HAL Id: dumas-01690147
https://dumas.ccsd.cnrs.fr/dumas-01690147
Submitted on 22 Jan 2018
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
L’iChemExplorer : fonctions et évaluations
Romain Pierre
To cite this version:
Romain Pierre. L’iChemExplorer : fonctions et évaluations. Chemo-informatique. 2016. �dumas-01690147�
Conservatoire National des Arts et Métiers
Paris
Mémoire
Présenté en vue d’obtenir
Le diplôme d’ingénieur du CNAM
SPECIALITE CHIMIE
Par
M Romain PIERRE
L’iChemExplorer® : Fonctions et Évaluations
Soutenu le 29 septembre 2016
JURY
PRÉSIDENT : Pr. Marc PORT, CNAM
MEMBRES : Pr. Clotilde FERROUD, CNAM
Dr. Maité SYLLA, CNAM
Dr. Gilles OUVRY, Galderma R&D
Dr. Craig HARRIS, Galderma R&D
I
Remerciements
Tout d’abord, je souhaite remercier le directeur du département de Recherche Mr. Laurent Hennequin de m’avoir permis d’effectuer mon stage au sein du département de Recherche de Galderma R&D.
Je remercie également la directrice du service Chimie de Recherche Mme Claire Bouix-Peter pour l’ensemble des moyens mis à ma disposition pour effectuer mon stage dans les meilleures conditions.
Je tiens à témoigner toute ma reconnaissance à Gilles Ouvry, mon maître de stage, pour sa disponibilité ainsi que ses conseils avisés tout au long de mon stage et de mon cursus au CNAM.
Je tiens également à remercier mes encadrants en laboratoire Craig Harris et Loïc Tomas pour leur disponibilité, leur aide précieuse en synthèse organique ainsi que le savoir qu’ils m’ont transmis.
Je remercie également ma responsable Laurence Clary de son soutien et de son aide.
Je remercie aussi chaleureusement Grégoire Mouis, Michelle Aurelly et Ghizlane El-Bazbouz, pour leur aide en chimie analytique et leur support.
Je tiens également à remercier l’ensemble de l’équipe de Chimie de Recherche Céline Bonneaud, Karine Bouquet, Bénédicte Dréan, Laurence Dumais, Frédéric Gaigne, Jing Jing, Laura Gauthier, Guillaume Lafitte, Corinne Millois, Véronique Parnet, Catherine Raffin, Sandrine Talano ainsi que les designers Yushma Bhurruth-Alcor, Jean-François Fournier et Branislav Musicki, et l’équipe du développement chimique, Jean-Marie Arlabosse, Jean-Guy Boiteau, Sébastien Daver, Thibaud Gerfaud, Cédric Martin, Christine Moureou, Franck Muller, Nicolas Rodeville et Samuel Tabet pour la bonne ambiance et l’esprit d’équipe que nous partageons depuis bientôt 9 ans.
Je souhaite aussi remercier le personnel du CNAM spécialité chimie et plus particulièrement les professeurs Ferroud, Guy, Port et Sylla pour leur précieux conseils ainsi que leur disponibilité et leur aide tout au long de ma formation au CNAM.
Enfin je souhaite remercier mes proches qui m’ont aidé, soutenu dans les moments les plus compliqués de ma formation et encouragés à poursuivre dans cette voie.
II
Table des matières
1 INTRODUCTION 1
2 MATERIEL 4
2.1 DESCRIPTION DE L’ICHEMEXPLORER® 4 2.2 DESCRIPTIONS DES METHODES ANALYTIQUES 7 2.3 DESCRIPTION DE LA METHODE DE TRANSFERT DE L’UHPLC VERS L’HPLC PREPARATIVE 9
3 ÉVALUATION DE L’ICHEMEXPLORER® 14
3.1 OPTIMISATION DE CONDITIONS REACTIONNELLES 15 3.1.1 OPTIMISATION DES CONDITIONS DE SYNTHESE DE L’OXAZOLIDINONE 1 15 3.1.2 OPTIMISATION DES CONDITIONS DE SYNTHESE DE L’AMINOALCOOL 2 19
3.2 ÉTUDE CINETIQUE 22
3.3 APPLICATION A LA CHIMIE COMBINATOIRE 24 3.4 SUIVI CINETIQUE CHIRAL EN TEMPS REEL 32 3.5 L’UTILISATION DES PALIERS DE TEMPERATURE 35
4 LA SYNTHESE D’ACIDES HYDROXAMIQUES 35
4.1 BIBLIOGRAPHIE 35
4.1.1 HYDROXYLAMINOLYSE DES ESTERS A L’AIDE D’UNE BASE INORGANIQUE 37 4.1.2 HYDROXYLAMINOLYSE DES ESTERS CATALYSEE PAR LE CYANURE DE POTASSIUM 39 4.1.3 REACTION D’HYDROXYLAMINOLYSE AVEC LES BASES ORGANIQUES 41
4.2 PARTIE EXPERIMENTALE 43
4.2.1 OPTIMISATION DES CONDITIONS 43
4.2.2 OPTIMISATION DE LA STŒCHIOMETRIE 50
4.2.3 FORMATION D’ESTERS ACTIVES ET DE COMPOSES A BASE DE SQUARATES POUR
CATALYSER LA REACTION D’HYDROXYLAMINOLYSE 52
4.2.4 UTILISATION DU TRIFLUOROETHANOL 55
4.2.5 CONDITIONS OPTIMISEES 56
4.2.6 UTILISATION DE BASE SUPPORTEE 56
III
4.2.8 REPRODUCTIBILITE DE LA METHODE AVEC D’AUTRES NUCLEOPHILES 64 4.2.9 VERIFICATION DE L’OPTIMISATION DE LA REACTION D’HYDROXYAMINOLYSE AVEC LE
LOGICIEL MODDE® 67
5 PROTECTION DES URACILES 68
5.1 BIBLIOGRAPHIE 68
5.2 PARTIE EXPERIMENTALE 72
5.2.1 CHOIX DU SOLVANT 73
5.2.2 CHOIX DE LA BASE 74
5.2.3 OPTIMISATION DE LA STŒCHIOMETRIE DES REACTIFS ET APPORT D’UN AJOUT DE DMAP 76
5.2.4 OPTIMISATION DES CONDITIONS DE FONCTIONNALISATION DE NOTRE URACILE 78 5.2.5 APPLICATION DE NOTRE MODE OPERATOIRE A D’AUTRES URACILES SUBSTITUES 80
6 ARBRE DE DECISION : AVANTAGES ET INCONVENIENTS DE
L’ICHEMEXPLORER® 87
7 CONCLUSION ET PERSPECTIVES 90
8 PARTIES EXPERIMENTALES 91
ANALYSES ET CARACTERISATIONS DES COMPOSES SYNTHETISES 91 8.1 EXEMPLE DE SYNTHESE DU COMPOSE 1 92 8.2 EXEMPLE DE SYNTHESE DU COMPOSE 2 92 8.3 EXEMPLE DE SUIVI CINETIQUE DE DEGRADATION DU COMPOSE 22 93 8.4 EXEMPLE DE SYNTHESE DU COMPOSE 26-1 93
8.5 SYNTHESE DU COMPOSE 85 94
8.6 SYNTHESE DU COMPOSE 81 94
8.7 EXEMPLE DE SYNTHESE DU COMPOSE 82 95
8.8 SYNTHESE DU COMPOSE 124 96
8.9 SYNTHESE DU COMPOSE 128 96
8.10 MODE OPERATOIRE GENERALE POUR L’OPTIMISATION DE LA SYNTHESE DES COMPOSES
119,125,141. 97
8.11 EXEMPLE DE SYNTHESE DES COMPOSES 137 ET 139 98
IV
8.13 EXEMPLE D’ESSAI DE FORMATION DU COMPOSE 125 EN UTILSANT LE TRIFLUOROETHANOL
99
8.14 SYNTHESE DES MOLECULES 149 ET 150 100
8.15 EXEMPLE DE SYNTHESE DU COMPOSE AVEC LA BASE SUPPORTE 101
8.16 SYNTHESE DU COMPOSE 152 102
8.17 SYNTHESE DU COMPOSE 153 102
8.18 SYNTHESE DU COMPOSE 158 103
8.19 SYNTHESE DU COMPOSE 155 104
8.20 MODE OPERATOIRE GENERALE POUR L’OPTIMISATION DE LA SYNTHESE DU COMPOSE 192
104
8.21 MODE OPERATOIRE DE LA SYNTHESE DU COMPOSE 194 105
V
Index des figures
Figure 1. Gammes de produits commercialisés dans 80 pays par GALDERMA ... 1
Figure 2 Description de l’iChemExplorer® ... 4
Figure 3. L'iChemExplorer® et le module de contrôle de la température par le groupe thermostaté ... 5
Figure 4. Chaine HPLC Agilent 1100 ... 5
Figure 5. Flacons avec filtre intégré ... 6
Figure 6. "iSample" programmation de la séquence ... 6
Figure 7. Partie "iGraph" retraitement des données dans le logiciel de l'iChemExplorer® ... 7
Figure 8. HPLC préparative Waters ... 9
Figure 9. Chromatogramme de l’analyse UHPLC de la coumarine ... 10
Figure 10. Chromatogramme pour la détermination du retard gradient de l'UHPLC Agilent 1290 ...11
Figure 11. Modèle de rétention pour les molécules neutres de S. Heinisch ... 13
Figure 12. Chromatogramme de la purification de la coumarine par HPLC prépartaive ... 14
Figure 13. Oxazolidinone 1 et amino-alcool 2. ... 15
Figure 14. Exemple de réaction de couplage d’Ullamn et de Buchwald développées par l'équipe de S.Cacchi ...16
Figure 15. Produit de départ pour la formation des composés 1 et 2 ... 16
Figure 16. Exemple de substitution nucléophile aromatique sur les oxazolidinones ... 17
Figure 17. Synthèse par substitution nucléophile aromatique par l’équipe de Lamb et de Zhu ... 20
Figure 18. Chromatogramme de la substitution nucléophile aromatique en présence de carbonate de potassium ...20
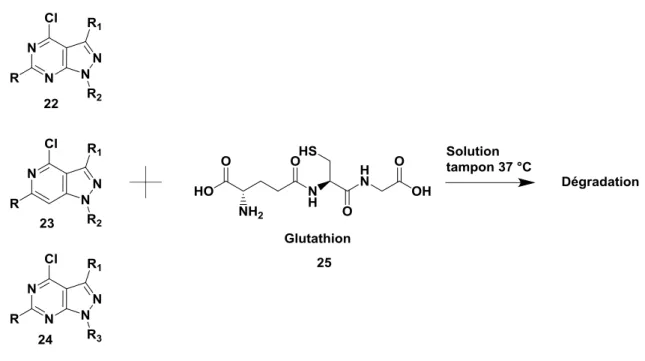
Figure 19. Étude de la dégradation entre les composés 22, 23 et 24 et le glutathion ... 23
