HAL Id: tel-03122766
https://tel.archives-ouvertes.fr/tel-03122766
Submitted on 27 Jan 2021HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Étude et identification des états transcriptionnels dans
le mélanome uvéal primaire
Charlotte Pandiani
To cite this version:
Charlotte Pandiani. Étude et identification des états transcriptionnels dans le mélanome uvéal pri-maire. Biologie moléculaire. Université Côte d’Azur, 2020. Français. �NNT : 2020COAZ6012�. �tel-03122766�
1
Etude et caractérisation des états
transcriptionnels dans le mélanome
uvéal primaire
Charlotte PANDIANI
Centre Méditerranéen de Médecine Moléculaire (C3M), INSERM U1065
Présentée en vue de l’obtention
du grade de docteur en Sciences de la Vie et de la Santé - Mention Interactions
moléculaires et cellulaires d’Université Côte d’Azur
Dirigée par : Corine BERTOLOTTO Soutenue le : 14 septembre 2020
Devant le jury, composé de :
Stéphanie BAILLIF, PU-PH, CHU Nice Corine BERTOLOTTO, DR INSERM, C3M Eleonora LEUCCI, PR, KU-Leuven Belgique Lukas SOMMER, PR, UZH Suisse
2
Etude et identification des états transcriptionnels
dans le mélanome uvéal primaire
Jury :
Présidente du jury :
Pr. Stéphanie BAILLIF, PU-PH, Hôpital Pasteur - Université Nice-Côte d’Azur
Rapporteurs :
Pr. Eleonora LEUCCI, Professeur, Université de KU-Leuven, Belgique
Pr. Lukas SOMMER, Professeur, Institute of Anatomy - Université de Zurich, Suisse
Directrice de thèse :
3 Etude et identification des états transcriptionnels dans le mélanome uvéal primaire
Le mélanome uvéal (MU) est la tumeur intraoculaire la plus fréquente chez l’adulte. C’est une tumeur agressive qui dérive de la transformation maligne des mélanocytes. Les traitements du MU primaire reposent sur des techniques de radiothérapies, et sur la chirurgie. Malgré le succès du traitement primaire, plus de 50% des patients développent des métastases qui vont principalement envahir le foie, suggérant l’existence d’une sous-population cellulaire, responsable de la dissémination métastatique. Il n’existe aujourd’hui aucun traitement systémique pour soigner le MU métastatique. A ce stade, 80% des patients meurent dans un délai de 1 an. Il est donc capital de mieux comprendre la biologie du MU, afin de découvrir de nouvelles cibles thérapeutiques et ainsi, à terme, pouvoir proposer un traitement efficace aux patients.
L’hétérogénéité intra-tumorale est reconnue pour être impliquée dans la résistance aux traitements et la dissémination métastatique dans de nombreux cancers, mais n’a encore jamais été démontrée dans le MU.
Au cours de ma thèse, grâce au séquençage à l’échelle de la cellule unique, nous avons démontré l’existence d’une hétérogénéité intra-tumorale aux niveaux génomique et transcriptomique dans des MU primaires. Nous avons mis en évidence une nouvelle signature, associée à un mauvais pronostic, comprenant des gènes n’ayant encore jamais été décrits dans le MU. Nous avons également identifié trois états transcriptionnels distincts, exprimés par différentes sous populations cellulaires, associés à la différenciation cellulaire, à la prolifération ou à l’invasion. La signature génique que nous avons établie est fortement associée à l’état transcriptionnel invasif, et des cellules exprimant cette signature ont été détectées au sein de chacune des tumeurs analysées, même dans celles classées de bon pronostic par les pathologistes. Cette découverte peut avoir un impact capital sur le pronostic et le suivi des patients.
Grâce à nos analyses bio-informatiques, nous avons identifié le facteur de transcription HES6 comme nouveau marqueur de mauvais pronostic du MU. L’expression hétérogène de HES6 au sein des tumeurs, même celles classées de bon pronostic, a été validée par des expériences de RNAscope. J’ai montré que l’inhibition d’expression de HES6 entraine une diminution de la prolifération, de la migration, et de l’invasion dans des lignées de MU primaires, et du développement métastatique in vivo, dans un modèle d’embryon de poulet. De plus, l’inhibition de HES6 bloque aussi la prolifération et la migration des cellules de MU métastatiques.
L’ensemble de mes résultats met en évidence le rôle essentiel de HES6 dans la progression du MU, et fait de HES6 une nouvelle cible pour bloquer la croissance et les capacités motiles des cellules de MU.
4 Study and identification of transcriptional states in primary uveal melanoma
Uveal melanoma is an aggressive and deadly neoplasm, which develops from melanocytes in the choroid. Treatments of primary UM treatments rely on radiotherapy techniques and surgery. Despite successful treatment of the primary tumor, metastases, that display a pronounced liver tropism, develop in 50% of patients. This implies the existence of a cellular subpopulation, that disseminate early during tumor progression dissemination. Currently, there is no systemic treatment for metastatic UM. At this stage, 80% of patients die within one year. It is therefore essential to better understand the biology of UM, in order to discover new therapeutic targets and, ultimately, to develop effective treatments.
Intra-tumor heterogeneity is known to be involved in metastatic spread and treatment resistance in many cancers, yet its role in UM remains to be elucidated.
During my thesis, through single cell sequencing, we demonstrated the existence of intra-tumor heterogeneity at genomic and transcriptomic levels in primary UM. We highlighted a new gene signature, associated with a poor prognosis, including genes that have never been reported in the UM. We also identified three distinct transcriptional cell states, associated with cell differentiation, proliferation or invasion. The invasive transcriptional state strongly associates with the poor prognosis gene signature that we have established. Cells expressing this signature were detected within all six tumors that we analyzed, even in those classified by pathologists as of good prognosis. These observations can have a major impact on patients’ prognosis and follow-up.
Through our bioinformatics analysis, we identified the transcription factor HES6 as a new marker of poor prognosis in UM. Heterogeneous expression of HES6 in tumors, even in those classified as of good prognosis, was validated by the RNAscope approach. We showed that the inhibition of HES6 expression leads to decrease in proliferation, migration, and invasion of primary UM cell lines, and metastatic development in vivo, in a chicken embryo model. In addition, HES6 inhibition also impairs the proliferation and migration of metastatic UM cell lines, suggesting that HES6 has also a critical role in the metastatic settings.
