Caractérisation d’IGFBP-2 comme biomarqueur
intégrateur de la santé cardiométabolique
Thèse
Sophie Carter
Doctorat en Pharmacie
Philosophiae doctor (Ph.D.)
Québec, Canada
© Sophie Carter, 2015
RÉSUMÉ
La protéine de liaison aux facteurs de croissance analogues à l’insuline (IGFBP)-2 est une protéine circulante fortement associée à la résistance à l’insuline qui module les effets métaboliques d’IGF-I et IGF-II en s’y associant directement, et qui exerce aussi des actions IGF-indépendantes via sa liaison à la matrice extracellulaire et aux intégrines. Chez l’homme, de faibles niveaux d’IGFBP-2 sont associés à un profil lipidique délétère, ainsi qu'à une augmentation de la masse grasse et de la résistance à l’insuline. Les travaux décrits dans cette thèse montrent chez l’humain et la souris que les niveaux d’IGFBP-2 sont associés de manière indépendante aux composantes du risque cardiométabolique. Chez l’homme, de faibles niveaux d’IGFBP-2 sont associés à la dyslipidémie athérogène. Une valeur seuil d’IGFBP-2 de 221.5 ng/mL a permis de discriminer entre les sujets métaboliquement sains et ceux répondant aux critères du syndrome métabolique. En plus de son association avec la résistance à l’insuline et les composantes du profil lipidique, de faibles niveaux d’IGFBP-2 sont associés à une fonction cardiaque diminuée chez les patients atteints de sténose aortique, tel qu’évaluée par le volume d’éjection indexé, un indice de fonction global du ventricule gauche qui intègre la fonction pompe et le remodelage du tissu. Chez l’homme, des niveaux d’IGFBP-2 élevés sont associés à un tissu adipeux brun plus volumineux ainsi qu’à une activité métabolique plus importante de ce dernier. Ces observations, telles qu’évaluées par PET/CT, sont aussi validées chez les souris surexprimant la forme humaine d’IGFBP-2. Nos travaux démontrent que les niveaux d’IGFBP-2 sont fortement associés au métabolisme des lipoprotéines et des lipides, à la fonction cardiaque ainsi qu’à l’activité du tissu adipeux brun. L’influence des niveaux d’IGFBP-2 par différentes altérations métaboliques menant à l’augmentation du risque cardiométabolique pourrait faire de ce dernier un biomarqueur précoce et intégrateur. Les travaux exposés dans la présente thèse soulignent aussi un rôle mécanistique potentiel pour IGFBP-2 dans la protection contre certaines altérations du métabolisme.
ABSTRACT
Insulin-like growth factor binding protein (IGFBP)-2 is a circulating protein strongly associated with insulin resistance. IGFBP-2 modulates the metabolic actions of IGF-I and IGF-II by direct binding, but can also exert IGF-independent effects through extracellular matrix and integrin binding. In humans, lower IGFBP-2 levels are associated with a deleterious lipid profile, increased fat mass and decreased insulin sensitivity. Causal links between IGFBP-2 levels and surrogate markers of cardiometabolic risk and the potential of IGFBP-2 as a biomarker of metabolic alteration have been scarcely studied. The work presented herein shows in humans and mice that IGFBP-2 levels are independently associated with components of the metabolic syndrome. In men, low IGFBP-2 levels are associated with atherogenic dyslipidemia. A cut-off value for IGFBP-IGFBP-2 at IGFBP-2IGFBP-21.5 ng/mL allowed us to identify subjects with or without the metabolic syndrome. In addition to its association with insulin resistance and the components of the lipoprotein-lipid profile, low IGFBP-2 levels are linked to decreased cardiac function in aortic stenosis patients, as assessed by stroke volume index, a global marker of left ventricle function and remodeling. In men, high IGFBP-2 levels are associated with a more important volume of brown adipose tissue and an increased activity. These observations, as assessed by PET/CT, were also confirmed in mice overexpressing the human form of IGFBP-2. Our results show that IGFBP-2 levels are strongly associated to lipid metabolism, cardiac function and brown adipose tissue activity. The combined influence of different metabolic alterations on IGFBP-2 levels could make it an early and integrative biomarker. The work presented here highlights a potential mechanistic role for IGFBP-2 in the protection against certain metabolic alterations.
TABLE DES MATIÈRES
RÉSUMÉ ... iii
ABSTRACT ... v
TABLE DES MATIÈRES ... vii
LISTE DES TABLEAUX ... xiii
LISTE DES FIGURES ... xv
LISTE DES ABRÉVIATIONS ET SIGLES ... xvii
REMERCIEMENTS ... xix
AVANT PROPOS ... xxiii
ARTICLES PRÉSENTÉS ... xxv
INTRODUCTION ... 1
1.0 Les maladies sociétales chroniques ... 3
1.1 Maladies sociétales chroniques ... 3
1.1.1 Facteurs de risque ... 3
1.2 Le vieillissement ... 4
1.2.1 Épidémiologie ... 4
1.2.2 Bases biologiques du vieillissement ... 5
1.2.3 Concept du vieillissement en santé ... 6
1.2.4 Modèles de vieillissement en santé ... 7
1.3 L’obésité ... 7
1.3.1 Épidémiologie ... 8
1.3.2 Causes de l’obésité ... 8
1.4 Désordres connexes entre l’obésité et le vieillissement ... 9
1.4.1 Accumulation et redistribution de la masse grasse ... 9
1.4.2 Inflammation ... 10
1.4.3 Résistance à l’insuline et à la leptine ... 10
1.4.4 Dyslipidémie ... 13
1.4.5 Modification de l’activité du tissu adipeux brun ... 14
1.5 Syndrome métabolique et risque cardiométabolique ... 16
2.0 Le système IGF/IGFBP ... 19
2.1 Les facteurs de croissance analogues à l’insuline ... 19
2.1.1 IGF-I 20 2.1.1.1 Régulation d’IGF-I ... 20
2.1.1.2 Activités physiologiques d’IGF-I ... 21
2.1.1.3 Dysrégulations d’IGF-I dans un contexte de maladies métaboliques ... 24
2.1.2 IGF-II ... 25
2.1.2.1 Régulation d’IGF-II ... 26
1. 2.1.2.2 Activités physiologiques d’IGF-II ... 27
2.1.2.3 Dysregulations d’IGF-II dans un contexte de maladies métaboliques ... 28
2.2 Les récepteurs aux IGF ... 29
2.2.1 Voie de signalisation ... 31
2.3 Les protéines de liaison aux facteurs analogues à l’insuline (IGFBP) ... 34
2.3.1 Introduction ... 34
2.3.2 Structure ... 35
2.3.3 Modifications post-traductionnelles ... 35
2.3.4 Rôles physiologiques ... 36
2.3.5 Interactions des IGFBP avec des ligands IGF-indépendants ... 37
2.3.6 Translocation des IGFBP au noyau ... 38
2.3.7 Dégradation des IGFBP ... 38
2.4 IGFBP-1 ... 39
2.4.1 Structure, expression et régulation ... 39
2.4.2 Modèles murins ... 39
2.4.3 IGFBP-1 et le risque cardiométabolique chez l’humain ... 40
2.5 IGFBP-3 ... 41
2.5.1 Structure, expression et régulation ... 41
2.5.2 Modèles murins ... 42
2.5.3 IGFBP-3 et le risque cardiométabolique chez l’humain ... 43
2.6 IGFBP-4 ... 44
2.6.2 Modèles murins ... 44
2.6.3 IGFBP-4 et le risque cardiométabolique chez l’humain ... 45
2.7 IGFBP-5 ... 46
2.7.1 Structure, expression et régulation ... 46
2.7.2 Modèles murins ... 46
2.7.3 IGFBP-5 et le risque cardiométabolique chez l’humain ... 47
2.8 IGFBP-6 ... 48
2.8.1 Structure, expression et régulation ... 48
2.8.2 Modèles murins ... 48
2.8.3 IGFBP-6 et le risque cardiométabolique chez l’humain ... 49
3.0 IGFBP-2 ... 51
3.1 Structure et expression d’IGFBP-2 ... 51
3.1.1 Structure ... 51
3.1.2 Expression d’IGFBP-2 ... 52
3.2 Modulation d’IGFBP-2 ... 55
3.2.1 Facteurs modulant l’expression d’IGFBP-2 ... 55
3.2.1.1 Description de la région promotrice du gène IGFBP-2 ... 55
3.2.1.2 Contrôle d’IGFBP-2 par l’axe GH/IGF ... 56
3.2.1.3 Régulation d’IGFBP-2 par les suppresseurs de tumeur ... 56
3.2.1.4 Régulation d’IGFBP-2 par l’hypoxie ... 58
3.2.1.5 Contrôle d’IGFBP-2 par la diète ... 58
3.2.1.6 Contrôle hormonal d’IGFBP-2 ... 59
3.2.1.7 Régulation centrale d’IGFBP-2 ... 60
3.2.2 Protéolyse d’IGFBP-2 ... 61
3.3 Actions cellulaires d’IGFBP-2 ... 62
3.3.1 Liaison à la matrice extracellulaire ... 63
3.3.2 Actions induites par les récepteurs des intégrines ... 64
3.3.3 Translocation nucléaire d’IGFBP-2 ... 65