Figure 20. Graphique de la dégradation du composé 22 ... 23
Figure 21. Librairie pour la chimie combinatoire ... 24
Figure 22. Synthèse finale de la série de composés 26 développée au laboratoire ... 25
Figure 23. Appareil de séchage Génévac® HT6 ... 25
Figure 24. Mécanisme réactionnel proposé pour la synthèse du composé 76 ... 30
Figure 25. Mécanisme réactionnel proposé de la dégradation du composé 26-16... 31
Figure 26. Synthèse du composé 87 avant optimisation ... 32
VI
Figure 28. Synthèse optimisée du composé 82 ... 34
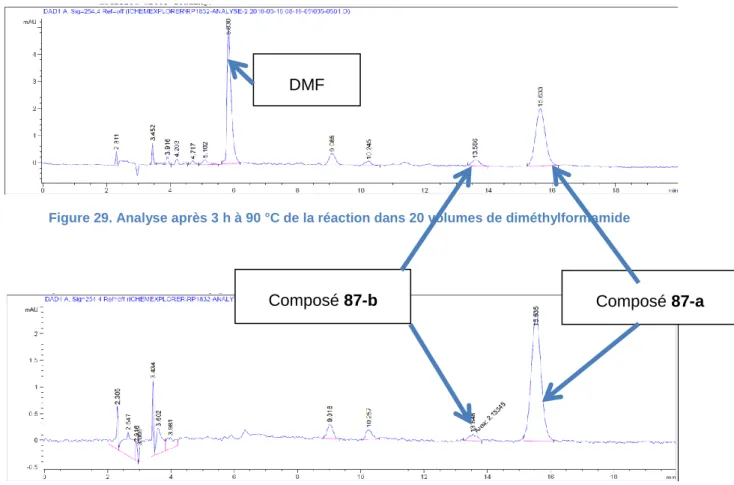
Figure 29. Analyse après 3 h à 90 °C de la réaction dans 20 volumes de diméthylformamide ... 34
Figure 30. Analyse après 3 h à 90 °C de la réaction dans 60 volumes d'acétonitrile ... 34
Figure 31. Formule générale et pKa de la fonction acide hydroxamique ... 35
Figure 32. Structure de produit sur le marché possédant une fonction acide hydroxamique . 36 Figure 33. Synthèse du Panobinostat® ... 37
Figure 34. Schéma réactionnel suggéré de la formation d'acide hydroxamique par action d'une base inorganique ...38
Figure 35. Schéma du système de chimie de flux... 38
Figure 36. Synthèse du Vorinostat ® par chimie de flux ... 39
Figure 37. Schéma réactionnel hydrolyse d'un ester en milieu basique ... 40
Figure 38. Schéma réactionnel suggéré pour la synthèse d'hydroxylaminolyse avec l'utilisation de cyanure de potassium catalytique ...41
Figure 39. Formes isoélectriques des amidines et des guanidines ... 42
Figure 40. Exemple de bases organiques ... 42
Figure 41. Molécule outil 118 et conditions pour l'optimisation de la synthèse d'acide hydroxamique ...43
Figure 42. Chromatogramme de la synthèse d'acide hydroxamique dans les conditions du Panobinostat® ...43
Figure 43. Proposition de schéma réactionnel pour l'hydroxylaminolyse en présence de TBD ... 46
Figure 44. Réaction d'hydroxyaminolyse sur le composé 124 ... 46
Figure 45. Cinétique de la réaction d'hydroxylaminolyse en utilisant le TBD comme base ... 47
Figure 46. Cinétique de la réaction d'hydroxylaminolyse en utilisant le DBU comme base ... 47
Figure 47. Diagramme représentant l'excès d'acide hydroxamique versus l'acide carboxylique en fonction du solvant utilisé au bout de 16 h ...48
Figure 48. Synthèse d'une nouvelle molécule outils pour l'optimisation de la stœchiométrie 50 Figure 49. Évolution de la formation de l'acide hydroxamique en fonction du temps et de la stœchiométrie de la base ...51
Figure 50. Évolution de la formation de l'acide hydroxamique en fonction du temps et de la stœchiométrie de l'hydroxylamine ...52
Figure 51. Structure des précurseurs d’esters activés ... 53
VII
Figure 53. Synthèse des composés 137 et 139 selon la méthode de Taylor ... 54
Figure 54. Formation d'ester activé dans la réaction d'hydroxyaminolyse ... 54
Figure 55. Exemple de formation d'ester activé avec le trifluoroéthanol selon les travaux de Jamieson ...55
Figure 56. Résine de Merrifield avec un TBD greffé ... 57
Figure 57. TBD greffé sur un support de silice ... 58
Figure 58. TBD greffé sur un support de JandaJel ... 58
Figure 59. Chromatogramme de la réaction d'hydroxylaminolyse du composé 128 au bout de 3 jours ... 59
Figure 60. Synthèse de nouvelles molécules outils permettant d’avoir une cinétique plus courte ...59
Figure 61. Chromatogramme réaction avec le réactif non supporté au bout de 5 minutes .... 60
Figure 62. Chromatogrammes après 16H de la réaction d'hydroxylaminolyse en présence des différentes bases supportées à notre disposition ...60
Figure 63. Chromatogramme de la réaction d'hydroxyaminolyse en présence de base supportée sans reactivation entre les deux réactions ...61
Figure 64.Chromatogramme de la réaction d'hydroxyaminolyse en présence de base supportée avec réactivation par une solution d'ammoniaque 7 N dans le méthanol entre les trois réactions ...61
Figure 65. Structure de la résine du Tentagel ... 62
Figure 66. Représentation des hydrogènes en α des esters des composés 170 et 171 ... 62
Figure 67. Réactions entre les composés 149 et différents nucléophiles en présence de 2 équivalents de TBD dans 5 volumes de méthanol et structure de l’impureté principale ...65
Figure 68. Exemple d’une représentation de résultat avec MODDE® ... 67
Figure 69. Plan d'expérience définit par MODDE® pour la synthèse d'hydroxyaminolyse .... 68
Figure 70. Schéma d'un uracile ... 68
Figure 71. Exemples de produits sur le marché comportant un motif uracile ... 69
Figure 72. Exemple d'alkylation direct en N3 développé par le groupe de Zhu ... 69
Figure 73. Voie de synthèse de la fonctionnalisation d'un uracile via une protection en N3 ... 70
Figure 74. Déprotection du groupement benzhydryle ... 70
Figure 75. Exemple de protection et alkylation du 6 amino uracile ... 71
Figure 76. Exemple de synthèse du groupe de Nomura ... 71
Figure 77. Exemple de di-protection par un groupement Boc suivi par la mono déprotection ... 72
VIII
Figure 78. Voie de synthèse envisagée pour la fonctionnalisation de l'uracile ... 72
Figure 79. Protection du composé 192 ... 73
Figure 80.Cinétique de la formation du composé 192 ... 74
Figure 81. Cinétique de la formation du composé 192 en fonction de la base ... 75
Figure 82. Intermédiaire activé par la DMAP ... 77
Figure 83. Chromatogramme de la réaction d'alkylation avec l'iodométhane au bout de 16 h ... 78
Figure 84. Uraciles 216 à 224... 80
Figure 85. Schéma réactionnel de l'étape de protection ... 84
IX
Index des Tableaux
Tableau 1. Gradient mis en place sur la chaine analytique HPLC Agilent® 1100 ... 8
Tableau 2. Gradient mis en place pour la chaine analytique UHPLC Agilent® 1290 ... 8
Tableau 3. Gradient mis en place pour les produits polaires sur la chaine analytique HPLC Agilent® 1100 ... 9
Tableau 4. Gradient appliqué pour la purification HPLC préparative de la coumarine ... 14
Tableau 5. Conversion en composé 1 et 2 en fonction de la base ... 18
Tableau 6. Conversion en composé 1 et 2 en fonction du solvant et de la température ... 19
Tableau 7. Conversion en composé 2 en fonction de la base et du solvant ... 21
Tableau 8. Série des anilines et des composés synthétisés par chimie combinatoire ... 29
Tableau 9. Résultats de la pureté optique du composé 82 en fonction du solvant et du volume de ce dernier ...33
Tableau 10. Comparaison de la formation d'acide hydroxamique avec et sans KCN catalytique réalisée par l’équipe de Ho ...40
Tableau 11. Résultats de la réaction d'hydroxyaminolyse en fonction de la base ... 45
Tableau 12. Propriétés des solvants organiques ... 49
Tableau 13. Rendement de la réaction d’hydroxyaminolyse en fonction du nombre d'équivalent de base ...51
Tableau 14. Rendement de la réaction d’hydroxyaminolyse en fonction du nombre d'équivalent d'hydroxylamine ...52
Tableau 15. Conversion en acide hydroxamique 141 en fonction du volume de trifluooéthanol ... 56
Tableau 16. Puretés optiques en fonction des conditions utilisées pour le composé 149 ... 63
Tableau 17. Résultats des puretés optiques en fonction des conditions utilisées pour le composé 150 ...64
Tableau 18. Conversion et rendement de la réaction entre le composé 149 et différents nucléophiles en présence de TBD ...66
Tableau 19. Résultats de l'évaluation du solvant pour la protection sélective ... 74