Together, my findings highlight the essential role of HES6 in the progression of UM, and identify HES6 as new target to prevent UM growth and motile ability.
5
6
TABLE DES MATIERES
LISTE DES ABREVIATIONS ... 8
TABLE DES ILLUSTRATIONS ... 12
INTRODUCTION ... 13
CHAPITRE 1 : Le mélanome uvéal, un cancer dérivant des mélanocytes oculaires ... 14
1) Structure et fonction de l’œil ... 14
2) Origine des mélanocytes ... 16
3) Facteurs cellulaires et moléculaires impliqués dans le développement mélanocytaire... 18
a) Signalisation cellulaire impliquée dans le développement mélanocytaire ... 19
b) Facteurs de transcription régulant la lignée mélanocytaire ... 20
4) Rôle des mélanocytes ... 21
CHAPITRE 2 : Le mélanome uvéal, un cancer totalement différent du mélanome cutané ... 23
1) Epidémiologie ... 25
2) Facteurs de risque et prédisposition génétique ... 25
3) Facteurs de risques acquis : Altérations génétiques ... 27
a) Mutations somatiques activatrices ... 27
b) Mutations secondaires associées au pronostic ... 29
4) Voies de signalisation ... 32
5) Altérations cytogénétiques ... 37
6) Propriétés histologiques ... 40
7) Diagnostic, classification et pronostic ... 42
a) Diagnostic ... 42
b) Classification des mélanomes uvéaux et pronostic... 42
8) Traitements ... 49
a) Tumeur primaire ... 49
b) Mélanome uvéal métastatique ... 50
CHAPITRE 3 : Le facteur de transcription HES6 ... 55
1) La famille des facteurs Hes ... 55
a) Structure des facteurs Hes ... 55
b) Mécanismes de répression transcriptionnelle ... 56
2) Les facteurs Hes et la voie de signalisation Notch ... 58
7
4) Rôle de la voie de signalisation Notch dans le mélanome uvéal ... 61
CONTEXTE ET OBJECTIFS DES RECHERCHES, RESUME DES RESULTATS ... 63
RESULTATS ... 66
DISCUSSION ET PERSPECTIVES ... 141
1) Identification d’une hétérogénéité tumorale dans le MU ... 142
a) Hétérogénéité inter-tumorale ... 142
b) Hétérogénéité intra-tumorale (HIT) ... 144
2) Identification de HES6 comme nouveau marqueur de mauvais pronostic ... 148
a) Rôle de HES6 ... 148
b) Signalisation associée à HES6 ... 148
8
LISTE DES ABREVIATIONS
ADN Acide désoxyribonucléique
APC Analyse en Composantes Principales ARF6 ADP-ribosylation factor 6
ARN Acide ribonucléique
ASAP1 ArfGAP With SH3 Domain, Ankyrin Repeat And PH Domain 1
BAP1 BRCA1 associated protein-1 BET Bromodomain and Extra-Terminal bHLH basic Helix-Loop-Helix
bHLH-LZ basic-Helix Loop Helix-Leucin-Zipper
BRAF B-Raf Proto-Oncogene, Serine/Threonine Kinase BRCA1 Breast cancer 1
CDKN2A Cyclin-dependent kinase inhibitor 2A CGH Comparative Genomic Hybridization
CN Crête neurale
CNN Cellule de la crête neurale CNV Copy Number Variation
CTLA4 Cytotoxic T-lymphocyte-associated protein 4 CYSLTR2 Cysteinyl leukotriene receptor 2
D3 Disomie du chromosome 3
DAG Diacylglycerol
DCT Dopachrome tautomérase DDR Damage Repair/Response Dll Delta-like ligand
DMLA Dégénérescence maculaire relative à l’âge DNMTi DNA methyltransferase inhibitor
EDN3 Endothéline 3
EDNRB Endothéline récepteur B
EGFR Epithelial Growth Factor Receptor
EIF1AX Eukaryotic Translation Initiation Factor 1A X-Linked EPR Epithélium Pigmentaire Rétinien
9 GEF Guanine nucleotide Exchange Factor
GEP Gene Expression Profile
GNA11 Guanine Nucleotide-binding protein subunit Alpha-11 GNAQ Guanine Nucleotide-binding protein subunit Alpha-Q
HDAC Histones Désacétylases
HE Hématoxyline/éosine
Hes Hairy Enhancer of Split
HES6 Hes Family bHLH Transcription Factor 6 HEY1 Hes-related with YRPW motif1
HGF Hepatocyte Growth Factor HIT Hétérogénéité intratumorale HMG High Mobility Group
HT hydroxytryptamine
HTR2A 5-Hydroxytryptamine Receptor 2A HTR2B 5-Hydroxytryptamine Receptor 2B
ICGC International Cancer Genome Consortium IP3 Inositol trisphosphate
IP3R3 Inositol 1,4,5-trisphosphate Receptor, type 3
LAG3 Lymphocyte Activating 3
LEF/TCF Lymphoïd Enhancer Factor/T Cell Factor lncRNA longs ARN non codants
LOH Loss of Heterozygosity
M3 Monosomie du chromosome 3
MAPK Mitogen-activated protein kinase MBD4 Methyl-CpG Binding Domain Protein 4
MC Mélanome cutané
MC1R MelanoCortin 1 Receptor
MEK Mitogen Activated/Extracellular Signal Regulated Protein Kinase MEKi Inhibiteur de MEK
MI Index mitotique
miRNA microARN
MITF Microphtalmia-associated Transcription Factor MLPA Multiplex Ligation-dependent Probe Amplification MRD Minimale Residual Disease
MU Mélanome de l’uvée
10 ncBAF non canonical BAF
NICD Notch Intra Cellular Domain NK Natural Killer
OxPhos Oxidative phosphorylation
PAX3 Paired box gene 3
PD Paired Domain
PD-1 Programmed cell death 1 PDX Patient Derived Xenografts PI3K Phosphoinositide 3-kinase
PIP2 Phosphatidylinositol(1,4,)bis-phosphate PIP3 Phosphatidylinositol(1,4,5)tris-phosphate PLCβ4 Phospholipase C β4
PRAME Preferentially expressed antigen in melanoma PR-DUB Polycomb-Repressive deubiquitylase
RCPG Récepteur couplé aux protéines G
RE Réticulum Endoplasmique
ROS Reactive Oxygen Species
RT-qPCR Reverse transcription-quantitative Polymerase Chain Reaction
SCF Stem Cell Factor
