UNIVERSITE DES SCIENCES PAUL SABATIER -TOULOUSE III
UFR SVT - Ecole doctorale : Biologie-Santé-Biotechnologies
Présentée en vue de l’obtention du grade de
Docteur de l’Université Paul Sabatier
Spécialité Cancérologie
GSK3
β : clé de voûte des mécanismes de résistance
au stress contrôlés par les intégrines dans les
leucémies aiguës myéloïdes
par Fabienne DE TONI
Soutenue le 14 novembre 2007 devant le jury composé de :
Pr. Catherine Muller IPBS/CNRS (Toulouse) Présidente Dr. Corinne Albigès-Rizo INSERM U823 (Grenoble) Rapporteur Dr. Fawzia Louache INSERM U362 (Villejuif) Rapporteur Dr. Jean-Antoine Girault INSERM U536 (Paris) Examinateur Dr. Claire Racaud-Sultan INSERM U563 (Toulouse) Directrice de thèse
INSERM U563 - CPTP - Equipe B. Payrastre « Phosphoinositides et signalisation dans les cellules hématopoïétiques », CHU Purpan, Toulouse, FRANCE
A Olivier, mon compagnon et futur mari,
A mes parents,
A mon parrain regretté,
A mes amis,
A mes grands-parents.
A tous ceux qui liront ou feuillèteront cette thèse,
qu’elle leur soit agréable à lire
tout comme j’ai eu plaisir à l’écrire.
Tout d’abord, je tiens à remercier les membres du jury qui m’ont fait l’honneur de lire et de juger ce travail ainsi que pour tout le temps qu’ils y ont consacré :
Madame le Professeur Catherine Muller qui m’a fait l’honneur de présider ce jury de thèse et qui m’a encadré lors de mes premiers pas dans la recherche, en stage de maîtrise. Je la remercie également de m’avoir donné le goût de la recherche.
Madame le Docteur Corinne Albigès-Rizo pour avoir accepté le lourd travail de rapporteur et pour les pistes intéressantes qu’elle nous a conseillé pour l’article sur les mécanismes d’activation de GSK3β en aval des intégrines α4β1 et α5β1.
Madame le Docteur Fawzia Louache pour avoir accepté d’évaluer ce travail de thèse en qualité de rapporteur et pour ses questions pertinentes. Je la remercie encore d’avoir lu et jugé ces 1,2 kilos de thèse !!
Monsieur le Docteur Jean-Antoine Girault qui a accepté d’évaluer ce travail de thèse et ce avec beaucoup de gentillesse. Je le remercie également pour ses remarques pertinentes et ses corrections sur la partie neuroscience de GSK3β.
Madame le Docteur Claire Racaud-Sultan pour son encadrement scientifique et ses conseils tout au long de ces trois ans de thèse. Je tiens également à la remercier pour son enthousiasme et sa passion communicante pour la recherche. Je me souviendrai toujours de notre premier entretien où j’ai senti que ce sujet de thèse serait passionnant (je ne me suis pas trompée !!) et qu’elle était une chercheuse passionnée au plus haut point. Merci donc pour tout, pour nos discussions qui m’ont permis de me remettre toujours en question scientifiquement, d’élargir ma réflexion sur mon sujet et de m’ouvrir sur de nombreux horizons scientifiques. Je vous souhaite beaucoup de bonheur scientifique, de nombreux articles, des résultats palpitants et des hypothèses de plus en plus passionnantes !!
travail de thèse, pour leur confiance et leur soutien tout au long de ces trois années.
Ainsi, je remercie vivement tous les membres de l’équipe du docteur Bernard Payrastre.
Bernard pour m’avoir accueilli dans son laboratoire et pour m’avoir fait confiance dès le
début. Merci également pour votre disponibilité et pour votre esprit critique qui m’a permis de m’améliorer et de réaliser ce travail.
Je tiens également à remercier tous les autres statutaires de l’équipe : Stéphane (pour ses conseils de manip à mon arrivée au labo et à ceux pour l’oral de ma thèse), Marie-Pierre (pour sa bonne humeur), Monique (pour ses coups de gueule pro-féministes, merci pour nous toutes), Hélène (pour m’avoir fait découvrir les « vivaspin » et la biotinylation des anticorps, et pour nos discussions scientifiques ou non), et Fred (merci pour tes corrections pour mon projet de thèse en septembre 2004, alors que tu ne me connaissais pas, et pour ton esprit critique tout au long de ma thèse ; je sais que c’était pour mon bien.)
Mathieu pour ces deux années de fous rires et pour nos discussions scientifiques. Merci
d’avoir choisi le sujet sur FAK pour ton DEA. Je me souviendrai longtemps de nos manips à quatre mains, et notamment celles commençant dans l’animalerie le matin et se finissant le soir au FACS. Bref de longues journées à travailler ensemble mais aussi à se marrer pendant les pauses.
Sonia et Audrey pour m’avoir tracé la voie de la fin de thèse, merci pour tous vos conseils et
votre expérience qui m’ont aidé et rassuré dans les moments d’écriture. A très bientôt pour un week-end in England pour faire la fête !! Gros bisous à toutes les deux.
Sophie pour toutes nos pauses clopes et sans clopes (depuis que j’ai arrêté), avec Sonia et
Audrey, qui me permettaient de repartir de plus belle. Je te souhaite une très très belle année 2008, avec une thèse réussie et un mariage magnifique. Plein de calins.
Anne V pour avoir été à mes côtés dans le bureau pendant ces trois années. Je te souhaite bon
courage pour la fin de ta thèse, je sais que tu vas y parvenir et ce de façon brillante.
Loïc pour m’avoir appris les rudiments de la CAM-DR et m’avoir soutenu et aidé pour mon
projet de thèse en septembre 2004. Merci.
Je voudrais remercier tous ceux du bureau des étudiants : Jessica (qui reprend le flambeau GSK3β, je te souhaite bon courage mais aussi du plaisir et de bons résultats. Je pense que GSK3β a bien d’autres surprises à nous réserver !!), Cédric (plus qu’une année et toi aussi tu auras fini ta thèse, bon courage pour la dernière ligne droite), Nathalie (même si tu n’es arrivée qu’à la fin de ma thèse, je voudrais te remercier pour tes conseils pour l’oral de thèse),
bon rétablissement).
Je n’oublie pas les autres étudiants : Damien (je te souhaite une bonne fin de thèse et une bonne année 2008 avec un beau mariage) et Junior (bon courage, je sais que tu vas faire une jolie thèse) du bureau phosphoinositides, Valérie et Marie-Cécile des plaquettes (merci pour votre calme et votre bonne humeur ; bon courage pour vos thèses).
Je tiens également à remercier tous les membres des autres équipes de l’étage, JJF, GL et
GD. Merci pour votre disponibilité (notamment pour les répétitions de l’oral de thèse) et votre
aide lorsque j’étais en manque de certains produits ou que j’avais besoin de conseils sur des techniques ou des concepts. Je ne vous cite pas tous car cela serait trop long, mais vous avez tous ma gratitude. Je voudrais également dire un grand merci à Sophie Allart pour m’avoir appris le fonctionnement du microscope confocal, à Fatima pour ces explications lors de mes manips de FACS et à Joël pour sa gentillesse et sa disponibilité (tu as sauvé beaucoup de mes western blots en me dépannant de marqueur de taille !!).
Enfin, je tiens à remercier toute ma famille et mes amis, qui même s’ils n’ont pas contribué directement à ce travail de thèse, m’ont permis de mener à bien cette thèse grâce à leur patience et à leur amour. Tout d’abord, merci à toi, Olivier, pour m’avoir soutenu tout au long de ma thèse, mais également pendant mon DEA. Je sais que l’écriture de ma thèse n’a pas été un moment très plaisant pour toi et je tiens donc à te remercier pour toute ta patience, pour ta présence réconfortante et ton amour.
Je remercie mes parents, Catherine et Michel, qui m’ont suivi pendant toutes mes études et qui ont vécu avec moi mes succès et mes échecs, merci d’avoir toujours cru en moi, de m’avoir redonné confiance dans les moments les plus difficiles et d’avoir été toujours là. Merci et encore merci à vous deux !!
Je voudrais remercier également la famille d’Olivier, sa mère Nicole, ses grands-parents, Paul
et Honorine, sa soeur Nathalie et son compagnon Nicolas pour leur soutien et leur
gentillesse.
A mes amis qui ont su me soutenir et me réconforter (Roxane, Sophie, Caroll, Christel et
Isa) et qui ont su me laisser « hiberner dans ma tanière » lors de l’écriture de cette thèse
(Caroll, Jo), merci pour votre patience. Un grand merci aussi pour les soirées qui m’ont permis de décompresser et rire (pas de liste, mais un gros merci et de gros bisous à tous)!! Une petite dédicace spéciale pour Sophie et Roxane qui vont bientôt passer leur thèse, la première en Math, et la seconde en bio. Bon courage et plein de bonheur à vous deux !!