Tableau 20.Résultat de l'évaluation de la base pour la protection sélective ... 75
Tableau 21. Résultats de la variation de la stœchiométrie de la base ... 76
Tableau 22. Résultats de la variation de la stœchiométrie du chlorure de pivaloyle ... 77
Tableau 23. Effet de l'ajout de la DMAP sur la réaction ... 78
Tableau 24. Optimisation de l'alkylation d'iodométhane... 79
X
Tableau 26. Résultat de la réaction de protection des composés 199 à 202 ... 83 Tableau 27. Résultat de la réaction de protection des composés 219 et 220 ... 83 Tableau 28. Résultats de la réaction d'alkylation ... 86
XI
Abréviations
ACE : Enzyme convertissant l’angiotensine
ACN : Acétonitrile
AcOEt : Acétate d’éthyle
ADN: Acide désoxyribonucléique
BEMP: 2-tert-Butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine
BiPy: 2,2-bipyridine
Boc: Tert-butoxycarbonile
n-BuOH: n-Butanol
CMR : Cancérigène, mutagène et reprotoxique
Cs2CO3: Carbonate de césium
CuI: Iodure de cuivre
Cu(OAc)2 : Acétate de cuivre II
DBU: 1,8-Diazabicyclo [5.4.0] undéc-7-ène
DBN: 1,5-Diazabicyclo [4.3.0] non-5-ène
DCM: Dichlorométhane
DEDL : Détecteur évaporatif à diffusion de lumière
DIPEA: Diisopropyléthylamine DMAP : Diméthylaminopyridine DMF : Diméthylformamide DMSO : Diméthylsulfoxyde équiv : Equivalent h : heures
XII HOAt: 7-aza-1-Hydroxybenzotriazole
HOBt: 1-Hydroxybenzotriazole
HPLC : Chromatographie liquide à haute performance
Imp : Impuretés
KCN : Cyanure de potassium
K2CO3 : Carbonate de potassium
KOH : Hydroxyde de potassium
MMP : Enzymes Matrix MétalloProtéases
min : minutes
Na2CO3 : Carbonate de sodium
NaH : Hydrure de sodium
NaOH : Hydroxyde de sodium
NaOtBu : tert butanolate de sodium
NEt3: Triéthylamine
NMP : N-méthyl-2-pyrrolidone
Oxyma: (2Z)-cyano(hydroxyimino)acétate d’éthyle
Pd2(dba)3 : Tris(dibenzylidèneacétone) dipalladium
pKa: Constante de dissociation du proton
R&D : Recherche et Développement
RMN : Résonance Magnétique Nucléaire
SAR : Relation structure activité
SFC : Chromatographie par Fluide Super Critique
TBAF : Fluorure de tétrabuthylammonium
XIII tBuOK : tert-butanolate de potassium
THF: Tétrahydrofurane
Tr: Temps de rétention
Troc : Formiate de trichloroéthyle
1
1
Introduction
La recherche pharmaceutique est un domaine extrêmement concurrentiel. L’innovation est le principal outil utilisé par les entreprises pour se démarquer des autres compagnies.
Dans ce milieu, le temps est une denrée rare. Chaque entreprise cherche à apporter la meilleure solution le plus rapidement possible pour répondre aux attentes des patients. De ce fait, il est nécessaire d’améliorer les procédés pour accélérer la découverte de nouveaux composés et leur mise sur le marché.
1.1
Présentation de Galderma R&D
Ce stage a été effectué au sein de l’entreprise Galderma R&D. Cette dernière est née de la joint-venture en 1981 de l’Oréal et Nestlé avec comme objectif de devenir le leader mondial de la dermatologie. Galderma R&D couvre principalement trois segments importants qui sont les médicaments de prescription (axe principal d’origine), l’automédication et les produits esthétiques et correctifs. Dans chacun de ces segments, Galderma R&D a développé une large gamme de produits commercialisés dans plus de 80 pays (Figure 1).
Figure 1. Gammes de produits commercialisés dans 80 pays par GALDERMA
Galderma R&D propose ainsi des traitements pour une large gamme de maladies telles que l’acné (Différine®, 1995 ; Epiduo®, 2008 ; Cétaphil®, 2010), la rosacée (Oracéa®, 2008 ; Soolantra®, 2015), le psoriasis (Vertical®, 2009), l’onychomycose (mycose des ongles-Locéryl Pro® anciennement Curanail), le cancer de la peau et la dégénérescence cutanée due au soleil (Metvix®, 2001). L’entreprise propose également des produits esthétiques pour le comblement des rides (Azzalure®, 2009 ; Restylane®, 2011) ou encore des produits de protection solaire (Daylong®).
2
Investisseur de premier plan en recherche dermatologique, Galderma R&D est aujourd’hui le leader de dépôt de brevets couvrant les maladies de la peau, des cheveux et des ongles, avec plus de cinquante demandes annuelles (55 dépôts en 2014). Elle s’appuie notamment sur un pôle R&D de pointe répartie sur cinq grands sites internationaux ainsi que trois sites de production :
Sophia-Antipolis (France), site spécialisé en recherche et développement de molécules bioactives. Il est aujourd’hui le plus grand centre de recherche dermatologique mondial avec plus de 500 collaborateurs.
Egerkingen (Suisse), site de recherche et de production, spécialisé sur les pathologies dues au soleil.
Uppsala (Suède), site de recherche, de développement et de production spécialisé dans les produits correctifs et esthétiques.
Forth Worth (États-Unis) et Tokyo (Japon), sites de développement spécialisés dans le développement clinique des produits de prescription.
Alby-sur-Chéran (France), la Baie d’Urfé (Canada) et Hortolândia (Brésil), sites de production.
En 2014, Nestlé ouvre sa branche Nestlé Skin Health en devenant le seul actionnaire de Galderma R&D. En 2015, Galderma R&D acquiert Innéov pour développer une gamme de produits sur le marché nutraceutique. Enfin, en 2016 Nestlé Skin Health et Guthy-Renker forment une joint-venture nommé « The Proactiv Company ». Cette entité est dédiée au traitement des peaux à tendance acnéique, ce qui permet à Galderma R&D d’augmenter sa présence sur le marché Nord-Américain.
La principale mission du service de Chimie de Recherche de Galderma R&D est de produire des molécules susceptibles d’interagir avec des cibles biologiques identifiées en amont. Ce service peut être divisé en quatre structures distinctes :
- Le Design : il s’agit de la conception des molécules, en tenant compte de différents critères biologiques et chimiques, comme la stabilité métabolique, la solubilité ou les interactions avec la cible.
- La Synthèse : celle-ci regroupe la partie identification des approches de synthèse et l’élaboration finale des composés.
- Le service d’Analyse et Structure : il s’agit d’un service support pour aider à la détermination de structure et à l’analyse des composés.
3
-Le service de gestion des molécules qui s’occupe de gérer et de distribuer les composés.
Définir la voie de synthèse d’une molécule nécessite souvent de nombreuses mises au point. Chez Galderma R&D, le temps médian nécessaire pour produire un composé est de 23 jours. Pour réduire ce délai, nous avons décidé d’évaluer l’iChemExplorer®. Cet appareil qui permet d’effectuer et d’analyser des synthèses en parallèle va contribuer à l’évaluation rapide de différentes conditions de synthèse et à sélectionner les plus efficaces.
1.2 Présentation du stage
Dans une première partie, nous décrirons l’iChemExplorer® et son environnement analytique. Dans une deuxième partie, nous évaluerons ses capacités et son apport au sein du service de chimie de recherche. Puis nous nous pencherons sur deux études approfondies qui nous ont permis d’optimiser la synthèse d’acide hydroxamique et la protection sélective d’uracile. Finalement, un arbre décisionnel sera établi pour définir l’utilisation et les limites de l’iChemExplorer®. Des opportunités d’évolution et d’amélioration seront aussi suggérées.