SF3B1 Splicing factor 3b subunit 1 snRNP Small nuclear Ribonucleoprotein
SRSF2 Serine And Arginine Rich Splicing Factor 2
TAZ Transcriptional coactivator with PDZ-binding motif TCGA The Cancer Genome Atlas
TEM Transition épithélio-mésenchymateuse TLE Transducine-like Enhancer
TRIO Trio Rho guanine nucleotide exchange factor TYRP1 Tyrosinase-related protein 1
U2AF1 U2 Small Nuclear RNA Auxiliary Factor 1
UV Ultraviolet
VEGF Vascular Endothelial Growth Factor
WES Whole Exome Sequencing
11 YAP Yes-Associated Protein
12
TABLE DES ILLUSTRATIONS
Figure 1 : Structure de l'œil ... 16
Figure 2 : Spécification des mélanocytes à partir des cellules de la crête neurale ... 18
Figure 3 : Localisations anatomiques des mélanomes uvéaux ... 24
Figure 4 : Principales altérations génétiques associées au mélanome uvéal ... 32
Figure 5 : Principales voies de signalisation impliquées dans le développement du MU ... 36
Figure 6 : Principales altérations cytogénétiques associées au pronostic dans le mélanome uvéal ... 39
Figure 7 : Caractéristiques histologiques associées au risque métastatique ... 41
Figure 8 : Signature génétique utilisée pour le pronostic des mélanomes uvéaux primaires 44 Figure 9 : Classification des MU selon le groupe de Harbour ... 46
Figure 10 : Classification des MU en quatre groupes moléculaires et cliniques distincts ... 48
Figure 11 : Structure et fonction des facteurs Hes ... 57
Figure 12: Voie de signalisation Notch ... 62
Figure 13 : L’inhibition de HES6 diminue l’activation de la voie Notch ... 150
Figure 14 : CHAC1 est une cible potentielle de HES6 ... 152
Figure 15 : FADS1 est associé à un mauvais pronostic dans le MU ... 153
13
14
CHAPITRE 1 : Le mélanome uvéal, un cancer dérivant des
mélanocytes oculaires
1) Structure et fonction de l’œil
L’œil est l’organe de la vision, permettant de coder l’information lumineuse perçue en information électrique interprétable par le cerveau. Il est composé de différentes couches et différents types cellulaires lui permettant d’assurer sa fonction (Figure 1).
La sclérotique est la couche la plus externe de l’œil. C’est un tissu conjonctif dense et peu vascularisé qui donne sa couleur blanche à l’œil en le recouvrant à 80%, et qui joue un rôle de protection de l’œil. Elle est entourée d'une membrane très fine et transparente, appelée conjonctive. La partie antérieure de la sclérotique est remplacée par la cornée, transparente, non vascularisée, et constituée de fibres nerveuses permettant de réagir au contact d’objets. La cornée est en contact direct avec l’air et correspond au point d’entrée des rayons lumineux dans le globe oculaire.
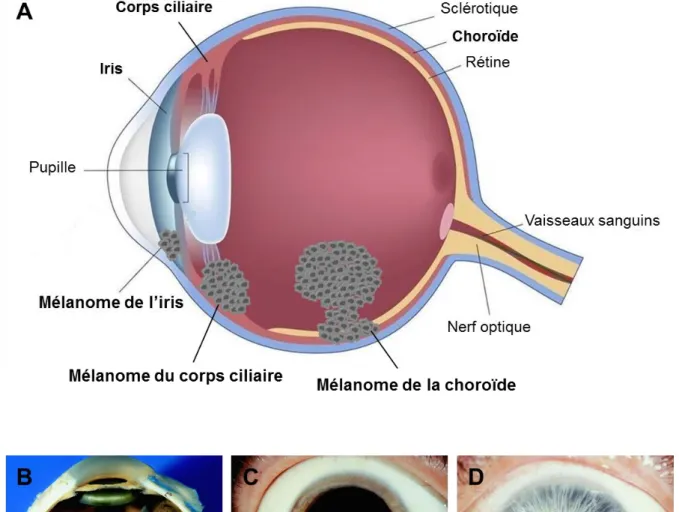
Sous la sclère se trouve l’uvée, constituée de la choroïde dans la partie postérieure de l’œil et par le corps ciliaire et l’iris dans la partie antérieure. La choroïde, le corps ciliaire et l’iris contiennent des mélanocytes issus de la crête neurale. Nous détaillerons plus tard le développement et le rôle de ces cellules à l’origine du développement tumoral.
- La choroïde tapisse la face interne de la sclérotique. C’est une couche cellulaire fortement vascularisée, assurant la nutrition de l’iris et des cellules photoréceptrices de la rétine. Elle est riche en mélanocytes, responsables de sa pigmentation, qui forment un écran opaque, absorbant les rayons lumineux inutiles et dangereux pour la vision.
- Le corps ciliaire est essentiellement formé de muscles lisses et de vaisseaux sanguins et assure plusieurs fonctions. Il permet d’une part la sécrétion des deux liquides composant l’œil ; le corps vitré, qui garantit la rigidité du globe oculaire et constitue 90% du volume de l’œil, et l’humeur aqueuse qui est un liquide nourricier dont le volume est à l'origine de la pression intra oculaire. D’autre part, grâce à la contraction de ses muscles lisses, il régule la courbure du cristallin qui permet la vision nette à différentes distances et participe donc au phénomène d’accommodation.
- L’iris est une membrane contractile situé dans l’humeur aqueuse entre la cornée et le cristallin, séparant ainsi la chambre antérieure de la chambre postérieure de l’œil. Il constitue la partie colorée visible de l'œil et est percé en son centre par la pupille. Il contient des mélanocytes contenant du pigment, et dont le nombre influence la couleur des yeux. L’iris est innervé par des fibres du système nerveux autonome qui
15 activent les muscles lisses permettant de contrôler la taille de la pupille, et donc les rayons lumineux pénétrant dans le globe oculaire. La contraction ou la dilatation de l'iris est un réflexe physiologique pour adapter la vision à la luminosité ambiante. Quand la luminosité ambiante est forte, l'iris se contracte, ce qui diminue l'intensité lumineuse qui vient frapper le centre de la rétine, et vice-versa. La contraction et la dilatation de l’iris sont contrôlées par deux muscles antagonistes, le muscle sphincter pupillaire et le muscle dilatateur de la pupille.