Un des problèmes majeurs rencontré actuellement dans le traitement des leucémies aiguës myéloïdes (LAM) est la récurrence de cette pathologie suite à la chimiothérapie, entraînant la rechute de 50 à 70 % des patients atteints de LAM post-chimiothérapie. Cette grave complication est due à un foyer de cellules leucémiques résistantes à la thérapie, qui sont localisées dans la moelle osseuse des os plats, où des éléments du microenvironnement tels que les ligands des intégrines et les morphogènes Wnt, régulent étroitement leurs fonctions de survie, de prolifération et de différenciation. Or, la sérine/thréonine kinase GSK3β (glycogen synthase kinase 3β) est un carrefour majeur des voies de signalisation contrôlant ces fonctions cellulaires. Nous avons recherché quel pourrait être l’impact d’une modulation de son activité dans la résistance au stress des cellules leucémiques, et quels étaient les mécanismes impliqués dans cette régulation.
Après avoir montré que l’adhésion sur fibronectine, via l’engagement des intégrines α4β1 et α5β1, et le blocage de la voie Wnt entraînaient la résistance des cellules de LAM à l’apoptose induite par la daunorubicine, différents inhibiteurs pharmacologiques et une stratégie de siRNA ont permis de mettre en évidence que cette voie de survie nécessitait l’activation de la GSK3β. De plus, dans notre modèle, GSK3β permet l’activation de NF-κB (nuclear factor κB), indépendamment de la voie de
régulation classique par IκB. La recherche des événements proximaux responsables de l’activation de GSK3β, en aval de l’engagement des intégrines α4β1 et α5β1, a permis de démontrer une régulation différentielle de cette kinase par les deux intégrines. En effet, alors que l’intégrine α4β1 régule une forme tyrosine phosphorylée active de GSK3β intranucléaire et associée à la tyrosine kinase Pyk2, l’intégrine α5β1 induit la translocation d’un complexe PP2A/GSK3β vers le compartiment cytoplasme/membrane et la déphosphorylation activatrice sur la sérine 9 de GSK3β. Enfin, nous avons révélé une régulation croisée entre les intégrines et la voie Wnt par le biais d’une sécrétion de l’antagoniste de Wnt, sFRP-1 (secreted Frizzled Related Protein-1), après adhésion des blastes leucémiques sur les ostéoblastes de la niche médullaire.
En conclusion, nos travaux ont permis de mettre à jour un mécanisme original de chimiorésistance des LAM, impliquant deux acteurs majeurs du microenvironnement médullaire, les molécules d’adhésion et les morphogènes Wnt. Ce processus de résistance au stress repose sur l’activation de la kinase GSK3β, dont nous révélons pour la première fois le rôle essentiel dans une voie de survie contrôlée par les intégrines. En perspectives, il reste à démontrer que GSK3β puisse représenter une cible thérapeutique différentielle permettant d’épargner les cellules souches hématopoïétiques normales, dans le traitement des LAM. Désormais, un grand champ d’investigations s’ouvre pour comprendre le rôle de l’activation de GSK3β, en tant qu’élément pro-oncogénique dans différents cancers.
Relapse after chemotherapy is the major problem found in acute myeloid leukemia (AML) and hits 50 to 70 % of AML patients. This serious complication is due to a pool of resistant leukemic cells. These tumoral cells are localized in bone marrow of flat bones, where some microenvironmental elements, such as integrin ligands and Wnt morphogens, tightly control their functions of survival, proliferation and differentiation. Now, the serine/threonine kinase GSK3β (glycogen synthase kinase 3β) is a crucial crossroad of signaling pathways that control these cellular functions. We sought the impact of a GSK3β activity modulation in stress resistance of leukemic cells, and mechanisms involved in this regulation.
After showing that adhesion on fibronectin, via α4β1 and α5β1 integrins engagement, and that blockade of the Wnt pathway promoted AML cell resistance against apoptosis mediated by daunorubicin, different pharmacological inhibitors and a siRNA strategy allowed us to reveal that this pathway required GSK3β activation. Following thus, we showed that GSK3β leads activation of a transcriptional factor, NF-κB (nuclear factor κB), independent of the classical regulatory pathway by IκB. By researching th signalling events downstream α4β1 and α5β1 integrin engagement responsible for GSK3β activation, we demonstrated a differential regulation of this kinase by these two integrins. In fact, while that α4β1 integrin regulates GSK3β by tyrosine phosphorylation and its co-localization with the tyrosine kinase Pyk2 in nucleus, α5β1 integrin leads to translocation of the PP2A/GSK3β complex out of the nucleus and the dephosphorylation of GSK3β on serine 9. Finally, we have shown a cross-talk between integrins and the Wnt pathway by the way of secretion of a Wnt antagonist, sFRP-1 (secreted Frizzled Related Protein-1), after adhesion of leukemic blasts on osteoblasts of the medullar niche.
In conclusion, our work has allowed us to reveal a novel mechanism of AML chemoresistance, involving two major players in the medullar microenvironment, adhesive molecules and Wnt morphogens. This process of stress resistance depends on the GSK3β kinase activation, which we demonstrate for the first time has a crucial role in a survival pathway controlled by integrins. In perspective of my thesis work, it is important to determine if GSK3β might represent a novel therapetic target, specific to leukemic stem cells, allowing preservation normal hematopoietic stem cells, in AML treatment. Furthermore, a huge field of investigation opens henceforth to understand the role of GSK3β activation in AML physiopathology, and as a pro-oncogenic element in different cancers.
Sommaire
RESUME
... 1ABSTRACT
... 2SOMMAIRE
... 3ABREVIATIONS
... 7REVUE BIBLIOGRAPHIQUE
... 11Chapitre I : L’hématopoïèse physiologique vs l’hématopoïèse leucémique
.... 12A. L’hématopoïèse normale ou physiologique... 12
1. Le concept de cellules souches... 12
2. Les cellules souches hématopoïétiques (CSH)... 13
2.1. La quiescence et la résistance au stress des CSH... 15
a). La quiescence des CSH... 15
b). La résistance au stress des CSH...17
2.2. L’autorenouvellement des CSH...20
a). La voie morphogène Wnt... 24
b). Les antagonistes de la voie Wnt... 28
2.3. La division asymétrique et la différenciation des CSH... 29
2.4. Des souris et des hommes... 33
B. Le rôle du microenvironnement médullaire... 34
1. La niche hématopoïétique... 35
1.1. La niche ostéoblastique ou endostéale... 35
a). Les ostéoblastes... 35
b). Les composants de la matrice extracellulaire... 38
c). Le calcium extracellulaire... 39
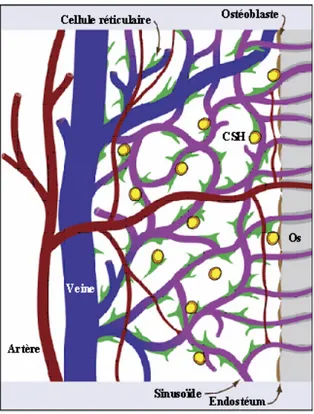
1.2. La niche vasculaire... 39
2. Les autres interactions entre le microenvironnement médullaire et les CSH ... 41
2.1. Les cellules souches mésenchymateuses (CSM)... 41
2.2. Les cellules immunitaires... 42
2.3. Les adipocytes... 43
3. Les autres mécanismes de régulation de l’hématopoïèse... 44
3.1. L’hypoxie et le stress oxydant... 44
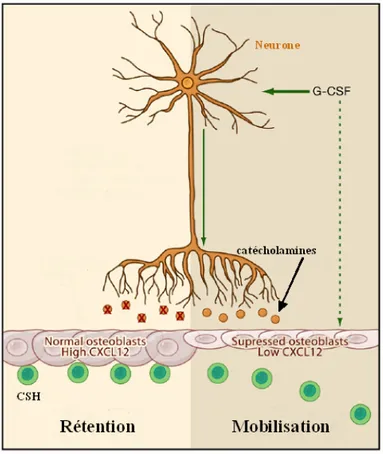
3.2. Le système adrénergique... 46
C. L’hématopoïèse leucémique... 48
1. La leucémie aiguë myéloïde (LAM)... 48
2. Les cellules souches leucémiques (CSL)... 51
2.1. Les CSH et les progéniteurs : cibles de la transformation... 52
2.2. Les anomalies de survie, d’autorenouvellement et de différenciation dans la leucémogenèse... 54
4. Les changements dans le microenvironnement médullaire leucémique... 57
5. Les traitements actuels et leurs conséquences... 58
D. Conclusions et hypothèses préliminaires sur les éventuelles similitudes et différences des CSH et des CSL dans la réponse au stress... 60
Chapitre II : Les intégrines
... 62A. Les structures et les fonctions des intégrines... 62
1. Généralités sur les intégrines... 62
2. Les intégrines et le contrôle de la survie cellulaire... 67
3. Les intégrines et le stress oxydant... 76
B. Les intégrines exprimées par les cellules hématopoïétiques et leur implication dans l’hématopoïèse physiologique et leucémique... 77
1. Le rôle général des intégrines dans l’hématopoïèse... 77
2. Le rôle critique des intégrines de type β1 dans l’hématopoïèse... 78
2.1. La distribution des intégrines β1 et de leurs ligands dans le système hématopoïétique... 79
2.2. La modulation de l’activité des intégrines β1 dans les CSH... 79
a). Les signalisations « inside-out» et «outside-in» des intégrines... 80
b). Les facteurs de croissance hématopoïétiques... 80
c). Les chémokines... 81
d). Les autres récepteurs d’adhérence... 81
e). Les cations métalliques divalents... 84