4
2
Matériel
Dans un souci constant d’amélioration de nos procédés de synthèse, nous avons décidé d’investir dans un appareil permettant de suivre plusieurs réactions en parallèle et ainsi d’optimiser rapidement des conditions réactionnelles. Après avoir évalué différentes propositions des fournisseurs, nous avons sélectionné l’iChemExplorer® tant pour ses capacités plus développées que pour son coût raisonnable.
Dans ce chapitre, nous décrirons dans un premier temps l’appareil, puis les méthodes analytiques utilisées au cours du stage. Enfin nous détaillerons la mise en place d’une méthode de transfert de l’UHPLC vers l’HPLC préparative.
2.1
Description de l’iChemExplorer®
L’iChemExplorer® (Figure 2) est un module qui permet d’effectuer un suivi analytique et d’étudier la cinétique des réactions de manière automatique.
Figure 2 Description de l’iChemExplorer®
Il permet une agitation magnétique ainsi qu’un chauffage ou un refroidissement linéaire ou par palier des milieux réactionnels à l’aide d’une plaque thermoélectrique ou d’un groupe thermostaté (Huber®). Pour ce dernier, une programmation préalable de l’iChemExplorer® est effectuée afin de piloter la variation de température. Les températures sont fonction des capacités du système de chauffage et de refroidissement ainsi que du liquide employée dans ce dernier. (Figure 3).
Réglage de l’agitation magnétique Plaque chauffante thermoélectrique Affichage de la température Échantillons
5
Figure 3. L'iChemExplorer® et le module de contrôle de la température par le groupe thermostaté
L’iChemExplorer® s’insère dans une chaine HPLC Agilent® 1100 (Figure 4) ou dans une UHPLC Agilent® 1260 ou 1290. Ces chaines possèdent un plateau d’échantillonnage fixe compatible avec le module. De plus, son logiciel ne peut piloter à ce jour que des appareils Agilent®.
.
Figure 4. Chaine HPLC Agilent 1100
Un maximum de 48 expériences, avec des flacons de 2 mL, ou 16 expériences, avec des flacons de 6 mL, peuvent être effectuées en parallèle. Pour les milieux réactionnels hétérogènes comme les réactions catalysées par les métaux, il existe des flacons de 2 mL avec un filtre intégré (Figure 5). Ainsi on évite toute précipitation dans l’injecteur ou sur la colonne de l’HPLC. Plaque chauffante ou refroidissante piloté par le groupe thermostaté
6
Figure 5. Flacons avec filtre intégré
Le logiciel pilote est simple d’utilisation et comporte de trois modules. Le premier, « iSample », permet de programmer la séquence. Les informations relatives aux réactions, à la méthode analytique utilisée et à la durée du suivi y sont renseignées. Puis en sélectionnant « transfert to chemstation », le logiciel transfère la séquence créée directement sur la chaine HPLC ou UHPLC. (Figure 6)
Figure 6. "iSample" programmation de la séquence
Le deuxième élément « iHeat » permet de piloter le chauffage et le refroidissement du milieu réactionnel au cours du temps. Le dernier élément du logiciel « iGraph » permet de retraiter les données. Les analyses et les résultats sont traités directement sur l’appareil et exportés pour une utilisation externe. Les pics d’intérêt peuvent être identifiés et les impuretés exclues via la fonction treshold (« Tresh ») et tolérance «Tol » (Figure 7).
7
Figure 7. Partie "iGraph" retraitement des données dans le logiciel de l'iChemExplorer®
De nombreux modèles de rapports spécifiques générés automatiquement sont à notre disposition en fonction de l’étude menée (dégradation ou cinétique). Ces rapports permettent d’accéder directement aux chromatogrammes et aux données mathématiques comme l’aire d’un pic et son évolution au cours du temps. Les données sont générées dans un tableur Exce®lpermettant leur exploitation sous d’autres formats.
À ce jour, seule la détection UV-visible est à notre disposition mais d’autres types de détecteurs tels qu’un spectromètre de masse ou un DEDL peuvent être utilisés. Ce dernier permet d’analyser les composés sans chromophore.
Pour ce stage, les réactions ont été effectuées sur une HPLC Agilent® 1100 pour la première partie du stage, puis, dans une deuxième partie, sur une UHPLC Agilent® 1290 couplée avec un détecteur UV.
2.2
Descriptions des méthodes analytiques
Au cours de mon stage trois méthodes analytiques différentes ont été mises en place. La première méthode est une méthode HPLC (Méthode A). Elle utilise une colonne analytique Kinetex® C18 2.6µM 50*4.6 mm de Phénomenex® à particules partiellement poreuses. Cette colonne permet d’avoir une perte de charge plus faible et donc des débits plus élevés, des temps d’analyses plus courts et des résolutions équivalentes à des équipements UHPLC. Le gradient est composé d’un mélange eau / acétonitrile et 0.1% d’acide formique comme phase mobile avec un débit de 1 mL / min. Un gradient de 5 minutes allant de 5 à 95% d’acétonitrile permet d’avoir une résolution suffisante. Il est suivi
8
par une purge de la colonne et un rééquilibrage de celle-ci. En tout, la méthode utilisée dure 10 minutes (Tableau 1).
Temps (min) % d’acétonitrile
0 5
5 95
8 95
8.10 5
10 5
Tableau 1. Gradient mis en place sur la chaine analytique HPLC Agilent® 1100
Une méthode UHPLC (Méthode B) a ensuite été utilisée permettant de réduire le temps d’analyse de 10 à 2,2 minutes. Une colonne analytique Acquity® CSH C18 1.7 µM 2.1*50 mm est utilisée avec comme phase mobile un mélange eau / acétonitrile et 0.1% d’acide formique à un débit de 0.8 mL / min (Tableau 2). Cette méthode a été utilisée pour effectuer un transfert de méthode de l’UHPLC vers l’HPLC préparative.
Temps (min) % d’acétonitrile
0 5
1.0 95
1.5 95
1.6 5
2.2 5
Tableau 2. Gradient mis en place pour la chaine analytique UHPLC Agilent® 1290
Les produits faiblement retenus dans les deux premières méthodes ont été analysés avec une méthode différente (Méthode C). Une colonne analytique Atlantis® 3 5 µM de waters® 3*150 mm permettant de retenir d’avantage les produits polaires en phase inverse a été utilisée. La durée de l’analyse est plus longue car elle ne possède pas la même taille de particules que la colonne précédente. Le gradient est composé d’un mélange eau / acétonitrile et 0.1% d’acide formique à un débit de 1 mL / min (Tableau 3).
9
Temps (min) % d’acétonitrile
0 0 2 0 9 95 12 95 12.2 0 15.0 0
Tableau 3. Gradient mis en place pour les produits polaires sur la chaine analytique HPLC Agilent® 1100
Plusieurs méthodes spécifiques ont donc été mises en place afin de permettre d’analyser l’ensemble des composés étudiés dans ce stage. Nous avons ensuite travaillé à la mise en place d’une méthode de transfert de l’UHPLC vers l’HPLC préparative.
2.3
Description de la méthode de transfert de
l’UHPLC vers l’HPLC
préparative
Afin de simplifier la purification des composés, une méthode de transfert a été mise en place de l’UHPLC vers l’HPLC-préparative. L’HPLC préparative est une chaine HPLC préparative Waters® gradient-system (Figure 8).
Figure 8. HPLC préparative Waters
Pour limiter les risques d’erreurs, la phase mobile, la phase stationnaire et la température de la colonne sont les mêmes pour les deux appareils. Les purifications ont été effectuées sur une colonne d’HPLC préparative Acquity® CSH C18 5 µM OBD 30*150 mm de Waters® équivalente à la colonne de l’UHPLC. Les débits utilisés sur les deux appareils sont les optimums des colonnes soit 0.8 mL / min pour l’analytique et 50mL / min pour celle de l’HPLC préparative.
10
Une analyse UHPLC du produit à purifier est d’abord effectuée. Le but est de déterminer la concentration d’élution Ce. Elle correspond à la concentration en acétonitrile à laquelle le
produit d’intérêt est élué. Cette donnée nous permet de déterminer une méthode d’élution isocratique où l’on estime avoir le meilleur rapport entre la séparation des composés et le temps de purification.
Pour mettre au point cette méthode la coumarine est utilisée comme produit de référence. Le temps de rétention sur l’analyse UHPLC de ce dernier est de 1.30 minutes soit 78 secondes (Figure 9).
Figure 9. Chromatogramme de l’analyse UHPLC de la coumarine
L’équation 1 permet de déterminer la constante d’élution Ce. 1
(
) ( ( ) ( ) ( ))
Ce : Concentration d’élution du produit désiré en acétonitrile Ci : Concentration initiale en acétonitrile de la méthode analytique Cf : Concentration finale en acétonitrile de la méthode analytique. Tg : Temps du gradient.
Tr : Temps de rétention produit analysé.
T0 : Temps mort, il s’agit du temps passé par le produit entre l’injecteur et le détecteur hors colonne aussi appelé volume extra-colonne.
Td : Retard gradient : il s’agit du délai entre la programmation du gradient et sa réponse en temps réelle.