3.4 Effets d’IGFBP-2 in vivo ... 67
3.4.1 Modèles d’invalidation génétique d’IGFBP-2 ... 67
3.4.2.1 Modèles de surexpression complète ... 67
3.4.2.2 Modèles de surexpression d’IGFBP-2 avec domaines fonctionnels mutés. ... 69
3.4.2.3 Modèles murins de supplémentation en IGFBP-2 ... 71
3.5 IGFBP-2 et le risque cardiométabolique chez l’humain ... 73
3.5.1 Associations entre IGFBP-2, l’âge et l’obésité ... 73
3.5.2 IGFBP-2 et la résistance à l’insuline ... 74
3.5.3 IGFBP-2 et les anomalies du métabolisme des lipides ... 74
3.5.4 IGFBP-2 et les maladies cardiovasculaires ... 75
3.5.5 IGFBP-2 et l’inflammation ... 75
3.5.6 IGFBP-2 et l’activité du tissu adipeux brun ... 75
CHAPITRE 1 ... 81
AVANT-PROPOS ... 83
RÉSUMÉ 85 ABSTRACT ... 87
INTRODUCTION ... 89
RESEARCH DESIGN AND METHODS ... 91
RESULTS 95 DISCUSSION ... 102 ACKOWLEDGEMENTS ... 105 REFERENCES ... 106 CHAPITRE 2 ... 111 AVANT-PROPOS ... 113 RÉSUMÉ 115 ABSTRACT ... 117 INTRODUCTION ... 119
RESEARCH DESIGN AND METHODS ... 121
RESULTS 124 DISCUSSION ... 131
CONCLUSION ... 135
ACKOWLEDGEMENTS ... 135
CHAPITRE 3 ... 141
AVANT-PROPOS ... 143
RÉSUMÉ ... 145
ABSTRACT ... 147
BRIEF REPORT ... 149
RESEARCH DESIGN AND METHODS ... 158
REFERENCES ... 163
DISCUSSION GÉNÉRALE ... 167
Perspectives ... 189
Conclusion ... 191
LISTE DES TABLEAUX INTRODUCTION
Tableau 1. Critères diagnostiques permettant l’identification des patients atteints du
syndrome métabolique selon le NCEP ATP III ... 17
Tableau 2. Liens unissant les différents IGFBP et le risque cardiométabolique ... 50
Tableau 3. Valeurs de références pour les niveaux d’IGFBP-2 ... 55
Tableau 4. Modulateurs positifs et négatifs de la transcription d’IGFBP-2 ... 61
Tableau 5. Effets de la surexpression d’IGFBP-2 dans des modèles murins ... 71
Tableau 6. Observations liées à la supplémentation en peptides d’IGFBP-2 ... 72
CHAPITRE 1 Table 1. Characteristics of the sample of 379 men ... 96
Table 2. Relationships between IGFBP-2 and features of the lipoprotein-lipid profile .. 100
Table 3A: Correlates of VLDL triglycerides ... 101
Table 3B: Correlates of HDL cholesterol ... 101
CHAPITRE 2 Table 1: Baseline Characteristics of the Study Population ... 126
Table 2. Correlates of Stroke Volume Index ... 128
STable 1. Independent association between IGFBP-2 and SVi after adjustment for concomitant metabolic factors ... 129
LISTE DES FIGURES INTRODUCTION
Figure 1. Schématisation du nombre d’individus représentant chaque strate d’âge en 2010 et prévisions pour 2050 ... 5 Figure 2. Représentation schématique de l’espérance de vie en santé lors du vieillissement
normal et lors du vieillissement en santé ... 6 Figure 3. Représentation schématique des conséquences de la résistance à la leptine et à
l’insuline ... 12 Figure 4. Représentation schématique de l’ensemble des facteurs regroupés sous le terme
risque cardiométabolique ... 18 Figure 5. Représentation schématique des effets métaboliques d’IGF-1 dans différents
organes cibles ... 24 Figure 6. Représentation schématique des récepteurs d’IGF-I et IGF-II ... 31 Figure 7. Représentation schématique simplifiée de la voie de signalisation en aval du
récepteur IGF-IR ... 33 Figure 8. Représentation schématisée de la forme mature d’IGFBP-2 retrouvée en
circulation ... 52 Figure 9. Patron d’expression d’IGFBP-2 dans différents tissus chez l’humain grâce à
l’utilisation de la sonde 202718_AT ... 54
Figure 10. Représentation schématisée des principaux effets IGF-dépendants et
indépendants d’IGFBP-2 ... 66 CHAPITRE 1
Figure 1. Excursion of plasma glucose and insulin during an OGTT according to the quantiles of IGFBP-2 levels………98 Figure 2. Levels of cardiometabolic risk variables across subgroups of subjects classified
on the basis of their quantiles of IGFBP-2 concentrations ... 99 CHAPITRE 2
Figure 2. Integrative representation of the impact of fat accretion and insulin sensitivity status on IGFBP-2 levels and potential mechanisms by which IGFBP-2 could exert cardioprotective effects ... 130 CHAPITRE 3
Figure 1. Effect of IGFBP-2 levels on BAT activity in humans. ... 151 Figure 2. Effect of cold exposure on BAT activity and gene expression in humanized
IGFBP-2 Tg mice………..152
Figure 3. Effect of cold and CL exposure on Igfbp-2 expression and modulation by the
melanocortin system……….154
DISCUSSION
Figure 1. État des connaissances avant la présente thèse sur les associations entre les niveaux d’IGFB-2 et les facteurs de risque cardiométabolique ... 170 Figure 2. État des connaissances actuelles sur les associations entre les niveaux d’IGFB-2
et les facteurs du risque cardiométabolique ... 171 Figure 3. Thématiques de recherches futures proposées sur IGFBP-2 ... 190
LISTE DES ABRÉVIATIONS ET SIGLES
Akt Protéine kinase B
ALS Sous-unité acide labile
AMPc Adénosine monophosphate cyclique
BAD Protéine associée à Bcl-2 induisant l'apoptose BAT Tissu adipeux brun
bFGF Facteur de croissance fibroblastique basique C- Carboxyl
C/EBPα CAAT/enhancer binding protein α
CCN Facteurs de croissance des tissus riches en cystéine du néphroblastome CD36 Translocase d'acides gras
CETP Protéine de transfert des esters de cholestérol CMV Cytomégalovirus
CNS Système nerveux central CRP Protéine C-réactive
ERK-1 Kinase régulée par les signaux extracellulaires ERα Récepteur de l'estrogène
FAK Kinase d'adhésion focale
G1 Phase Gap 1
GDP Guanosine diphosphate GFAP Protéine acide fibrillaire gliale GLP-2 Peptide analogue au glucagon 2
Grb2 Protéine 2 liant le récepteur des facteurs de croissance GTP Guanosine triphosphate
HBD Domaine de liaison à l'héparine HBD2 Domaine de liaison à l'héparine 2
HIF-1α Facteur de transcription inductible par l'hypoxie Hox Famille des homeobox
Icv Intra-cérébroventriculaire
IGF Facteur de croissance analogue à l'insuline IGFBP Protéines de liaison aux IGF
IGFBP-rP Protéines apparentées aux IGFBP IGF-IIR IGF-II récepteur
IGF-IR Récepteur des IGF IL Interleukine
IMC Indice de masse corporelle
IRS Substrat du récepteur de l'insuline LDL Lipoprotéine de faible densité M6PR Récepteur du mannose 6-phosphate
MAPK Protéine kinase activée par la mitogenèse MetS Syndrome métabolique
MMP Matrixines
mTOR Cible mécanistique de la rapamycine MyoD Facteur de différenciation myogénique N- Amino
NFkB Facteur nucléaire kappa B NLS Signal de localisation nucléaire
PAI1 Inhibiteur de l'activateur de plasminogène PAPA1 Protéine 1 associée à Pim1
PDK1 Kinase dépendante de 3-phosphoinositide Phase S Phase de réplication d'ADN
PI3K Phosphoinositide 3-kinase
PIP2 Phosphatidylinositol (4,5)-biphosphate PIP3 Phosphatidylinositol (4,5)-triphosphate pp120 Protéine phosphorylée de 120 kDa PPARα Proliférateurs de peroxysomes α Ptch1 Patched 1
PTEN Homologue du phosphate et de la tensine QTL Locus de trait quantitatif
RAS Sarcome de rat RGD Motif Arg-Gly-Asp
RPTPβ Récepteur des protéines tyrosines phosphatase beta SA Sténose aortique calcifiante
sdLDL LDL petites et denses
SH2 Domaine homologue de Src2
SHBG Globuline liant les hormones sexuelles
SHC Substrat cytosolique proximal homologue au collagène TGFβ Facteur de croissance transformant beta
TNFa Facteur de nécrose tumorale alpha TSC Tuberine/hamartine
UCP-1 Protéine découplante 1
VEGF Facteur de croissance vasculaire endothélial VG Ventricule gauche
VLDL Lipoprotéine de très faible densité Wnt Protéines de site d'insertion sans ailes
REMERCIEMENTS
On dit qu’il faut un village pour élever un enfant. Dans le cas de la réalisation d’études de 3e cycle, ce village devient indispensable. Les travaux réalisés dans le cadre de
la présente thèse ont nécessité de nombreuses collaborations et le support d’un grand nombre de personnes, sans qui jamais je n’aurais pu réussir. Je souhaite donc prendre quelques instants pour remercier les membres de mon village.
