Aqueous reactivity of glassy industrial byproducts in
alternative cementitious systems
by
Hugo Jake Uvegi
Bachelor of Science, The Johns Hopkins University (2015)
Submitted to the Department of Materials Science and Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Materials Science and Engineering
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
September 2020
© Massachusetts Institute of Technology 2020. All rights reserved.
Author . . . .
Department of Materials Science and Engineering
August 7, 2020
Certified by. . . .
Elsa A. Olivetti
Esther and Harold E. Edgerton Associate Professor in Materials Science
and Engineering
Thesis Supervisor
Accepted by . . . .
Frances M. Ross
Chair, Department Committee on Graduate Studies
Aqueous reactivity of glassy industrial byproducts in
alternative cementitious systems
by
Hugo Jake Uvegi
Submitted to the Department of Materials Science and Engineering on August 7, 2020, in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy in Materials Science and Engineering
Abstract
Alkali-activated, geopolymeric, and other novel binders offer an opportunity to curb
the carbon footprint associated with ordinary Portland cement (OPC). CO2
emis-sions inherent to source-material processing (i.e., firing of limestone at 1450 C) and annual OPC production volumes of 4.1 billion metric tons cause an estimated 5– 11% of global annual greenhouse gas (GHG) emissions. Material substitution with lower-footprint resources is therefore necessary for GHG impact mitigation. Glassy silica-, alumina-, lime-, and/or alkali-rich industrial byproducts (IBs) exhibit the properties necessary to achieve emissions reductions while preserving final product attributes expected of cementitious binders. Research and industry have both fo-cused primarily on metakaolin and IBs such as blast furnace slag and coal fly ash as supplementary and alternative cementitious precursors. Given projected limitations in such IB supply, it is imperative that we efficiently expand the materials search to other useful precursor candidates. This thesis focuses on chemical characterization and kinetic reactivity analysis of lesser-studied glassy materials through a combined experimental-computational approach, resulting in (1) physicochemical drivers for material aqueous reactivity and (2) a framework for evaluating new materials. First, I describe laboratory experiments involving reaction of a siliceous mixed-feedstock Indian biomass ash in aqueous sodium hydroxide solutions with selectively present lime and alumina sources. These experiments respectively yield tobermoritic calcium silicate hydrate products (Ca/Si ⇡ 0.6–1) and semi-crystalline zeolite / geopolymer products (Si/Al ⇡ 1); shown compositional ratios are known to be relevant to final material properties. Through this work, I demonstrate a novel approach to calculat-ing reaction product composition uscalculat-ing spectroscopic solution analysis of dissolution / precipitation experiments. Subsequently, I describe computational efforts to mine literature-reported data for potential precursor materials. This results in a database of material compositional and physical property data represented by a SiO2-Al2O3-CaO ternary diagram. Finally, I employ supervised and semi-supervised computa-tional models, which confirm log-linear relationships between glass dissolution rates (i.e., log10(rate)) and pH, inverse temperature (1/K), and glass connectivity (i.e.,
non-bridging oxygens per tetrahedron). While less interpretable, black-box models are observed to be more robust to the presence of additional features. Throughout the research program, reactivity is understood via material dissolution in aqueous solutions.
Thesis Supervisor: Elsa A. Olivetti
Title: Esther and Harold E. Edgerton Associate Professor in Materials Science and Engineering
Acknowledgments
These past five years have been quite a whirl. To say there were ups and downs would be putting it lightly. The support of my advisor, mentors, lab mates, my friends, girlfriend, and, of course, my family were key to making it through with my sanity intact.
Would I trade my time at MIT for anything else? Probably not. Would I do it again? Debatable.
Am I the person I am today because of it? Definitely.
The Ph.D. journey has taught me more about myself than I could have ever imagined at the outset, and I would like to thank the following people for making it the journey it was.
First and foremost, I would like to thank my advisor, Professor Elsa Olivetti. I cannot tell you how much I respect and admire you. I still remember our first interactions—you emailed congratulating me for my acceptance to MIT. As you may remember, I had previously learned of your work from one of my undergraduate professors, Prof. Tim Mueller. It was quickly apparent why he spoke so highly of you. A few conversations and an offer to conduct research trips to India later, and I joined your lab as your first Ph.D. student focusing on experimental research. New to both cement science and MIT, I was anxious and excited to get to work. Whenever I felt uncertain, your ability to problem solve and your action-oriented mentality helped me push through both research and personal obstacles along this path. Where some might have pushed away, you approached interactions directly and honestly, working with me to achieve both of our goals. Ever patient, ever insightful, and always finding a way to fit me (and any student!) into your schedule, from the bottom of my heart, I would like to thank you for your mentorship over these past five years.
Professors Krystyn Van Vliet and Cem Taşan, as members of my Ph.D. thesis committee, you were instrumental in guiding me down the most intriguing path. You helped me formulate important questions and made sure I was able to verify any claims. Cem and Krystyn, thank you for your kindness and ease of communication
over the past few years. It made my time at MIT that much more pleasant.
When I arrived at MIT, I had the pleasure of joining the Olivetti group simulta-neous with a then-budding post-doc, now budding-professor by the name of Piyush Chaunsali. Piyush, sharing an office and lab with you for the first three years of my Ph.D. did more to set me on the right track than you know. I will never forget our first experiments in Building 1—the surprise on your face at the extent of time nec-essary to mix brick formulations; the race between pressing bricks and assembling / disassembling molds—it was a blast. Not to mention our four research trips together to Muzaffarnagar. Thank you for your always-calm, always-jovial demeanor, your strict in-the-office work ethic, and your love of teaching. You helped me get started in a field that was at-first completely new to me.
To Brian Traynor, Tunahan Aytaş—the two remaining members of the brick mafia; Zach Jensen—hereby an honorary member (you spent enough time in brick meetings to deserve the title); and all past brick mafiosos—Rachel Osmundsen, Josh Dennison, Bassel Tarabay, Ciara Mulcahy, and Sam Wilson; I deeply appreciate all of the time you each spent assisting me with experiments, computation, and simply discussing any random thought or uncertainty I had over the years. There were a lot of those. Your support and patience were instrumental in helping me not only achieve this body of work, but also in developing my comprehension along the way.
To the other members of the Olivetti Group with whom I overlapped, both past and present—Jonathan Krones, Eddie Kim, Xinkai Fu, Alex van Grootel, Jiyoun Chang, Luly Alcaraz, Pat Ford, Weitong Liu, Adriano Polli, Kevin Huang, Rubayyat Mahbub, Haihao Liu, Olivia Pfeiffer, Basuhi Ravi, John Ryter, Jacqueline Baidoo, and Aubrey Toland—thank you not only for your insightful feedback and interesting presentations during group meetings but also for the great times we enjoyed together outside the walls of MIT. The work you all do and the joy you all bring to the group has made this process that much more fulfilling.
To John Ochsendorf, thank you for all of your insight during brick meetings through my first two years at MIT. It was a privilege to be able to learn from you on a weekly basis.
To Rupert Myers, I am so glad you came to work with Elsa for those few months and that we were able to spend the ICCC conference together in Prague. Your knowl-edge of cement and alkali-activation science is beyond impressive, and I appreciate having you for a mentor.
To my mentors and colleagues at the Tata Center—Professors Rob Stoner and Chintan Vaishnav, and all of the fellows and post-docs—thank you for making my first two years at MIT all the more remarkable. The field work in India and intimate seminars will stay with me forever. Additionally, thank you for funding my first three years of research at MIT. The work would not have been possible without your generosity.
To Thomas Poinot, Mike Laracy, thank you for setting the ground work for my research and setting myself and Piyush off on the right foot when we arrived at MIT. To Pankaj Aggarwal, thank you for coordinating my research efforts while visiting in Muzaffarnagar and for so graciously welcoming me into your city. To Ankur Garg, thank you for allowing me and my colleagues to use your facilities at Deepak Ceramic & Allied Products Pvt. Ltd in Muzaffarnagar.
To Dr. Soumen Maity, thank you for your collaboration and interest in our re-search. Use of the facilities at Development Alternatives in New Delhi and your knowledge of industrial waste streams in India was invaluable to my research efforts. To Dr. Charlie Settens, Tim McClure, Patrick Boisvert, and Dr. Shiahn Chen, thank you for your assistance with the shared experimental facilities at the MIT Ma-terials Research Lab. To Dr. Caitlin Quinn at The University of Delaware, thank you for conducting the solid state nuclear magnetic resonance spectroscopy measurements. To Dr. Richard Goodwin and Dr. Nghia Hoang, thank you for your and IBM’s collaboration over the past few months as we work to model the dissolution of alkali silicate and more complex glasses. It has been an incredible boon to our work and my understanding of both glass science and machine learning.