11
Afin d’obtenir la concentration d’élution, les différentes constantes de l’équation 1 doivent être déterminées. Les concentrations initiales et finales, ainsi que la durée du gradient sont données dans la méthode analytique B (Tableau 2). Le retard gradient et le temps mort sont des valeurs spécifiques à chaque appareil HPLC et mesurables. Pour déterminer le temps mort de l’appareil, une injection d’acétone sans colonne sur l’appareil est effectuée. L’acétone étant UV-visible, le temps mis par ce produit correspond au temps mort entre l’injecteur et le détecteur. Ainsi le volume extra-colonne peut être déterminé à l’aide du débit machine.Puis le retard gradient Td est mesuré. Pour cela une solution eau / acétone (1/1) est préparée. Une analyse est réalisée sans colonne sur l’appareil où l’on effectue un gradient par palier avec comme éluant l’eau et le mélange eau / acétone. Le retard gradient correspond à la différence de temps entre le démarrage du gradient planifié et celui observé. Ici la mesure du gradient est une moyenne réalisée sur plusieurs paliers (Figure 10).
Figure 10. Chromatogramme pour la détermination du retard gradient de l'UHPLC Agilent 1290
La même opération est effectuée avec la colonne. La différence entre les deux mesures nous permet de déterminer les volumes morts de la colonne. Le même travail est effectué sur l’HPLC préparative. Pour nos systèmes, les résultats sont les suivants :
T0 (analytique) = 10 secondes
Td (analytique) = 20 secondes
T0 (Preparative) = 1.2 minutes
En utilisant ces valeurs mesurées, la Ce suivante pour la coumarine est obtenue à partir de
l’équation 1.
( ) ( ) ( ( ) ( ) ( )) ( ) %
12
Une fois la concentration d’élution du produit définie, la concentration finale à appliquer dans la méthode de purification est obtenue par l’équation 2.
( ) [ ]
Cfinal isocratique : Concentration d’acétonitrile défini pour que le produit ait un temps de rétention optimal en HPLC préparative.
[%MeCN] : Il s’agit d’une constante de correction à ajouter pour avoir un temps de rétention optimal en HPLC préparative.
Équation 2. Formule pour obtenir la concentration finale à appliquer
Cette différence de concentration est donnée par la formule du modèle de rétention (Équation 3).
( ) ( ) [ ]
Ki : Facteur de rétention défini pour les molécules neutres.
S : Coefficient de pente dépendant de l’environnement chromatographique pour les molécules neutres. K : Facteur de rétention du produit.
[%MeCN] : La différence de concentration à ajouter pour avoir un temps de rétention optimal en HPLC préparative.
Équation 3. Formule du modèle de rétention en mode d’élution isocratique 2
La valeur de la pente S pour les molécules neutres varie entre 0.02 et 0.04. Le facteur de rétention initiale Ki varie entre 1 et 1.5 en fonction des composés d’après le modèle de S. Heinisch 3 (Figure 11). Afin de simplifier le modèle, nous avons effectué une approximation en fixant la valeur de Ki à 1 et celle de la pente à 0.03. Afin d’avoir un modèle plus précis, il faudra étudier le facteur de rétention K de chaque molécule. Ce travail est en cours au sein du laboratoire pour déterminer les pentes des composés acides, basiques et amphotères, ainsi que leur facteur de rétention initiale Ki afin d’améliorer notre modèle.
13
Figure 11. Modèle de rétention pour les molécules neutres de S. Heinisch
Pour obtenir la valeur du facteur de rétention K de notre produit, l’équation 4 est utilisée.
( ) ( ) ( )
Tr : Temps rétention du produit en HPLC préparative. T0 prep : Temps mort de l’HPLC préparative. K : Facteur de rétention du produit.
Équation 4. Formule du facteur de rétention en mode d’élution isocratique
Le temps de la méthode en HPLC préparative est fixé à 15 minutes et un temps de rétention de nos produits d’environ 6 minutes est visé. Le facteur de rétention suivant est déduit de la formule suivante:
( ) ( ) ( )
La valeur de K est ensuite appliquée dans l’équation 3.
[ ] ( ) ( )
Cette valeur est ajoutée à la concentration d’élution Ce de l’équation 2 ce qui donne un
Cfinal isocratique de 57% acétonitrile pour la coumarine.
( ) [ ]
Un léger gradient d’élution de plus ou moins 4 points encadrant la valeur définie est mis en place en HPLC préparative. Ce gradient permet d’obtenir des pics en HPLC préparative de largeur plus fine et constante (Tableau 4).
14
Temps (min) % d’acétonitrile
0 53 2 53 9 61 12 61 12.5 95 15 95
Tableau 4. Gradient appliqué pour la purification HPLC préparative de la coumarine
Une injection en HPLC préparative de 50mg de coumarine est effectuée. Le temps de rétention de notre produit est de 5.5 minutes (Figure 12).
Figure 12. Chromatogramme de la purification de la coumarine par HPLC prépartaive
Cette méthode a été appliquée pour la purification des composés étudiés dans ce rapport mais aussi étendue aux autres projets de Chimie de Recherche de Galderma R&D.
Une fois ces différentes méthodes analytiques paramétrées, il est aussi possible de transférer les méthodes UHPLC analytique vers une méthode préparative et ainsi faciliter la purification.
Nous allons maintenant étudier et évaluer les capacités de cet appareil pour nos activités de recherche.
3
Évaluation de l’iChemExplorer®
À ce jour, peu de publications cite l’utilisation de l’iChemExplorer® 456, mais il est facile d’imaginer des applications possibles avec ce type d’appareil : l’optimisation de conditions
15
réactionnelles, l’étude de cinétique, ou encore une application à la chimie combinatoire. Nous avons donc voulu évaluer ces différentes fonctions en intégrant l’iChemExplorer® aux projets actifs au sein du laboratoire de Chimie de Recherche.
3.1
Optimisation de conditions réactionnelles
Afin d’évaluer les capacités de l’iChemExplorer®, nous avons effectué des optimisations de conditions sur les synthèses des composés 1 et 2 (Figure 13).
Figure 13. Oxazolidinone 1 et amino-alcool 2.
3.1.1 Optimisation des conditions de synthèse de l’oxazolidinone 1
Dans le cadre d’un programme de recherche en cours chez Galderma R&D l’accès au synthon 1 devait être développé.
De manière classique, ces synthons sont obtenus par réaction d’Ullman ou de Buchwald (Figure 14).78 Nous avons décidé de déprioriser ces approches car le manque de sélectivité entre un iode ou un brome et le chlore activé en ortho du groupement nitro pouvait être problématique. De plus les exemples développés, ci-dessous, par Cacchi et al. présentaient un encombrement stérique plus faible en alpha de l’azote.
16
Figure 14. Exemple de réaction de couplage d’Ullamn et de Buchwald développées par l'équipe de
S.Cacchi
La disponibilité et le faible coût du composé 10 (<5€/g) nous a incité à nous tourner vers une approche par SNAr (Figure 15).
Figure 15. Produit de départ pour la formation des composés 1 et 2
À nouveau, nous étions conscients que la réaction pouvait avoir lieu au niveau du chlore activé au cours d’une substitution nucléophile aromatique. Plusieurs auteurs (Natesan et al.9, Capraro et al.10 ont démontré la possibilité d’effectuer cette réaction. Les différences se situent au niveau de la base utilisée, des solvants et de la température de chauffage. (Figure 16).
17
Figure 16. Exemple de substitution nucléophile aromatique sur les oxazolidinones
En nous basant sur ces travaux, deux essais de substitution ont été testés au laboratoire. Un premier essai a été effectué au micro-onde à 170 °C pendant 15 min en présence DIPEA comme base et solvant. Cependant nous n’avons pas observé de conversion. Puis dans un deuxième essai nous avons utilisé l’hydrure de sodium comme base dans le DMF à température ambiante pendant 3 jours. Nous avons alors obtenu une faible conversion de 30%.