Je souhaite tout d’abord remercier mon directeur de recherche Dr Frédéric Picard, pour sa contribution importante à mon développement tant scientifique que personnel. Merci d’avoir pris une chance, il y a 7 ans, en m’acceptant comme stagiaire d’été dans ton laboratoire et d’avoir cru en moi, parfois plus que moi-même. Le lien de confiance qui s’est installé m’a permis d’apprendre à voler de mes propres ailes et m’a donné l’opportunité de découvrir mes habiletés. Merci de m’avoir impliqué dans les différentes facettes de la recherche, incluant la rédaction et la soumission d’articles et l’élaboration de demandes de subventions.
Je tiens aussi à remercier les gens avec qui j’ai eu la chance de travailler pendant mes études graduées. Je désire en premier lieu remercier les gens de l’équipe Picard pour leur contribution à mes travaux et pour leur ardeur à l’ouvrage. Je souhaite particulièrement remercier Stéphanie, qui m’a non seulement enseigné pratiquement tout ce que je sais faire aujourd’hui, mais qui m’a aussi donné l’exemple de la quantité de travail nécessaire pour réussir.
Je souhaite remercier les membres des nombreuses équipes qui m’ont entourée lors de la réalisation de cette thèse. Au Dr Mathieu Laplante et à son équipe (Blandine, Étienne, Inan, Christian et Yves), merci de m’avoir permis de participer à vos rencontres de laboratoire pendant la dernière année et de m’avoir inclus comme membre honoraire de votre équipe. Malgré le fait que je connaisse ses opinions sur les compliments, je ne peux rédiger ces remerciements sans adresser un merci tout spécial à Yves Gélinas. Merci Yves de ta joie de vivre, de ta disponibilité et de ta générosité. C’est toujours un plaisir de travailler à tes côtés, que ce soit pour tes blagues vulgaires où parce que c’est plus facile de
voler tes pipettes (oups!). Merci de me corrompre dans le vice avec tes coupons McDo, ça me fait toujours un bien fou!
Je tiens aussi à remercier tous les membres de l’équipe du Dr Denis Richard passés et présents, en particulier Alex, Sébastien, Kanta, Damien, Marie-Claude, Julie et Pierre. Merci de votre aide, de vos conseils et de votre support technique toujours professionnel. Un grand merci aussi à tous les membres de l’équipe de l’animalerie (Audrey, Tony, Sébastien, Nicholas et bien sûr Justin) pour votre aide technique, votre assistance et votre sourire contagieux, je vais m’ennuyer de vous!
Je désire remercier les membres des équipes des Dr Pibarot et Després pour leur aide précieuse à l’élaboration des travaux présentés dans cette thèse. Un merci tout spécial à Romain et Isabelle avec qui j’ai eu la chance de travailler de plus près.
Merci aux filles (et Maxime, et JC) de l’équipe du Dr Yohan Bossé pour votre sourire, votre joie de vivre et votre intérêt scientifique. Particulièrement, merci à Cyndi; on se connait déjà depuis 9 ans, ça passe vite! Merci d’avoir été toujours là dans les moments l’fun et dans ceux qui l’ont moins été. Merci pour tous ces diners où je parlais beaucoup et toi tu écoutais beaucoup.
Je désire aussi particulièrement remercier le Dr Yves Deshaies. Je crois que beaucoup de gens comprendront si je vous dis : merci d’être vous. Vous possédez des qualités de scientifique et de mentor exceptionnelles ainsi qu’une bonne humeur contagieuse. Vous êtes toujours de bon conseil et de bonne compagnie.
Un grand merci à tous les autres qui ont fait/font partie de mon quotidien. Merci à Renée, Jolyane, Nancy, Dominique, Pierre-Gilles, Thomas et les membres du GRV pour les discussions scientifiques et les moins scientifiques…..
Un merci tout spécial aux membres fondateurs et invités des FisherMark’s Friends (Alex, Marc, Christian, Marie-Josée), merci pour toutes ces activités «hors programme».
Rien de mieux que d’aller se valoriser au quiz night de temps à autre. On ne gagne pas souvent, mais on rit tout le temps! Merci à Christian pour ton calme, ta compréhension et ton écoute. Merci à Marie-Josée pour ton attitude pétillante et ta volonté de nous écouter parler de science pendant des heures. Merci Marc, il y tellement de choses pour lesquelles je voudrais te dire merci, mais en particulier merci de ton humanisme et merci pour tous ces courriels de fin d’après-midi m’offrant des beignes à la cardamome avec un glaçage à l’orange ou la saveur du jour du resto chez Marc.
Un grand merci à ma famille, qui n’a pas toujours compris où je m’en allais, mais qui m’a offert son soutien inconditionnel. Merci à mes parents de s’informer de mes cellules et de mes souris et d’être fiers de moi. Quand on se connait depuis 25 ans, on tombe dans la catégorie de la famille, alors merci à ma meilleure amie Maude pour son soutien inconditionnel et son positivisme. Merci d’être toujours disponible (avec un avis de 30 jours) et d’apporter du vin quand tu viens.
Je souhaite aussi remercier mon conjoint Alex. Deux scientifiques dans la famille des fois c’est complexe, mais la plupart du temps c’est valorisant. Merci de me pousser à être une meilleure personne et une meilleure scientifique, ça me motive. Merci aussi pour ton support et ta présence, je t’aime! Je ne pourrais terminer ces remerciements sans remercier la personne la plus importante dans ma vie, Olivia. Merci ma bibitte pour ton amour inconditionnel et ton beau sourire. Merci de m’avoir appris que, des fois, c’est mieux de colorier que de travailler. J’espère que la prochaine étape va nous permettre d’avoir une belle vie ensemble.
AVANT PROPOS
Le présent ouvrage est déposé à la Faculté des études supérieures de l’Université Laval pour l’obtention du diplôme de Philosophae Doctor ès Sciences (Ph.D.). Généralement, cette thèse porte sur la caractérisation de la protéine circulante IGFBP-2 en tant que marqueur du risque cardiométabolique. Les études décrites dans cet ouvrage s’attardent spécifiquement à l’identification des liens qui existent entre les niveaux d’IGFBP-2 et le métabolisme des lipides, la fonction cardiovasculaire et l’activité du tissu adipeux brun. La thèse contient une introduction générale rédigée en français portant sur le risque cardiométabolique, le système IGF/IGFBP et sur IGFBP-2. Les chapitres 1 à 3 constituent le corps de l’ouvrage et relatent en détail les travaux de recherche réalisés pour cette thèse. Ces chapitres sont rédigés en anglais sous forme d’articles scientifiques tels qu’ils ont été ou seront présentés en vue de leur publication, selon les exigences éditoriales. Finalement, une conclusion termine cette thèse en résumant les principaux résultats obtenus, en analysant leurs liens respectifs et en discutant des voies futures à explorer pour l’utilisation d’IGFBP-2 comme biomarqueur du risque cardiométabolique.
ARTICLES PRÉSENTÉS
Les articles présentés dans cette thèse sont le fruit de mes travaux de doctorat qui ont été réalisés au cours des cinq dernières années dans le laboratoire du Dr Frédéric Picard. Chacune des études présentées implique mon entière participation; de la mise en place des protocoles à la rédaction finale des manuscrits. Au cours de mes études doctorales, j’ai eu la chance de collaborer à de nombreux projets de recherche en marge de mes travaux. Les collaborations que j’ai entretenues avec mes collègues ou avec d’autres équipes de recherche ont conduit, ou conduiront sous peu, à la publication de plusieurs manuscrits. La liste qui suit résume les articles découlant de tous mes travaux et collaborations. Les articles précédés d’un cercle vide sont ceux qui seront présentés dans cette thèse.