3. Les grandes voies de signalisation en aval des intégrines β1... 85
3.1. La famille des Focal Adhesion Kinases : le rôle particulier de Pyk2... 85
a). Son expression tissulaire... 87
b). Son activation... 88
c). Son rôle en oncogenèse... 89
d). Son rôle dans la survie cellulaire... 90
3.2. Les phosphatases PP2A... 91
a). Leurs structures et leurs régulations... 91
b). Leurs multiples implications dans les fonctions cellulaires... 92
c). Leur lien avec les intégrines β1... 94
4. Les intégrines α4β1 et α5β1 dans l’hématopoïèseleucémique... 95
4.1. L’intégrine α4β1... 95
a). Ses fonctions dans les blastes leucémiques... 95
b). Son ciblage thérapeutique... 95
4.2. L’intégrine α5β1... 96
a). Ses fonctions dans les blastes leucémiques... 96
b). Son ciblage thérapeutique... 97
4.3. La coopération entre les intégrines α4β1 et α5β1... 97
C. Conclusions et hypothèses préliminaires sur le rôle des intégrines α4β1 et α5β1 dans les CSH et les CSL... 98
Chapitre III : La glycogène synthase kinase 3β (GSK3β)
... 100A. Les glycogène synthase kinases 3... 100
2. Les knock-out de GSK3... 101
B. La régulation de GSK3β... 102
1. Par sa phosphorylation... 102
1.1. L’autophosphorylation de GSK3β... 102
1.2. La phosphorylation de GSK3β par des kinases et phosphatases... 105
1.3. L’état de phosphorylation des substrats de GSK3β...108
2. Par sa localisation intracellulaire et celle de ses partenaires spécifiques.. 110
2.1. Dans le cytosol... 110
a). La voie insulinique... 111
b). Les voies morphogènes Wnt et Shh... 111
c). Le cytosquelette... 114
2.2. Dans le noyau... 116
2.3. Dans la mitochondrie... 118
C. Le rôle de GSK3β dans les processus physiologiques et pathologiques... 119
1. GSK3β et ses fonctions cellulaires... 119
1.1. L’inhibition pharmacologique de GSK3β... 119
1.2. Dans le métabolisme énergétique... 119
1.3. Dans la survie cellulaire... 122
1.4. Dans l’architecture et la migration cellulaires... 128
1.5. Dans la prolifération... 130 1.6. Dans la sénescence... 130 2. GSK3β et le microenvironnement médullaire... 131 2.1. L’inflammation et l’immunité... 131 2.2. L’hypoxie... 131 3. GSK3β et les cancers... 133
3.1. Dans les hémopathies... 133
3.2. Dans les autres cancers... 134
D. Conclusions et hypothèses préliminaires sur le rôle de GSK3β dans les CSH et les CSL... 135
OBJECTIF DES TRAVAUX
... 136RESULTATS EXPERIMENTAUX
... 137Partie I. Le rôle des intégrines et de la voie Wnt dans la chimiorésistance des LAM : Mise en évidence du rôle clef de GSK3β... 138
1. Introduction... 138
2. Résultats... 139
Partie II. Les voies adhésives contrôlant l’activation de GSK3β dans les LAM... 143 1. Introduction... 144 2. Matériels et méthodes... 146 3. Résultats... 150 3.1. Résultats expérimentaux... 150 3.2. Figures... 156
3.3. Légendes des figures... 167
4. Conclusion et discussion... 169
CONCLUSION GENERALE ET PERSPECTIVES
... 175REFERENCES BIBLIOGRAPHIQUES
... 180Abréviations
AES : amino-terminal enhancer of split AGM : aorta-gonad-mesonephros ALDH : aldéhyde déshydrogénase Ang-1 : angiopoïétine-1
APC : adematous polyposis coli ATM : Ataxia Telangiectasia Mutated BMP : bone morphogenic proteins
BMPR : bone morphogenic protein receptor
BMPRIA : bone morphogenic protein receptor type IA CADTK : calcium-dependent tyrosine kinase
CAK-β : cell adhesion kinase β
CAM-DR : cell adhesion mediated-drug resitance CaR : calcium-sensing receptor
cdk : cyclin-dependent kinase
CFU-GM : colony forming unit-granulocyte macrophage CFU-S : colony forming unit-spleen
Ci : cubitus interruptus CK : caséine kinase Cos : costal2
CREB : Cyclic AMP response element binding protein CSE : cellule souche embryonnaire
CSH : cellule souche hématopoïétique CSH-LT : CSH à long terme
CSH-CT : CSH à court terme
CSM : cellule souche mésenchymateuse DISC : death-inducing signaling complex Dkk : Dickkopf
Dsh : dishevelled
E-cadhérine : cadhérine épithéliale EGF : Epidermal growth factor
ERK : extracellular signal-regulated kinase
FAB : French-American-British
FADD : Fas-associated-death-domain protein FAK : focal adhesion kinase
FAT : focal adhesion targeting
FERM : protein 4.1, ezrin, radixin and moesin homology Fg : fibrinogène
Fn : fibronectine Flt-3L : Flt-3 ligand
FOXO ou FoxO : Forkhead box subgroup O Frat : Frequently arranged in T cell lymphomas FRNK : FAK-related non-kinase
Fus : fused FX : facteur X
Fzl-R : récepteur Frizzled
G-SCF : granulocyte colony-forming stimulating factor GEF : guanine nucleotide-exchange factor
GM-CSF : granulocyte macrophage-colony-stimulating factor GRB2 : growth-factor-receptor-bound-2
GSK3 : Glycogen Synthase kinase 3 HDAC : histone deacetylase
Hh : Hedgehog
HoxB4 : Homeo box B4
ICAM : intercellular cell adhesion molecule IGF1/2 : insuline-like growth factor 1/2 IL : interleukine
ILK : integrin-linked kinase IRS : insulin receptor substrate
JNK : Jun amino-terminal kinase
KL : kit ligand
L-IC : leukemia-initiating cells
LAL : leucémie aiguë lymphoblastique Lam : laminine
LAM : leucémie aiguë myéloïde LDL : low-density lipoprotein
LEF/TCF : Lymphocyte enhancer factor/T cell factor LFA-1 : lymphocyte function-associated antigen-1 LIF : leukemia inhibitory factor
LLC : leucémie lymphoïde chronique LMC : leucémie myéloïde chronique
LMCT : leucine carboxyl methyltransferase LPA : lysophosphatidic acid
LRP5-LRP6 : lipoprotein related-receptor proteins 5 and 6 LSK : Lin- Sca1+ c-Kit+
LTC-IC : long-term culture initiating capabilities M-CSF : monocyte colony-forming stimulating factor MAD1 : Myc-antagonist
MadCAM-1 : mucosal adressin cell adhesion molecule-1 MAPK : Mitogen activated protein kinase
MGF : mast cell growth factor MEC : matrice extracellulaire
MICA/B : MHC class-I-related chain A and B MMP : matrix metalloproteinase
MnSOD : manganese superoxide dismutase MO : moelle osseuse
MRD : minimal residual disease MRM : maladie résiduelle minimale NAC : N-acétyl-L-cystéine
NADP : nicotamide adenine dinucleotide phosphate
NADP(H) : nicotamide adenine dinucleotide phosphate sous forme réduite Nck2 : non-catalytic (region of) tyrosine kinase adaptor protein 2
NES : nuclear export sequence NFκB : nuclear factor κ B
NICD : Notch intracellular domain NK : natural killer
NLS : nuclear localization sequence
NOX : NAD(P)H oxidase Nrf2 : NF-E2-related factor 2
NRTK : non-receptor tyrosine kinase Opn : ostéopontine
PDK : 3’-phosphoinositide-dependent kinase PEM : progéniteur érythro-myéloïde
PEr : progéniteur érythroïde
PGM : progéniteur granulo-myéloïde PI-3K : phosphatidylinositol-3 kinase
PINCH : particularly interesting new cysteine-histidine rich protein
PKB : protein kinase B = Akt
PLC : progéniteur lymphoïde commun PMc : progéniteur mégaryocytaire PMC : progéniteur myéloïde commun PME-1 : phosphatase methylesterase 1 pO2 : pression en oxygène
PP2A : protein phosphatases 2A Pro-NK : pro-natural killer PPR : PTH/PTHrP receptor PRNK : Pyk2-related non-kinase PSGL-1 : P-selectin glycoprotein-1 Ptc : patched
PTEN : phosphatase and tensin homologue PTH : parathyroid hormone
PTHrP : PTH-related protein
PTP : protéines tyrosine phosphatases Pyk2 : proline-rich tyrosine kinase 2
RAFTK : related adhesion focal tyrosine kinase RE : réticulum endoplasmique
RI : récepteur à l’insuline
RIP : receptor-interacting protein ROS : reactive oxygen species
RTK : récepteur à activité tyrosine kinase SAPK : stress-activated protein kinase SCF : Stem cell factor
SDF-1 : stromal-cell-derived factor 1 ser : sérine
SFK : Src-family kinases
sFRP : secreted frizzled related protein SH2 : Src-homology-2
Shh : Sonic hedgehog
siRNA : small interfering RNA Smo : smoothened
SNO : spindle shape N-cadherin+ osteoblast SOD : superoxide dismutase
SOS : son-of-sevenless SP : side population
TCF/LEF : T cell factor / lymphocyte-enhancer-binding factor TGFβ : transforming growth factor-β
TLR : Toll-like receptor Tn-C : ténacine-C
TNF : tumor necrosis factor tyr : tyrosine
VCAM : vascular cell adhesion molecule VDAC : voltage dependent anion channel
VE-cadhérine : cadhérine endothéliale vasculaire VEGF : vascular endothelial growth factor VIH : virus de l’immunodéficience humaine VLA : very late antigen
Vn : vitronectine
VWF : facteur von Willebrandt Wnt : wingless
Chapitre I : L’hématopoïèse physiologique versus
l’hématopoïèse leucémique
A. L’hématopoïèse normale ou physiologique
La production permanente de cellules sanguines par la moelle osseuse (MO) est essentielle au transport de l’oxygène par les hématies, à l’hémostase par les plaquettes et aux réponses immunes, ainsi qu’aux fonctions réparatrices tissulaires probablement. Cette pérénité de l’hématopoïèse, tout au long de la vie d’un organisme, est assurée par des cellules souches.