Le cristallin se situe juste derrière l’iris et est suspendu au corps ciliaire par la zonule. Il constitue la lentille principale de l'œil, et est responsable de l'accommodation ; fonction fondamentale de la vision permettant la mise au point des objets, en modifiant sa courbure par tension ou relâchement de la zonule, sous l’effet des muscles lisses du corps ciliaire. Les rayons lumineux qui traversent l'iris parviennent au noyau central du cristallin qui absorbe une partie des ultraviolets (UV) en protégeant ainsi la rétine.
La rétine est le tissu nerveux tapissant la face interne de l’œil et correspond à la région directement sensible à la lumière. Elle est composée de cellules en cônes et
en bâtonnets qui sont les cellules photoréceptrices de l’œil, permettant aux rayons lumineux d’être captés, puis traduits en message nerveux pour gagner le cerveau via le nerf optique. La lumière doit traverser les trois milieux transparents ; humeur aqueuse, cristallin et corps vitré, avant d’atteindre la rétine où se forme l’image. La rétine est constituée de deux tissus distincts, la rétine neurosensorielle, ou neurorétine, et l’épithélium pigmentaire, ou rétine pigmentaire (EPR) (Land 1999).
L’EPR est la plus externe des couches rétiniennes. C’est une monocouche de cellules pigmentées contenant de la mélanine, localisée entre la rétine neurosensorielle et la choroïde. Les cellules épithéliales pigmentées de la rétine sont issues de neuroépithelium optique, et sont donc à différencier des mélanocytes. L’intégrité de l’EPR est essentielle pour le fonctionnement normal de la rétine, en particulier des photorécepteurs avec lesquels il est en contact. L’EPR est impliqué dans la phagocytose des segments externes usagés des photorécepteurs et est donc indispensable pour la survie des cellules photoréceptrices puisqu’elle permet un renouvellement rapide et permanent de leurs segments externes. L’EPR synthétise et stocke la mélanine. Ce pigment absorbe l’excès de photons qui arrive au niveau de la rétine, et limite ainsi la quantité de lumière réfléchie et diffractée à l’intérieur de l’œil.
16
2) Origine des mélanocytes
Un mélanocyte est une cellule qui se caractérise par une production de mélanine. Les mélanocytes sont issus de la crête neurale.
La crête neurale (CN) est une structure embryonnaire transitoire qui se forme à partir de l’ébauche neurale primitive. Les crêtes neurales sont induites pendant la gastrulation à partir de la région dorsale du tube neural et sont à l’origine de toutes les cellules pigmentées de l’organisme, à l’exception des cellules épithéliales de la rétine, qui proviennent directement du neuroectoderme. Les mélanocytes dérivent donc des cellules de la crête neurale (CNN), cellules progénitrices pluripotentes. Les CNN subissent une transformation morphogénétique appelée la transition épithélio-mésenchymateuse (TEM), grâce à laquelle elles acquièrent des propriétés motiles et s’engagent dans la migration (Le Douarin et al. 1992). La trajectoire migratoire des cellules est déterminante pour leur devenir et leur
Figure 1 : Structure de l'œil
L’œil se compose de différentes tuniques, d’un cristallin et de liquides lui permettant de capter la lumière et de la transformer en signal électrique interprétable par le cerveau. Les mélanomes oculaires dérivent des mélanocytes de la conjonctive et des mélanocytes de l’uvée comprenant la choroïde, l’iris et le corps ciliaire.
17 fonctionnalité. Il a été démontré que la migration des mélanoblastes, cellules indifférenciées précurseur des mélanocytes, s’effectuait de façon très précise en migrant entre le dermomyotome et l’ectoderme selon un trajet dorso-latéral. Plusieurs jours après le début de la migration, les mélanoblastes qui parviennent dans leur site définitif entrent dans une phase de prolifération intense pour s’y différencier en mélanocytes dendritiques matures, capables de synthétiser les pigments mélaniques (Figure 2). Les mélanoblastes migrent jusqu’à différents sites de l’organisme tels que l’épiderme, le bulbe pileux et l’œil, mais aussi dans le cœur, les méninges et l’oreille interne. Les mélanocytes oculaires se situent dans la conjonctive et dans l’uvée, localisant alors dans le stroma de l’iris, du corps ciliaire et de la choroïde. Les mélanocytes uvéaux diffèrent des mélanocytes cutanés par certains aspects. Tout d’abord, les mélanocytes cutanés synthétisent et transfèrent la mélanine aux kératinocytes, alors que les mélanocytes uvéaux ne la transfèrent pas à d’autres cellules in vivo. Ensuite, les mélanocytes cutanés prolifèrent in vivo et répondent à différents facteurs tels que les radiations UV ou l’inflammation, alors que les mélanocytes uvéaux sont relativement stables in vivo et ne répondent généralement pas aux différents facteurs environnementaux. Ces différences peuvent s’expliquer par le fait que les microenvironnements de l’œil et de la peau sont différents (Hu 2000).
La pigmentation de l’iris est initiée au cours du 4ème mois de gestation.
L’embryogénèse de toutes les cellules pigmentées de l’organisme, exceptées les cellules épithéliales de la rétine, est identique, et suit le schéma de la figure 2. Seule la migration vers le site définitif est différente (Thomas and Erickson 2008; Cichorek et al. 2013).
18
3) Facteurs cellulaires et moléculaires impliqués dans le développement
mélanocytaire
Au cours du développement embryonnaire, les mélanoblastes prolifèrent et se différencient tout en migrant vers leur site définitif. La compréhension des mécanismes impliqués dans la migration des mélanoblastes au cours du développement s’appuie sur l’étude de différents modèles animaux : souris, poulet, poisson zèbre et xénope. Ces processus sont finement régulés par de multiples voies de signalisation (WNT, NOTCH, KIT) et des facteurs de transcription tels que MITF, SOX10, PAX3, SLUG et SNAIL. Ces facteurs impliqués dans le développement mélanocytaire ont essentiellement été découverts dans Figure 2 : Spécification des mélanocytes à partir des cellules de la crête
neurale
Les cellules de la crête neurale migrant le long de la voie dorso-latérale sont spécifiées et se différencient en mélanoblastes, puis en mélanocytes.