Miard S, Dumbrowski L, Carter S, Boivin L, Picard F, Aging alters PPARgamma
in rodent and human adipose tissue by modulating the balance in SRC-1. Aging
Cell, 8 : 449-459, 2009
Carter S, Miard S, Roy-Bellavance C, Li Z, Pibarot P, Mathieu P, Picard F. Sirt1
inhibits resistin expression in aortic stenosis. PLoS ONE, 7(4):e35110, 2012
Carter S*, Caron A*, Richard D, Picard F. Role of leptin resistance in the
development of obesity in older patients. Clin Interv Aging. 2013;8:829-44
o *Carter S*, Li Z*, Lemieux I, Alméras N, Tremblay A, Bergeron J, Poirier P, Deshaies Y, JP Després, Picard F. Circulating IGFBP-2 Levels Are Incrementally
Linked to Correlates of the Metabolic Syndrome and Independently Associated with VLDL Triglycerides. Atherosclerosis. 2014 Dec;237:645-51
Meloche J, Courchesne A, Barrier M, Carter S, Bisserier M, Paulin R, Breuils-Bonnet S, Tremblay È, Briardel S, Racine C, Courture C, Breuils-Bonnet P, Majka S.M, Deshaies Y, Picard F, Provencher S, Bonnet S. Critical Role for the Advanced
Glycation Endproducts Receptor in Pulmonary Arterial Hypertension Etiology.
Journal of the American Heart Association, 2013. Jan 16;2(1):e005157.
o Carter S*, Capoulade R*, Arsenault M, Bédard É, Dumesnil JG, Mathieu P,
Pibarot P. Picard F. Relationship Between Insulin-like Growth Factor Binding
Protein-2 and Left Ventricular Stroke Volume in Patients with Aortic Stenosis.
Canadian Journal of Cardiology, Sous presse; http://dx.doi.org/10.1016/j.cjca.2015.04.024.
Carter S, Lemieux I, Li Z, Alméras N, Tremblay A, Bergeron J, Poirier P, Deshaies Y, Després JP, Picard F. Longitudinal changes in IGFBP-2 induced by a 1 year
life-style modification program are associated with improvements in ApoB and cardiorespiratory fitness. En préparation
o Carter S, Labbé SM, Caron A, Laplante M, Carpentier A, Richard D, Picard F.
Upregulation of brown adipose tissue activity by insulin-like growth factor binding protein 2 in rodents and humans. En préparation
Caron A, Labbé S, Carter S, Roy MC, Lecomte R, Ricquier D, Picard F, Richard D. Molecular imaging to investigate the role of uncoupling protein 2 in brown
adipose tissue substrate metabolism. Soumis au Journal of Nuclear Medicine.
Carter S, Miard S, Caron A, St-Pierre P, Anhê F, Sallé S, Charland E, Drolet MC, Lefebvre J, Bossé Y, Laplante M, Rivest S, Marette A Marsolais D, Richard D, Picard F. Loss of OcaB prevents age-associated fat accretion and insulin resistance
through browning of white adipose tissue and increased sympathetic drive. En préparation
1.0 Les maladies sociétales chroniques
1.1 Maladies sociétales chroniques
On regroupe aujourd’hui sous le vocable maladies sociétales chroniques un ensemble de conditions non-transmissibles qui affectent de façon importante la mortalité et la morbidité. Comme leur nom l’indique, ces maladies sont de longue durée et connaissent une évolution lente. Les quatre principaux types de maladies regroupées dans cette catégorie par l’Organisation Mondiale de la Santé sont : les maladies cardiovasculaires, les cancers, les maladies respiratoires chroniques et le diabète [1]. Ces conditions trônent en tête de liste des principales causes de mortalité avec un total de plus de 36 millions de décès par année [1].
1.1.1 Facteurs de risque
Ces affections chroniques partagent des facteurs de risque communs qui sont classés dans la catégorie des facteurs modifiables et comprennent: le tabagisme, la sédentarité, l’abus d’alcool et une mauvaise alimentation. Malheureusement, le nom donné à ce groupe de facteurs de risque est trompeur. Malgré leur caractère modifiable, la plupart de ces facteurs de risque comprennent une composante addictive non négligeable rendant la modification des habitudes de vie difficile et souvent éphémère.
Il existe aussi des facteurs de risque non modifiables, certains nous rendant inégaux face aux possibilités de développer des maladies sociétales chroniques. D’une part, l’avancement de l’âge est un facteur de risque incontrôlable important qui s’accompagne souvent d’une augmentation de l’incidence de cancers, d’une accumulation de masse grasse et d’une diminution des défenses immunitaires [2, 3]. D’autre part, des différences génétiques peuvent aussi nous rendre individuellement plus susceptibles au développement de pathologies. Conséquemment, de subtiles mutations peuvent prédisposer au développement de certains cancers [4] ou amplifier les effets d’une diète riche en gras [5].
Ces facteurs de risque, modifiables ou non, n’augmentent pas seulement les risques de développement de maladies sociétales chroniques, ils en sont souvent la cause.
Dans ce contexte, nos recherches s’intéressent particulièrement à l’obésité et au vieillissement. Il s’agit là de deux facteurs de risque importants qui mènent à des altérations métaboliques souvent similaires. Nous croyons que l’étude de ces altérations communes, tant chez l’animal que chez l’humain, nous permettra d’identifier des cibles pharmacologiques potentielles et des biomarqueurs permettant la prise en charge des maladies sociétales chroniques et leurs facteurs de risque.
1.2 Le vieillissement
D’un point de vue individuel, le vieillissement réfère à un processus multidimensionnel qui inclut bien sûr des processus physiques, mais aussi psychologiques et sociétaux. Le vieillissement est une partie intégrante de chaque société, reflétant à la fois des changements biologiques et culturels. L’avancement de l’âge est le facteur de risque connu le plus important, en effet plus de 100 000 personnes meurent chaque jour dans le monde de causes reliées à l’âge [6].
1.2.1 Épidémiologie
A la fin du 21e siècle, les tendances démographiques convergeront vers un déclin
des naissances, une stabilisation de la population et un vieillissement de la population à l’échelle mondiale [7]. La composition de la population subira elle aussi des altérations avec une augmentation de l’âge médian et la continuité du décalage entre les jeunes et les personnes âgées (Figure 1). Déjà à l’an 2000, on comptait en Europe plus de citoyens de plus de 60 ans que de moins de 15 ans, une tendance qui sera suivie par l’Amérique du Nord en 2030 et par l’Asie en 2040 [7]. Dès lors, on estime que plus de gens vivront pour voir leur 80e anniversaire. En effet le nombre d’octagénaires devrait passer de 69 millions à
379 millions en 2050, où 10% de la population mondiale sera âgée de plus de 80 ans. Seule valeur divergente dans le tableau mondial : l’Afrique, dont la population continuera de
croitre et de demeurer jeune avec un tiers de sa population sous la barre des 15 ans en 2050 [7].
Figure 1. Schématisation du nombre d’individus représentant chaque strate d’âge en 2010 et prévisions pour 2050. Les bandes de couleur rouge représentent la population active. Modifié de Harper, 2014 [7].
1.2.2 Bases biologiques du vieillissement
Bien que le vieillissement soit inévitable, peu de processus biologiques causant le vieillissement ont été identifiés. Neuf caractéristiques présentes dans les organismes vieillissants ont toutefois été récemment mises à jour [8] et impliquent : l’instabilité génomique, le raccourcissement des télomères, les modifications épigénétiques, les altérations de l’homéostasie des protéines, la dysrégulation des nutriments, la dysfonction mitochondriale, la sénescence cellulaire, l’épuisement des cellules souches et l’altération de la communication cellulaire [8]. Chacune de ces caractéristiques se manifeste lors du vieillissement normal. Pour la plupart, l’aggravation expérimentale de ces phénomènes en
accélère le cours tandis que la compensation de ces phénomènes retarde le processus de vieillissement normal et favorise le vieillissement en santé.
1.2.3 Concept du vieillissement en santé
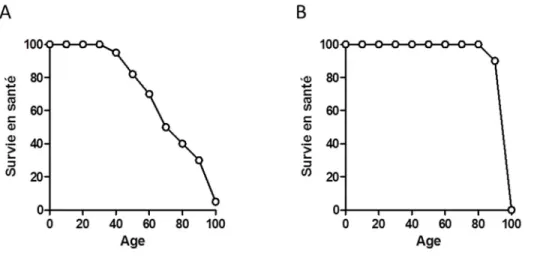
Le concept de vieillissement en santé est central à la recherche sur le vieillissement. En effet, plusieurs croient à tort que ce type de recherche vise à prolonger l’espérance de vie tandis qu’il tente en réalité de prolonger l’espérance de vie en santé, sans nécessairement affecter la longévité (Figure 2).
Figure 2. Représentation schématique de l’espérance de vie en santé lors A) du vieillissement normal et B) lors du vieillissement en santé.
Le vieillissement en santé est bien sûr favorisé par une bonne hygiène de vie, incluant l’activité physique, une alimentation saine et une salubrité limitant les infections, et entraîne une diminution de la prévalence des comorbidités affectant la qualité de vie. Le concept du vieillissement en santé cible une gestion des facteurs de risque en vue de conserver l’autonomie et l’activité des personnes âgées jusqu’à leur décès. Ceci vise à diminuer la portion de leur espérance de vie passée avec une qualité de vie réduite.