To my DMSE cohort and the rest of the DMSE community, thank you for being such a welcoming, interesting, curious, smart group of people. I will cherish all of the conversations we had and all of the studying we did together.
Outside of the lab, being able to spend time with wonderful roommates and friends has been one of the highlights of my time in Cambridge. To my roommates at 9 Seckel—PJ Santos, Quantum Wei, Scott Hunter, and Bitzy Flamholz—thank you for the countless fun times and never-ending motivation. To my roommates at 26 Columbia—Daniel Duane, Sarah Walker, and Xiaoyu Wu—thank you for welcoming me with open arms. I am sad our year together got cut short (thanks COVID-19), but I greatly enjoyed our time together. To the rest of the Potlucks Crew, thank you for coming together (almost) every Sunday over the past four years. You taught me to try my hand at cooking and to bring more than clementines to a potluck. I will forever remember our Friendsgiving celebrations. Also, thank you to the best team at Thirsty Ear Trivia for not getting mad at me when I would occasionally ditch you for other activities.
To my friends in the MIT Bitcoin Club and the DCI, thank you for the incredible conversations about cryptography, computation, money, power, and decentralization and for allowing me to be involved in some of the largest events I have had the pleasure of organizing.
To my friends from Heschel and Johns Hopkins, thank you for making me the person I am and for always being there for a laugh, a phone call, or to hang out—love you all. Jonah, Ellen, Bernard, and Ian, thank you for teaching me proper research etiquette and for your mentorship at JHU and beyond.
To my girlfriend, Elle Braun, when I moved to Cambridge, I never thought I would meet and fall in love with someone as wonderful as you. Thinking back to how we met half-way through my second year, it feels like you have been on this journey with me from the beginning. Reminding myself that I was in the midst of writing the proposal for this thesis when we met, in a way you have been. When you were admitted to law school in New York and we realized that long distance was in our future—3 years ago—many would have called it quits. Quite the contrary, it has made the weekends we have been able to spend together some of the highlights of my Ph.D. Thank you for your incessant support, love, and affection—I could not have done it without you. Mom, Dad, Gab, Nance, Dave, Fred, Brighton, Shnellers, Nanny, Poppy—I could
not ask for a more loving and supportive family. Mom and Dad, from the very beginning, you have always encouraged my curiosity and yearning for knowledge, signing me up for every after-school program in the book. Thank you for all of the love you have shown me and for reassuring me that it would continue regardless of any individual exam score (my own pressure, not yours). Gab, sharing a room with you growing up has shaped me into a more loving and caring person. I thank you for all of your social insights over the years, and love the woman you have become. Nance and Dave, you are the best, most encouraging aunt and uncle a nephew could ask for. Fred and Brighton (and Georgie and Lala), I will never forget all of the long drives we took up from D.C./Baltimore during my undergrad days, and all of the wonderful times since. Shnellers—Shiffy, Udi, Avital, Chesky, Daniel, Einat, Noam, and more—I love you all so much and cannot wait to celebrate with you in Israel soon. Nanny and Poppy, while you are both no longer with us, your love and support are imprinted on me forever. I will never forget when I told you both I was switching from pre-med to engineering in undergrad. Poppy asked, "What can you do with an engineering degree?" That question was short-lived, and I felt nothing but adoration and care before and since. I love you all and cannot thank you enough for providing me with the foundation necessary to achieve this milestone.
Professional acknowledgments
I would like to further acknowledge all of the financial support that went towards funding this research, or from which this research otherwise benefited. Thank you to the Tata Center for Technology and Design and the Environmental Solutions Ini-tiative, both at The Massachusetts Institute of Technology (MIT), for your financial support for this work. I also acknowledge support from NSF CAREER #1751925, as well as from the Government of Portugal through the Portuguese Foundation for International Cooperation in Science, Technology, and Higher Education, through the MIT Portugal Program and from IBM through the MIT-IBM Watson AI Lab. Thank you to Mr. Pankaj Agrawal of Bindlas Duplux LTD. (Muzaffarnagar, India)
for arranging the procurement of the biomass ash used in this thesis and Advanced Cement Technologies for providing the metakaolin used in this thesis. This work made use of the MRSEC Shared Experimental Facilities at MIT, supported by the National Science Foundation under award number DMR-1419807. NMR experiments were carried out by Dr. Caitlin Quinn at the University of Delaware NMR Facility. XRF measurements were conducted by Wyoming Analytical Laboratories, Inc.
Contents
Abstract 3
Acknowledgments 5
List of Figures 15
List of Tables 24
1 Alternative cementitious materials: Novel, low-footprint products
for an increasingly developed world 25
1.1 Ordinary Portland Cement: The building material of yore . . . 26
1.1.1 A brief history . . . 26
1.1.2 OPC-based calcium silicate hydrate binder chemistry . . . 28
1.2 Geopolymers and alkali-activated materials: Potential solutions to the cement crisis . . . 29
1.2.1 General overview . . . 29
1.2.2 Raw materials . . . 31
1.2.3 Activating solutions . . . 38
1.2.4 Geopolymeric and alkali-activated binder chemistry . . . 40
1.3 Material reactivity . . . 44
1.3.1 Glass chemistry and structure . . . 45
1.3.2 Glass dissolution . . . 49
2 Materials of interest and methods employed 55
2.1 Experimental materials . . . 56
2.1.1 Indian biomass ash . . . 56
2.1.2 Hydrated lime (Ca(OH)2) . . . 59
2.1.3 Metakaolin . . . 60
2.2 Experimental methods . . . 62
2.2.1 Inductively coupled plasma optical emission spectrometry (ICP-OES) . . . 62
2.2.2 X-ray diffraction (XRD) . . . 64
2.2.3 Fourier transform infrared spectroscopy (FTIR) . . . 64
2.2.4 Solid state27Al and29Si magic angle spinning nuclear magnetic resonance spectroscopy (MAS-NMR) . . . 64
2.2.5 Scanning electron microscopy (SEM) . . . 65
2.2.6 Selective dissolution . . . 66
3 Kinetic studies of Indian biomass ash dissolution 69 3.1 Sample preparation . . . 72
3.2 Multi-faceted dissolution and product characterization . . . 73
3.3 Byproduct supply / utility prediction . . . 83
3.4 Reactivity / dissolution of raw materials . . . 84
3.5 Precipitation of reaction products . . . 85
3.6 Utility of ICP-OES . . . 86
3.7 Chapter Summary . . . 89
4 Applying new methods to dissolution and geopolymerization of metakaolin with Indian biomass ash 91 4.1 Sample preparation . . . 93
4.2 Dissolution and precipitation as measured through liquids analysis . . . 95
4.2.1 Separate dissolution of Metakaolin (MK) and silicate ash (SA) 97 4.2.2 Solution chemistries resulting from SA-MK interactions . . . . 99
4.2.3 Calculation of Si/Al from ICP-OES . . . 101
4.3 Solids analysis: XRD, 27Al and 29Si MAS-NMR, SEM, and selective dissolution . . . 103
