Simulation de systèmes chimique et physiologique
Texte intégral
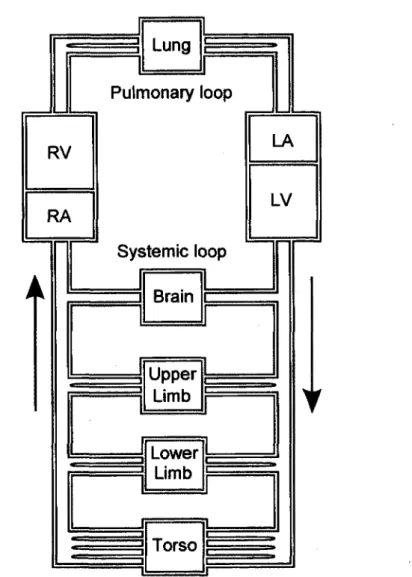
Figure


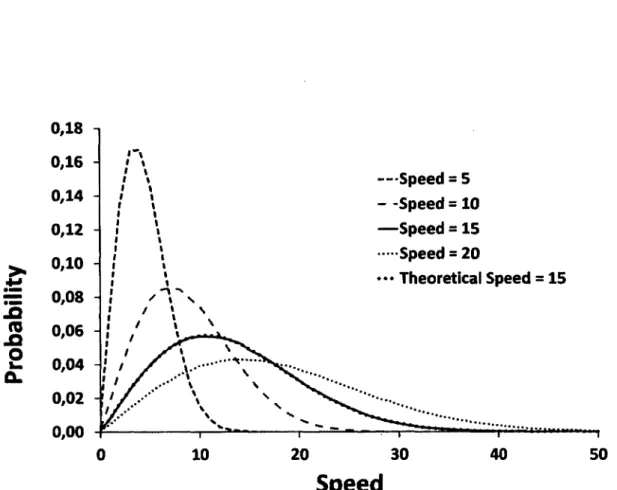
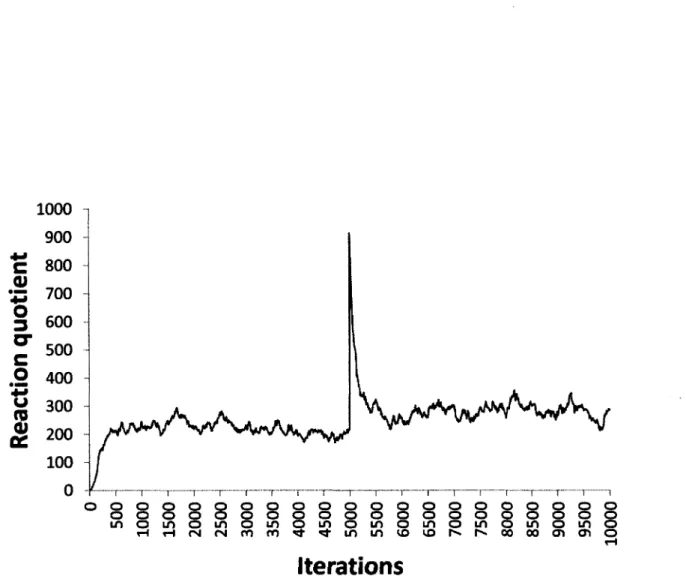
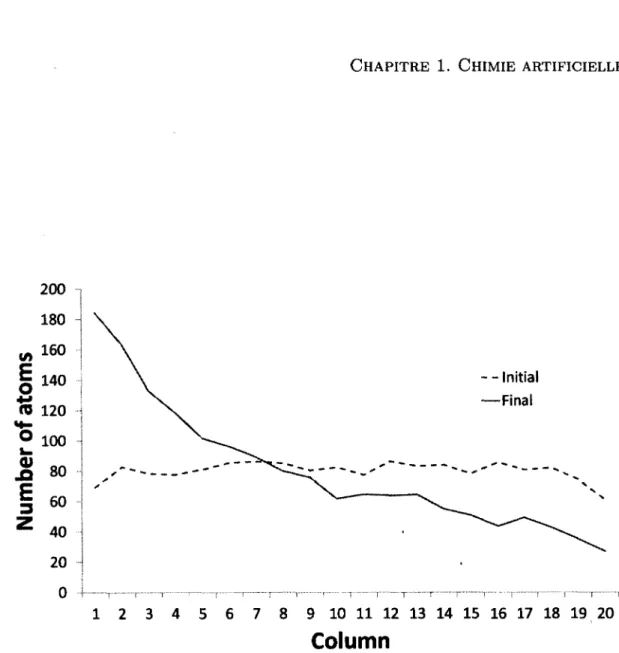

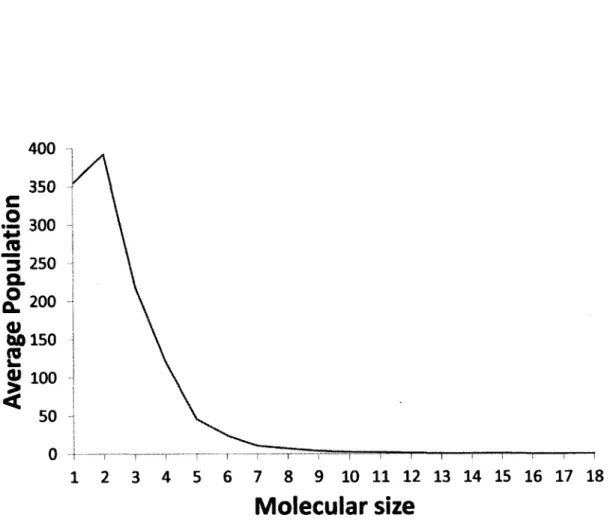


Documents relatifs
Keywords: Behavioural Science, Behavioural Economics, Health Promotion, Public Health, Nudge.. David McDaid is Senior Research Fellow at LSE Health and Social Care and at
French Charter of 1814 contains the only explicit mention of the term octroi in a constitutional document of the considered period.. Regarding all the other
In section 3 we define a new operator for action negation therefore give arise to a new dynamic logic.. In Section 4 we apply the new logic to the
[r]
The high-reliability keyboard is also integrated with main logic, and can generate all 128 ASCII characters (upper and lower case, numerics, punc- tuation, and control)..
A two-dimensional representation is either irreducible or has an invariant one-dimensional subspace, which corresponds to a common eigenvector of all transformations representing
S everal years ago, at the urging of some nonmedical friends, a small group of physicians and our spouses created a new board game called “Diagnosis.” Each player was a
2 Until a refrigerator-stable vaccine becomes available, however, varicella vac- cine will not be incorporated into the recommend- ed immunization schedule in Canada, as most
