrat. Cette différence est importante pour le modèle de translocation/déroulement pour la famille SF1A proposée plus bas.
• L’hélicase UvrD
Une étude cristallographique plus récente a résolu la structure de la protéine UvrD complexée avec de l’ADN (contenant également une région duplex et une région simple-brin) et différents intermédiaires d’hydrolyse d’ATP (Lee and Yang, 2006). La structure du monomère UvrD lié à son substrat et les changements conformationnels conséquents à l’hydrolyse du nucléotide sont identiques à ceux décris pour PcrA et Rep. En revanche, les auteurs ont montré que l’étape de déroulement du duplex est distincte de l’étape de translocation de l’enzyme le long de l’ADN simple-brin. Dans un premier temps, la liaison d’une molécule d’ATP entraîne le déroulement d’une paire de base à la jonction double-brin/simple-brin, et dans un second temps, l’hydrolyse du nucléotide provoque la translocation de la protéine UvrD en direction de la paire de base déroulée. En définitive, l’hydrolyse d’une molécule d’ATP entraîne le déroulement d’une paire de base.
(b)
Le modèle « chenille » de déroulement de l’ADNFigure 5 : Les différentes étapes de déroulement d’un duplex ADN pour la famille SF1A. Représentation des différents intermédiaires conformationnels de la protéine UvrD durant un cycle de déroulement d’une paire de base. Le domaine 1A sert de référence dans l’espace sur ce schéma. Les domaines 1A, 1B, 2A et 2B sont colorés en vert, beige, bleu foncé et bleu ciel respectivement. L’ATP et « hélice porte » sont représentés respectivement en rouge et en mauve. Le brin d’ADN sur lequel la protéine se déplace est représentée en jaune, le brin complémentaire en orange. Pour plus de détails, voir dans le texte. De (1) à (5), changements conformationnels conséquents à la liaison à l’ADN simple-brin. Liaison (6), hydrolyse (7) et relargage (8) de l’ATP. En (9), la structure du monomère UvrD est revenue à celle représentée en (5), mais cette fois-ci la protéine a déroulé une paire de base et a transloqué d’une base.
20
(Dillingham et al., 1999; Dillingham et al., 2001; Soultanas et al., 1999; Soultanas et al., 2000) ont permis de proposer un modèle pour le déroulement d’un duplex d’ADN catalysé par les enzymes de la famille SF1A. Un schéma récapitulatif est proposé Figure 5. Le cycle commence avec l’interaction entre l’enzyme et l’ADN simple-brin, qui induit un changement conformationnel de l’enzyme, similaire à celui observé entre les deux conformères de Rep (Korolev et al., 1997). Cette transition structurale consiste en une rotation de 130° du domaine 2B qui va établir une nouvelle surface d’interaction entre d’une part, la surface des domaines 1B et 2B et d’autre part, l’ADN double-brin (Figures 5-1 à 5-5). Ensuite la liaison à l’ATP cause une rotation du domaine 1A de 20° contre le domaine 2A de façon à enfermer le nucléotide entre ces deux domaines (Figures 5-6). En se refermant ainsi, le domaine 1A entraîne avec lui les domaines 1B et 2B. Le domaine 2B fonctionne alors comme une « cale » qui s’appuie sur la partie duplex et provoque le déroulement d’une paire de base à la jonction simple-brin/double-brin.
La région de liaison à l’ADN simple-brin est comprise entre les domaines 2A et 2B. Dans la conformation ATP-lié, la dernière hélice du domaine 2B appelée « hélice porte » (représentée en rose Figure 5) bloque l’ADN simple-brin entre les deux domaines 2A et 2B. L’hydrolyse de l’ATP (Figure 5-7) provoque une ouverture de cette hélice porte permettant à l’ADN simple-brin de transloquer à l’intérieur de son domaine d’une base en direction de la paire de base déroulée. Le relargage des produits de la dégradation ADP + Pi (Figure 5-8) provoque le dernier changement conformationnel de l’enzyme que revient dans une structure ATP-libre (comparer Figure 5-5 et Figure 5-9).