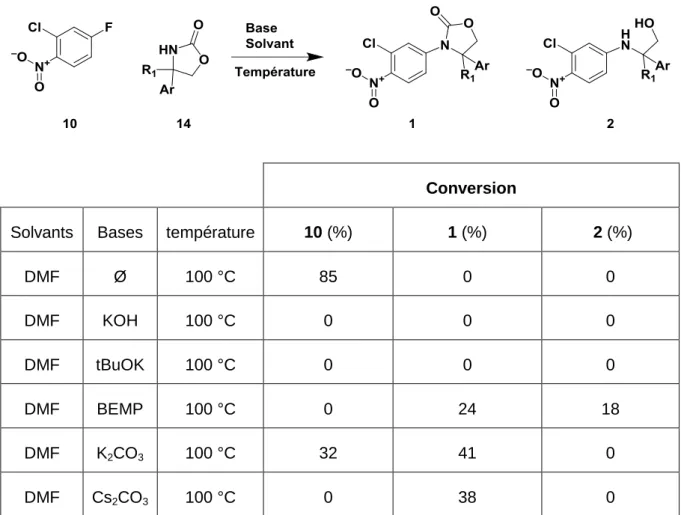
En nous basant sur les résultats des premiers essais, une exploration de conditions a été mise en place. Dans un premier temps nous allons évaluer l’impact de la base. Ces réactions ont été lancées dans 10 volumes de DMF et avec 2 équivalents de bases à 100 °C pendant 16 h. Grace au traitement des données sur l’iChemExplorer®, la meilleure base est identifiée rapidement. La réaction sans base ne permet pas d’obtenir de produit désiré. Par ailleurs, les réactions avec KOH et t-BuOK dégradent totalement le produit de départ. La réaction avec la base BEMP conduit à une conversion totale. Cependant seuls 24% du produit 1 se forment. Le produit secondaire majoritaire observé (18%) est l’aminoalcool 2 issu de l’ouverture de l’oxazolidinone in situ. Ceci pourrait être expliqué par une instabilité du produit 1 en présence d’une base forte et soluble dans le milieu réactionnel. Les réactions avec le K2CO3 et le Cs2CO3 montrent la formation respectivement de 41% et 38% de
composé 1, la différence se situant au niveau de la disparition totale du produit de départ dans le cas du Cs2CO3. Cela pourrait s’expliquer à nouveau par la solubilité accrue du
18
Conversion
Solvants Bases température 10 (%) 1 (%) 2 (%)
DMF Ø 100 °C 85 0 0 DMF KOH 100 °C 0 0 0 DMF tBuOK 100 °C 0 0 0 DMF BEMP 100 °C 0 24 18 DMF K2CO3 100 °C 32 41 0 DMF Cs2CO3 100 °C 0 38 0
Tableau 5. Conversion en composé 1 et 2 en fonction de la base
Nous avons décidé de continuer l’optimisation en fixant la base (Cs2CO3 pour avoir
une conversion totale) et en faisant varier le solvant et la température. Les réactions ont été lancées dans 10 volumes de solvant avec 2 équivalents de Cs2CO3 à 100 °C puis 80 °C pour
les solvants les plus intéressants pendant 16 h.
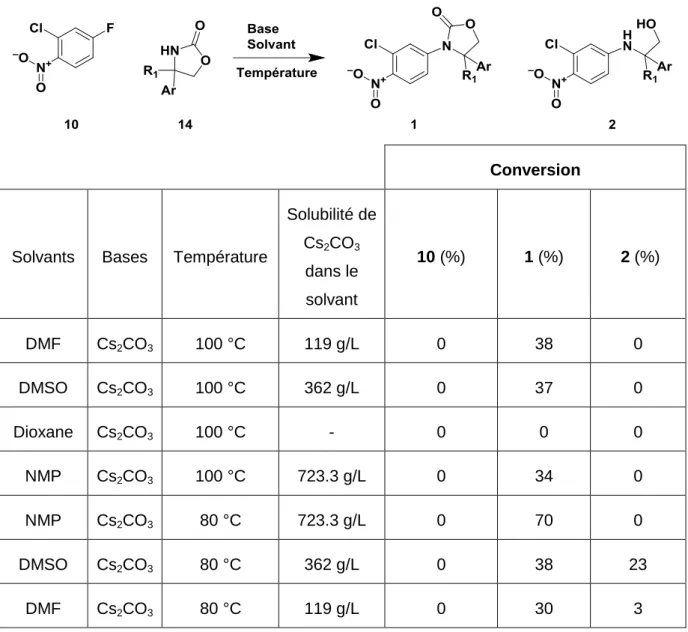
On observe que les réactions à 100 °C dans le NMP, le DMF et le DMSO donnent des résultats comparables, une formation de produit 1 variant entre 34% et 38%. A contrario la réaction dans le dioxane ne permet pas la formation du produit 1. Ce résultat pourrait être expliqué par la solubilité accrue du carbonate de césium dans les solvants protiques polaires (DMF, DMSO, et NMP). Les réactions dans les solvants protiques polaires ont été relancées à 80 °C. Le NMP montre les résultats les plus intéressants à cette température avec une conversion totale et la formation de 70% de composé 1 (Tableau 6).
19
Conversion
Solvants Bases Température
Solubilité de Cs2CO3 dans le solvant 10 (%) 1 (%) 2 (%) DMF Cs2CO3 100 °C 119 g/L 0 38 0 DMSO Cs2CO3 100 °C 362 g/L 0 37 0 Dioxane Cs2CO3 100 °C - 0 0 0 NMP Cs2CO3 100 °C 723.3 g/L 0 34 0 NMP Cs2CO3 80 °C 723.3 g/L 0 70 0 DMSO Cs2CO3 80 °C 362 g/L 0 38 23 DMF Cs2CO3 80 °C 119 g/L 0 30 3
Tableau 6. Conversion en composé 1 et 2 en fonction du solvant et de la température 11
Une autre approche en deux étapes a été explorée. Une étape de substitution nucléophile aromatique nous donnerait le composé 2 et une étape de cyclisation nous permettrait d’obtenir le composé 1 à partir cet aminoalcool.
3.1.2 Optimisation des conditions de synthèse de l’aminoalcool 2
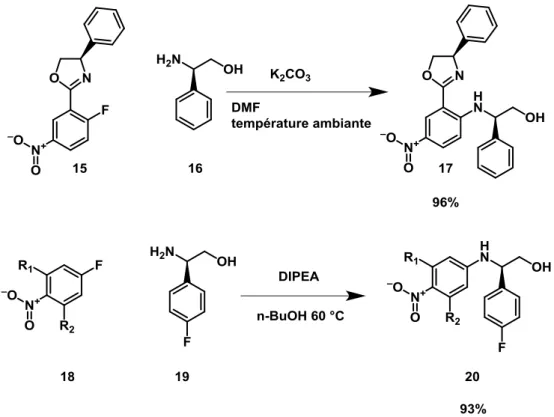
Cette approche a été inspirée des travaux du groupe Rengaraju et al. 12 L’étape de substitution reste l’étape clef, les conditions doivent permettre d’obtenir une bonne sélectivité entre la O-alkylation et la N-alkylation. Zhu et al. ont démontré la possibilité d’effectuer cette substitution nucléophile aromatique sélectivement sur l’azote, dans le DMF en présence de carbonate de potassium.13 Lamb et al. ont aussi décrit cette réaction de substitution dans le butanol en présence de diisopropyléthylamine à 60 °C(Figure 17). 14
20
Figure 17. Synthèse par substitution nucléophile aromatique par l’équipe de Lamb et de Zhu
Ces conditions ont été testées au laboratoire. La réaction avec la DIPEA n’a pas permis d’observer le produit. En présence de carbonate de potassium une conversion de 50% dans un mélange 2/1 de N- et de O-alkylation a été observée (Figure 18).
Figure 18. Chromatogramme de la substitution nucléophile aromatique en présence de carbonate de potassium
Produit de départ
N-alkylé
21
En nous basant sur les résultats du premier essai et le plan d’expérience du composé 1, une sélection de trois bases et cinq solvants, nous ont permis d’optimiser rapidement nos conditions. Cette fois-ci, nous avons décidé de faire varier le solvant et la base en parallèle et non pas en séquence. Le Cs2CO3 et la DIPEA ont été évalués avec le DMF, la NMP et le
DMSO, tandis que le BEMP a été évalué dans des solvants aprotiques et apolaires (DCM et Toluène). Le produit désiré a été formé dans toutes les conditions testées et nous n’avons pas vu de trace de l’isomère O-arylé (Tableau 7).
Solvant ε Base Température 10 (%) Imp (%) 2 (%)
DMF 36.7 Sans base 100 °C 81 19 0 DMF 36.7 Cs2CO3 100 °C 45 13 43 DMSO 46.7 Cs2CO3 100 °C 20 14 66 NMP 32.6 Cs2CO3 100 °C 39 11 49 DMF 36.7 DIPEA, 100 °C 67 10 23 DMSO 46.7 DIPEA 100 °C 78 6 16 NMP 32.6 DIPEA 100 °C 75 7 17 Toluène 2.4 BEMP 100 °C 47 33 19 DCM 8.9 BEMP 30 °C 54 9 37
Tableau 7. Conversion en composé 2 en fonction de la base et du solvant 15 16
Une fois les conditions optimales identifiées (surlignées dans les tableaux 6 et 7), les deux réactions ont été effectuées sur 500 mg de composé 10 et ont permis d’isoler 722 mg soit 63.5 % de rendement de l’oxazolidinone 1 et 495 mg soit 47% de rendement de l’aminoalcool 2.
22
L’iChemExplorer® a permis de rapidement tester différentes conditions opératoires en jouant sur les réactifs, les solvants et la température. Son impact en temps réel sur la résolution d’une problématique dans un projet actif a pu ainsi être démontré.
3.2
Étude cinétique
Avec des prélèvements HPLC automatiques et programmables, l’iChemExplorer® est un outil idéal pour suivre des cinétiques réactionnelles. Une série de composés développés dans un programme récent a montré un métabolisme particulier avec le déplacement d’un chlore électrophile par du glutathion dans des hépatocytes humains. Nous avons voulu vérifier si cette réaction avec le glutathion nécessitait une enzyme ou si elle pouvait se produire en présence de glutathion seul.
Le glutathion est un petit peptide composé de 3 acides aminés qui sont la glycine, la cystéine, et l’acide glutamique (Figure 19). Selon Mindell, il s’agit du « maître antioxydant ». En effet, il est présent dans 90% des cellules de notre organisme et permet de le protéger de tous les oxydants. 17
De nombreux auteurs ont travaillé sur le glutathion et sa capacité, à travers son groupement thiol, à se lier de façon covalente à certaines fonctions électrophiles comme le groupement nitrile, les accepteurs de Michaël ou encore les hétéroaromatiques pauvres en électron. 18192021 Cette réactivité peut dans certains cas être liée à une toxicité aigüe.