1.2.4 Modèles de vieillissement en santé
Grâce au développement de la transgénèse, il a été possible d’identifier différentes façons de favoriser un vieillissement en santé. Bien sûr, la modulation de différentes voies de signalisation clés, telles les voies des facteurs de croissance analogues à l’insuline (IGF) et de l’insuline [9] permettent de conserver une sensibilité à l’insuline avec l’avancement de l’âge, évitant ainsi les impacts négatifs de la résistance à l’insuline sur l’accumulation de masse grasse et le métabolisme du glucose. Le tissu adipeux semble jouer un rôle important dans ce phénomène puisque l’invalidation du récepteur de l’insuline spécifique au tissu adipeux rétablit la sensibilité à l’insuline et augmente la longévité [10]. De plus, les souris hétérozygotes pour le récepteur des IGF (IGF-IR) sont moins grasses, ce qui est associé à une augmentation de la longévité moyenne de 26% [11]. Il est toutefois possible d’influencer le tissu adipeux sans modifications génétiques. Dans des études charnières, Barzilai et al. ont démontré que l’ablation chirurgicale du tissu adipeux viscéral augmente la longévité et améliore l’insulino-résistance [12, 13]. D’un autre côté, il est possible de limiter l’accrétion de masse grasse via la restriction calorique sans malnutrition. Cette modification de l’apport calorique entraîne une augmentation de la longévité accompagnée d’une perte de tissu adipeux viscéral chez les rongeurs [14], mais se transpose difficilement chez l’humain.
1.3 L’obésité
L’obésité se caractérise par une accumulation anormale ou excessive de masse grasse qui peut affecter négativement la santé [15]. L’indice de masse corporelle [16] est un index simple, utilisé pour classifier le surpoids et l’obésité chez l’adulte, qui se défini comme le poids en kilogrammes divisé par la taille en mètre au carré (kg/m2). Les gens en
surpoids sont caractérisés par un IMC supérieur ou égal à 25 tandis que les sujets ayant un IMC supérieur ou égal à 30 sont considérés obèses [17]. Bien qu’imparfait, l’IMC est l’outil qui fournit les données les plus utiles d’un point de vue populationnel puisque son calcul reste le même chez les adultes indépendamment du sexe et de l’âge.
1.3.1 Épidémiologie
En 2014, on dénombrait plus de 1.9 milliard d’adultes en surpoids. De ce nombre, 600 millions étaient obèses [18]; plus près de chez nous, au Canada, 53% de la population avait un IMC de plus de 25 en 2013 [19]. De façon inquiétante, la prévalence mondiale d’obésité a plus que doublé entre 1980 et 2014 [18]. Malheureusement, cette dernière est appelée à poursuivre son augmentation dans les prochaines années. Une des causes de cette augmentation constante est l’apparition de l’obésité infantile. En effet, bien qu’il s’agissait autrefois d’un problème spécifique aux pays riches, l’obésité infantile touche maintenant les pays en développement et les économies émergentes [18]. Parallèlement, au Canada, 20% des jeunes de moins de 18 ans sont en surpoids. Ces données sont d’autant plus inquiétantes que chez ces jeunes, les comorbidités associées à l’obésité se feront sentir plus tôt et influenceront négativement leur qualité de vie sur une période plus longue, suggérant ainsi que la génération des enfants obèses et en surpoids pourrait vivre moins longtemps que celle de ses parents.
1.3.2 Causes de l’obésité
La cause principale de l’obésité est un débalancement de la balance énergétique qui est un équilibre fin entre l’apport et la dépense énergétique. L’apport calorique est constitué des calories ingérées tandis que la dépense comprend le métabolisme basal, l’activité physique et la thermogenèse [20]. De façon à maintenir un poids stable, l’apport calorique doit être équivalent à celui de la dépense énergétique. Dans le cas d’un apport plus important que la dépense, le corps accumule l’excédent d’énergie sous forme de masse grasse. Si ce phénomène est chronique, l’embonpoint et l’obésité s’installent.
Plusieurs observations suggèrent que la variance de l’IMC observée dans la population peut être expliquée par la génétique. Cette héritabilité est estimée entre 40-70% [21, 22]. De façon intéressante, il existe de fortes corrélations entre l’IMC de jumeaux ayant été élevés séparément [23] ou ayant été soumis à une suralimentation [24], ce qui
souligne l’importance de l’activité physique dans la prévention de l’obésité puisqu’il s’agit de la seule composante modifiable de la dépense énergétique.
Un faible pourcentage de la population souffre aussi de mutations monogéniques conduisant à l’obésité. La plupart de ces mutations affectent les récepteurs des mélanocortines et de la leptine et ont des effets sur la prise alimentaire et la dépense énergétique [25]. Malgré tout, l’identification de causes polygéniques de l’obésité demeure encore difficile. Ceci suggère peut-être que d’une façon générale, l’obésité est un problème environnemental plus que génétique.
1.4 Désordres connexes entre l’obésité et le vieillissement
Malgré des différences se situant dans l’initiation et la pathophysiologie de ces deux conditions [26], l’obésité et le vieillissement résultent fréquemment en des altérations métaboliques similaires.
1.4.1 Accumulation et redistribution de la masse grasse
En condition de vieillissement et d’obésité, le tissu adipeux est soumis à de nombreux changements. Le vieillissement est associé avec une redistribution importante du tissu adipeux. Le poids corporel augmente habituellement jusqu’à la cinquantaine et décline par la suite [27]. De façon contrastante, la masse grasse totale maximale est atteinte entre 40-70 ans, ce qui résulte en une augmentation du pourcentage de gras corporel [28]. Cette augmentation est la conséquence de l’augmentation de la masse grasse, mais aussi de la diminution de la masse maigre, principalement provenant du muscle squelettique et des os, rencontrée chez les personnes âgées. Dans ce contexte, une redistribution des dépôts adipeux est observée en faveur du dépôt intra-abdominal viscéral [29].
En condition d’obésité, le tissu adipeux sous-cutané apparait être le tissu de prédilection pour l’entreposage des graisses de manière à limiter leurs conséquences
métaboliques délétères. Effectivement, les animaux chez lesquels le tissus adipeux sous-cutané peut accumuler des lipides presqu'indéfiniment conservent un profil métabolique sain [16]. Néanmoins, cette capacité demeure limitée par différents facteurs chez l’humain. Une fois le tissu adipeux sous-cutané surchargé et dysfonctionnel, les graisses sont redirigées vers le dépôt viscéral. Tant en contexte d’obésité que de vieillissement, l’incapacité du tissu adipeux sous-cutané à accumuler les graisses favorise l’entreposage dans les dépôts viscéral et ectopiques (i.e. le muscle, le foie et la moelle osseuse) [29]. Cette accumulation de graisse ectopique participe à la résistance à l’insuline pan-corporelle.
1.4.2 Inflammation
Longtemps, le tissu adipeux a été vu comme un simple organe d’entreposage, mais est aujourd’hui reconnu en tant qu’organe endocrinien dynamique. Les modifications du tissu adipeux exposées précédemment affectent la sécrétion des différentes cytokines. Les niveaux circulants de cytokines pro-inflammatoires tel le facteur de nécrose tumoral (TNF) α, la protéine C-réactive (CRP) et l’interleukine (IL)-6 sont élevés à la fois au cours du vieillissement et de l’obésité [30]. Le tissu adipeux est un site de relargage pour de nombreuses cytokines et chemokines qui ont le pouvoir d’attirer les cellules immunitaires comme les lymphocytes T et les macrophages. Tant chez les sujets obèses qu’âgés, on observe aussi une augmentation de l’accumulation des macrophages proinflammatoires dans le tissu adipeux viscéral [31, 32].
1.4.3 Résistance à l’insuline et à la leptine
Les progrès actuels réalisés par la recherche en obésité et en vieillissement ont permis l’identification d’un système intégré responsable du maintien de l’homéostasie en réponse à la disponibilité des nutriments et à la dépense énergétique [33]. Ce système comprend l’hypothalamus comme centre intégrateur du système nerveux central (CNS), les adipokines en tant que signal afférent vers le CNS et les fibres sympathiques qui permettent
une régulation entre les tissus métaboliques tels le foie, le pancréas, le muscle, le tissu adipeux et le cerveau. En temps de suralimentation, la leptine joue un rôle critique comme hormone anti-obésogène. Toutefois, lors d’une surabondance constante de nutriments, ce système devient surchargé, ce qui mène à la résistance à la leptine, et à l’insuline de même qu’à l’obésité [34, 35]. Des niveaux élevés de leptine sont observés à la fois chez les rongeurs [36, 37] et les humains vieillissants [38, 39], suggérant que le vieillissement est aussi un état de résistance à la leptine.