4.4 Chapter Summary . . . 107
5 Computational study of glass dissolution 111 5.1 Computational methods . . . 115
5.1.1 Data extraction . . . 116
5.1.2 Data engineering and normalization for dissolution rate analysis 118 5.1.3 Thermodynamic considerations . . . 120
5.1.4 Dissolution rate modeling . . . 122
5.2 Expanding the precursor pool: Ternary diagram . . . 126
5.3 Dissolution rate modeling . . . 129
5.3.1 Supervised model . . . 129
5.3.2 Semi-supervised model . . . 135
5.4 Chapter summary . . . 138
6 Future work 141 6.1 Summary of findings . . . 141
6.2 Open questions and recommended future directions . . . 143
A Appendix: Glossary and Notation 147 A.1 Glossary . . . 147
A.2 Notation . . . 149
A.2.1 Cement notation . . . 149
A.2.2 Connectivity notation . . . 150
B Appendix: Supplementary experimental data 151 B.1 Chapter 3 ICP data . . . 151
C Appendix: Supplementary computational data 159
C.1 DOI search keyword list . . . 159
C.2 Table keyword search list . . . 161
C.3 Dimensional analysis of normalization equations . . . 168
C.4 Connectivity metrics . . . 170
C.4.1 Optical basicity . . . 170
C.4.2 Non-bridging oxygens per tetrahedron (NBO/T) . . . 171
List of Figures
1-1 (a) 14 Å tobermorite structure. Silicate groups are represented by
purple tetrahedra, with oxygen atoms in red. Ca2+ ions are shown
in light blue, and charge balancing OH are shown in black. P and B denote the position of bridging and paired tetrahedra characteristic of dreierketten chains. Reproduced from Richardson (2008) [26]. (b) A schematic of tobermorite-based C-S-H structure according to Stade
and Wieker’s 4 layer model. Reproduced from Stade (1980) [31] . . . 28
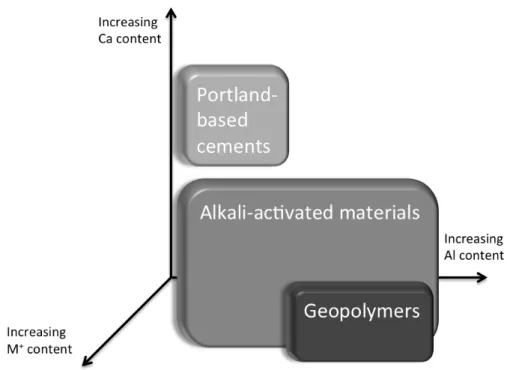
1-2 Diagram depicting relative concentrations of Ca, Al, and M+ (Na+ and/or K+) in each system—shading corresponds to increased alkali
concentration. Adapted from Provis and van Deventer, 2014 [45] . . . 31
1-3 (a) Crystalline structure of kaolinite. Reproduced from Fernandez, et al. (2011) [51], (b) Reorganization of kaolinite upon dehydroxylation
to form metakaolinite. Adapted from Davidovits (2015) [44] . . . 32
1-4 Proposed reaction mechanism for geopolymer formation. Reproduced from Duxson, et al. (2007) [47] . . . 42 1-5 Generalized structure of C-(N)-A-S-H type gels composed of cross
linked and non-cross linked type tobermorite structures. Reproduced from Provis and van Deventer (2014) [45] with the permission of the artist. . . 44 1-6 Schematic reactions showing introduction of (A) alkali and (B) alkali
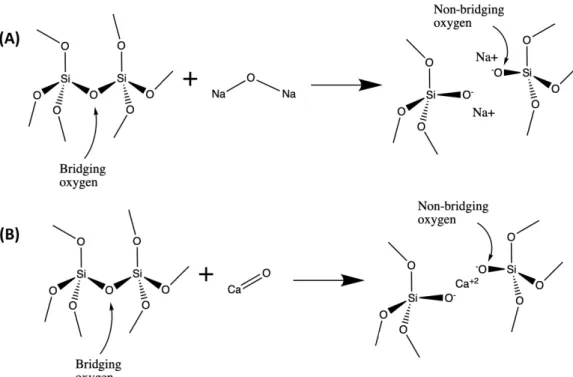
earth species into the silicate glass network, creating NBOs in the process as labeled. . . 47
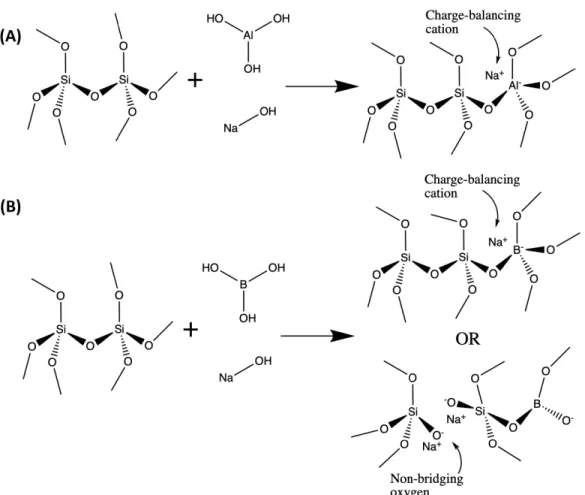
1-7 Schematic reactions showing introduction of (A) aluminum and (B) boron an alkali silicate glass network . . . 48 1-8 Schematic depicting material flow from industry through dissolution
and finally to reaction product precipitation. Graphics used in the third section are reproduced from Koleżyński, et al. (2018) [231], The IZA Database of Zeolite Structures [232], Richardson (2008) [26], and Myers, et al. (2013) [35], as labeled. . . 52
2-1 Scanning electron micrograph of Silverton biomass ash . . . 57
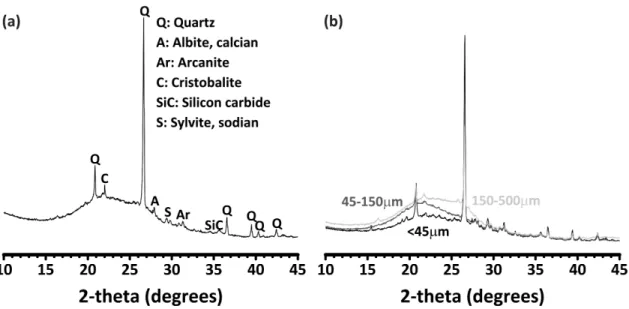
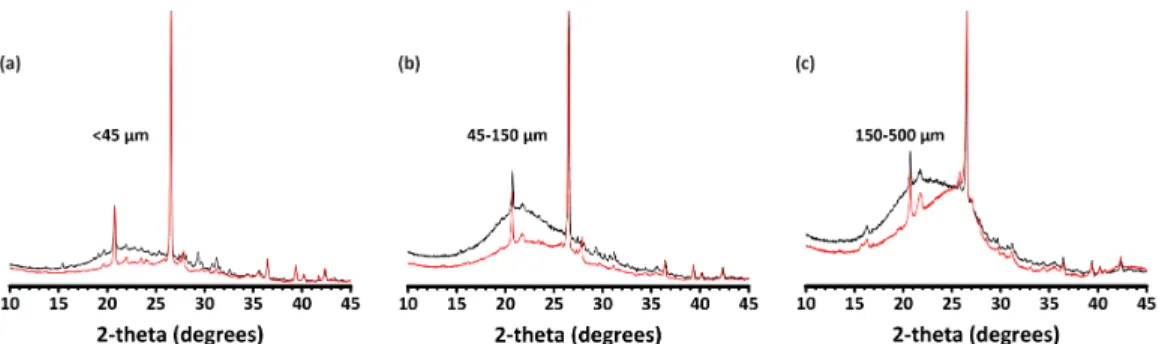
2-2 (a) XRD pattern of raw biomass ash (<500 µm) sourced from Silverton Pulp & Papers Pvt. Ltd. (b) XRD patterns of ash fraction: <45 µm,
45-150 µm, and 150-500 µm . . . 59
2-3 (a)-(c) Normalized XRD patterns comparing ash fractions, as labeled, pre- and post-672-hour dissolution in aqueous 1M NaOH, respectively shown in black and red. . . 60 2-4 (a) X-ray diffraction pattern of metakaolin and (b) Comparison of full
XRD patterns (5 -70 2✓) of metakaolin and Silverton biomass ash . . 62
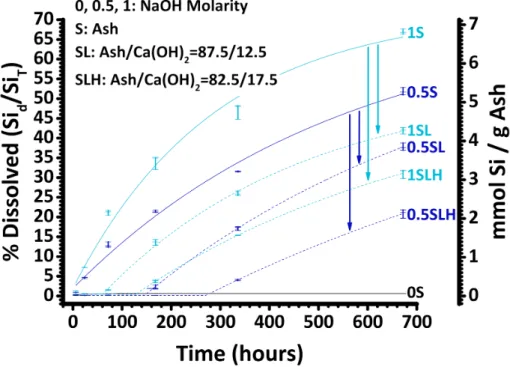
3-1 Extent of Si dissolution from SA as a function of time at L/S of 25. 0S, 0.5S, and 1S fit asymptotically with solid lines. SL and SLH sample data fit qualitatively for two regions (dotted lines): (1) apparently flat dissolution (reaction) followed by (2) asymptotic dissolution. . . 75 3-2 Normalized XRD patterns of SA particles 150–500 µm in size before
and after 672 hours (28 days) of dissolution. Identical to Figure 2-3(c). 76 3-3 Concentration of Ca (millimolarity) in aqueous 0.5 mol/L and 1 mol/L
NaOH systems as a function of time. Dotted lines denote literature values for Ca solubility in such solutions [237, 238]. . . 77 3-4 Normalized FTIR absorbance spectra of four samples for comparison
of Ca(OH)2 consumption (peak at 640 cm 1) with C-S-H production
3-5 XRD patterns of silicate ash (SA), 6 hour, 24 hour, 72 hour, 168 hour, and 672 hour samples of ash dissolved in 1 mol/L NaOH in the
pres-ence of Ca(OH)2 at an ash/lime ratio of 87.5/12.5. Note: diminution
of portlandite peaks (Ca(OH)2), evolution of C-S-H peak (specifically associated with tobermorite-like phase), and decrease in intensity of
amorphous hump with time. . . 79
3-6 Normalized FTIR absorbance spectra of silicate ash (SA), 6 hour, 24 hour, 72 hour, 168 hour, and 672 hour samples of ash dissolved in
1 mol/L NaOH in the presence of Ca(OH)2 at an ash/lime ratio of
87.5/12.5. Note: disappearance of Ca(OH)2 peak at 3640 cm 1 and
emergence of C-S-H (Q2) peak with time. . . . 80
3-7 (a)-(c) 29Si MAS-NMR of raw SA, 1SL168, and 1SL672, respectively,
along with their deconvolution into Gaussian curves (in gray) and sum of the separate curves (in red) and (d) the three spectra stacked for comparison. Referenced externally to hexamethylcyclotrisiloxane at -9.00 ppm. . . 83
4-1 Schematic of experimental program for SA and metakaolin experi-ments, detailing dissolution (control) and reaction/precipitation ex-periments. Note: Scheme 1 represents a 1-step reaction scheme, while Scheme 2 is carried out in 2 steps. . . 93
4-2 Solution chemistry of samples over time, presented in millimolarity (mM) and % dissolved (Xdissolved/XTotal). mM represent the total con-centration of Si or Al in the filtrate at each time point, while % dis-solved represents the fraction of initial total solid Si or Al of the pre-cursors found in solution. Each time point exhibits samples taken in triplicate. (a)-(d), (e)-(h), and (i)-(l) respectively represent dissolu-tion/reaction in 1M, 3M, and 6M NaOH-based solutions, with the blue horizontal dotted lines indicating initial concentrations of Al and Si in the Na-silicate systems. MK, SA, MK+SA, and MK+Si(-SA) samples are described in Table 4.1. . . 96 4-3 Solution concentration in mM of Al and Si for (a) the MK+Si(-SA)
system and (b) the MK+SA system. . . 104 4-4 (a)-(d)27Al and (e)-(f)29Si solid state MAS-NMR spectra of indicated
samples. Time points are specified in hours. Peaks are labeled accord-ing to the center point as fit by MestreNova software. Stars indicate spinning side bands. . . 105
4-5 27Al solid state MAS-NMR specra of 1M and 3M samples after 672
hours of reaction, indicating that even in these samples, only Al(IV) peaks remained. . . 106 4-6 XRD patterns of (a) 6-hour and 672-hour 6M samples and (b) 672-hour
1M and 3M samples. . . 107 4-7 SEM Micrographs of (a)-(b) MK+SA and (c) MK+Si(-SA). Notice
surface-driven precipitation in (a) and existence of two morphological species in (c) as compard to one in (b) . . . 108