1. Le concept de cellules souches
Les cellules souches sont définies comme des cellules qui ont la capacité de se perpétuer elles-mêmes via le processus d’autorenouvellement et de générer des cellules matures d’un tissu particulier via la différenciation. Dans la plupart des tissus, les cellules souches sont rares. A cause de cela, elles doivent être identifiées et soigneusement purifiées afin d’étudier leurs propriétés. Chaque tissu de l’organisme adulte possèderait donc des cellules souches spécifiques, mais leur identification n’a été réalisée que dans peu de cas. A l’heure actuelle, elles ont été mises en évidence dans le système hématopoïétique, le cerveau, la peau, l’intestin (Weissman I.L. et al., 2001), le sein, la prostate et identifiées potentiellement dans le pancréas et le foie (Burke Z.D. et al., 2007). Les cellules souches de la MO (les cellules souches hématopoïétiques et mésenchymateuses), de l’intestin et du sein présentent des marqueurs spécifiques, mais les études de transcriptome et de protéome montrent que certaines familles de gènes/protéines sont exprimées dans toutes ces cellules souches. Or, ces gènes et ces protéines sont notamment en relation avec l’adhésion, l’hypoxie et le métabolisme, et régulent donc un contexte microenvironnemental nécessaire pour la pérénité des cellules souches (Unwin R.D. et al., 2006), bien que les niches tissulaires correspondantes soient différentes. Les cellules souches (notamment mésenchymateuses) ont le potentiel d’être utilisées en thérapie cellulaire dans le futur, mais seules les cellules souches
hématopoïétiques (CSH) - cellules souches les mieux caractérisées (Kondo M. et al., 2003) - ont déjà été largement utilisées dans le cadre de greffe de MO.
2. Les cellules souches hématopoïétiques (CSH)
Les CSH ont été originellement identifiées fonctionnellement par Till et McCulloch, par leur transplantation dans une souris irradiée permettant ainsi d’obtenir des colonies dans la rate, appelées CFU-S (Colony forming unit-spleen), constituées de plusieurs lignées cellulaires et correspondant chacune à une cellule souche injectée au départ (Becker A.J. et
al., 1963). Les CSH représentent 0,01 à 0,05 % des cellules de la MO. C’est une petite
population cellulaire responsable de la production de toutes les lignées sanguines et donc du maintien de l’homéostasie au cours de la vie d’un individu, grâce à diverses propriétés spécifiques : la quiescence à long terme, une forte résistance au stress, l’autorenouvellement, la multipotence et la division asymétrique.
La balance entre la quiescence et la prolifération des CSH est vraisemblablement critique pour l’hématopoïèse à long terme, pendant toute la durée de vie d’un adulte, et dépend des besoins de l’organisme au niveau du système sanguin ou immunitaire (Glimm H. et al., 2000 ; Passegue E. et al., 2005). La prolifération des CSH entraîne soit un autorenouvellement simple permettant de maintenir leur nombre et leur multipotence, soit une division asymétrique qui engendre à la fois une CSH identique à la CSH mère (par le processus d’autorenouvellement), et une cellule plus mature qui sort de la niche permettant ainsi son expansion et sa perte de multipotence. Face à un stress hématologique, tel qu’une hémorragie, une infection ou une exposition à des agents cytotoxiques, les CSH doivent reconstituter tout le système sanguin de l’organisme, ce qui provoque leur sortie de quiescence, suivie de leur expansion et leur différenciation en tous les lignages du sang par ce processus de division asymétrique. Or, pour proliférer et donc entrer dans le cycle cellulaire, les CSH doivent sortir de la niche et donc perdre leur capacité de se greffer à la niche. En effet, lors de la division asymétrique, une polarité s’installe dans la niche au niveau des contacts adhésifs et semble essentielle pour cette maturation des CSH. De plus, il a été récemment prouvé que ce mécanisme de division asymétrique est caractérisé par une répartition génique différente de celle constatée pour la CSH quiescente, grâce à une puce à ADN du transcriptome humain (Wagner W. et al., 2004).
Le compartiment des CSH peut être divisé en trois populations différentes : (i) les CSH à l’autorenouvellement à long terme (CSH à long terme ou CSH-LT), seules capables de péréniser l’hématopoïèse durant toute la vie d’un organisme, (ii) les CSH à l’autorenouvellement à court terme (les CSH à court terme ou CSH-CT), qui sont un peu plus différenciées que les précédentes et donc ne peuvent reconstituer l’hématopoïèse que pendant une durée limitée (quelques semaines), et (iii) les progéniteurs multipotents sans potentiel d’autorenouvellement détectable (Morrison S.J. & Weissman I.L., 1994 ; Morrison S.J. et al., 1997). Ces populations constituent un lignage dans lequel les CSH à long terme donnent naissance aux CSH à court terme, qui donnent lieu à leur tour aux progéniteurs multipotents (Morrison S.J. et al., 1997) (figure 1).
Figure 1 : Les cellules souches hématopoïétiques. Le compartiment des CSH se
compose de trois populations cellulaires : les CSH à long terme avec un fort pouvoir d’autorenouvellement (flèche rouge pleine), les CSH à court terme avec un plus faible pouvoir d’autorenouvellement (flèche rouge en pointillés) et les progéniteurs multipotents sans ce potentiel d’autorenouvellement mais plus actifs mitotiquement. Ce sont les CSH-LT qui permettent de reconstituer une hématopoïèse à long terme, lors d’une greffe de MO dans une souris NOD-SCID (Nonobese diabetic - Severe
combined immunodeficiency) au préalable irradiée, alors que les CSH-CT et les
progéniteurs multipotents reconstituent l’hématopoïèse de souris irradiées pour moins de huit semaines. (D’après Reya T. et al., 2001).