Ce modèle actif de déroulement d’un duplex ADN (voir avant-propos) est appelé dans la littérature « modèle chenille » en référence au mode de déplacement de l’animal le long d’une branche. Ce modèle est très similaire pour l’ensemble des enzymes appartenant aux familles SF1 et SF2. Il implique que l’enzyme procède au déroulement d’une paire de base à la fois, avec un couplage stoechiométrique d’un ATP hydrolysé par paire de base déroulée. Ce couplage stoechiométrique est en accord avec celui observé pour la translocation du monomère de PcrA (Dillingham et al., 2000) ou du monomère de UvrD (Tomko et al., 2007) le long d’un ADN simple-brin.
(c)
Implication sur l’état oligomérique de la forme active des hélicases SF1A Le modèle « chenille » implique que l’état monomérique soit suffisant pour produire un moteur protéique actif. Curieusement, alors qu’un mutant de Rep déplété du domaine 2BFigure 6 : Structure et mécanisme d’action du complexe RecBCD. (A) Structure de la sous-unité RecD. Les domaines 2 et 3 sont équivalents aux domaines 1A et 2A des enzymes de la famille SF1A. (B) Structure du complexe RecBCD. L’ADN lié est représenté en rose, les sous-unité RecB, RecC et RecD sont représentés en orange, bleu et vert respectivement. (C) Vue en coupe du complexe recBCD en représentation encombrement stérique, mettant en évidence les canaux empruntés par l’ADN. Les sous-domaines des trois sous-unités sont indiqués. Le code couleur est le même qu’en (B). (D) Modèle illustrant la translocation des deux moteurs protéiques du complexe RecBCD et les conséquences du passage par la séquence !. Voir le texte pour les détails. Le complexe RecBCD est un complexe hélicase bipolaire avec deux sous-unités transloquant sur les deux brins opposés de la molécule d’ADN. Avant la rencontre avec la séquence !, RecD est le moteur protéique du complexe. Le mouvement des deux protéines motrices à des vitesses différentes génère une boucle simple-brin en avant de la sous-unité RecB. Après le passage sur la séquence !, le moteur RecD est inhibé et la protéine RecB devient le moteur qui conduit le complexe. Notez l’apparition d’une extrémité 3’ sortante en (6). Cette extrémité est essentielle pour l’initiation de la recombinaison homologue. Les flèches indiquent la direction et les vitesses de translocation relatives des sous-unités motrices.
(A), (B) et (C) D’après Singleton et al. (2004) Nature. 432, 187-193. (D) D’après Spies et al. (2007) Cell. 131, 694-705.
21
est un moteur protéique actif à l’état monomérique (Brendza et al., 2005), la protéine pleine taille requiert une oligomérisation pour induire une activité hélicase (Cheng et al., 2001), suggérant un rôle régulateur de ce domaine. Les monomères de PcrA et de UvrD sont capables de transloquer le long d’un ADN simple-brin (Dillingham et al., 2000; Fischer et al., 2004). Par conséquent, la forme enzymatique la plus probable agissant activement à une jonction simple-brin/double-brin est certainement une forme monomérique. Ceci n’exclue pas que plusieurs monomères peuvent coopérer pour induire une activité hélicase plus efficace dans des conditions in vitro, comme cela a été montré pour la l’hélicase UvrD (Ali et al., 1999).
2)
Les hélicases SF1B
Les enzymes de cette famille les plus connues sont l’hélicase bactérienne RecD et l’hélicase du bactériophage T4 Dda. Il existe des membres de cette famille chez les eucaryotes comme les protéines Pif1 et Rm3 (Ivessa et al., 2002) mais elles restent très mal décrites.