5-1 Ternary SiO2-Al2O2-CaO diagram depicting different material gories of interest to the cement community. All chemistry and cate-gory data extracted from tables in the literature. The numbers below each category label reflect the number of samples over the number of unique DOIs (i.e., Samples/DOIs). Data points shown in yellow are as yet unlabeled, reflecting an additional 15,500 samples . . . 118 5-2 Schematic diagram of our semi-supervised learning analysis that spans
multiple domains of data. Unlabeled and labeled data (X) were first entered into a probabilistic encoder P(Z|X) that maps to a space of la-tent feature representation (Z), which were re-directed to a probabilis-tic decoder P(X|Z) that generates a standardized data summarization (X*). The distributional discrepancy between X and X* (conditioned on Z) is then used as an unsupervised loss on information summariza-tion while the latent representasummariza-tion Z that corresponds to the labeled data is used to optimized for a latent predictor that maps from Z to the corresponding outcome Y. All component parameterizations in the above pipeline are customized following the guiding principles in (Kingma, 2014) [298], which can be optimized end-to-end via gradient backpropagation. . . 126 5-3 (A) Cumulative density functions of normalized oxide content (CaO,
SiO2, and Al2O3) for all extracted samples; (B)-(L) Respective fre-quency densities of normalized oxide content as labeled. The sum of the three oxides (CaO, SiO2, and Al2O3) was normalized to 100%. . . 128 5-4 (A) Plot of log rate as a function of pH for all samples extracted from
the literature (n = 580). (B) Same plot for samples from experiments conducted at temperatures in the range of 20 T ( C) 35 (n = 228) with schematic lines included displaying disparate trends in acidic and basic pH. Different colors represent distinct DOIs. Trendlines are schematic to show opposing trends in acidic and basic regions. . . 130
5-5 Data from Figure 5-4 (n = 580) replotted with overlaid thermal in-formation. Temperatures trend from low to high with dark to light colors. Trendlines again schematically show opposing trends in acidic and basic regions. . . 130
5-6 A) Decision tree regression trained on samples from [78, 209, 210, 212, 220–222, 225–229, 287, 288, 299, 300] (472 samples) and all features as listed in Table 5.2. Testing the regression on samples from [211, 223, 224, 230, 286] (108 samples) with a max depth of 8 yielded RMSE = 1.26 and R2 = 0.52. (B) Decision tree regression based on the same train/test split but with only 3 features: pH, 1/Temperature (1/K), and NBO/T, yielding RMSE = 1.23 and R2 = 0.54. . . 132
5-7 Linear regression trained on all samples from [78, 209, 210, 212, 220– 222, 225–229, 287, 288, 299, 300] and tested on samples from [211, 223, 224, 230, 286]. (A) Train (n = 472) and test (n = 108) sets included all samples and resulted in respective RMSE values of 0.97 and 1.22; (B) Train (n = 322) and test (n = 71) sets included samples from
experiments with pH 7 and resulted in respective RMSE values of
0.68 and 0.94. . . 133
5-8 Graphs of predicted vs true values of log10(dissolution rate) for data with pH 7. In (A) the model is trained on a single data set as listed (cement, nuclear, or geochem), while in (B) the opposite is true—the model is trained on 2/3 sets and tested on the third set as listed. In
(C), the model is trained and tested on B2O3-containing or -absent
glasses as labeled. In (D) the model is trained and tested on batch or flow experiments as labeled. Number of samples in training set (n) and RMSE scores of prediction on test sets are listed on each plot. . . 134
5-9 (A) RMSE of machine learning models as a function of number of training epochs. Input consisted of full feature set as defined by Table 5.2, with size of training set varied as labeled. (B) RMSE of machine learning models, where input consisted of only 3 key features (pH, 1/temperature (1/K), and NBO/T) and size of training set varied as labeled. (C) Comparison between RMSE of four input sets: (1) All fea-tures, all samples, (2) 3 feafea-tures, all samples, (3) All feafea-tures, samples
List of Tables
2.1 Average chemical (oxide wt%) and mineralogical (phase wt%) compo-sitions of Silverton biomass ash samples as determined by XRF and quantitative Rietveld XRD, respectively. Uncertainty in oxide wt%
represents one standard deviation of measurements from four samples. 58
2.2 Amorphous percent and loss on ignition values for different size frac-tions of ash (as-received) . . . 59 2.3 Chemical (oxide wt%) and mineralogical (phase wt%) compositions of
"PowerPozzTM (HRM)" metakaolin as determined by XRF and quan-titative Rietveld XRD, respectively. XRF data provided by Advanced Cement Technologies. . . 61
2.4 Dilution procedure for ICP-OES calibration standards. . . 63
2.5 Solution compositions for selective dissolution techniques (per 1 g of sample) . . . 67
3.1 Sample identification. Liquid/Solid ratio for all samples = 25 . . . 74
3.2 Band assignments for FTIR spectra of SA-based binder samples [89,265]. 81
3.3 29Si MAS-NMR deconvolution and quantification. . . . 82
3.4 Ca/Si ratios of solid SL and SLH products as calculated from ICP-OES results at 7, 14, and 28 days. Uncertainty represents 95% confidence interval when calculating differences in Si levels between ash dissolution
and reaction with horizontal and vertical aggregation analysis. . . 87
3.5 Ca/Si of solid 1SLH sample after 336 hours of reaction as calculated from all techniques (XRD, NMR, ICP-OES, SAM, and EDTA/NaOH). 88
3.6 Na adsorption after 672 hours of sample evolution, in mmol Na/g solid. 89 4.1 Ash and metakaolin experimental program—solution set-up and
de-scriptions (Silicate ash = SA, metakaolin = MK) . . . 94
4.2 Initial elemental concentrations in Na-silicate used for MK+Si(-SA) systems . . . 99 4.3 Si/Al ratios of solid reaction products as calculated from ICP-OES
results. Calculated values for 6–168 hours maintained for illustrative purposes; 336, 672 hours reflect reasonable product chemistries, as corroborated by solids analysis in Section 4.3. . . 103 4.4 Si/Al of solid reaction product samples after 672 hours of reaction
as calculated from all techniques (XRD, NMR, ICP-OES, and HCl dissolution). . . 109 5.1 Calculated equilibrium constants and temperature correction terms for
relevant dissolution and aqueous reactions. . . 123 5.2 Full feature lists for regression and machine-learning analyses. . . 124 5.3 Statistical parameters for log(rate) of B2O3-containing and B2O3-absent
glasses . . . 135 C.1 Search keywords for discovering relevant dissolution rate-focused papers.160 C.2 Search keywords for classifying tables by caption (indicating included
data). . . 161 C.4 Optical basicity values of relevant oxides, from Rodriguez, et al. (2011)
Chapter 1
Alternative cementitious materials:
Novel, low-footprint products for an
increasingly developed world
Ordinary Portland cement (OPC), the most manufactured material by mass and the binding phase (read: glue) in the world’s most consumed industrial product— concrete—is estimated to be responsible for anywhere from 5–11% of global annual greenhouse gas (GHG) emissions [1–9]. While the general public often assumes GHG emissions to be primarily associated with the burning of fossil fuels, in the case of
OPC, more than 50% of emitted CO2 is inherent to feedstock material processing.