Nous nous sommes inspirés des conditions décrites dans les références citées ci-dessus afin d’étudier la probable dégradation de nos composés. Les molécules 22, 23 et 24 ont été mises en solution en présence de glutathion dans un milieu tampon phosphate à pH 7 et à 37 °C. Le suivi HPLC de la dégradation de ces composés a été ensuite effectué sur l’iChemExplorer®. Afin d’avoir un temps de départ commun à chaque molécule et une courbe de cinétique juste, le glutathion est ajouté juste avant l’injection (Figure 19).
23
Figure 19. Étude de la dégradation entre les composés 22, 23 et 24 et le glutathion
Les premiers essais n’ont pas été concluants. La faible solubilité des composés dans un milieu tamponné a entraîné un problème au niveau de la répétabilité des injections. Les analyses générées n’ont de ce fait pas été représentatives.
Dans l’optique de s’affranchir de ce problème de répétabilité, un deuxième suivi a été effectué dans un mélange de méthanol et de DMSO (9/1). La solubilité des produits a été ainsi fortement augmentée. Le suivi cinétique a clairement montré une dégradation des composés. Pour illustrer les résultats, seul l’exemple du composé 22 est présenté dans la Figure 20 et montre la diminution du produit de départ, ainsi que l’apparition et l’augmentation de nombreuses impuretés au cours du temps
24
Un inconvénient majeur observé au cours de cette expérience est le problème de solubilité des échantillons. Aucune dilution n’étant possible avant l’injection, les milieux réactionnels doivent être homogènes pour permettre une analyse fiable.
3.3
Application à la chimie combinatoire
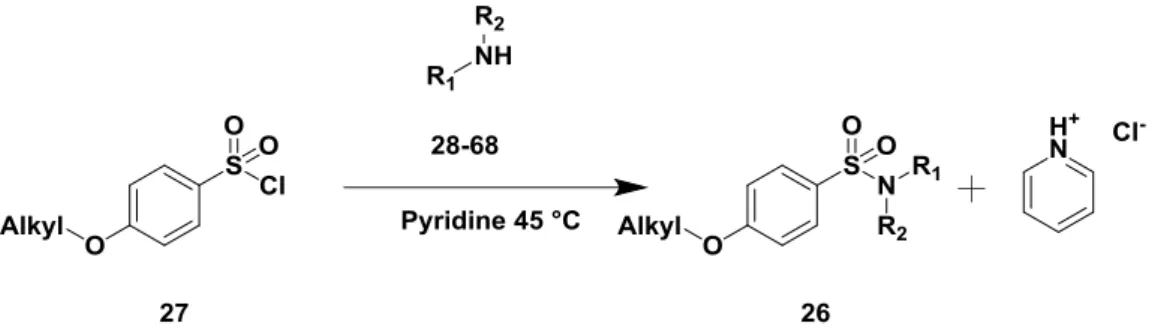
Dans l’optique de tester les capacités de l’iChemExplorer® en chimie combinatoire et de vérifier la méthode de transfert de l’UHPLC vers l’HPLC préparative, la synthèse des molécules suivantes a été envisagée (Figure 21). Dans un projet de chimie de Recherche en cours, nous avons eu besoin d’explorer la Relation Structure-Activité autour de la position sulfonamide pour des composés de type 26. Pour cela une série de 41 molécules a été sélectionnée.
Figure 21. Librairie pour la chimie combinatoire
Il existe différentes méthode de synthèse de sulfonamides. Les travaux de Sparks et al. ont montré la possibilité de coupler des anilines directement en présence de chlorure de sulfonyle. La réaction s’effectue en présence de triéthylamine dans le dichlorométhane, en utilisant de la DMAP comme catalyseur.22 D’autres auteurs ont montré que l’utilisation de la pyridine comme base dans le dichlorométhane ou le THF permettait aussi d’obtenir les sulfonamides désirés. 2324
Au laboratoire, nous avons optimisé les conditions opératoires en nous basant sur les travaux décrits précédemment. Par ailleurs nous avons voulu simplifier le traitement de la réaction pour qu’une simple filtration et une injection en HPLC préparative permette de purifier les composés.
Dans notre étude les groupe R1 et R2 représentent différents groupements aryles,
alkyles ou même hydrogène. La quantité de produit nécessaire pour les tests biologiques n’excédant pas 20 mg, les réactions ont été réalisées sur 50 mg du composé 27 dans un flacon de 2 mL.
L’aniline ou l’amine sélectionnée ont été ajouté au composé 27 préalablement solubilisé dans la pyridine, et le milieu réactionnel chauffé à 45 °C (Figure 22). Ces
25
conditions nous ont paru suffisamment générales pour être utilisées avec l’ensemble de nos amines / anilines.
Figure 22. Synthèse finale de la série de composés 26 développée au laboratoire
Toutes les réactions ont été démarrées en même temps en utilisant l’appareil proche de sa capacité maximale. Après 16 h de réaction, l’analyse des chromatogrammes sur le logiciel a permis de rapidement interpréter les résultats. Une analyse préalable du composé 27, nous a permis et de connaître son temps de rétention. Au total, 39 produits sur 41 ont été formés. Une analyse UHPLC couplée avec un spectromètre de masse a été effectuée afin de vérifier si les composés formés correspondaient aux produits attendus. Puis toutes les réactions ont été traitées et les bruts réactionnels purifiés par HPLC-préparative en se basant sur la méthode de transfert. Les produits purifiés ont ensuite été séchés dans le Génévac® HT6. Il s’agit d’une centrifugeuse qui permet une évaporation sous vide (Figure 22).
Figure 23. Appareil de séchage Génévac® HT6
Les produits obtenus ont alors été analysés dans les conditions standards d’analyse de produits finaux en Chimie de Recherche : une analyse sur UHPLC couplée avec un
26
spectromètre de masse avec une méthode analytique de 12 minutes en utilisant un gradient d’élution plus lent que la méthode analytique de l’iChemExplorer®. Cette dernière permet d’obtenir une meilleure résolution chromatographique. Une analyse RMN est aussi nécessaire pour confirmer la structure et vérifier la pureté. Pour les molécules qui ne possèdent pas de chromophores, une analyse RMN est indispensable pour déterminer la pureté.
On obtient 39 produits avec des rendements variant de 15 à 84% et une pureté en moyenne supérieure à 90% (Tableau 8).
N° Aniline Puretés (%) Rendement (%) Commentaires
26-1 99.2 78.3 - 26-2 - 15.0 Mélange de 2 composés 1/1 et 26-3 99.7 55.5 - 26-4 99.9 57.0 - 26-5 100 57.0 - 26-6 100 41.0 - 26-7 100 44.0 - 26-8 100 69.0 -
27
N° Aniline Puretés (%) Rendement (%) Commentaires
26-9 98.8 80.0 - 26-10 100 75.5 - 26-11 - 0 - 26-12 96.6 69.0 - 26-13 100 21.0 26-14 100 19.0 - 26-15 89.5 28.0 26-16 68.0 55.7 30% du produit ci-dessous 26-17 99.5 70.0 - 26-18 100 75.0 - 26-19 100 37.0 - 26-20 100 80.0 -
28
N° Aniline Puretés (%) Rendement (%) Commentaires
26-21 100 47.0 - 26-22 99.0 56.0 - 26-23 99.5 83.6 - 26-24 100 79.0 - 26-25 97.6 61.0 - 26-26 100 18.0 -
26-27 - 0.0 Réaction complète mais perte du
produit lors du traitement
26-28 100 57.0 -
26-29 99.2 69.0 -
26-30 100 79.0 -
26-31 100 65.8 -
29
N° Aniline Puretés (%) Rendement (%) Commentaires
26-33 100 65.0 - 26-34 100 64.0 - 26-35 100 79.1 - 26-36 100 67.2 - 26-37 - 0.0 Dégradation 26-38 100 81.0 - 26-39 97.4 79.6 - 26-40 100 72.2 - 26-41 100 72.4 -
Tableau 8. Série des anilines et des composés synthétisés par chimie combinatoire
Lors de la réaction entre les composés 40 et 27, le produit désiré n’a pas été obtenu. En se référant aux travaux de Shinsaku et al.,25 nous suggérons la structure 71, confirmée par RMN, pour ce produit. Un mécanisme possible pourrait être l’ouverture de la pyrrolidine par un ion chlore suivie d’une substitution nucléophile du dérivé chloré par la pyridine (Figure 24).
30
Figure 24. Mécanisme réactionnel proposé pour la synthèse du composé 76
Lors du séchage du composé 26-16 au Génévac® en sortie d’HPLC préparative, nous avons observé que ce dernier se dégradait. L’hydrolyse du composé 26-16 par l’action de l’acide formique contenu dans la phase éluante conduirait ainsi à une ouverture de cycle et l’obtention du composé 73. En se basant sur les travaux de Dixon et al.26 le mécanisme
réactionnel suivant peut être proposé (Figure 25) : l’ouverture de l’hémiacétal suivie par une perte d’une molécule de benzaldéhyde.