(a)
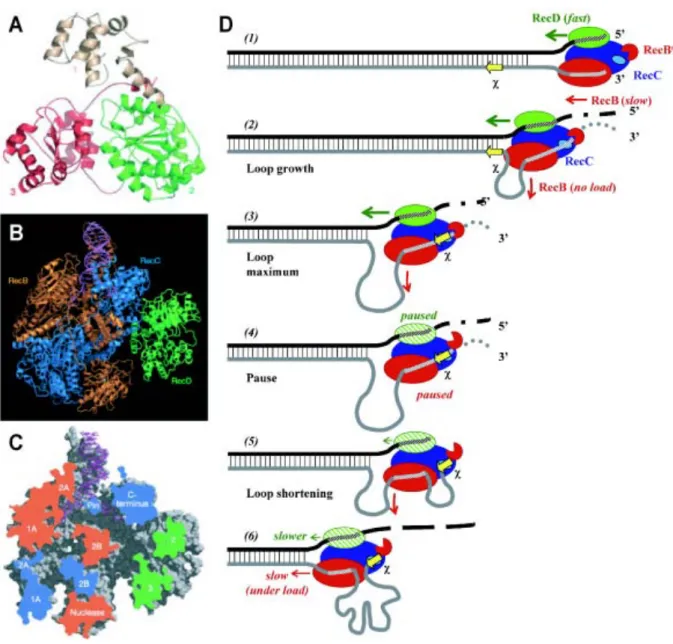
L’hélicase RecD au sein du complexe RecBCD.Les seules données structurales disponibles pour la famille SF1B viennent de la sous-unité RecD dont la structure a été obtenue au sein du complexe RecBCD (Singleton et al., 2004). Les caractéristiques biochimiques de la protéine RecD sont bien décrites mais toujours au sein du complexe RecBCD, ce qui rend impossible une analyse mécanistique de la protéine sans ces partenaires.
L’hélicase RecD agit ainsi à l’état monomérique mais dans le contexte d’un hétérotrimère (Nanduri et al., 2002; Singleton et al., 2004). La structure de RecD révèle que les enzymes SF1B possèdent, comme les hélicases SF1A, deux modules « RecA-like » avec un domaine de liaison à l’ATP situé à l’interface de ces deux modules (Figure 6A). La protéine RecD possède par ailleurs un domaine N-terminal supplémentaire, qui forme l’interface avec la sous-unité RecC et qui n’est pas conservé dans la protéine Dda. Enfin, la structure du complexe RecBCD montre que la protéine RecD ne lie pas l’ADN double-brin au niveau de la jonction simple-brin/double-brin, cette liaison étant assurée par la sous-unité RecB (Figures 6B et 6C).
Chez la bactérie, le complexe RecBCD protéique initie le processus de réparation par RH en convertissant les cassures d’ADN bicaténaires en extrémités simple-brin 3’ sortantes.
22
Pour cela, l’hétérotrimère s’assemble sur des bouts francs d’ADN double-brin. La sous-unité RecB reconnaît l’extrémité 3’ d’ADN tandis que la protéine RecD se lie à l’extrémité 5’. La molécule d’ADN duplex est déroulée par l’association de deux activités hélicases de polarité inverse. Alors que RecB déroule l’ADN dans le sens 3’-5’, RecD le déroule dans le sens opposé 5’-3’ (Dillingham et al., 2003). Les molécules simple-brin produites sont digérées en aval de la translocation par la sous-unité RecB qui possède également une activité nucléase. Ces trois activités sont étroitement régulées par une séquence nucléotidique appelée Chi (ou #, pour « cross-over hot-spot instigator »). Cette séquence se retrouve dans le génome bactérien dans des zones de RH très élevée.
Le complexe RecBCD transloque à une vitesse de l’ordre de 600 paires de bases par seconde. À la rencontre d’une séquence #, il effectue une pause de quelques secondes qui se traduit par une atténuation de son activité nucléase, la translocation redémarre ensuite mais à une vitesse plus lente de l’ordre de 300 paires de bases par seconde (Dixon and Kowalczykowski, 1993; Spies et al., 2003). Récemment, l’équipe de Kowalczykowski a montré comment cette séquence # régule les activités hélicases des deux moteurs RecB et RecD (Spies et al., 2007), expliquant ainsi la différence de vitesse observée. Les auteurs ont montré que la protéine RecD transloque deux fois plus rapidement que la protéine RecB, et donc que la sous-unité RecD est le moteur protéique du complexe RecBCD. Ceci est vrai jusqu’à la rencontre avec la séquence #. Après le passage de cette séquence, l’activité motrice de déroulement du duplex est relayée à la sous-unité RecB (Figure 6D).