The calcination of limestone (CaCO3)—the primary calcareous feedstock for OPC—
yields fixed CO2 emissions at one mole per mole of fired carbonate (0.44 g/g). Add
to that the fuel necessary to reach processing temperatures of 1450 C and you arrive at average estimates of 0.8–0.9 tons CO2 per ton of cement [1, 2,9, 10]. On a per-ton basis, OPC is, in fact, a lower-emissions material than other widely used materials, such as primary raw steel (up to 3 tons CO2/ton) and aluminum (up to 15 tons CO2/ton) [10]. However, the sheer volume of global cement production—estimated at 4.1 billion metric tons annually as compared to 1.9 billion and 64 million metric tons for primary raw steel and aluminum, respectively [11]—results in quite massive
an opportunity to decrease this volume by taking advantage of the hydraulic and cementitious natures of other natural and industrial materials [9,12–14]. This chapter serves as an introduction to the materials of focus throughout the thesis, providing a background on ordinary Portland cement, and delving into novel alternatives and the precursor materials of interest.
1.1 Ordinary Portland Cement: The building
mate-rial of yore
1.1.1 A brief history
One of humanity’s most important inventions, cement-based concrete has been de-veloped as a "just add water" building solution over centuries of chemical research and discovery. Evolving from a mortar-like binder primarily based on the pozzolanic reaction of lime (shown in Equation 1.3) [15, 16], cement is now one the most used building materials in the world, and concrete—the cement-based composite—is often cited as the second most consumed material by mass after water. The initial discov-ery of lime resulted from limestone (CaCO3) calcination at elevated temperatures to produce calcium oxide (CaO). While as-mined limestone varies in its composition, incorporating impurities in the form of siliceous, aluminous, and ferrous phases, its primary constituent is calcium carbonate (CaCO3). When heated, it releases carbon dioxide gas (CO2), yielding solid CaO and contributing to the high carbon footprints of lime and OPC. The resulting CaO, often converted to the relatively less caustic hydrated lime (Ca(OH)2), readily reacts with silicon containing species in aqueous en-vironments to produce calcium silicate hydrate (C-S-H1) binder phases. A simplified reaction scheme illustrating this process is shown in Equations 1.1-1.3.
CaCO3 heat! CaO + CO2 (1.1)
1Letters here refer to oxides (C = CaO, S = SiO
2, H = H2O), with dashes denoting ill-defined
stoichiometry, as per typical cement notation. Full details regarding such notation is included in Appendix A.
CaO + H2O ! Ca(OH)2 (1.2)
Ca(OH)2+Si(OH)4 ! CaH2SiO4 · 2H2O (1.3)
This method of calcining limestone and the observation of its propensity to form hydrated binder phases in the presence of silicon and water eventually led to the production of ordinary Portland cement (OPC)—named for its similar appearance to Portland stone [15]. While developed from pozzolanic materials (i.e., those which require external sources of calcium to form a cementitious binder), OPC itself is actually classified as a hydraulic material, as it is manufactured to contain both reactive calcium and silicon oxides and can self-react in the presence of water to form C-S-H binder products.
While for years concrete systems were produced solely from OPC clinker, water, and aggregate, today’s concrete often incorporates various supplementary cementi-tious materials (SCMs). Hailed as a means of reducing the environmental footprint of OPC, the addition of SCMs to the cement or concrete mixture also often results in increased physical properties. Highly siliceous materials (e.g., silica fume), and aluminosilicate materials (e.g., coal fly ash and metakaolin), have been of interest to concrete researchers for decades due to their beneficial effects on the system’s phys-ical and structural properties, such as flowability, setting time, and compressive and flexural strengths [16–18].
While SCMs continue to be explored as a means of curtailing OPC use, research in this field still focuses on primarily (>50%) OPC-based mixtures. More recently, scientists have realized the possibility of creating OPC-free mixtures based upon slightly different solution chemistries and using the so-called SCMs as alternative cementitious materials (ACMs) instead. ACMs capitalize on observed hydraulic and pozzolanic natures of different materials to produce hardened binders in the absence of OPC. These materials often require caustic, alkali-rich solutions to enhance reactivity, as slow reaction kinetics or insufficient reaction energies hinder their hydraulic or pozzolanic activities in water. These materials will consistute the bulk of this thesis, and their chemistry will be explored in more depth in Section 1.2.
1.1.2 OPC-based calcium silicate hydrate binder chemistry
Similar to earlier lime-pozzolan mortars, OPC binders are composed primarily of poorly crystalline calcium silicate hydrate (C-S-H) gel. Although OPC has been in use for over a century and its reaction to form C-S-H has long been discussed on a purely chemical basis, the intimate details of the C-S-H structure are still being explored and clarified [19–30]. Models often debate the level of organization present in what is typically termed a S-H gel, and while much literature maintains C-S-H’s predominantly amorphous nature, many researchers model C-S-H as tending toward two approximate chemistries and crystal structures—one similar to 14 Å to-bermorite (Ca4H4Si6O18· 8H2O, Ca/Si = 0.66 [24]; Ca5Si6O16(OH)2· 7H2O, Ca/Si = 0.83 [26]) and another similar to jennite (Ca8H4Si6O18(OH)8·6H2O, Ca/Si = 1.33 [24]; Ca9Si6O18(OH)6· 8H2O, Ca/Si = 1.50 [26]) [19, 21, 24, 26]. Still, a range of potential structures have been proposed, with most based on some arrangement of inter-layered silicate tetrahedral units (SiO4), ionically bound Ca-O and OH units, and structural H2O. Examples of this are depicted in Figure 1-1.
Figure 1-1: (a) 14 Å tobermorite structure. Silicate groups are represented by purple tetra-hedra, with oxygen atoms in red. Ca2+ ions are shown in light blue, and charge balancing
OH are shown in black. P and B denote the position of bridging and paired tetrahe-dra characteristic of dreierketten chains. Reproduced from Richardson (2008) [26]. (b) A schematic of tobermorite-based C-S-H structure according to Stade and Wieker’s 4 layer model. Reproduced from Stade (1980) [31]
.
Regardless of the model employed, most cement scientists are in agreement as to the "dreierketten" structure of C-S-H, where silicon chains are kinked such that two
tetrahedra are connected directly to the calcium ion layer, while a third tetrahedron acts as a bridge between two successive dimers [19, 24, 26]. Due to the complex and only semi-crystalline nature of C-S-H, most cement scientists utilize the Ca/Si ratio as a placeholder for full empirical formulae when investigating system properties. While idealized cases of 14 Å tobermorite and jennite yield respective Ca/Si ratios of 0.66– 0.83 and 1.33–1.50, models accounting for defects typical of C-S-H structures have accounted for Ca/Si ratios ranging from 0.66–2.3, all of which have been observed experimentally depending on precursor composition and system age [19,24,26,27,32]. Recent advances in cement science have centered primarily on the use and effect of supplementary cementitious materials (SCMs) on the physical properties of the final concrete products [16, 18, 33, 34] as well as the substitution of aluminum for silicon and uptake of alkalis in modified C-S-H materials (i.e., C-(A)-S-H and (N,K)-C-A-S-H) [35–39]. As aluminum is typically incorporated as a substitute for silicon in the described tetrahedral units, it is important to note that any incorporated cationic species first go to balancing the associated residual charge before otherwise altering the network structure (i.e., Al3+ in tetrahedral coordination with oxygen results in a net negative charge of -1, which must be balanced by the positive charges associated with introduced cations, such as Na+, K+, Ca2+, Mg2+, etc.).
1.2 Geopolymers and alkali-activated materials:
Po-tential solutions to the cement crisis
1.2.1 General overview
Geopolymers (GPs) and alkali-activated materials (AAMs), both classes of alternative cementitious materials, pose a significant opportunity for the reduction of construc-tion materials-related greenhouse gas emissions.