Actuellement, il existe plusieurs méthodes pour isoler et purifier des CSH humaines ou murines (Bonnet D., 2005). A partir de la MO de souris, on peut réaliser un tri cellulaire se composant de plusieurs étapes. D’abord, par l’utilisation de nanoparticules ferro-magnétiques, une sélection négative sur le lignage suivie d’une sélection positive sur le marqueur de surface Sca1 est effectuée, puis grâce à un trieur cellulaire (FACS), les cellules positives pour le récepteur c-Kit sont isolées. Cette technique permet donc d’obtenir des cellules LSK (Lin -Sca1+ c-Kit+), considérées comme les CSH murines (voir la partie suivante Des souris et des
hommes). En ce qui concerne les CSH humaines, une sélection positive sur le marqueur de
surface CD34 puis une négative sur le CD38, par l’utilisation également de nanoparticules ferro-magnétiques, permet d’isoler des cellules hématopoïétiques immatures CD34+ CD38-. Pour enrichir cette population en CSH, d’autres tris complémentaires peuvent être réalisés sur d’autres marqueurs de surface, considérés comme spécifiques de la souchitude (CD90, CD123, CD133, ..). Une autre procédure, pour purifier des CSH à partir de la MO humaine, est basée sur la capacité de ces cellules à effluer un composé coloré fluorescent, le Hoechst 33342. Les cellules faiblement colorées par le Hoechst 33342, ainsi isolées, sont appelées la «side population» (SP) et ont été montrées comme ayant le même phénotype que des cellules Lin- Sca1+ CD34-, indépendamment identifiées dans le compartiment des CSH adultes murines (Pearce D.J. et al., 2004). Enfin, une autre méthode a été développée à partir de l’utilisation de l’aldéhyde déshydrogénase (ALDH), une enzyme cytosolique qui est responsable de l’oxydation des aldéhydes intracellulaires. Des taux élevés d’ALDH ont été constatés dans les cellules progénitrices murines et humaines, comparés aux autres cellules hématopoïétiques (Hess D.A. et al., 2004).
2.1. La quiescence et la résistance au stress des CSH a). La quiescence des CSH
Les CSH adultes existent dans un état quiescent, dans la niche médullaire (Morrison S. J. & Weissman I. L., 1994 ; Bradford G.B. et al., 1997 ; Cheshier S.H. et al., 1999). Le maintien de la quiescence des CSH implique à la fois des mécanismes extrinsèques et intrinsèques. Ainsi, un nombre de gènes qui codent pour des régulateurs du cycle cellulaire tels que p21cip1/waf1, p27Kip1, β-caténine/axine, cycline D1, PTEN (Phosphatase and tensin
homolog), c-Myc et MEF/ELF4 (Cheng T. et al., 2000a ; Cheng T. et al., 2000b ; Wilson A. et al., 2004 ; Walkley C.R., 2005 ; Zhang J. et al., 2006 ; Lacorazza H.D. et al., 2006), ont été
montrés comme régulant les programmes intrinsèques de quiescence des CSH. Par exemple, chez des souris déficientes en p21cip1/waf1 (p21-/-), régulateur du point de contrôle (checkpoint) G1 et inhibiteur de cdk (cyclin-dependent kinase), la prolifération et le nombre absolu de CSH augmentent dans des conditions homéostasiques normales (Cheng T. et al., 2000a). Au contraire des souris p21-/-, le nombre des CSH est normal dans les souris déplétées pour le gène de la protéine p27Kip1 (p27-/-), alors que ces souris ont une augmentation du nombre de cellules progénitrices hématopoïétiques (Cheng T. et al., 2000b). Ainsi, les deux inhibiteurs de cdk, p21 et p27 gouvernent plus spécifiquement la quiescence des cellules souches et des progéniteurs hématopoïétiques, respectivement.
De plus, les interactions entre les CSH et leur microenvironnement médullaire dans des zones anatomiquement et fonctionnellement spécifiques, appelées « niches » (un paragraphe sera consacré à celle de la MO ultérieurement), ont permis de faire avancer les connaissances dans le maintien de la quiescence des CSH (Li L. & Xie T., 2005). En effet, comme les CSH ont besoin de se détacher de la niche où elles sont maintenues dans un état quiescent pour entrer en division asymétrique et en expansion, il semble donc que l’interaction des CSH avec le microenvironnement médullaire et le maintien de l’état de quiescence soient étroitement interconnectés. Ainsi, on peut suggérer que les signaux extrinsèques de la niche hématopoïétique, tels que les BMP (bone morphogenic proteins), le calcium (Ca2+), les ligands Notch, et l’angiopoïétine-1 (Ang-1) (Calvi L.M. et al., 2003 ; Zhang J. et al., 2003 ; Arai F. et al., 2004 ; Adams G.B. et al., 2006), et les interactions adhésives entre la matrice extracellulaire (MEC) médullaire et les CSH (Jiang Y. et al., 2000) pourraient réguler de façon harmonieuse l’état quiescent des CSH. Malgré tout, les mécanismes coordonnant la régulation du cycle cellulaire des CSH restent obscurs.
De façon intéressante, les interactions entre les CSH et leur microenvironnement reposent sur une polarisation et une organisation du cytosquelette, qui peuvent influencer l’entrée cellulaire en mitose et son déroulement. L’équipe de Zheng s’est intéressée à Cdc42, un membre de la famille des GTPase Rho, exprimée de façon ubiquitaire et impliquée dans la régulation de multiples fonctions cellulaires, comme la polymérisation de l’actine, l’adhésion cellule-cellule ou cellule-MEC, et la transcription génique (Etienne-Manneville S. & Hall A., 2003). Lors d’expériences préliminaires, ils ont montré, dans un modèle murin de gain d’activité de Cdc42, que l’augmentation constitutive des formes de Cdc42-GTP entraînait une apoptose accrue des progéniteurs hématopoïétiques, la désorganisation de la structure de l’actine et un défaut dans la prise de la greffe sans affecter le statut du cycle cellulaire (Wang L. et al., 2006). Grâce à un modèle murin d’invalidation conditionnelle du gène Cdc42 dans les cellules hématopoïétiques, cette équipe a également mis en évidence le rôle de Cdc42 dans le maintien de la quiescence des CSH et dans leur localisation au sein de la niche médullaire. Il est aussi montré que Cdc42 régule l’expression d’un grand nombre de régulateurs clés du cycle cellulaire (incluant c-Myc et p21Cip1), de molécules d’adhésion cellulaires (telles que l’intégrine β1 et la N-cadhérine) et la structure de l’actine (Yang L. et al., 2007 ; Yang L. & Zheng Y., 2007). Par contre, leurs résultats prouvent que Cdc42 n’est pas nécessaire à la survie des CSH. Quant à Rac1 et Rac2, deux autres membres de la famille des GTPases Rho, elles sont importantes pour la rétention des CSH dans la niche hématopoïétique et pour leur
survie et leur progression dans le cycle cellulaire, mais elles ne sont pas impliquées dans ce maintien de l’état de quiescence des CSH (Cancelas J.A. et al., 2005).
b). La résistance au stress des CSH
C’est grâce à leur état quiescent et à leur capacité à métaboliser les radicaux oxygénés, que les CSH peuvent résister à différents stress microenvironnementaux de la niche hématopoïétique et ainsi assurer le processus de l’hématopoïèse, tout au long de la vie des individus. Or, dans la CSH-LT quiescente, le suppresseur de tumeur PTEN est actif et réprime la signalisation PI-3K (phosphatidylinositol-3 kinase)/Akt (appelée aussi PKB) conduisant à l’inhibition ou à l’activation des composés distaux, tels que mTOR et FoxO (Forkhead box
subgroup O), respectivement (figure 2).
Figure 2 : Régulation de la quiescence des CSH par PTEN, FoxO et ATM. Le
suppresseur de tumeur PTEN (Phosphatase and tensin homolog) contrôle les facteurs de transcription FoxO (Forkhead box subgroup O) et ATM (Ataxia Telangiectasia
Mutated). Même s’il n’existe encore aucun lien expérimental entre ces deux protéines,
FoxO et ATM sont, l’une comme l’autre, capables de promouvoir la détoxification des ROS (reactive oxygen species), et d’inhiber le cycle cellulaire des CSH. Tous ces mécanismes permettent donc de maintenir les CSH en quiescence. (D’après Coffer P.J. & Burgering B.M.T., 2007).
Or, les facteurs de transcription FoxO actifs programment les CSH à rester quiescentes par une répression du cycle cellulaire et une induction de la quiescence, mais aussi permettent leur survie face à un stress oxydant, en les orientant vers la néoglucogenèse et un métabolisme des acides gras, avec élimination des reactive oxygen species (ROS). Bien qu’aucun lien entre FoxO et ATM (Ataxia Telangiectasia Mutated) n’ait encore été découvert expérimentalement, ATM a été aussi impliquée dans la fonctionnalité des CSH, en ce qui concerne leur entrée dans le cycle cellulaire et la concentration intracellulaire en ROS (figure 2).
PTEN a été démontrée comme régulant le maintien des CSH, à travers une restriction de leur prolifération (Yilmaz O.H. et al., 2006 ; Zhang J. et al., 2006). En effet, ces deux équipes ont montré que, dans des souris invalidées pour PTEN, la prolifération des CSH était augmentée et le compartiment souche se réduisait progressivement et de façon très importante. L’ablation de PTEN provoque, en outre, la phosphorylation et l’inhibition des isoformes de FoxO, grâce à l’activation accrue d’Akt/PKB, permettant ainsi au stress oxydant de s’installer de manière permanente. Ces données sont très intéressantes, car PTEN est située en amont des facteurs de transcription FoxO et ATM.