31
Figure 25. Mécanisme réactionnel proposé de la dégradation du composé 26-16
Le produit attendu de la réaction entre les composés 27 et 64 n’a pas été observé. Les dérivés de benzoxazoles pouvant parfois être des composés peu stables, nous avons supposé que le réactif n’était pas stable dans ces conditions. 27 28
Les 39 produits obtenus ont ensuite été répartis dans deux flacons pour les tests biologiques. Ces produits ont permis de répondre rapidement à une hypothèse de l’équipe de design sur la relation structure / activité avec la cible biologique.
Ainsi, l’iChemExplorer® permet un gain de temps essentiellement lors du suivi de la réaction. Le logiciel permet de savoir rapidement si telle réaction a fonctionné ou non. Une bonne organisation est cependant nécessaire afin d’éviter les erreurs de manipulation et les contaminations croisées. La méthode de transfert de l’UHPLC vers l’HPLC préparative a donné les résultats attendus et permis de purifier tous les composés en 48 h. Ainsi nous avons pu synthétiser et purifier nos composés en moins de 72h. La caractérisation des produits et le double flaconnage a pris plus d’une semaine.
32
Mais la chimie combinatoire n’est clairement pas l’application principale pour cet appareil. Le gain de temps reste minime comparé à une approche plus traditionnelle.
3.4
Suivi cinétique chiral en temps réel
Dans l’optique d’évaluer toutes les capacités de l’iChemExplorer®, nous avons voulu coupler l’appareil avec une HPLC chirale. L’uracile 82 (Figure 26) représentait un intermédiaire clef pour un des projets de la Chimie de Recherche. Une synthèse efficace avait été développée à partir de l’intermédiaire optiquement pur 81. Malheureusement, si le produit désiré avait obtenu avec un bon rendement, c’était au détriment de la pureté énantiomérique. Nous avons décidé d’utiliser l’iChemExplorer® pour trouver des conditions minimisant cette racémisation.
Figure 26. Synthèse du composé 87 avant optimisation
Pour effectuer le suivi chiral, il a été nécessaire de trouver des conditions compatibles avec l’iChemExplorer®, les conditions opératoires et le produit désiré. L’analyse est effectuée sur une colonne Chiralpak® Ic 4.6*250mm 5µM à température ambiante avec comme éluant un mélange heptane / éthanol (90 / 10) et 3% d’acide trifluoroacétique à un débit de 1mL / min. En tout l’analyse prend 30 minutes.
Le composé 81 est préparé à partir de l’ester aminé 83. Une première étape consiste à former l’urée en ajoutant l’isocyanate 84. L’urée est ensuite mise en réaction avec l’orthoformiate d’ethyle 86 et le 2-nitroacétate d’éthyle 87 dans le toluène à 100 °C pour obtenir le composé 81 (Figure 27). Parallèlement, la même voie de synthèse est effectuée afin d’obtenir le produit racémique. Ce dernier nous a permis de travailler sur les conditions d’analyse et de déterminer l’excès énantiomérique de notre composé final.
33
Figure 27. Synthèse multi-étapes du composé 81
L’étape de cyclisation s’effectue en milieu basique en présence de 2 équivalents de carbonate de césium à 90 °C dans 20 volumes de DMF pendant 3 h. Dans ces conditions, une pureté optique de 56% est obtenue. Pour réduire l’épimérisation in situ, nous avons fait varier la concentration de notre solution de 20 à 60 volumes afin de réduire la concentration de la base dans le milieu réactionnel. Nous avons aussi documenté l’impact de l’acétonitrile comme solvant (Tableau 9).
Solvant Volume Durée Rapport Composé 82 a/b
DMF 20 30 min 91/9
ACN 20 1 h 94/6
ACN 40 2 h 95/5
ACN 60 3 h 99/1
Tableau 9. Résultats de la pureté optique du composé 82 en fonction du solvant et du volume de ce dernier
34
Les meilleures conditions sont obtenues dans 60 volumes d’acétonitrile (Figure 28).
Figure 28. Synthèse optimisée du composé 82
La pureté optique s’améliore en diminuant la concentration mais cela entraine aussi une cinétique plus longue. Comme on peut le voir sur les analyses (Figures 29 et 30) la réaction est beaucoup plus propre dans l’acétonitrile.
Figure 29. Analyse après 3 h à 90 °C de la réaction dans 20 volumes de diméthylformamide
Figure 30. Analyse après 3 h à 90 °C de la réaction dans 60 volumes d'acétonitrile
Ces travaux représentent, à notre connaissance, la première utilisation de l’iChemExplorer® associé à une HPLC chirale. La durée des analyses dans ces conditions peut cependant être un problème si les cinétiques de réactions sont courtes.
Composé 87-a Composé 87-b
35
3.5
L’utilisation des paliers de température
Une des fonctions de l’iChemExplorer® que nous n’avons pas exploitée est la possibilité de contrôler précisément la température d’une réaction et de la faire varier automatiquement au cours du temps d’une réaction. Le fournisseur suggère par exemple d’étudier la dégradation de composés en fonction de l’évolution de la température au cours du temps soit par palier soit de manière linéaire.
Nous avons pu voir que l’iChemExplorer® est un outil multifonctionnel qui permet d’effectuer différentes opérations comme une optimisation de conditions de synthèse, un suivi cinétique ou chiral ou la synthèse d’une petite chimiothèque. Nous avons ensuite voulu utiliser cet appareil plus en profondeur pour développer deux réactions fréquemment utilisées à Galderma R&D.
4
La synthèse d’Acides Hydroxamiques
Deux projets en cours chez Galderma R&D ont pour cible des composés comportant des fonctions acides hydroxamiques. Nous avons utilisé l’iChemExplorer® pour optimiser une nouvelle approche à la synthèse de ce groupe fonctionnel.
4.1
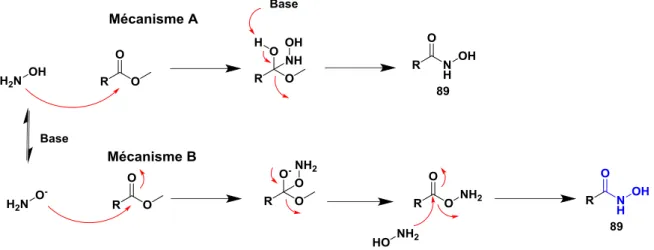
Bibliographie
Depuis 1869 et leur découverte par Lössen, le nombre de leurs applications dans différents domaines, et particulièrement dans l’industrie pharmaceutique n’a fait que croître.
Les acides hydroxamiques sont une famille de composés organiques particuliers. De par leur structure, on observe deux valeurs de pKa sur cette fonction (Figure 31) et des
propriétés spécifiques. 29 En effet, ce sont des ligands très répandus en chimie et biochimie. Un des rôles physiologiques particuliers des acides hydroxamiques est leur capacité chélatante avec différents métaux de transition, notamment le fer et le zinc.
Figure 31. Formule générale et pKa de la fonction acide hydroxamique
Dans le milieu pharmaceutique, cette fonction se retrouve fréquemment dans les inhibiteurs d’enzymes contenant un métal. Les protéines HDAc par exemple sont des cibles de choix. Le but est d’inhiber une enzyme capable de dé-acétyler les histones sur lesquelles
36
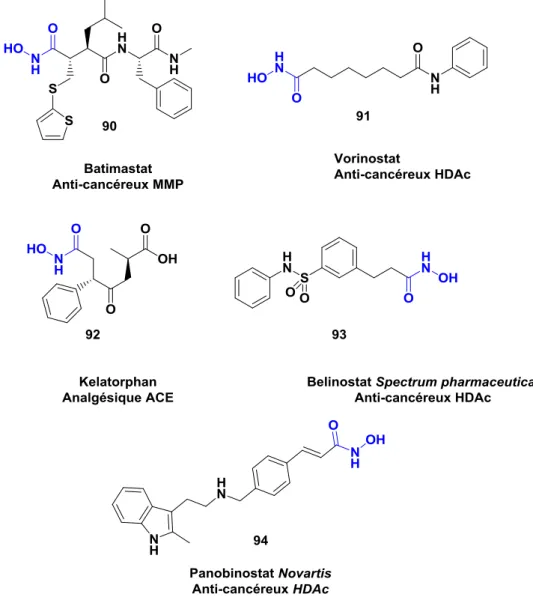
l’ADN s’enroule et empêcher ainsi la transcription de ce dernier. Cette approche a été validée par l’arrivée sur le marché du Vorinostat® (90) approuvé depuis 2006 et du Belinostat® (93) en 2014 pour le traitement des cancers cutanés des cellules lymphocytes T. Plus récemment, le Panobinostat® (94) a été approuvé en février 2015.30 Le Kelatorphan® (92) [26] est un inhibiteur d’enzyme ACE présentant la fonction acide hydroxamique. Cette enzyme est responsable de la formation de l’octapeptide angiotensine II à partir de l’angiotensine I. Cette transformation provoque notamment la diminution de la pression sanguine, ce qui en a fait une cible de choix pour l’industrie pharmaceutique. 31 Le Batimastat® (90) 32 est un exemple d’inhibiteur de MMP. Ce composé inhibe plusieurs MMP ce qui lui procure des propriétés anticancéreuse. Les MMP sont des enzymes possédant du zinc (Figure 32). 33
Figure 32. Structure de produit sur le marché possédant une fonction acide hydroxamique
Il existe de nombreuses synthèses d’acide hydroxamique à partir d’acide carboxylique et de différents agents de couplage. Dans la revue bibliographique ci-dessous,