TN : Tube neural ; TEM : Transition épithélio-mésenchymateuse ; M : Mélanocyte ; K : Kératinocyte. Adapté de Domingues et al., 2013
19 des mélanocytes cutanés car il existe très peu de données concernant les mélanocytes oculaires. Les facteurs décrits ci-dessous sont les mêmes pour tous les mélanocytes.
a) Signalisation cellulaire impliquée dans le développement mélanocytaire
La signalisation de la voie Wnt/β-caténine est essentielle pour l’induction des cellules de la crête neurale et le développement des mélanoblastes. Au niveau membranaire, la caténine peut interagir avec les cadhérines présentes à la membrane plasmique. La β-caténine peut également agir comme facteur de transcription lorsqu’elle est transloquée dans le noyau et interagit alors avec les co-activateurs LEF/TCF permettant l’activation des gènes cibles spécifiques du lignage mélanocytaire (Larue et al. 2003). Il a été montré qu’une invalidation conditionnelle du gène codant pour la β-caténine lors de la différenciation de la crête neurale dans l’embryon de la souris, empêche la génération de mélanoblastes. Des mélanoblastes exprimant MITF, facteur clé du développement mélanocytaire, sont retrouvés dans l’œil des embryons contrôles, mais pas dans les embryons déficients pour la β-caténine (Hari et al. 2002).
La voie de signalisation KIT est impliquée dans le développement et la pigmentation des mélanocytes, via la régulation positive du facteur de transcription MITF (Microphthalmia-associated transcription factor), que nous décrirons plus tard (Li and Hou 2018). c-KIT est le récepteur du SCF (Stem Cell Factor) ou Kitl (KIT-ligand), et les mutations qui inactivent ce récepteur sont associées aux taches blanches dépigmentées que l’on retrouve dans le piébaldisme chez l’homme et chez la souris mutante « Dominant White Spotting ». Les travaux chez la souris et l’existence de mutants présentant un phénotype de taches blanches au niveau du pelage ont permis d’identifier les gènes et les protéines essentiels à la première étape de migration des mélanoblastes, qui suit leur délamination à partir du tube neural. Le ligand Kitl produit dans le dermomyotome, le derme et le follicule pileux, se lie au récepteur à activité tyrosine kinase c-KIT exprimé par les mélanoblastes. Seuls les mélanoblastes exprimant le récepteur c-KIT et le facteur MITF peuvent répondre aux signaux attractifs et répulsifs contrôlant la migration (Domingues et al. 2013).
Le développement embryonnaire de l’œil est lié à la formation du système nerveux central (SNC). Une structure importante dans la formation de l’œil est l’épithélium ciliaire, qui régule la production de l’humeur aqueuse. L’épithélium ciliaire de l’œil adulte contient des cellules souches neurales, et c-KIT a été identifié comme régulateur du renouvèlement et de la différenciation des cellules souches neurales de l’épithélium ciliaire (Das et al. 2004).
Enfin, les modèles mutants murins ont montré que l’endothéline 3 (EDN3) et le récepteur de l’endothéline B (EDNRB) jouent un rôle majeur lors du développement des mélanocytes à partir des précurseurs des cellules de la crête neurale. Le ligand EDN3 produit par l’ectoderme se lie au récepteur EDNRB exprimé par les mélanoblastes. Les mutations ou délétions des gènes EDN3 et EDNRB chez la souris donnent respectivement des phénotypes
20 appelés « piedbald » et « spotting » présentant des anomalies pigmentaires (Saldana-Caboverde and Kos 2010). L’endothéline est présente dans l’œil, et plus précisément dans l’humeur aqueuse, l’iris, le corps ciliaire et la rétine, et pourrait jouer un rôle dans la physiopathologie de l’œil (Minton et al. 2012; Salvatore and Vingolo 2010).
b) Facteurs de transcription régulant la lignée mélanocytaire
MITF est connu comme étant le facteur de transcription essentiel au développement des mélanocytes. C’est un facteur de transcription de type « hélice-boule-hélice » à répétition de leucines (bHLH-LZ). Les facteurs de transcription de la famille bHLH-LZ se lient généralement sur une séquence d’ADN consensus appelés boite E : CANNTG. Il a été montré que MITF se lie spécifiquement aux séquences CATGTG et CACGTG (Goding and Arnheiter 2019a). MITF est nécessaire au maintien des cellules souches mélanocytaires. De plus, MITF régule les gènes impliqués dans la mélanogénèse, spécifiques aux mélanocytes (TYR, TYRP1, DCT/TYRP2), pour promouvoir la synthèse de mélanine dans les mélanocytes (Hou et al. 2006). Les sites de liaison de MITF sur les promoteurs des enzymes de la mélanogénèse sont définis par une séquence plus étendue que la boite E, appelée boite M (Bertolotto et al. 1998).
Des mutations de MITF sont retrouvées chez l’homme et entrainent des troubles de la pigmentation impliquant les mélanocytes de la choroïde. C’est le cas notamment dans le syndrome de Waardenburg (WS) de type 2a, qui se caractérise par une hypopigmentation des yeux, des cheveux et de la peau, et une perte auditive, ainsi que dans le syndrome de Tietz. Chez la souris, les mutations hétérozygotes de Mitf peuvent entrainer une absence de pigmentation due à une perte totale des mélanocytes, ainsi qu’une microphthalmie et une ostéopétrose (Tassabehji et al. 1994).
L’expression tissu-spécifique de l’isoforme M de MITF dans les mélanocytes est coordonnée par plusieurs facteurs de transcription tels que SOX10 (SRY-box related), PAX3 (paired box gene 3), CREB, LEF1/TCF et POU3F2 (D’Mello et al. 2016).