La résistance à la leptine qui s’installe en condition de vieillissement et d’obésité s’accompagne de modifications de différents systèmes clés dans la régulation du métabolisme (Figure 3). Brièvement, cet état entraîne une diminution de la sensibilité à l’insuline et de l’oxydation des acides gras, ainsi qu’une augmentation de la lipolyse dans le foie, le muscle et le tissu adipeux blanc. Les mécanismes sous-jacents de la résistance à la leptine demeurent élusifs, mais il a été postulé que 1) des incapacités de la leptine à rejoindre ses cibles dans le cerveau, 2) une diminution de l’expression du récepteur de la leptine et 3) une inhibition des voies de signalisations de certaines régions spécifiques du cerveau pourraient participer au développement de ce phénomène [35, 40, 41].
L’obésité et le vieillissement sont aussi associés au développement de l’insulino- résistance. En effet, en condition d’accumulation viscérale et ectopique de masse grasse, certains types cellulaires, particulièrement les adipocytes, les cellules musculaires et les hépatocytes deviennent résistants aux effets de l’insuline. L’incapacité des adipocytes et des cellules musculaires à capter le glucose entraîne une élévation de la glycémie. Dans ces conditions, l’inhibition de la néoglucogenèse par l’insuline dans le foie est aussi altérée. L’augmentation des niveaux sanguins de glucose s’accompagne d’une augmentation de la sécrétion d’insuline et peut éventuellement mener à l’épuisement des cellules pancréatiques et au diabète de type 2 [35]. Cette résistance à l’insuline semble principalement associée à une augmentation des niveaux circulants de glucose induite par une incapacité du récepteur à l’insuline à transmettre une réponse appropriée au signal hormonal [42].
Figure 3. Représentation schématique des conséquences de la résistance à la leptine et à l’insuline. Modifié de Carter, Caron et al. [35]
Légende : A) Chez les individus de jeune âge ou de poids santé, les adipocytes blanc (particulièrement ceux du tissu
sous-cutané) sécrètent des niveaux normaux de leptine. En périphérie, la leptine contribue à la sensibilité à l’insuline et à l’oxydation des acides gras par le foie, les muscles et le tissu adipeux. Au niveau central, la leptine traverse la barrière hématoencéphalique via un système de transport actif saturable pour aller rejoindre ses cibles. La liaison de la leptine à son récepteur dans l’hypothalamus induit une augmentation de POMC et une diminution des niveaux de NPY/AgRP. Cette modulation de populations neuronales spécifiques induit une activation du SNS, qui entraîne une augmentation de la transcription d’UCP-1 et la thermogenèse du BAT. B) En condition de vieillissement ou d’obésité, le tissu adipeux est redistribué et les adipocytes viscéraux produisent une grande quantité de leptine. En périphérie, la résistance à la leptine s’installe dans le foie, le muscle et le tissu adipeux pour causer une diminution de la sensibilité à l’insuline, de l’oxydation des acides gras et une augmentation de la lipolyse. Centralement, des altérations de la barrière hémato-encéphalique diminuent le transport de la leptine, ce qui entraîne une diminution de POMC qui induit l’atrophie du BAT et diminue du fait même l’expression d’UCP-1 et la thermogenèse. Abréviations: BBB : Blood brain barrier, CVO : Circum ventricular organ, CNS : Central nervous system, POMC : Pro-opiomelanocortin, NPY : Neuropeptide Y, AgRP : Agouti-related protein, SNS : Sympathetic nervous system, BAT : Brown adipose tissue, UCP-1 : Uncoupling protein 1.
1.4.4 Dyslipidémie
La dyslipidémie précède souvent l’apparition du diabète de type 2 de plusieurs années, ce qui indique que la dysfonction du métabolisme des lipoprotéines est un des premiers évènements dans la chaîne des complications cardiovasculaires entrainées par le diabète de type 2 [43]. La surproduction de lipoprotéines de très faible densité (VLDL) est une altération fréquente de la résistance à l’insuline hépatique [44]. D’un point de vue moléculaire, l’insuline inhibe normalement l’assemblage des VLDL en empêchant le recrutement des triglycérides lors de la phase de maturation [45]. En conséquence, dans le foie des patients diabétiques, la synthèse et l’entreposage des triglycérides sont augmentés et l’excès de triglycérides est sécrété sous forme de VLDL. Cette condition est un facteur de risque majeur pour les maladies cardiovasculaires et est caractérisée par ce qu’on appelle la triade lipidique:
• Niveaux élevés de triglycérides plasmatiques
• Niveaux bas de cholestérol dans les lipoprotéines de haute densité (HDL) • Apparition de lipoprotéines de faible densité (LDL) petites et denses (sdLDL)
La diminution du catabolisme périphérique est aussi une caractéristique de la dyslipidémie diabétique, en partie causée par une diminution d’activité de la lipoprotéine lipase [44, 46]. Ce phénomène mène à l’augmentation du temps de résidence des particules VLDL en circulation et à l’enrichissement des LDL et HDL en triglycérides. La formation de sdLDL très athérogènes est associée de près à la résistance à l’insuline et à l’hypertriglycéridémie [47]. Deux enzymes clés sont impliquées dans la production de sdLDL: la protéine de transfert des esters de cholestérol (CETP) et la lipase hépatique. L’augmentation des VLDL circulants diminue leur catabolisme intravasculaire, ce qui les rend susceptibles à l’action de la CETP, dont l'action favorise les échanges de triglycérides des VLDL vers les LDL. Les LDL riches en triglycérides qui en résultent deviennent un substrat de choix pour la lipase hépatique et leur hydrolyse subséquente résulte en la formation de sdLDL [48]. L’augmentation des niveaux de sdLDL influence aussi la
composition des HDL, encore une fois via l’action de la CETP et de la lipase hépatique. Ceci mène à la formation de petites HDL denses facilement dégradables [49].
1.4.5 Modification de l’activité du tissu adipeux brun
Le BAT est un dépôt adipeux spécialisé qui possède un potentiel thermogénique puissant [50, 51]. Jusqu’à récemment, il était estimé que, chez l’humain, le BAT disparaissait après la naissance et ne jouait qu’un faible rôle physiologique chez l’adulte [52, 53]. Néanmoins, de récentes études basées sur l’imagerie médicale sont venues ébranler ce dogme et ont démontré la présence de tissu adipeux brun métaboliquement actif chez l’humain [54]. La présence et l’activité du BAT sont fortes à un jeune âge et chez l’adulte, mais décroissent rapidement chez les patients âgés [55, 56]. En effet, l’âge a été identifié comme étant le déterminant négatif le plus fortement associé à la présence de BAT [57].
Parallèlement, une diminution de la prévalence du BAT est aussi observée en condition d’obésité [58, 59]. D’un point de vue thermogénique, le BAT des souris obèses ou vieilles est quiescent [60]. Ces observations suggèrent un rôle protecteur pour le BAT lors de l’accumulation de masse grasse. Ce déclin serait attribuable à une réduction graduelle de la quantité de BAT actif suivi d’une diminution de sa fonction et de la sensibilité aux signaux métaboliques [57]. De façon intéressante, la capacité et l’activité du BAT sont diminuées à la fois chez les vieilles souris minces et obèses, mais particulièrement chez les obèses [61]. Ces observations suggèrent qu’il existe une synergie entre le vieillissement et l’obésité dans un contexte de thermogenèse du BAT.
1.4.6 Maladies cardiovasculaires
Les maladies cardiovasculaires sont la principale cause de décès au niveau mondial. Les ramifications du vieillissement de la population et de l’épidémie d’obésité sont indéniables : on estime qu’en 2030, plus de 23,3 millions de décès seront attribuables aux
maladies cardiovasculaires [1]. Les principales maladies cardiovasculaires associées au vieillissement et à l’obésité incluent l’hypertension, l’athérosclérose et l’infarctus du myocarde [62, 63]. Ceci s’explique facilement par le fait que tant l’obésité que le vieillissement s’accompagnent de morbidités qui sont des facteurs de risque importants dans le développement de maladies cardiovasculaires.
D’un point de vue cardiovasculaire, le vieillissement et l’obésité s’accompagnent d’altérations pathologiques qui incluent l’hypertrophie, les altérations de la fonction diastolique du ventricule gauche (VG), l’augmentation de la rigidité artérielle et l’altération de la fonction endothéliale [62, 64]. En milieu de vie, le temps de remplissage du VG en diastole connait un déclin qui est compensé par une augmentation de la contraction artérielle de façon à préserver le volume d’éjection [65, 66]. Toutefois, avec la progression de l’âge, la contractilité, la fraction d’éjection ainsi que la capacité à répondre à la stimulation adrénergique sont diminuées. Une réduction du débit cardiaque associée à cette diminution de la fonction amène le myocarde à compenser par une augmentation de la masse musculaire, ce qui mène à l’hypertrophie. Bien que ce processus soit profitable à court terme, l’hypertrophie à long terme entraîne une diminution de la fonction cardiaque [67]. De la même façon, une surcharge pondérale s’associe à une diminution de la compliance du VG, une augmentation de la masse du VG et à du remodelage concentrique [68]. De nombreuses maladies cardiovasculaires sont associées à une diminution de la fonction cardiaque, incluant la sténose aortique dont le facteur de risque principal est l’âge [69].