Cette étude permet ainsi de mieux comprendre comment une étroite régulation des différentes activités du complexe RecBCD par la séquence #, conduit à la génération d’une extrémité 3’ sortante nécessaire pour initier le processus de RH. Cependant, ce travail n’aborde pas les différents changements conformationnels induits par la séquence # sur les deux moteurs protéiques RecB et RecD au sein du complexe RecBCD.
(b)
La protéine DdaPlusieurs études cinétiques mettent en évidence que la protéine Dda est capable de dérouler un duplex ADN à l’état oligomérique (Morris et al., 2001; Nanduri et al., 2002). À l’inverse, des travaux plus récents ont clairement révélé une coopérativité de plusieurs monomères de Dda dans l’activité de déroulement d’un duplex ou dans sa capacité à rompre une liaison biotine/streptavidine (Byrd and Raney, 2004, 2005, 2006).
Pour expliquer ces résultats, les auteurs ont proposé, pour la protéine Dda, un mécanisme d’action à l’échelle moléculaire qui est une variation du modèle « chenille »
23
proposé pour la famille SF1A (Figure 5). Dans ce modèle, les monomères fonctionnent indépendamment durant la translocation le long de l’ADN simple-brin comme il a été proposé dans le modèle « chenille ». Par contre, lorsqu’un monomère rencontre un obstacle comme un ADN duplex ou une protéine liée à la chaîne monocaténaire, la probabilité de passer à travers cet obstacle augmente proportionnellement avec le nombre de monomères présents sur le substrat simple-brin. La coopérativité serait alors associée à des interactions transitoires entre les monomères liés au même substrat d’acide nucléique, et transloquant dans la même direction. Des données structurales font pour l’instant défaut pour mettre en évidence une telle coopération entre deux monomères de Dda.
B)
La superfamille SF2
Les enzymes de la superfamille SF2 sont une très vaste famille d’hélicase impliquées dans des processus cellulaires très variés. La famille SF2 est découpée en plusieurs sous-familles très largement étudiées, comme les hélicases ARN à boîte DEAD (leur nom fait référence à la séquence de leur boîte de Walker A) (Cordin et al., 2006; Rocak and Linder, 2004), les hélicases ADN RecQ-like (Bennett and Keck, 2004) et les enzymes de la famille Swi2/Snf2 (Flaus et al., 2006; Flaus and Owen-Hughes, 2004). La fonction biochimique du coeur catalytique des enzymes SF2 est probablement la translocation ATP-dépendante unidirectionnelle le long d’un acide nucléique simple-brin ou double-brin. Cependant, plusieurs membres agissent en appliquant une force mécanique non-processive qui altère la conformation de la protéine et de l’acide nucléique de manière ATP-dépendante.
Pendant ces dix dernières années, un nombre considérable de structure tridimensionnelle a été publié pour les hélicases SF2 (à ce jour, 12 structures sont disponibles pour la seule sous-famille des ARN hélicases). Le but de ce manuscrit n’étant pas de décrire de manière exhaustive l’ensemble de ces cristaux, ne seront donc détaillés à titre d’exemple, que certains de ces travaux apportant des données sur la mécanistique d’action à l’échelle moléculaire des enzymes SF2.
1)
La famille SF2!
(a)
Structure de la protéine virale NS3La protéine du virus de l’hépatite C NS3 est probablement l’enzyme SF2! la mieux caractérisée. La protéine NS3 peut dérouler des duplex ADN ou ARN de longue taille de
Figure 7 : Structure et mécanisme des hélicases SF2!. Exemple de la protéine du virus de l’hépatite C NS3. (a) Structure primaire indiquant la position et les fonctions des quatre domaines. Notez que le domaine protéase est absent de la structure présentée. (b) Complexe ADN/NS3 représenté avec le même code couleur qu’en (a), l’oligonucléotide est représenté en rose. L’ion sulfate indique la position de la poche de liaison au nucléotide situé à l’interface des domaines RecA-like. (c) Représentation des huit motifs conservés, regroupés autour de la poche de liaison au nucléotide. Les résidus clés, qui ne font pas partie des motifs hélicases, mais qui sont impliqués dans le mécanisme de translocation sont indiqués (W501 et V432, notés W et V respectivement) et colorés en noir. (d) Représentation en encombrement stérique utilisant le même code couleur qu’un (c).