La famille SOX se compose d’environ 20 facteurs de transcription contenant un domaine HMG (High Mobility Group) permettant une liaison à l’ADN séquence-spécifique, et joue un rôle important dans l’embryogénèse. L’expression de SOX10 est restreinte aux cellules de la crête neurale et leurs dérivés non ectomésenchymateux (Kelsh 2006). SOX10 représente l’un des acteurs clés dans le lignage mélanocytaire en régulant directement l’expression des gènes nécessaires à la survie, la migration et la différenciation (Wegner 2005). SOX10 joue un rôle capital dans la biologie de la crête neurale, le maintien des cellules souches et dans la différenciation cellulaire. En effet, l’invalidation de Sox10 dans la lignée germinale de la souris ou du poisson zèbre est létal aux premiers stades du développement embryonnaire (Potterf et al. 2000; Dutton et al. 2001). SOX10 contrôle la transcription de MITF, qui à son tour contrôle le répertoire de gènes essentiels à la mélanogénèse, incluant TYRP2, PMEL et TYRP1. En l’absence de liaison de SOX10 à MITF, MITF ne peut pas induire l’expression de la tyrosinase (Hou et al. 2006; Potterf et al. 2001).
21 Des mutations de SOX10 sont retrouvées chez les patients atteints de WS de types 2 et 4, associées à la perte de pigmentation des cheveux, de la peau et des yeux (Yu et al. 2020)
Les protéines PAX, codant pour des régulateurs de la transcription, sont caractérisées par la présence d’un domaine paired (PD) permettant leur liaison à l’ADN. Les gènes PAX jouent un rôle clé dans la spécification et le maintien des cellules progénitrices et conduisent à la formation des tissus et organes au cours de l’embryogénèse (Thompson and Ziman 2011). PAX3 (Paired box gene 3) est exprimé dans les stades précoces lors du développement de la crête neurale et coordonne la différenciation de la crête neurale en différents types cellulaires tels que les mélanocytes (Watanabe et al. 1998). PAX3 régule un grand nombre de gènes importants pour la différentiation de la crête neurale, comprenant des gènes de la pigmentation : MITF, TYRP1 et DCT (Kubic et al. 2008) et des gènes de la survie tels que c-MET, PTEN et BCLXL. Chez l’homme les mutations de PAX3 sont associées aux WS de types 1 et 3, présentant une perte de pigmentation des cheveux, de la peau et des yeux (Yu et al. 2020).
PAX6, un autre membre de la famille PAX, joue un rôle capital dans la formation de l’œil et est requis pour la spécification de multiples lignages oculaires. La perte de l’expression de PAX6 empêche la formation de l’œil chez la drosophile, la souris et l’homme (Ashery-Padan and Gruss 2001; Kozmik 2005).
4) Rôle des mélanocytes
Le rôle et la fonction des mélanocytes sont bien établis dans la peau mais peu connus dans les autres organes. Les mélanocytes sont les cellules responsables de la synthèse de la mélanine dans leurs organelles, appelés mélanosomes, à travers une cascade enzymatique impliquant la tyrosinase, la protéine 1 liée à la tyrosinase (Tyrosinase related protein 1 : TYRP1) et la dopachrome tautomérase (TYRP2/DCT). Deux types de pigments sont produits ; l’eumélanine, pigment noir/marron et la phéomélanine pigment jaune/orangé. La proportion de ces types de mélanine définit la couleur de la peau, des cheveux et des yeux. Le ratio eumélanine/phéomélanine est plus élevé chez les personnes ayant la peau et les yeux foncés que chez celles à la peau et aux yeux clairs.
Dans l’épiderme, les mélanocytes transfèrent la mélanine produite aux kératinocytes avoisinants, assurant ainsi une pigmentation homogène et une protection de la peau contre les effets nocifs des rayonnements UV du soleil (Brenner and Hearing 2008). La mélanine a un rôle photoprotecteur puisqu’elle a la propriété essentielle d’absorber le rayonnement qui n’a pas été réfléchi à la surface de la peau.
À l’inverse des mélanocytes cutanés, les fonctions des mélanocytes oculaires restent mal comprises. Le rôle des mélanocytes de la conjonctive est d’ailleurs totalement inconnu. Contrairement aux mélanocytes cutanés, les mélanocytes uvéaux sont quiescents après le stade embryonnaire, et ce, tout au long de la vie et n’ont plus la capacité de fabriquer de la mélanine, c’est pourquoi la couleur des yeux ne change pas après une exposition au soleil. La
22 présence de mélanine dans les mélanocytes uvéaux contribue à la protection de l’œil contre des maladies oculaires comme le mélanome de l’uvée (Sarna 1992). Les mélanocytes de l’iris absorbent et bloquent la lumière visible et les rayonnements UV, protégeant ainsi le reste de l’œil des effets délétères de ces longueurs d’onde. La mélanine uvéale, notamment dans le corps ciliaire et la choroïde, peut également protéger les mélanocytes contre le stress oxydatif et réduire la transformation maligne des mélanocytes uvéaux (Wakamatsu et al. 2008). Les mélanocytes des yeux foncés contiennent une plus grande quantité de mélanine que les yeux clairs, assurant ainsi une meilleure protection, ce qui est cohérent avec une plus grande incidence de mélanome uvéal chez les individus aux yeux clairs (Yu et al. 2006).
Les mélanocytes sont également retrouvés à d’autres endroits de l’organisme, tels que dans la cochlée de l’oreille interne où ils sont impliqués dans la production de l’endolymphe et les échanges ioniques essentiels pour l’audition (Tachibana 1999; Barrenäs and Lindgren 1990), mais aussi au niveau des leptoméninges du cerveau où ils fonctionneraient comme des cellules endocriniennes et protègeraient des dommages oxydatifs (Zecca et al. 2008). Enfin, les mélanocytes cardiaques sont indispensables au développement correct du cœur lors de l’embryogénèse (Mjaatvedt et al. 2005), au bon fonctionnement des valves cardiaques (Carneiro et al. 2015) et sont impliqués dans la fibrillation atriale (M.D. et al. 2009).
23
CHAPITRE 2 : Le mélanome uvéal, un cancer totalement différent du
mélanome cutané
Les mélanomes oculaires sont classés en fonction de leur site d’origine, ainsi nous retrouvons le mélanome de la conjonctive et le mélanome de l’uvée (MU). La majorité des mélanomes oculaires sont d’origine uvéale (95%) alors que les mélanomes de la conjonctive sont beaucoup plus rares et ne constituent que 5% des mélanomes oculaires. Le mélanome de la conjonctive se présente généralement sous la forme d’un nodule pigmenté dans la partie bulbaire de la conjonctive (Kaštelan et al. 2018). Il a été montré que le mélanome de la conjonctive présente des anomalies génétiques communes au mélanome cutané, notamment la mutation de BRAF, ce qui pourrait impliquer une tumorigénèse et une réponse clinique similaires (Griewank et al. 2013). Le mélanome uvéal est quant à lui, complètement différent des mélanomes de la conjonctive et cutanés. Dans ce travail de thèse nous nous sommes concentrés sur le mélanome uvéal que nous décrirons en détails.