Emerging initially from research into the cementitious properties of blast furnace slag—a byproduct of the iron smelting industry—alkali-activated systems were dis-covered over a century ago, albeit explored only in a limited capacity until the last few
decades. The reaction between an alkali source and an aluminosilicate solid was first documented by German chemist Hans Kühl in his 1908 patent [40]. Kühl’s material, although slow to set, was deemed comparable in its final physical properties to hard-ened Portland cement, which by that point had been in use for over 50 years [15]. This work was expanded through empirical and theoretical studies carried out by British scientist A.O. Purdon in 1940 and Ukrainian scientist V.D. Glukhovsky in 1959, re-spectively [41, 42]. In 1979, the term "geopolymer" was first coined by Davidovits, who published extensively about the mechanisms of reorganization when inorganic aluminosilicates were introduced into to high-pH, alkali-rich aqueous solutions. As his original intention was to formulate a nonflammable alternative to organic polymers, he specifically described geopolymers using terminology typically reserved for organic systems, thereby demonstrating the similarities between these inorganic systems and their organic counterparts [43, 44].
While the term geopolymer has come to refer specifically to low-calcium alkali aluminosilicate network solids, alkali-activated materials can be more broadly defined as gel / network solids with varying levels of silica, alumina, and lime, with alkali cations incorporated as charge-balancing species similar to what was described at the end of Section 1.1.2 above [44, 45]. A diagram depicting the typical chemistries of Portland-based cements, alkali-activated materials, and geopolymers is shown for reference in Figure 1-2.
Most initial geopolymer systems were synthesized using metakaolin as the alu-minosilicate source, with Si/Al ratios controlled using a high-pH alkali silicate solu-tion [43,46]. Since their incepsolu-tion, however, geopolymer and alkali-activated systems have enjoyed the integration of a variety of aluminous, siliceous, and calcium-rich pre-cursor materials, as well as a number of alkali activating sources, typically limited to sodium and potassium-based solutions. Significant research has focused specifically on metakaolin, coal fly ash, and blast furnace slag systems due to their relatively well-understood and documented chemistries and extensive use as SCMs [45, 47, 48]. Their use to date is reviewed in the following sections. Still, there exist a plethora of other natural and industrial waste materials that may prove useful in such systems
Figure 1-2: Diagram depicting relative concentrations of Ca, Al, and M+ (Na+ and/or K+) in each system—shading corresponds to increased alkali concentration. Adapted from Provis and van Deventer, 2014 [45]
and which this research hopes to inform [49].
1.2.2 Raw materials
1.2.2.1 Metakaolin
Used as both an SCM and an AAM precursor, metakaolin is formed through the calcination of kaolinitic clay at temperatures ranging from 400–800 C [50]. Kaoli-nite, a layered aluminosilicate mineral with empirical formula Al2Si2O5(OH)4 (i.e., 2SiO2 · Al2O3 · 2H2O), has strongly bound hydroxide groups which exist in interlayer positions within the clay structure, as shown in Figure 1-3(a) [51].
2Al2Si2(OH)4 heat! 2Al2Si2O7 + 4H2O (1.4)
Upon dehydroxylation, kaolinite undergoes the reaction seen in Equation 1.4 and schematically in Figure 1-3(b), and reorganizes to form the highly reactive
metakaoli-Figure 1-3: (a) Crystalline structure of kaolinite. Reproduced from Fernandez, et al. (2011) [51], (b) Reorganization of kaolinite upon dehydroxylation to form metakaolinite. Adapted from Davidovits (2015) [44]
nite2. Due to the placement of the OH groups in the original kaolinite structure,
the Si-O network remains relatively intact during the transformation, while the Al-O network undergoes significant reorganization—converting from predominantly 6-coordinated Al, to majority 4- and 5-6-coordinated Al [44, 52, 53], (i.e., metakaolinite is generally not quite as ordered as is depicted in Figure 1-3(b)). The reactivity of the final metakaolin has been linked to the crystallinity of the initial kaolin sam-ple, with lesser initial crystallinity yielding more reactive metakaolin upon calcina-tion [54]. It has further been shown that dehydroxylacalcina-tion at lower temperatures produces metakaolin with higher specific surface area, which has also been observed to increased reactivity [55].
As both an SCM and a precursor for AAMs, metakaolin plays distinct roles in high and low-calcium environments. Whereas it acts as a highly pozzolanic material in high-calcium systems, readily reacting with calcium to form calcium aluminum silicate hydrate (i.e., C-A-S-H) products with varied levels of aluminum incorporation, it geopolymerizes in high pH, low-calcium environments to form alkali aluminosilicate
2As natural clays are seldom pure in their mineralogy, while kaolinite refers specifically to the
mineral, kaolin refers to the clay composed primarily of kaolinite. The same distinction holds for metakaolinite and metakaolin.
i.e., N-A-S-(H)) networks with high strength [56].
As a highly-reactive aluminosilicate material, it is important to note that the geopolymerization of metakaolin is often studied in isolation. Given the presence of both silica and alumina, metakaolin can dissolve, gel, and geopolymerize without requiring anything other than a caustic, alkali-rich solution [43,44,57]. Furthermore, as one of the prime precursors for geopolymerization, many studies have focused on its reaction in such solution, exploring thermodynamics [58], kinetics [59–63], nano- and micro-structure [57,64–66], Si/Al ratio [67,68], reaction in the presence of calcium [69, 70], and the role of water [71], to name a few subjects.
1.2.2.2 Coal fly ash
Coal fly ash is most commonly produced by the controlled combustion of coal at ther-mal power plants operating at temperatures exceeding 1400 C [16]. Coal’s inherent compositional and physical variability plays a direct role in the variability of its fly
ash3. Due to the high rate of use within the cement and concrete industries as an
SCM, coal fly ash has been categorized into different classes based on oxide content as determined by X-ray fluorescence and as defined by ASTM C618 [72]. Class F fly ash, typically a product of anthracite or bituminous coal combustion, is, among other things, defined as having a minimum primary oxide content of 70 wt% (i.e.,
SiO2+ Al2O3+Fe2O3 70 wt%), while Class C fly ash, often a product of lignite or
sub-bituminous coal combustion, must contain a minimum of 50% primary oxides. While CaO content is not explicitly defined by the ASTM standard, Class C fly ash typically contains higher levels of CaO—as high as 30–40 wt%, compared to Class F’s 1–12 wt%—resulting in self-cementing, hydraulic properties [16]. In other words, while both Classes C and F fly ashes exhibit pozzolanicity in the presence of calcium-rich solutions, due to Class C’s high Si and Ca content, upon submersion in water it will often form C-S-H phases without the addition of other constituents. Still, both
3Fly ash differs from bottom ash in its location of capture. Due to its small size and light mass,
fly ash is typically carried by flue gases, from which it is filtered before gases exit the chimney, typically via electrostatic precipitation. In contrast, heavier bottom ash falls to the bottom of the boiler, and is typically larger, more varied in shape and size, and often has higher levels of unburnt carbon.
classes of fly ash typically require caustic solutions to destroy the network and bring about reaction. With that in mind, Class F fly ash is more commonly studied in GP and AAM systems, often in conjunction with some level of blast furnace slag (further described below) added as a controlled source of calcium [73]. Other fly ashes exist, but do not experience high market demand, as their properties are not as well defined. While Class C and other, lower-quality fly ashes are not the focus of this thesis, they do pose an opportunity for further research.
Class F fly ash typically exhibits a SiO2:Al2O3 ratio of �2:1 and is composed of a mix of vitreous and crystalline material, depending on processing conditions [74]. Due to the high temperatures at which thermal power plants operate, contained inorganic material sometimes crystallizes, commonly forming phases such as quartz, mullite, hematite, and others [75, 76]. That being said, crystalline material within the fly ash generally does not participate in the reactions taking place during alkali activation, and instead remains as an aggregate phase contributing to final binder strength [76–78]. Therefore, observations have shown ashes with higher amorphous content to exhibit greater reactivity.
Similar to metakaolin, Class F coal fly ash has been very well studied, with avail-able literature focusing on reactivity [79–82], kinetics [63, 83–86], structural organi-zation [87–89], reaction in conjunction with metakaolin or blast furnace slag [90–94], the role of water [95], and other topics [74, 96, 97]
1.2.2.3 Blast furnace slag
Blast furnace slag (BFS), also often referred to as ground granulated blast furnace slag (GGBFS), is a vitreous byproduct of the iron smelting process. Initially formed as a liquid, this slag is typically rapidly quenched in water, causing it to form a primarily glassy structure with few crystalline inclusions [45]. As with metakaolin and coal fly ash, it has been utilized by the cement and concrete industries for years as an SCM. Unlike metakaolin and Class F fly ash, BFS is usually rich in reactive calcium, often reaching concentrations of �40 wt%, making BFS inherently hydraulic [56, 98–101]. In his original work, Kühl described the hydraulic nature of BFS, addressing the fact
that though rich in calcium, BFS required a caustic solution to accelerate the rate of reaction and setting [40].