En absence de stimulation par des facteurs de croissance (notamment par la voie insuline/PI-3K/Akt), ou dans des conditions de stress, les facteurs de transcription de la famille FOXO se trouvent dans le noyau et entraînent l’expression de gènes impliqués dans l’arrêt du cycle cellulaire, l’apoptose et la résistance au stress (Tothova Z. & Mercher T., 2007). A l’heure actuelle, il a été démontré par plusieurs équipes que selon le type de stress, déprivation en nutriments et/ou blocage de la voie insulinique, le type de FOXO activé est différent, ainsi que les gènes des superoxide dismutase (SOD), cibles de FOXO impliquées dans la détoxification des ROS (Kops G.J. et al., 2002 ; Essers M.A. et al., 2005). Les FOXO (pour les gènes humains ou FoxO pour les formes murines) représentent une sous-famille des facteurs de transcription Forkhead et sont remarquablement conservés de la levure à l’homme. Cette famille comporte quatre membres principaux chez les mammifères, identifiés comme des orthologues de la protéine DAF-16 (Dauer formation defective) de
Caenorhabditis elegans : FOXO1 (FKHR), FOXO3a (FKHRL1), FOXO4 (AFX), FOXO6.
Seuls FOXO1, FOXO3a et FOXO4 sont exprimés de façon ubiquitaire, ont des rôles partiellement redondants, et ont été étudiés dans le système hématopoïétique (Anderson M.J.
et al., 1998 ; Furuyama T. et al., 2000 ; Biggs W.H. et al., 2001). Ainsi, pour s’affranchir de
cette redondance partielle, Tothova et al. ont réalisé récemment l’invalidation conditionnelle simultanée des gènes FoxO1, FoxO3a et FoxO4 (Tothova Z. et al., 2007), dans le système hématopoïétique de souris adultes. Les souris déficientes en FoxO (FoxO1/3/4- /-) montrent
notamment une expansion du lignage myéloïde et une diminution marquée du compartiment LSK (lignage négatif Lin-, Sca1+ , c-Kit+), qui contient les populations de CT et CSH-LT. La MO déficiente en FoxO a un défaut dans l’activité de repopulation à long terme (expériences de greffe de MO), ce qui corrèle avec un épuisement des capacités d’autorenouvellement des CSH, suite à d’intenses cycles de divisions cellulaires, et de leur apoptose massive due à une accumulation intracellulaire de ROS. De plus, un traitement in
vivo avec un agent anti-oxydant, la N-acétyl-L-cystéine (NAC), résulte en une réversion du
phénotype des CSH déficientes en FoxO, s’accompagnant d’une inactivation de la Mitogen
activated protein kinase (MAPK) p38. Les protéines FoxO, et notamment FoxO3a (Yamazaki
S. et al., 2006), jouent donc un rôle majeur dans la réponse au stress oxydatif physiologique et par conséquent participent de manière active dans le maintien de la quiescence et la résistance au stress du compartiment des CSH, une fonction nécessaire pour leur potentiel de régénération à long terme.
Nous avons vu précédemment que les facteurs de transcription FOXO exercent des effets sur le devenir des CSH, qui semblent liés à une augmentation du stress oxydatif (c’est-à-dire des ROS) dans les CSH, résultant d’un défaut d’expression des gènes normalement impliqués dans leur détoxification. De plus, dans cette étude, ils ont observé que l’expression d’ATM et d’une de ses cibles, p16INK4a, est modifiée dans les souris déficientes en FoxO (souris FoxO1/3/4-/-), suggérant que le facteur ATM pourrait être une cible de FoxO (Tothova Z. et al., 2007), bien qu’aucun lien direct entre les deux n’ait été décrit à ce jour. Or, l’équipe d’Ito a montré précédemment que l’inhibition du stress oxydant par ATM est nécessaire à l’autorenouvellement des CSH. En effet, dans cette étude, les auteurs nous révèlent qu’ATM a un rôle essentiel dans la capacité de reconstitution des CSH, mais n’est pas importante pour la prolifération ou la différenciation des progéniteurs (Ito K. et al., 2004). En outre, des souris déficientes pour ATM (souris ATM-/-) âgées de 24 semaines montrent une défaillance de la MO résultant d’un défaut dans la fonction des CSH qui est associé à une concentration élevée de ROS et à une activation de p38. Or, dans ces études, un traitement avec des agents anti-oxydants, tels que la NAC et la catalase, ou avec un inhibiteur de la MAPK p38, restaure la capacité de reconstitution d’une hématopoïèse des CSH ATM-/-, en empêchant la défaillance de la MO, indiquant que l’inactivation de p38 protège les CSH contre la perte de leur capacité d’autorenouvellement induite par les ROS (Ito K. et al., 2004 ; Ito K. et al., 2006). Grâce à ces résultats, on peut en conclure que la capacité d’autorenouvellement des CSH dépend de l’inhibition du stress oxydant par le biais d’ATM. De plus, tous ces travaux sur FoxO et ATM montrent que ces protéines sont majeures pour maintenir des faibles taux intracellulaires
d’oxydants dans les CSH et suggèrent donc qu’elles joueraient un rôle essentiel dans la prévention du vieillissement prématuré.
Au vu de ces résultats, prouvant que PTEN, FoxO et ATM sont nécessaires à la quiescence des CSH, au maintien de leur pool et à leur résistance au stress, et notamment au stress oxydant par des mécanismes d’élimination des ROS, nous avons résumé, dans la figure 3, les effets des invalidations géniques de ces trois protéines sur le devenir des CSH murines.
Figure 3 : Rôles de PTEN, FoxO et ATM sur la quiescence/résistance au stress des CSH murines, démontrés par des invalidations de ces gènes chez la souris.
2.2. L’autorenouvellement des CSH
A l’opposé de l’état de quiescence/résistance au stress des CSH, l’autorenouvellement les fait basculer dans un état dynamique de prolifération, impliquant des acteurs de signalisation spécifiques. Ce processus est une division cellulaire permettant d’aboutir à la formation d’au moins une cellule fille, identique à la CSH mère, c’est-à-dire ayant conservé intactes ses capacités d’autorenouvellement et de multipotence.
Bien que les propriétés phénotypiques et fonctionnelles des CSH aient été largement abordées (Weissman, I. L., 2000), la question cruciale de savoir comment l’autorenouvellement est régulé reste sans réponse précise. Dans la plupart des cas, les combinaisons de facteurs de
croissance qui peuvent induire une prolifération efficace ne peuvent empêcher la différenciation des CSH en culture à long terme. Bien que des progrès aient été réalisés dans l’identification des conditions de culture qui maintiennent l’activité des CSH (référencées, par exemple, dans les papiers de Miller C. L. & Eaves C. J., 1997 ; Ema H. et al., 2000 ; Sauvageau G. et al., 2004), des difficultés ont été constatées pour identifier les combinaisons de facteurs de croissance qui provoquent une expansion significative des CSH ayant conservé leur activité de régénération hématopoïétique après une greffe. Ainsi, toutes ces expériences ont permis de déterminer que le granulocyte macrophage-colony-stimulating factor (GM-CSF) joue un rôle majeur - tout comme l’interleukine-3 (IL-3) - dans la survie, la prolifération et la différenciation des CSH multipotentes en cellule souches plus matures et restreintes en terme de lignages (Olofsson T.B., 1991). Quant à l’insuline-like growth factor (IGF), ce facteur de croissance permet d’inhiber l’apoptose programmée des progéniteurs hématopoïétiques, de façon dépendante de la PI-3K, et l’expression de son récepteur est nécessaire pour la survie et la prolifération de ces cellules. Donc, IGF-1 joue à la fois un rôle de facteur de croissance et de survie pour enrichir les populations de progéniteurs hématopoïétiques primaires (Kelley K.W. et al., 1998). Cependant, l’équipe de Weissman a montré que les acteurs du cycle cellulaire responsables de l’autorenouvellement des CSH sont différents de ceux qui régulent la prolifération des progéniteurs hématopoïétiques (Passegue E. et al., 2005).
Des voies impliquées dans l’embryogenèse, les voies morphogènes Wnt, Sonic hedgehog (Shh) et Notch, se sont révélées comme essentielles dans le contrôle de la balance quiescence/autorenouvellement des CSH (figure 4). Grâce à des carrefours de signalisation communs (voir plus loin le chapitre sur la glycogène synthase kinase 3β), ces voies vont dialoguer et influencer les CSH vers un phénotype quiescent (via la voie Notch) ou d’autorenouvellement (via les voies Wnt et Shh).