Le mélanome uvéal est une tumeur résultant de la transformation maligne des mélanocytes de l’œil, situés dans l’uvée postérieure, impliquant la choroïde dans 90% des cas, ainsi que dans l’uvée antérieure avec le corps ciliaire (6%) et l’iris (4%) (Shields et al. 2012) (Figure 3). Il peut survenir de novo ou par dégénérescence d’un nævus choroïdien. Il s’agit d’une tumeur relativement rare mais très agressive, au potentiel métastatique élevé, formant des métastases chez la moitié des patients, en dépit de la réussite du traitement de la tumeur primaire. Seulement 1% à 3% des patients présentent des lésions extra oculaires lors du diagnostic, alors que 50% développent des métastases, impliquant la présence de micro métastases établies des années avant le diagnostic. La choroïde est fortement vascularisée en raison de sa fonction primaire consistant à nourrir les membranes voisines telles que la sclère et la rétine, mais aussi l’iris. Le mélanome de l’uvée se propage donc principalement par circulation sanguine (Dithmar et al. 2000) et métastase dans 90% des cas dans le foie, mais aussi dans les poumons et les os (Rietschel et al. 2005a; Singh et al. 2005). Le taux de survie n’est alors que de 2 à 6 mois au stade métastatique, pouvant augmenter jusqu’à 19 à 28 mois lorsque les métastases n’atteignent pas le foie, et 80% des patients meurent dans l’année qui suit (Diener-West et al. 2005).
24 Les raisons pour lesquelles le MU présente un tropisme aussi important pour le foie ne sont pas encore claires. Certains facteurs peuvent cependant favoriser le développement des métastases au foie, comme la voie de dissémination utilisée par les cellules tumorales. En effet, la circulation sanguine pourrait favoriser la dissémination de la tumeur de l’œil vers le foie, qui est l’un des organes les plus vascularisés du corps. Il a également été montré que les exosomes dérivant de MU, exprimant les intégrines αVβ5, sont capturés par les cellules hépatiques pour préparer la niche pré métastatique et orienter le tropisme hépatique
Figure 3 : Localisations anatomiques des mélanomes uvéaux
Les mélanomes uvéaux (MU) dérivent de la transformation des mélanocytes de l’uvée.
(A) L’uvée est constituée de la choroïde, du corps ciliaire et de l’iris. Les MU sont classés selon leur site d’origine : (B) Mélanome de la choroïde, (C) Mélanome du corps ciliaire, (D) Mélanome de l’iris. Les flèches rouges indiquent la tumeur. De Eagle RC, Duane’s Ophtalmology, Foundation Volume 3, Chapter 20, Intraocular Tumors in Adults, 2013.
25 (Hoshino et al. 2015). De plus, le foie pourrait également fournir un environnement favorable pour le développement métastatique du MU, grâce à sa production de HGF (Hepatocyte Growth Factor) qui stimule la prolifération et la survie des cellules de MU exprimant à leur surface le récepteur c-Met (Bakalian et al. 2008).
1) Epidémiologie
Le mélanome de l’uvée est une maladie rare mais représente la tumeur intraoculaire la plus fréquente chez l’adulte, devant le lymphome intraoculaire. Environ 5% de tous les mélanomes affectent l’uvée, faisant du MU le mélanome le plus fréquent après le mélanome cutané. L’incidence du MU est de 4,3 cas par million d’habitants par an aux Etats-Unis (Aronow et al. 2018). Environ 8 000 nouveaux cas de MU sont diagnostiqués dans le monde par an, contre 230 000 nouveaux cas de mélanomes cutanés. Comme la plupart des cancers, l’incidence du MU augmente fortement avec l’âge, et présente un pic d’incidence vers 70 ans (Amaro et al. 2017).
Les populations caucasiennes sont les plus exposées au risque de développer un mélanome uvéal, ainsi 97% des cas recensés apparaissent au sein des populations blanches (Singh and Topham 2003), avec une incidence qui augmente avec la latitude (Virgili et al. 2007).
2) Facteurs de risque et prédisposition génétique
L’étiologie du mélanome de l’uvée n’est pas encore bien comprise. Aucun facteur de risque ou d’exposition n’a réellement été identifié jusqu’à présent, rendant une prévention par dépistage à grande échelle impossible. Le rayonnement UV a clairement été identifié comme principal facteur de risque du mélanome cutané (Gandini et al. 2005), mais le rôle du rayonnement UV dans le développement du mélanome uvéal est toujours à l’étude (Mallet et al. 2014). Le rayonnement UV émis par le soleil est composé des UVC (100nm-280nm), des UVB (280nm-315nm) et des UVA (315nm-400nm). La cornée est la première membrane oculaire en contact avec les rayonnements UV du soleil et absorbe les longueurs d’onde inférieures à 295 nm. Les UVA et les longs UVB passent donc à travers la cornée et atteignent l’iris et le cristallin. L’iris est opaque aux rayons UV et le cristallin adulte absorbe toutes les longueurs d’onde inférieures à 370 nm, ne laissant passer que 2% des UVA entre 370 et 400 nm (Norren and Vos 1974). Il est intéressant de noter que dans le cristallin de l’enfant de moins de 8 ans, la formation de 3-hydroxykynurénine et ses glucosides qui absorbent les rayonnements UV entre 300 et 400 nm n’est pas complète (Dillon et al. 1990). Par conséquence, une partie de ces rayonnements est transmise à travers le cristallin, et la rétine des enfants est exposée aux UVA et UVB. Une exposition précoce au soleil pourrait donc conduire à la formation de dommages génotoxiques de l’ADN dans les parties des segments antérieur et postérieur de l’œil. Une surexposition solaire juvénile peut donc
26 constituer un facteur de risque au développement du MU, comme c’est le cas dans le mélanome cutané, mais cela reste encore à démontrer.