La sténose aortique calcifiante (SA) est actuellement la cause la plus commune de remplacement valvulaire chirurgical en Amérique du Nord [70]. De plus, il est estimé que la prévalence de la SA et des remplacements valvulaires doublera d’ici 2020 en réponse au vieillissement de la population [71]. Cette dernière découle d’un processus actif complexe qui implique le métabolisme des lipides, l’inflammation, le métabolisme minéral ainsi que le remodelage de la valve aortique et du VG [72]. Les patients souffrant de SA sont habituellement asymptomatiques pour une longue période malgré l’augmentation de la charge du VG. Suite à l’apparition des symptômes qui incluent l’angine, la dyspnée et la
syncope, une intervention chirurgicale rapide est nécessaire puisque l’espérance de vie moyenne sans remplacement valvulaire est de 1 à 5 ans avec un risque de mort subite très élevé [73]. Il n’existe présentement aucun traitement pharmacologique pour freiner ou faire régresser la progression de la SA; le seule traitement disponible est le remplacement valvulaire aortique, réalisé suite à l’apparition des symptômes [74]. Malheureusement, il est impossible de prévoir l’apparition de ces derniers puisque la SA progresse à une vitesse différente chez chaque patient. Il n’existe présentement aucun biomarqueur permettant de prédire la progression de la maladie.
1.5 Syndrome métabolique et risque cardiométabolique
Chacun des facteurs cités précédemment affecte de façon délétère la santé. Toutefois, plusieurs de ces conditions possèdent des bases communes et peuvent donc être concomitantes chez un même individu. Dans ce contexte, une synergie existe entre ces facteurs, ce qui augmente le risque pour la santé. C’est pourquoi un nombre important de ces facteurs ont été regroupés sous le terme syndrome métabolique (MetS), où plus récemment risque cardiométabolique.
Le MetS se caractérise par un regroupement de facteurs de risque métaboliques qui favorisent le développement de maladies cardiovasculaires. Il a été suggéré que la résistance à l’insuline soit la plateforme commune permettant le développement des autres facteurs de risques [75]. Chez l’homme, la sensibilité à l’insuline est profondément affectée par l’obésité et le vieillissement [76]. En effet, ces deux conditions sont associées à une prévalence plus importante du MetS [77]. Conséquemment, les patients atteints de MetS démontrent un risque élevé de développer certaines maladies qui affectent à la fois la morbidité et la mortalité, incluant les maladies cardiovasculaires et le diabète de type 2 [78]. Un groupe d’experts sur la détection, l’évaluation et le traitement de l’hypercholestérolémie a identifié un ensemble de critères permettant le diagnostic du MetS (tableau 1). Si au moins 3 des critères suivants sont identifiés chez un patient, un diagnostic de MetS peut être posé. Cette technique diagnostique implique toutefois une population hétérogène de patients atteints de MetS et un suivi individuel devient important.
Tableau 1. Critères diagnostiques permettant l’identification des patients atteints du MetS selon le NCEP ATP III [79].
Paramètre Valeur clinique
Obésité abdominale Tour de taille de ≥102 cm chez les hommes ou ≥88 cm chez les femmes Résistance à l'insuline Glycémie à jeun ≥5.6 mmol/l ou sous traitement hypoglycémiant
Dyslipidémie Athérogène
Triglycérides ≥1.69 mmol/l ou sous traitement visant à réduire les triglycérides, HDL cholestérol <1.03 mmol/l pour les hommes et <1.29 mmol/l pour les femmes
ou sous traitement visant à augmenter le HDL cholestérol. Hypertension Pression sanguine systolique ≥130 ou diastolique ≥85 mm Hg ou sous traitement antihypertenseur État pro-inflammatoire Aucun
État pro-thrombotique Aucun
Abréviations : NCEP ATP III : National Cholesterol Education Program Adult Treatment Panel III, HDL: High density
lipoprotein.
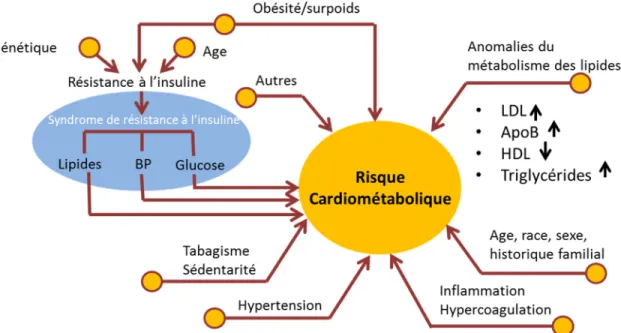
Le terme risque cardiométabolique quand à lui est un terme plus inclusif qui comprend les facteurs de risque du MetS, mais aussi de nombreux autres facteurs de risque incluant l’historique familial et l’âge (Figure 4). De plus, certains facteurs émergents pourraient s’ajouter à la liste, tel la diminution de l’activité du BAT [80].
Figure 4. Représentation schématique de l’ensemble des facteurs regroupés sous le terme risque cardiométabolique. Adapté de l’Association Américaine du Diabète.
Légende : Le risque cardiométabolique englobe une pléiade de facteurs de risques modifiables ou non qui jouent un rôle
central dans le développement des maladies cardiovasculaires. Abréviations : BP : pression sanguine, LDL : lipoprotéine de faible densité, ApoB : apolipoprotéine B, HDL : lipoprotéine de haute densité.
L’insuline semble jouer un rôle de premier plan dans la pathophysiologie du MetS et de ses conséquences cardiovasculaires. Il existe toutefois des protéines analogues à l’insuline qui peuvent être responsables des effets métaboliques similaires, ajoutant ainsi un niveau de complexité supplémentaire à la compréhension des mécanismes associés au développement du MetS et à l’augmentation du risque cardiométabolique.
2.0 Le système IGF/IGFBP
2.1 Les facteurs de croissance analogues à l’insuline
Les facteurs de croissance analogues à l’insuline IGF-I et IGF-II possèdent une homologie de structure similaire à celle de l’insuline. Cette dernière découle du fait que la proinsuline, IGF-I et IGF-II ont évolué d’un précurseur commun il y a 60 millions d’années [81]. Dans les organismes primitifs, cet unique précurseur agit en tant que signal visant à informer de la présence suffisante de nutriments, non seulement pour le métabolisme basal, mais pour la synthèse protéique et la prolifération. Avec l’apparition des vertébrés, ce système a été appelé à évoluer en un système plus complexe permettant l’entreposage des calories excédentaires sous forme de gras. Ces modifications ont entrainé l’apparition de l’hypophyse, de l’hormone de croissance (GH) et la division des fonctions de l’insuline et d’IGF-I. Ensemble, ces trois hormones régulent la disponibilité des nutriments en période de jeûne et de satiété et agissent comme signaux de croissance.
IGF-I et IGF-II appartiennent à la superfamille des peptides favorisant la croissance et sont parmi les facteurs de croissance les plus abondants et ubiquitaires [82]. Les IGF sont exprimés dans la plupart des tissus dès l’embryogenèse. Alors que les niveaux d’IGF-II sont très élevés in utero, IGF-I gouverne la croissance post-natale [83, 84]. Bien qu’ubiquitaire, l’expression des IGF est régulée de façon tissu-spécifique par des facteurs qui peuvent agir de manière autocrine ou paracrine [85]. Tant chez l’humain que chez les rongeurs, les IGF partagent environ 50% d’homologie avec la proinsuline [86]. Cette homologie leur confère une caractéristique unique parmi les autres facteurs de croissance : en plus de réguler la prolifération, la différentiation et l’apoptose, ils peuvent aussi mimer les effets métaboliques de l’insuline dans les tissus exprimant de hauts niveaux du récepteur des IGF de type 1, tel que le muscle squelettique et les cellules bêta du pancréas [87, 88].
2.1.1 IGF-I
IGF-I (aussi appelée la somatomedine C) est une protéine de 7 kDa qui est exprimée de façon ubiquitaire. Elle est produite de façon endocrine par le foie et de façon autocrine/paracrine par les autres organes [89]. Les transcrits produits par le gène IGF-I sont soumis à de nombreux évènements d’épissage alternatif. En effet, celui-ci donne naissance à différents pré-pro-IGF qui possèdent des peptides signaux variables. Toutefois, après conversion, toutes les isoformes convergent en la formation d’une seule et même protéine de 70 acides aminés [90]. La forme mature d’IGF-I est conservée chez différentes espèces et contient 4 domaines, nommés pour leur similarité à ceux de l’insuline B-C-AD [91]; le domaine B étant responsable de la liaison aux récepteurs des IGF [92]. Chez l’homme, les niveaux d’IGF-I plafonnent lors de la puberté et diminuent de 2.5 fois de 20 et 30 ans. Une diminution de 2 fois se produit aussi entre 30 et 80 ans [93].