24
manière processive mais requiert une extrémité monocaténaire 3’ sortante (Pang et al., 2002). Bien que plusieurs observations suggèrent une interaction fonctionnelle entre plusieurs monomères de NS3 (Levin et al., 2004; Tackett et al., 2005), une étude en molécule unique indique que la forme monomérique de NS3 est un moteur protéique actif pour dérouler un acide nucléique (Dumont et al., 2006).
La structure de l’hélicase NS3 a été obtenue seule ou complexée à un ADN simple-brin (Kim et al., 1998). Le coeur catalytique est composé des deux modules « RecA-like » (notés N- et C-core Figure 7a) similaires à ceux observés pour les hélicases SF1, associés à un domaine C-terminal appelé domaine 3, jouant un rôle dans les interactions NS3/NS3 (Mackintosh et al., 2006). Le domaine N-terminal de la protéine, absent de la structure, possède une activité protéase requise pour la maturation de la polyprotéine virale.
La poche de liaison au nucléotide est située à l’interface des modules « RecA-like » (comme pour la famille SF1), alors que le site de liaison à l’ADN est situé dans une seconde poche perpendiculaire à la première, à l’interface entre le coeur catalytique et le domaine 3 non-conservé (Figures 7b, c et d). L’ensemble des motifs hélicases caractéristiques de la famille SF2 se retrouve à la surface de ces deux poches (Figure 7d). Le cœur catalytique adopte une conformation ouverte avec six bases d’ADN liés le long de sa surface supérieure à une position similaire à celle décrite pour les enzymes SF1. Cependant, ces bases ne sont pas dans une conformation étirée comme cela a été observé pour la protéine PcrA. En effet, la protéine NS3 lie la chaîne d’ADN monocaténaire principalement via des interactions avec le squelette phosphate, l’ADN simple-brin se retrouve ainsi dans une structure en pseudo forme B. Les résidus impliqués dans cette liaison sont les motifs 1a, TxGx, 4 et 5. Des contacts additionnels impliquent deux résidus V432 et W501 qui prennent en « sandwich » cinq bases à l’intérieur du cœur protéique (Figure 7d). L’importance de ces deux résidus, qui ne font pas partie des motifs hélicases canoniques, a été montrée par mutagénése dirigée (Kim et al., 2003; Paolini et al., 2000; Tai et al., 2001).
(b)
Modèle par mouvement brownien de déroulement d’un acide nucléique double-brinLe modèle de translocation par mouvement brownien a été proposé par l’équipe de Patel suite à plusieurs études cinétiques réalisées sur la protéine NS3 (Levin et al., 2005; Levin et al., 2003). Les auteurs ont observé dans un premier temps que l’hélicase NS3 lie l’ADN simple-brin de manière beaucoup plus faible en présence d’un analogue non-hydrolysable de l’ATP. Ensuite, ils ont montré que la liaison de la protéine NS3 à un substrat
25
ADN contenant une partie monocaténaire et une partie bicaténaire, entraîne un désappariement de quelques paires de base à la jonction simple-brin/double-brin.
Dans ce modèle, en absence d’ATP, la protéine lie son substrat fortement. La liaison à l’ATP affaiblit l’interaction entre l’enzyme et l’acide nucléique, conduisant à un bref mouvement brownien aléatoire de l’hélicase. L’hydrolyse de l’ATP conduit à une nouvelle liaison de la protéine sur son substrat avec un biais énergétique dans la direction du mouvement (de 3’vers 5’ pour la protéine NS3).
La structure de NS3 suggère que le changement conformationnel se fait par l’intermédiaire du motif 1 (appartenant au domaine « RecA-like » N-terminal) et le doigt arginine contenu dans le domaine 6 (appartenant au domaine « RecA-like » C-terminal) qui en se liant à l’ATP entrainerait une fermeture des deux domaines « RecA-like » autour du nucléotide. La conséquence de cette transition se traduirait alors par une altération de la distance entre les résidus V432 et W501 et par conséquent, une baisse de l’affinité de l’enzyme pour son substrat simple-brin.