In the context of AAMs, the presence of calcium leads to different system chem-istry, with simultaneous formation of hydrate gels similar to those found in hydrated OPC systems and alkali aluminosilicate networks such as found in activated low calcium systems [98, 102, 103]. Studies have investigated the coexistence of these two types of products and whether or not they phase separate, but work still re-mains [94, 100, 104–107]. BFS used in alkali activation studies typically exhibits CaO/SiO2 ratios in the range of 0.5–2 and Al2O3/SiO2 ratios between 0.1–0.6 [45].
Again, similar to fly ash and metakaolin, there is ample research in the scientific literature focused on BFS, investigating topics such as reaction degree [108, 109], kinetics [63,110,111] microsctructure [91,112], compositional dependencies [113–116], surface chemistry [99, 117] and others [101, 118]
1.2.2.4 Biomass ash
Portions of this section have been adapted from:
[119] Piyush Chaunsali, Hugo Uvegi, Rachel Osmundsen, Michael Laracy, Thomas Poinot, John Ochsendorf, and Elsa Olivetti. Mineralogical and microstructural characterization of biomass ash binder. Cement and Concrete Composites, 89:41–51, 2018.
Biomass residues, rich in carbon due to their biogenic nature, have long been one of the most heavily utilized energy sources and are often consumed alongside coal and other fossil fuels as a feedstock for powering industry. Primary solid biofuels (i.e., plant matter used directly as fuel or converted into solid fuels) are responsible for approximately 9% of global energy production, while they play an even larger role in developing countries, contributing as much as 35% of total energy generation [120, 121]. Though traditional uses of biomass, such as in-home burning for heat, are not expected to grow significantly, large-scale industrial biomass incineration for combined heat and power is predicted to triple by 2035, compared with 2008 levels [122]. This expansion is triggered, in part, by regulations touting biomass as a renewable source of energy and its combustion, a CO2-neutral process [123–125]. While effective as
a method of converting waste to energy, combustion often results in significant ash production due to the complex chemical makeup of many feedstocks. The incineration process generally does not consume inorganic constituents, which remain as ash along with a percentage of unburnt carbon, dependent on process temperature and efficiency [126, 127].
Agricultural residues, a subset of biomass residue that includes straws, husks, and woods, have been studied by an array of disciplines due to their global prevalence and wide-ranging chemical compositions. The focus of soil and fuel scientists alike, agricultural residues have more recently received the attention of the cement industry and those interested in lower-emission alternatives. While the inorganic content of these residues is initially low, pre-combustion inorganic phases translate reliably to post-combustion ash composition. Therefore, materials such as rice husk ash (RHA) and sugarcane bagasse ash (SCBA) are gaining increasing attention due to their relatively high concentrations of silica (SiO2) [128,129]. Consequently, the number of studies exploring the pozzolanic activity of biomass ashes has seen a steady uptick in recent years.
While a number of studies have examined the chemical compositions of biomass residues and their ashes, few have done so with the expressed interest of their appli-cability to alkali-activated systems [119,130,131]. Biomass ashes have been found to exhibit extraordinarily variable compositions, with an array of physical and chemical properties often related to their original biological functions [126, 132]. RHA and SCBA are of particular interest in this thesis due to their high silica content. With
reported levels of SiO2 ranging from 65% to 95%, and global production exceeding 40
million metric tons annually [126,133–139], they are prime candidates for use in both alkali-aluminosilicate and calcium silicate hydrate systems. Through their dissolution in basic media, ashes such as these have been shown to contribute much of their silica to the formation of networked inorganic polymer and hydration products. The rate of this dissolution is highly dependent on the choice and concentration of solvent and is essential to the formation of strongly networked products [140].
temperature of the original agricultural residues is an important determinant of ash reactivity, as it often directly impacts both the particle size and crystallinity of the resulting ashes, as previously mentioned in the case of coal fly ash [141, 142]. For alkali-activated systems, high surface area, amorphous particles are known to be the most highly reactive. While high incineration temperatures can often result in smaller particles, they also tend to crystallize inorganic constituents, thereby diminishing par-ticle reactivity [141–143]. Mechanical activation of ashes can also have a beneficial impact on reactivity, as activation directly increases particle surface area [144, 145]. Finally, while single-feedstock, controlled-incineration ashes have been thoroughly investigated, mixed-feedstock ashes burned under less well-defined conditions are rel-atively under-studied and present an additional precursor stream.
1.2.2.5 Other materials
Many other materials with compositions entailing some combination of reactive silica, alumina, lime, and alkali oxides have been proposed as potential precursors for alkali activation. Bernal, et al. (2016) comprehensively reviewed the various "one-off" ex-periments exploring the aluminosilicate space, with particular focus on waste valoriza-tion [49]. Examples of highlighted materials of interest include municipal solid waste incineration ashes [146–148], construction and demolition wastes [149–151], waste glasses [152,153], clays [154], and mining and mineral processing wastes [155,156]. A major theme across studies is the utility of glassy phases as compared to their crys-talline analogues, with some researchers thermally treating and amorphizing their materials prior to alkali activation.
Further considerations when proposing use of various other materials include: • Locally available supply, including local availability of any necessary additives
or co-reactants
• Embodied energy and carbon footprint • Pre-processing requirements
• Material heterogeneities that might preclude use in certain circumstances Regarding the first point, it is important to stress the oft-ignored topic of re-source availability—either in absolute global or local quantity. Rere-source reactivity is important, but only insofar as material supply can satisfy demand. For a more comprehensive review of supplementary cementitious material-availability, the reader is directed to Snellings (2016) and Juenger, et al. (2019) [18, 33].
Regarding the last point, a recent study by Traynor, et al. indicated through dis-solution experiments on the major crystalline phases present in steel and copper slags that such slags might require further investigation before effective use as precursors at high pH [157, 158].
1.2.3 Activating solutions
Almost as important as the precursor materials themselves, activating solutions have also been observed to dictate product chemistry and structure [44, 45, 48]. While OPC has been engineered to react and set upon addition of only water, AAMs re-quire caustic, alkali-rich solutions to exhibit similar behavior and achieve desirable physical properties (e.g., high strength, long-term durability, etc.). For this reason, it is important to discuss the most commonly used activating solutions. This discussion will be brief, and the reader is directed to Provis and van Deventer (2014) [45], Provis (2009) [159], and Shi, et al. (2006) [160].
Furthermore, it should be noted that while GP and AAM precursors of interest are typically low-footprint wastes and industrial byproducts, the use of such activating solutions increases the embodied energy of final binder products. While quantification of this contribution to binder carbon footprints is beyond the scope of this thesis, the reader is directed to Habert, et al. (2016) [13] and Provis (2018) [14]
1.2.3.1 Alkali hydroxides
Alkali hydroxide solutions, such as aqueous sodium and potassium hydroxides (i.e., NaOH and KOH) are the most commonly used aqueous solutions for alkali activation
due to their ease of access and use. Such alkali hydroxides are highly soluble in water and can thus be used to create extremely caustic solutions, capable of breaking down many aluminosilicate network solids. That being said, very high alkali concentrations (up to 12 molar or higher under certain circumstances), producing solution pH values greater than 14, have been cited as necessary to destroy precursor networks and induce reaction and setting [48,60,78]. This is dependent on material structure and solubil-ity, with glassy silicates exhibiting satisfactory dissolution under low-concentration conditions and experiencing faster dissolution kinetics with increased alkali hydroxide concentrations, as observed in Chapters 3 and 4. In contrast, with some aluminosil-icate precursors, such as metakaolin and fly ash, use of higher molarity hydroxide solutions may be necessary, as in Chapter 4, where use of 6M NaOH is observed to be necessary to achieve sufficient reaction of metakaolin. Dissolution dependence on solution molarity will be further discussed in Chapters 3 and 4
1.2.3.2 Alkali silicates
Alkali silicate solutions, often again primarily sodium- or potassium-based and some-times referred to as water glass, are the second most common type of activating solu-tion. Such solutions provide an additional compositional lever defined by their mod-ulus; Ms = SiO2/M2O (M = Na, K). In this way, they introduce an additional degree of freedom, allowing for the production of alkali activated materials with externally-controlled concentrations of both silica and alkalis (i.e., silica in the final product higher than in the starting materials), where hydroxide solutions only provide control over alkali content. Most alkali silicate solutions exhibit buffered pH values in the range of 11–13.5 and higher viscosities than their silicate free, hydroxide analogues due to the silicate speciation in solution. Additionally, due to the buffering nature of the aqueous species in alkali silicate solutions, some researchers have observed silicate solutions to be more effective than hydroxide solutions at promoting geopolymeriza-tion and alkali activageopolymeriza-tion [45, 161].