Des études contradictoires ont examiné le rôle de Notch dans l’autorenouvellement des CSH. Ainsi, l’activation de Notch dans les CSH en culture via le ligand Jagged-1 augmente la quantité des progéniteurs primitifs qui peut être observée in vitro et in vivo, suggérant que l’activation de la voie Notch promeut l’autorenouvellement des CSH, ou au moins la maintenance de leur multipotentialité (Varnum-Finney B. et al., 2000 ; Karanu F. N. et al., 2000). A l’inverse de ces résultats, une autre équipe a analysé l’effet fonctionnel de la voie de signalisation Notch sur des cellules progénitrices hématopoïétiques humaines, par l’expression ectopique de Notch 1 constitutivement actif conduisant à un arrêt du cycle cellulaire et à l’apoptose des cellules CD34+ (Chadwick N. et al., 2007).
Figure 4 : Les trois grandes voies morphogènes régulant l’autorenouvellement des CSH. La voie Wnt (à gauche) est activée lorsqu’un ligand Wnt s’associe au récepteur
Frizzled (Fzd) couplé aux récepteurs LRP5/6, inhibant ainsi GSK3β qui ne peut plus exercer son rôle négatif sur le facteur de co-transcription, la β-caténine (β-cat), permettant alors à cette dernière d’induire la transcription, avec LEF/TCF (Lymphocyte
enhancer factor/T cell factor), de gènes impliqués dans l’autorenouvellement des CSH,
tels que la cycline D1. Pour la voie Sonic Hedgehog (Shh) (au milieu), la liaison du ligand Hedgehog sur le récepteur Patched (Ptc) lève l’inhibition que ce dernier exerçait sur le récepteur Smoothened (Smo) et permet ainsi au facteur de transcription Gli d’aller dans le noyau et d’induire la transcription de plusieurs gènes dont, entre autres, celui de Ptc. Pour activer la voie Notch (à droite), le récepteur Notch interagit avec son ligand (Delta 1,2 et 4 et Jagged 1 et 2), ce qui entraîne le clivage et la libération du domaine intracellulaire Notch 1 (NICD pour Notch intracellular domain) dans le noyau, qui en se liant au facteur de transcription CBF-1, permet la transcription de gènes comme celui de la protéine Hes-1. (D’après Reya T. et al., 2001).
Les auteurs expliquent cette contradiction des résultats par le fait que des taux élevés de Notch actif (comme ce qui est observé lors de leur transfection rétrovirale) résulteraient en une apoptose, alors que des taux faibles entraîneraient l’autorenouvellement des CSH. Quant à la voie Shh (cette voie sera plus détaillée, en terme de signalisation, dans le chapitre « La glycogène synthase kinase 3β »), elle a aussi été impliquée dans la régulation de l’autorenouvellement par la constatation expérimentale que les populations hautement enrichies en CSH humaines (Lin- CD34+ CD38-) arboraient un autorenouvellement accru en réponse à la stimulation de la voie Shh in vitro (bien qu’en combinaison avec d’autres facteurs de croissance), et ce via la régulation des BMP (Bhardwaj G. et al., 2001).
Comme nous l’avons abordé précédemment, les interactions adhésives, les facteurs de croissance et les trois voies morphogènes jouent un rôle critique dans le contrôle de la balance entre quiescence et autorenouvellement des CSH. Une de leurs cibles communes est le facteur
de transcription c-Myc (Satoh Y. et al., 2004). Ainsi, Wilson et al. proposent un modèle de régulation de l’hématopoïèse par c-Myc (figure 5 ; Wilson A. et al., 2004).
Figure 5 : Modèle hypothétique de régulation de l’hématopoïèse par c-Myc. (a) Le
système hématopoïétique physiologique (sauvage). (b) La déficience en c-Myc dans les CSH-LT. (c) La surexpression de c-Myc dans les CSH-LT. (D’après Wilson A. et
al., 2004).
Au cours de l’hématopoïèse, une sous-population des CSH-LT expriment c-Myc, entraînant ainsi une différenciation en CSH-CT puis, après la perte de la capacité de l’autorenouvellement, en cellules plus différenciées. Au début de la différenciation terminale, l’activité de la protéine c-Myc diminue pour permettre la sortie du cycle cellulaire et la progression vers la différenciation terminale. Dans des cas de déficience en c-Myc, les CSH-LT ne cessent de s’autorenouveler et ne peuvent plus se différencier, ce qui provoque leur accumulation dans la niche endostéale. De plus, les progéniteurs en amplification transitoire stoppent leur expansion. Par contre, quand c-Myc est surexprimée, les CSH-LT se
différencient au détriment de l’autorenouvellement, créant ainsi un épuisement en cellules souches et donc à terme la perte de toutes les cellules hématopoïétiques.
De façon intéressante, les modulations d’expression de c-Myc dans les CSH ont un impact totalement différent, selon que les cellules soient traitées in vitro ou in vivo. Cela signifie donc que c-Myc entretient un dialogue important avec le microenvironnement des CSH. Son impact, notamment sur la régulation de l’expression des molécules d’adhésion, sera détaillée dans la partie Le rôle du microenvironnement médullaire.
Enfin, la régulation épigénétique, qui est réalisée via des facteurs modulant la structure de la chromatine, joue un rôle clef dans le contrôle de l’autorenouvellement. Ainsi, une protéine Polycomb-like, Bmi-1, s’est révélée comme nécessaire pour le maintien de l’autorenouvellement des cellules souches et notamment des CSH (Park I.K. et al., 2003). Or, le proto-oncogène Bmi-1, un des composants clés du complexe répresseur Polycomb 1 (PRC1) - impliqué dans le maintien stable de gènes répresseurs -, est exprimé dans les CSH murines et humaines et est nécesssaire à la répression des gènes codant pour des inhibiteurs des cdk, comme p16Ink4a (responsable sinon du blocage du cycle cellulaire et donc de l’arrêt de prolifération). En effet, dans des souris déficientes pour Bmi-1, aucun autorenouvellement des CSH adultes n’est détecté et l’expression de cet inhibiteur du cycle cellulaire, p16Ink4a, dans des CSH normales, provoque un arrêt de leur prolifération (Park I.K. et al., 2003).
a). La voie morphogène Wnt
Les régulations croisées entre les voies morphogènes et les voies adhésives jouent très probablement un rôle clef dans le contrôle de la balance quiescence/autorenouvellement des CSH. Ce dialogue perpétuel a été partiellement étudié, dans différents modèles, entre la voie de signalisation Wnt et les voies d’adhésion en aval des intégrines ou des cadhérines. Voilà pourquoi, il nous semble nécessaire de détailler ici plus longuement la voie Wnt, ainsi que ses antagonistes.
Les protéines Wnt sont des molécules hydrophobes de signalisation intercellulaire (Nusse R. & Varmus H.E., 1992) qui contrôlent le développement de plusieurs organismes (Cadigan K. M. & Nusse R., 1997). La voie morphogène Wnt a aussi été montrée comme régulant à la fois l’autorenouvellement et l’oncogenèse, dans différents organes, et notamment dans le système hématopoïétique (Taipale J. & Beachy P.A., 2001 ; Reya T. et al., 2003). Les protéines solubles Wnt peuvent être produites par les CSH elles-mêmes, aussi bien que par le microenvironnement. Des études réalisées sur la MO humaine suggèrent que Wnt 2B, Wnt 5A, et Wnt 10B sont exprimées dans les cellules de la MO, alors que seule Wnt 5A est
exprimée dans les cellules CD34+ Lin- (population enrichie donc en progéniteurs et en CSH) (Van Den Berg D. J. et al., 1998). Plusieurs travaux démontrent la capacité des ligands Wnt à stimuler la prolifération des progéniteurs hématopoïétiques de foie fœtal murin ou de MO humaine (Austin T.W. et al., 1997 ; Van Den Berg D.J. et al., 1998) et l’autorenouvellement des CSH (Willert K. et al., 2003). De plus, un traitement avec du milieu conditionné en Wnt 5A, chez des souris NOD-SCID (Nonobese diabetic - Severe combined immunodeficiency), a permis d’augmenter la population des CSH de ces souris (Murdoch B. et al., 2003). Ce papier démontre donc le rôle in vivo de Wnt sur la régulation du développement et de la fonction des CSH, et ces résultats pourraient avoir un intérêt direct pour améliorer l’efficacité des greffes chez les patients.