Ce mécanisme est très similaire au modèle « chenille » proposé pour la famille SF1A. La différence majeure est l’absence d’un domaine de liaison double-brin qui permet à l’enzyme de rester « agripper » à son substrat pendant le changement conformationnel induit par l’hydrolyse de l’ATP (assuré par le domaine 2B pour les protéines PcrA, Rep et UvrD, Figures 3, 4 et 5 respectivement). Cependant, l’existence de ce domaine (responsable du déroulement « actif » de l’ADN) n’est pas à exclure pour les hélicases SF2!, puisque aucune structure n’a été obtenue en présence d’un substrat contenant à la fois une partie double-brin et une partie simple-brin.
(c)
Implication sur le pas des hélicases SF2!Aucune structure d’une protéine SF2! complexée à un acide nucléique dans les deux conformations ouverte et fermée n’est disponible à ce jour. En absence de cette donnée, il est difficile d’estimer le pas enzymatique associé à ce modèle. Cependant, il paraît assez improbable que ce mécanisme par mouvement brownien conduise à nombre élevé de paires de bases transloquées par ATP hydrolysé. Des études cinétiques sur la protéine NS3 ont pourtant révélé des pas assez grands (9, 11 et 18 paires de bases) ponctués éventuellement de courtes phases de déroulement de 4 paires de bases (Dumont et al., 2006; Levin et al., 2004; Serebrov and Pyle, 2004). Une autre étude a par ailleurs montré que l’hélicase SF2! NPH-II déroule l’ADN avec un pas de 6 paires de bases (Jankowsky et al., 2000). Ces valeurs sont difficilement conciliables entre elles, avec la structure de NS3 et avec le modèle par
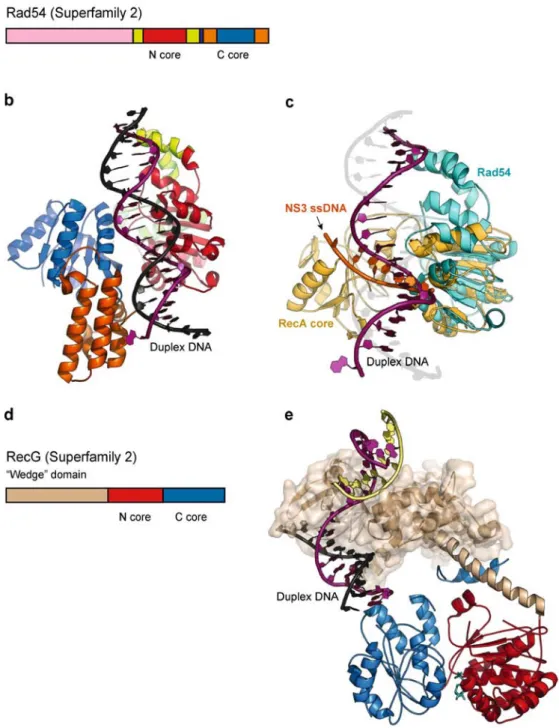
Figure 8 : Structures des enzymes SF2!. (a) Structure primaire de la protéine de Sulfolbus solfataricus Rad54. Une large portion N-terminal de la protéine (en rose) n’a pas été
déterminée dans les structures représentées en (b) et (c). Le module nommé RecA-like N-terminal (ou C-N-terminal) dans le texte est noté N-core (ou C-core). (b) Structure du complexe ADN duplex/Rad54 représentée avec le même code couleur qu’en (a). Le brin d’ADN représenté en mauve est le brin appelé 3’-5’dans le texte. C’est par ce brin que se fait la majeure partie des interactions avec le module RecA-like N-terminal de la protéine. (c) Superposition des domaines RecA-like, montrant la similarité des chemins choisis par l’ADN simple-brin lié à la protéine NS3 (en orange) et le brin 3’-5’ (en mauve) lié à la protéine Rad54 (en bleu ciel). (d) Structure primaire de la protéine de Thermotoga maritima RecG. (e) Structure du complexe ADN/RecG, révélant comment le domaine accessoire (noté « wedge ») agit comme un facteur spécifique pour les jonctions d’ADN à trois branches. Notez que l’ADN duplex représenté en mauve et noir et dirigé vers le cœur protéique de RecG dans une orientation similaire à celle observée pour le complexe ADN/Rad54.