1.2.3.3 Other alkali-rich solutions
While other solutions, such as alkali carbonates and sulfates, are not employed in this thesis, it is important to acknowledge their utility as activating solutions. Their use has been more limited, but promising nonetheless. For further information, the reader is directed to [160, 162–166].
1.2.4 Geopolymeric and alkali-activated binder chemistry
1.2.4.1 Low-calcium binders
GPs are typically defined as a specific subset of AAMs described as inorganic alu-minosilicate polymers consisting of low levels of calcium with variable amounts of silica and alumina and minimal bound water, as shown schematically in Figure 1-2. For such materials, researchers have observed product Si/Al ratio to be an important determinant of physical property development [44, 59, 67, 167]. Similar to alkali alu-minosilicate glasses, GPs generally lack long-range order, and are composed of the same tetrahedral silicate and aluminate building blocks—the same in fact as most alu-minosilicate solids including crystalline zeolites, feldspars, and many rock and clay
materials. More specifically, GPs consist of interconnected SiO4 and AlO4
tetrahe-dra4, as defined by their Si/Al ratios, linked by bridging oxygen atoms, with cations
necessary to balance the residual negative charge of Al3+ in tetrahedral
coordina-tion [43, 47].
To clarify the structural organization in GPs, geopolymer scientists have bor-rowed from other aluminosilicate literature, defining the interconnected nature of
the inorganic polymer network by through the following metric: Qn(mAl), where
0 m n 4, n is the coordination of a central silicon atom, and m is the
num-4For completeness, it is important to mention that these tetrahedral representations assume
oxygen atoms are shared between adjacent tetrahedra (i.e., each tetrahedral group is connected through bridging oxygens to four others). In a fully-connected network of silicate tetrahedra, such as quartz or pure silicate glass, this results in an empirical formula of SiO2. A standalone silicate
tetrahedron in water would exhibit an empirical formula of Si(OH)4, where H+ (or other) cations
are necessary for charge balancing. The same is true for the aluminate tetrahedron. While here it is schematically listed as AlO4, this charge is only accurate when the four oxygen atoms are shared
ber of those neighbors which happen to be aluminum [167, 168]. This helps define short-range T-O-T ordering, where T can be Al or Si5.
Given the overlapping compositional ranges and similar structural natures of GPs and zeolites, reactions with identical precursors have been observed to form crystalline zeolite phases upon hydrothermal curing and amorphous GPs at low to moderate tem-peratures. In fact, in a review of published data, Provis, et al. (2005) concluded that while GPs lack long-range order, evidence points to the presence of nanocrystalline zeolites caught in the GP network [64]. Other studies have concluded similarly, indi-cating that zeolitic crystal growth is kinetically limited at low temperatures, resulting in GPs that set and harden before crystallites are able to develop and converge [46]. The proposed mechanism for geopolymerization in low-calcium environments in-volves the dissolution of aluminosilicate precursors in alkali silicate or alkali hydroxide solutions followed by recombination, with intermediate gel precipitation and reorga-nization, forming a primarily amorphous inorganic network [43,44,46,47,73,171,172]. A schematic of the proposed reaction process is presented in Figure 1-4.
Although idealized geopolymers are composed entirely of an alkali charge-balanced aluminosilicate network as described, more general low-calcium AAMs, often pro-duced through the activation of metakaolin, Class F fly ash, or a mixture of the two, have been observed with moderate calcium substitution in charge-balancing sites, forming N,K-(C)-A-S-(H) gels. [48].
The final networked structure has been established through the combined efforts of X-ray diffraction (XRD), magic angle spinning nuclear magnetic resonance (MAS-NMR), and Fourier transform infrared (FTIR) spectroscopy experiments [46, 79, 81, 84, 167, 173, 174]. XRD experiments alone were found to be insufficient due to the amorphous nature of the aluminosilicate reaction products. When used in combina-tion, these techniques have enabled the characterization of geopolymer structure as
5Loewenstein’s rule states that Al-O-Al bonds are not energetically favorable enough to form in
tetrahedral aluminosilicate solids [169], indicating that fully polymerized materials with Al/Si>1 will not form under typical conditions. In other words, for any fully polymerized aluminosilicate solid, all aluminate tetrahedra will be surrounded by four silicate tetrahedra, and vice versa when m = n = 4. As with any rule, there are exceptions, and Al-O-Al bonds have, of course, been observed in geopolymeric systems, necessitating a more detailed study of this phenomenon [58, 167, 170].
Figure 1-4: Proposed reaction mechanism for geopolymer formation. Reproduced from Duxson, et al. (2007) [47]
well as the comparison with precursor materials, demonstrating that precursors often do not dissolve completely, but instead act as both reactants and fillers for the final matrix, giving the formed binders enhanced strength [46].
While debate exists over the role of water in geopolymers and low-calcium alkali-activated binder, in the past few years, experts have come to a consensus that in the absence of calcium, water is involved in the initial dissolution steps, but is present only as an adsorbed species in the final product [44, 48, 71, 95, 175]. This comes in contrast to typical C-S-H and C-A-S-H type gels present in high-calcium AAM systems described below, where water assumes a structural role [19, 26, 48, 176].
1.2.4.2 High-calcium binders
High-calcium AAMs, generally produced through the activation of BFS or through pozzolanic activation of siliceous ashes (with or without secondary components such as fly ash or metakaolin) exhibit structural similarities to OPC-based C-S-H. They often include C-S-H phases in conjunction with aluminum incorporated C-A-S-H phases as well as other alkali aluminosilicate phases similar to those found in low-calcium systems. However, the solubility limits and co-existence of these phases with each other are still being investigated [70,104,106]. The overall structure has been observed to predominantly exhibit a disordered tobermorite-like atomic arrangement, with lesser formation of secondary phases dependent on precursor and activator choice [35, 45, 177].
High calcium AAMs are described as exhibiting Ca/(Si + Al) ⇡ 1 [48], and while the presence of calcium leads to the formation of hydrate products similar to those found in conventional cements, the Ca/Si ratio is typically lower than that of hydrated OPC. Aluminum often substitutes the silicon position in the gel network, making the Ca/(Si+Al) ratio a more useful metric. Still, the exact ratios are highly dependent on precursor chemistry and activator type [45,178]. Where hydroxide activators typically produce more ordered structures, the increased Si availability in silicate-activated systems often yields a lower degree of order as well as a lower Ca/Si ratio [100]. It is interesting to note that the presence of calcium in AAMs has been observed to increase both the strength and durability of cured binder products due to its ability to fill space through the formation of hydration products [175].
Building on recent29Si nuclear magnetic resonance data, a 2013 model by Myers, et al. has shown C-(N)-A-S-H type gels to be a combination of 11 and 14 Å tobermorite-like structures, exhibiting "drierketten" units with both cross-linked and
non-cross-linked tetrahedra [35] as depicted in Figure 1-56. Myers’ model built on previous
C-S-H literature and models to inform a better understanding of the structure of high-calcium C-(N)-A-S-H gels, with specific interest in the Ca/Si ratio and mean
614 Å tobermorite does not typically exhibit cross-linking between adjacent silicate chains, while
chain length of formed gel products, as they are both important to the development of mechanical properties [179]. Furthermore, the model took into account recent
findings that some chemically bound Ca2+ incorporated into the gel network from
precursor materials can be replaced by Na+ from the activating solution (hence the
N in C-(N)-A-S-H) [35].
Figure 1-5: Generalized structure of C-(N)-A-S-H type gels composed of cross linked and non-cross linked type tobermorite structures. Reproduced from Provis and van Deventer (2014) [45] with the permission of the artist.
1.3 Material reactivity
As this thesis focuses primarily on glassy and amorphous silica-based materials, this section will briefly review the current understanding of such material structure and dissolution in the context of highly alkaline environments. For a broader discussion of glass science, the reader is directed to two specific textbooks, from which much of Section 1.3.1 was developed:
1. Fundamentals of Inorganic Glasses by Varshneya and Mauro [180] 2. Silicate Glasses and Melts by Mysen and Richet [181]
Crystalline analogues to described glassy materials may be referenced for com-parison; however, given the highly specific nature of dissolution mechanisms as they relate to well-defined mineral structures, a discussion of such mechanisms and rates is beyond the scope of this work. That being said, for silicate and aluminosilicate min-erals with compositional and structural similarities to glassy analogues, dissolution rates are typically observed to be significantly slower in the minerals as compared