La voie Wnt/β-caténine régule la prolifération, l’adhésion, la morphologie, la migration et les remodelages structuraux cellulaires. Cette voie se constitue des voies canoniques, dites classiques, et non-canoniques, appelées alternes (figure 6). Les voies de signalisation canoniques Wnt sont celles qui, en aval de la liaison d’un ligand Wnt sur son récepteur Frizzled, jouent sur le devenir de la protéine β-caténine. Par contre, les voies Wnt, qui n’incluent pas ce facteur de co-transcription, sont les voies Wnt appelées « alternes » ou « non canoniques » (Kühl M. et al., 2001). Les composés majeurs des voies Wnt sont les glycoprotéines Wnt, qui peuvent être codées par 19 gènes dans les génomes de Mammifères. Elles sont sécrétées, subissent des modifications lipidiques et se lient aux récepteurs Frizzled à la surface membranaire. Ces récepteurs sont couplés à d’autres récepteurs moléculaires, que sont les lipoprotein related-receptor proteins 5/6 (LRP5/6).
En absence de ligand Wnt, la β-caténine est associée, dans le cytoplasme, au complexe axine/APC (adematous polyposis coli)/GSK3β (Glycogen Synthase kinase 3β), où elle est phosphorylée par GSK3β et la casein kinase I (CKI), ce qui entraîne ainsi son ubiquitination par la β-transducin repeat-containing protein (βTrCP) et sa dégradation par le protéasome 26S (figure 6 ; Polakis P., 2002). L’activation de la voie Wnt/β-caténine canonique, par la liaison d’un agoniste Wnt, tel que Wnt 1, Wnt 3A et Wnt 8 (appelés également Wnt canoniques), permet de stimuler Dishevelled (Dsh) et Frequently arranged in T cell
lymphomas (FRAT), provoquant la libération de la β-caténine du complexe inhibiteur et
Figure 6 : Les voies Wnt canonique et alternes. La voie Wnt/β-caténine est la voie
canonique, c’est-à-dire classique, tandis que les autres voies schématisées ici, telles que la voie du Ca2+ et la voie induisant la polarité planaire cellulaire, sont les voies Wnt alternes. La voie Wnt/β-caténine est activée par des agonistes, comme le ligand Wnt 3A. D’autres ligands, comme Wnt 5A et Wnt 11, peuvent dans certaines conditions entraîner l’activation de voies alternes, comme la voie du Ca2+ et la voie de polarité planaire cellulaire.
La β-caténine peut alors s’accumuler dans le cytoplasme de la cellule. Elle s’associe, au niveau intracellulaire, à la membrane plasmique avec les cadhérines pour promouvoir l’adhésion cellulaire et avec les microfilaments d’actine du cytosquelette, via l’α-caténine, pour contrôler la forme cellulaire (Jamora C. & Fuchs E., 2002). La β-caténine peut aussi entrer dans le noyau où elle active la transcription, avec les facteurs de co-transcription LEF/TCF, de gènes impliqués dans la prolifération et l’autorenouvellement des CSH, comme la cycline D1 (figure 6 ; Cadigan K.M. & Nusse R., 1997). La répartition entres ses localisations membranaires et nucléaires n’est pas encore tout à fait bien comprise, mais
semble faire intervenir sa concentration cytoplasmique, son état de phosphorylation et ses partenaires spécifiques.
Il existe de nombreuses voies Wnt alternes, et vraisemblablement d’autres sont encore inconnues. Parmi ces voies non canoniques, nous pouvons citer et décrire la voie de la polarité planaire cellulaire et celle du Ca2+. Ainsi, certains ligands Wnt, comme Wnt 5A, peuvent inhiber la voie canonique, mais activer celle du Ca2+, via la protéine G hétérotrimérique du récepteur Frizzled. Cela va permettre de stimuler la phospholipase C (PLC) et donc d’augmenter la concentration en calcium, ce qui conduit à l’autophosphorylation de la kinase II dépendante de la calmoduline (CAMKII), et finalement à des processus cruciaux pour la cellule, comme l’adhésion dépendante du calcium. Quant à la voie de la polarité planaire cellulaire, la liaison d’un Wnt non canonique, comme Wnt 5A ou Wnt 11, active Dsh ce qui provoque l’activation de RhoA qui, par deux kinases différentes - la Jun amino-terminal kinase (JNK) ou la Rho-kinase - , peut jouer sur les microfilaments d’actine du cytosquelette et réguler par conséquent la polarité cellulaire.
En utilisant des CSH de MO, l’équipe de Weissman a montré que la surexpression de la β-caténine activée (acteur central de la voie de signalisation Wnt) accroît le pool de CSH transplantables caractérisées à la fois par le phénotype (Thy1.1low Lin-/low Sca1+ c-kit+) et la fonction (la capacité de reconstituer le système hématopoïétique in vivo) (Reya T. et al., 2003). De plus, l’expression ectopique de l’axine, un inhibiteur de la β-caténine, conduit à l’inhibition de la prolifération des CSH, à une augmentation de la mort des CSH in vitro, et à une réduction de la reconstitution hématopoïétique in vivo. Enfin, le fait que certains acteurs de la voie Notch soient sous la « houlette » de la β-caténine (Reya T. et al., 2003 ; Duncan AW & Reya T., observation non publiée) indique qu’il pourrait exister une hiérarchie moléculaire du contrôle de l’autorenouvellement des CSH.
Une équipe allemande, quant à elle, a montré qu’un inhibiteur des déacétylases des histones, l’acide valproïque, permettait d’augmenter la prolifération et l’autorenouvellement des CSH (Bug G. et al., 2005). Cet inhibiteur accélère la progression des CSH dans le cycle cellulaire, qui s’accompagne d’une inhibition de p21cip-1/waf-1. De plus, il inhibe GSK3β par phosphorylation sur sa sérine 9 (ser 9), ce qui induit une activation de la voie Wnt, ainsi qu’une suractivation de HoxB4, un gène cible de la voie β-caténine et un facteur clé dans la régulation de l’autorenouvellement et de la prolifération des CSH.
Ainsi, les ligands Wnt contribuent à l’autorenouvellement des CSH, mais aussi à celui de cellules souches d’autres tissus (pour revue Reya T. & Clevers H., 2005). Or, leur action
est étroitement contrebalancée par l’existence d’antagonistes des voies canoniques et alternes Wnt.
b). Les antagonistes de la voie Wnt
Outre les ligands Wnt, potentiellement antagonistes de la voie canonique de signalisation Wnt, comme Wnt 4, Wnt 5A et Wnt 11 - qui activent les voies Wnt alternes et sont nommés les Wnt non canoniques -, il existe des antagonistes extracellulaires de ces voies (Kawano Y. & Kypta R., 2003). Ils peuvent être divisés en deux grandes classes de molécules qui empêchent les interactions récepteur/ligand : (i) la première classe inclut la famille
secreted frizzled related protein (sFRP), le Wnt inhibitory factor 1 (WIF-1) et Cerberus, qui se
lient principalement aux protéines Wnt, altérant ainsi leur capacité de s’associer aux récepteurs Frizzled/LRP5/6, et (ii) la seconde classe comprend certains membres de la famille
Dickkopf (Dkk), qui eux se lient aux co-récepteurs de Frizzled, que sont les LRP5/6. Donc, en
théorie, les antagonistes de la classe sFRP peuvent inhiber à la fois les voies Wnt canoniques et alternes, tandis que ceux de la classe Dkk inhibent spécifiquement la voie classique.
Les sFRP, au nombre de huit, sont des antagonistes qui se lient directement aux glycoprotéines Wnt. Une nomenclature existe seulement pour cinq d’entre eux, de sFRP1 à sFRP5. Sur la base des homologies de séquence, sFRP1, 2 et 5 forment un sous-groupe, tout comme sFRP3 et 4. Bien que les sFRP soient sécrétés, notamment par les cellules leucémiques et les ostéoblastes (composés structurels et fonctionnels majeurs de la niche médullaire), il a été rapporté que des sFRP synthétisés par des cultures cellulaires étaient retrouvés dans la membrane plasmique et/ou dans la MEC. Des expériences ont permis de montrer que durant le développement, les sFRPs pourraient ne pas fonctionner tout le temps comme des antagonistes de la voie Wnt. De plus, ils sont très importants pour réguler la croissance cellulaire, notamment en cas de cancers, où cette dernière fonction devient accrue et non contrôlée pour les cellules tumorales.
La famille Dkk comprend quatre membres, de Dkk-1 à Dkk-4, et une protéine apparentée à Dkk-3, appelée Soggy (Sgy) ou Dkk-11. Le membre de cette famille le plus étudié est Dkk-1. Il pourrait être un important médiateur de l’apoptose induite par des stimuli variés. En outre, en inhibant la voie Wnt canonique, après une inactivation suivie d’une internalisation des LRP5/6, Dkk empêche la formation du complexe LRP5/6-Wnt-Frizzled. Ceci entraîne la phosphorylation et la dégradation de la β-caténine, et donc l’inhibition de la voie Wnt classique, alors que le complexe Wnt-Frizzled peut activer les autres voies alternes, comme celle induisant la polarité planaire cellulaire.