2.1.1.1 Régulation d’IGF-I
Les besoin évolutifs ayant mené à l’apparition d’IGF-I ont, sans surprise, mis l’expression de ce gène sous le contrôle des nutriments. L’expression d’IGF-I est régulée tant par l’apport calorique que par celui en protéines [94]. Les effets de l’apport calorique sont tels qu’une diminution de 50% de ce dernier entraîne une forte diminution de la sécrétion d’IGF-I. Les effets des protéines sont plus graduels; il a été estimé qu’une diminution de 25% de l’apport protéique entraîne une diminution équivalente des niveaux d’IGF-I [95]. Cette diminution des niveaux circulants d’IGF-I par l’apport calorique et protéique s’accompagne d’une diminution de la synthèse d’IGF-I au foie [96]. Les niveaux d’IGF-I peuvent aussi être régulés indirectement par la modification de l’apport en glucides. Ces effets sont concomitants aux changements dans la sécrétion d’insuline puisque l’insuline contrôle elle aussi l’expression d’IGF-I. Ce principe s’illustre bien chez les diabétiques de type I non traités chez qui une injection d’insuline entraîne une hausse substantielle des niveaux plasmatique d’IGF-I [97]. Chez les modèles animaux, il a aussi été démontré qu’une inhibition des actions de l’insuline au foie entraîne une diminution des niveaux plasmatiques d’IGF-I. L’apport en glucides agit donc non seulement en
augmentant la quantité d’énergie disponible résultant en une augmentation de la synthèse d’IGF-I, mais en régulant IGF-I au niveau transcriptionnel via un effet direct par l’insuline [98].
La GH est un second régulateur fort des niveaux d’IGF-I. Toutefois pour que le foie réponde à celle-ci en synthétisant des niveaux normaux d’IGF-I, l’état de nutrition doit être adéquat [81]. Ceci s’illustre chez les animaux déficients en GH chez qui l’administration de cette dernière entraîne une augmentation accrue de la transcription d’IGF-I au foie qui est associée avec une augmentation parallèle des niveaux circulants [99]. Cette augmentation rapide des niveaux d’IGF-I assure une boucle de rétrocontrôle négative sur l’hypophyse en augmentant l’expression de la GH, maintenant ainsi l’homéostasie [99].
D’autres hormones participent au contrôle d’IGF-I et comrennent : les glucocorticoïdes qui antagonisent les actions d’IGF-I et donc mènent à la diminution de ses niveaux plasmatiques, et les hormones thyroïdiennes qui sont nécessaires pour la synthèse d’IGF-I. L’œstrogène empêche quant à elle la stimulation de la production hépatique d’IGF-I par la GH [81]. Toutes ces hormones fonctionnent de façon coordonnée avec les modifications de l’apport en nutriments de façon à moduler la capacité d’IGF-I à influencer la croissance et le métabolisme.
2.1.1.2 Activités physiologiques d’IGF-I
2.1.1.2.1 Croissance et développement
IGF-I est une hormone de croissance anabolique possédant un rôle essentiel dans la prolifération cellulaire et la croissance [100, 101]. IGF-I joue un rôle prédominant lors de la croissance post-natale qui s’illustre bien par le retard de croissance important chez les animaux déficients en IGF-I [102, 103]. Néanmoins, ce dernier semble aussi jouer un rôle crucial dans la croissance fœtale. En effet, les animaux invalidés pour IGF-I démontrent un retard de croissance débutant au jour embryonnaire 13,5 [104] ainsi qu’une atrophie musculaire associée à une diminution de la viabilité périnatale [102, 103]. Ces observations
vont de pair avec le fait qu’IGF-I est nécessaire pour la prolifération cellulaire. Par conséquent, dans les fibroblastes de souris, IGF-I est requis pour passer de la phase Gap1 (G1) du cycle cellulaire à celle de la réplication d’ADN (phase S) [105, 106]. Un nombre
important de types cellulaires démontrent une réponse mitogénique suite à un traitement d’IGF-I comme les ostéoblastes, les myocytes, les cellules épithéliales et les chondrocytes. Ceci illustre bien pourquoi le temps de division cellulaire est doublé chez les fibroblastes embryonnaires dérivés de souris invalidés pour IGF-IR [107].
2.1.1.2.2 Effets métaboliques d’IGF-I
Bien qu’IGF-I soit classiquement considéré comme un facteur de croissance, il n’en demeure pas moins qu’il exerce des fonctions métaboliques importantes (Figure 5). Le rôle métabolique d’IGF-I s’articule principalement autour de la signalisation informant les cellules de la disponibilité des nutriments, évitant ainsi l’apoptose et favorisant la synthèse protéique et la division [81]. Même dans les types cellulaires cytostatiques comme les neurones, IGF-I fournit des signaux entraînant des changements du métabolisme cellulaire [81]. L’activation du récepteur ubiquitaire IGF-IR par IGF-I permet de coordonner le métabolisme des protéines, des glucides et du gras au sein de plusieurs types cellulaires. De façon importante, chacun de ces processus est modulé conjointement avec l’insuline et la GH.
Synthèse protéique
Tant in vitro qu’in vivo, IGF-I est un régulateur fort de la synthèse protéique. Cette réponse est modulée par l’activation de la cible mécanistique de la rapamycine (mTOR) via la voie de la phosphoinositide 3-kinase (PI3K). Dans le muscle squelettique, IGF-I stimule le transport des acides aminés et la synthèse protéique, mais inhibe le catabolisme des protéines [108], et ce tant en conditions normales qu’en situation d’apport protéique réduit. Lorsqu’IGF-I est administré à des volontaires sains, on observe une augmentation de la synthèse protéique, mais sans stimulation catabolique : ainsi peu d’effets sur la protéolyse sont observés [109]. De façon intéressante, l’administration de doses élevées d’IGF-I à des sujets ayant une mutation du récepteur de la GH induit la synthèse protéique [110]. Dans le
muscle, l’insuline peut inhiber la protéolyse à de très faibles concentrations, mais les concentrations requises pour stimuler la synthèse protéique sont beaucoup plus élevées [81]. Il serait donc raisonnable de conclure qu’IGF-I est le principal facteur responsable de l’anabolisme protéique dans le muscle suite à l’ingestion d’un repas.
Métabolisme des lipides
Bien que les adipocytes matures n’expriment pas IGF-IR, les préadipocytes expriment abondamment ce récepteur dont la liaison à IGF-I stimule la différentiation [111]. Une fois différentiés, leur expression d’IGF-IR diminue au profit du récepteur à l’insuline. Conséquemment, dans les dépôts adipeux, IGF-I ne possède pas la capacité d’initier des changements dans la synthèse des lipides ou la lipolyse. Dans le muscle, IGF-I est un stimulant fort du captage des acides gras libres et de l’oxydation. Une invalidation d’IGF-IR spécifique au muscle squelettique chez la souris entraîne l’apparition du diabète associée à l’incapacité d’IGF-I à stimuler le transport des acides gras libres [112]. Ces résultats sont d’ailleurs appuyés par l’observation que l'expression du transporteur d’acide gras CD36 rétablit ce phénotype [113]. Ceci amène la conclusion que ce sont les effets d’IGF-I sur le transport et l’oxydation des acides gras libres dans le muscle squelettique qui sont responsables de cette réponse et que l’inhibition de ce mécanisme entraîne la résistance à l’insuline.
Métabolisme du glucose
L’impact d’IGF-I sur le métabolisme du glucose s’appuie principalement sur la notion qu’IGF-I a la capacité de moduler les actions de l’insuline et de la GH. IGF-I réduit les niveaux plasmatiques de la GH et diminue par le fait même l’action directe de cette dernière sur la stimulation de la néoglucogenèse hépatique. Simultanément, IGF-I augmente le captage des acides gras par le muscle et favorise les actions hépatiques de l’insuline [114]. A très fortes concentrations, IGF-I peut stimuler le transport du glucose dans le muscle squelettique via IGF-IR ou les récepteurs hybrides insuline/IGF-I R [115, 116] et supprimer la néoglucogenèse rénale chez la souris [117]. Ces observations ont été



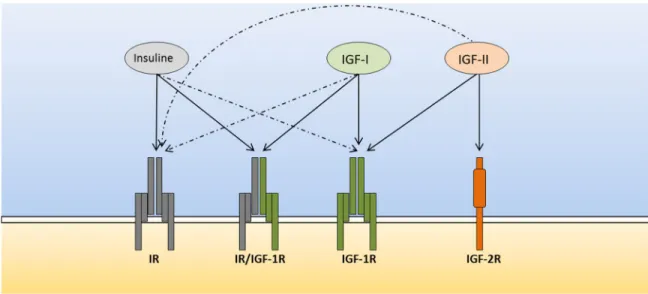



![Figure 9. Patron d’expression d’IGFBP-2 dans différents tissus chez l’humain obtenu grâce à l’utilisation de la sonde 202718_AT [333]](https://thumb-eu.123doks.com/thumbv2/123doknet/6367273.168245/80.918.210.582.131.396/figure-patron-expression-igfbp-tissus-humain-obtenu-utilisation.webp)
