Hyperparathyroïdie primaire et acromégalie non syndromique : une association fortuite ou un syndrome de prédisposition incluant le cancer de la thyroïde ?
Texte intégral
Figure
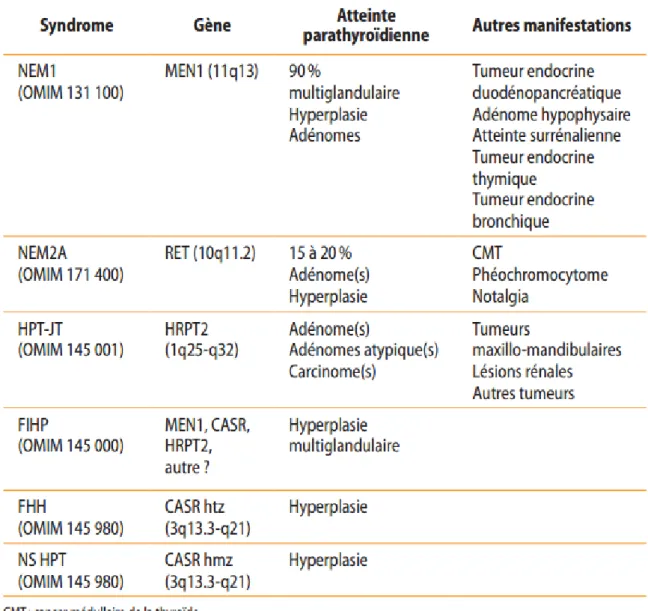
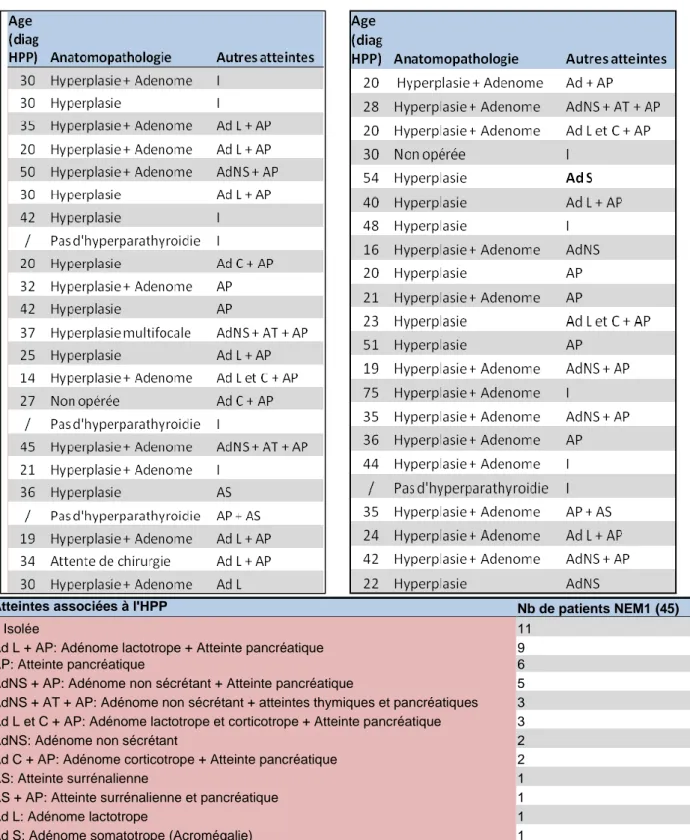
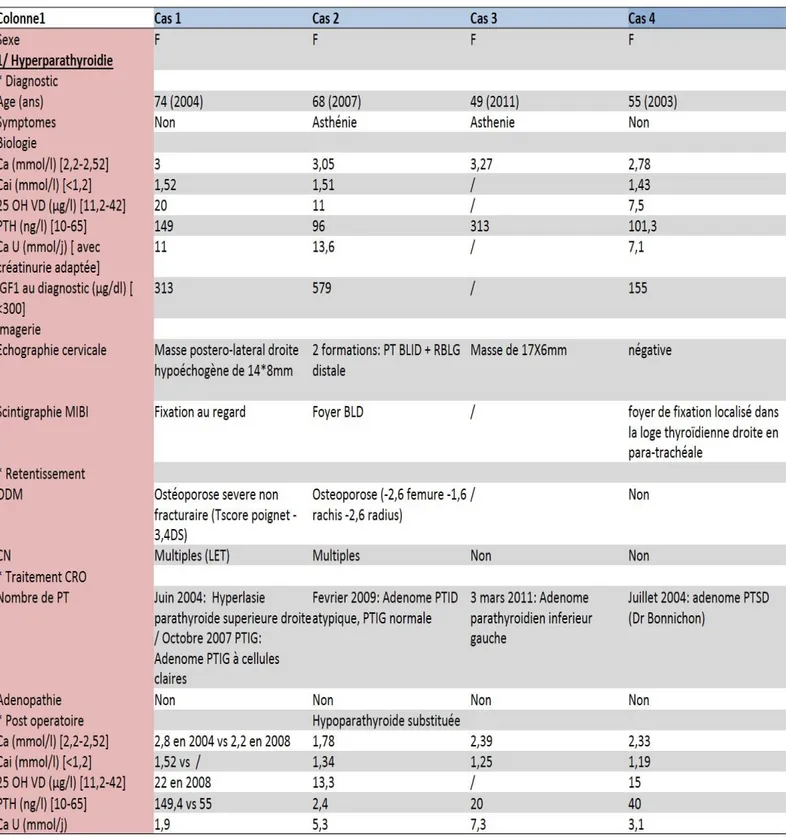
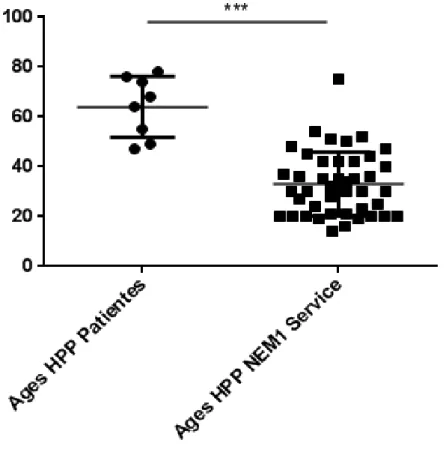
Documents relatifs
Placer dans un autre repère le nuage de points (t i ; y i ) et constater que sa forme allongée justifie un ajustement affine.. Commentez le
L’enseignement de la pensée systémique voit ici sa place dans l’enseignement obligatoire en Suisse et dans le monde confirmée, mais de nombreuses questions se posent encore : quelles
En cas de bilan de première ligne négatif (= scintigraphie et échographie cervicale ne retrouvant aucune lésion parathyroïdienne suspecte) ou discordant, une imagerie de seconde
Le contrat d'assurance-vie, comme le contrat de capitalisation, est très souvent utilisé en tant qu'instrument de garantie. Selon le type de contrat, la garantie porte
— Une exentération pelvienne avec résection urinaire a permis, dans notre expé- rience 64 % de contrôle local de la maladie (dont 90,5 % sur l’appareil urinaire) au prix
De nombreuses preuves expérimentales ont démon- tré que le système immunitaire, notamment les cellules T infi ltrant les tumeurs, joue un rôle majeur dans le contrôle de
In-lab software developed to predict the infrared heat flux on the top surface of the composite will be coupled with COMSOL in order to simulate the whole
A partir des éléments précédents, il ressort que le dispositif AIS facilite alors le développement de la recherche avec d’une part