Le MU survient 200 fois plus chez les populations caucasiennes que chez les populations noires, et le phénotype yeux, cheveux et peau clairs est un facteur de risque (Singh et al. 2004). Cette prédisposition ethnique pourrait être une preuve des effets cancérigènes du soleil dans l’étiologie du MU, comme dans le mélanome cutané. Cependant, les études épidémiologiques sont contradictoires et ne valident pas le rôle du soleil dans l’apparition du MU. En effet , ces études ne montrent pas de corrélation significative ni entre l’exposition aux rayonnements UVB et le risque de MU (Seddon et al. 1990; Vajdic et al. 2003b), ni entre les bains de soleil et le temps passé dehors et le risque de MU (Seddon et al. 1990; Tucker et al. 1985).
Les données moléculaires, quant à elles, semblent exclure l’exposition au soleil comme facteur de risque du MU. Dans le mélanome cutané, les rayonnements UV du soleil entrainent plusieurs types de dommages à l’ADN induisant des mutations UVB spécifiques dans le génome des cellules et une charge mutationnelle élevée. Cependant le MU est généralement caractérisé par l’absence de signature type UVB et une faible charge mutationnelle, contrairement au mélanome cutané. Cette signature UV apparait très clairement dans les gènes mutés du mélanome cutané (TP53, CDKN2A…)(Besaratinia and Pfeifer 2008; Pollock et al. 1995). Les principaux gènes mutés dans le MU sont les gènes GNAQ et GNA11 que nous décrirons plus tard. Les mutations de ces gènes apparaissent principalement dans le codon 209, mais aussi dans le codon 183. Furney et al. ont effectué un séquençage du génome entier de 12 mélanomes de l’uvée primaires et n’ont pas observé de mutations contenant la signature UV (Furney et al. 2013; Royer-Bertrand et al. 2016; Johansson et al. 2016). D’autres études ont également montré qu’aucune des mutations du codon 209 n’étaient des mutations de la signature UV, mais que celles du codon 183, retrouvées dans 3% des MU primaires et 5.9% des métastases contenaient la signature UV (Van Raamsdonk et al. 2010, 2009a). Le rôle des rayonnements UV du soleil dans l’apparition du MU n’est donc toujours pas clair.
La lumière bleue émise par le soleil, mais aussi par certaines sources lumineuses artificielles de plus en plus présentes dans notre quotidien, est un autre facteur de risque potentiel du MU. La partie de la lumière bleue tirant sur le violet, comprise entre 415 et 455 nm, est la forme de la plus énergétique après les UV, et est par conséquent susceptible d'endommager la rétine et d’autres composants de l’œil.
Des expériences in vitro et in vivo dans des modèles animaux ont montré que la lumière bleue affecte les cellules uvéales et augmente leur prolifération (Marshall et al. 2006; Di Cesare et al. 2009).
Outre les facteurs environnementaux, certains facteurs héréditaires et altérations génétiques germinales ont été corrélés à un risque plus important de développer un MU dans le cadre de syndromes de cancers familiaux, même si cela n’arrive que dans 1 à 2% des cas de MU (Krygier et al. 2001). Les principaux facteurs de risque comprennent un grand nombre de nævi dysplasiques, la présence d’une mélanocytose oculaire ou d’un nævus
27 d’Ota, des variations dans la région HERC2/OCA2 qui détermine la couleur des yeux chez l’homme (Ferguson et al. 2016) et des mutations peu fréquentes dans le gène du syndrome de prédisposition tumorale BAP1 (Harbour et al. 2010). Le gène MC1R qui influence le processus de pigmentation et le gène CDKN2A qui régule le cycle cellulaire sont impliqués dans le risque de mélanome cutané mais ne jouent pas de rôle dans la susceptibilité de développer un MU (Vajdic et al. 2003a). BAP1 était donc jusqu’à présent le seul gène de prédisposition du MU. Cependant, une étude très récente a identifié MBD4 (Methyl-CpG Binding Domain Protein 4) comme nouveau facteur de prédisposition du MU (Derrien et al. 2020). En effet, des mutations germinales de MBD4 ont été retrouvées dans des tumeurs de charge mutationnelle élevée. Ce gène est localisé sur le chromosome 3 et joue un rôle dans la réparation des dommages à l’ADN (Hendrich et al. 1999).
Les patients atteints de MU présentent un risque plus élevé de développer un mélanome cutané même si les mécanismes impliqués sont encore inexpliqués (Scélo et al. 2004).
3) Facteurs de risques acquis : Altérations génétiques
Des études à grande échelle de séquençages du génome entier (whole genome sequencing) et de l’exome entier (whole exome sequencing) de mélanomes uvéaux primaires ont confirmé les principales mutations somatiques préalablement identifiées et en ont mis en évidence de nouvelles (Figure 4).
a) Mutations somatiques activatrices
Les gènes GNAQ (Guanine Nucleotide-binding protein subunit Alpha-Q) et GNA11 (Guanine Nucleotide-binding protein subunit Alpha-11), deux gènes codant pour la sous unité α des protéines G hétérotrimériques, sont considérés comme les mutations activatrices majeures de la carcinogénèse du MU, survenant dans plus de 80% des MU (Van Raamsdonk et al. 2009a, 2010). GNAQ et son paralogue GNA11 partagent 90% d’homologie de séquence nucléotidique et représentent les gènes les plus fréquemment mutés du MU. Les mutations de GNAQ et GNA11 sont trouvés dans 45% et 32% des MU primaires respectivement, et 22% et 57% des MU métastatiques respectivement, suggérant que les tumeurs mutées GNA11 ont une tendance plus importante à métastaser (Griewank et al. 2014). Cependant, aucune corrélation significative n’a été faite entre le statut mutationnel de GNAQ et GNA11 et la survie des patients (Koopmans et al. 2013), ces mutations sont donc des mutations activatrices non significatives du pronostic. Les mutations de GNAQ et GNA11 apparaissent de manière exclusive sur la glutamine 209 (Q209) de l’exon 5 dans 97% des cas, et moins fréquemment sur l’arginine 183 (R183) de l’exon 4 (3%). Les substitutions p.Q209L ou p.Q209P entrainent une perte totale de l’activité GTPase intrinsèque, maintenant GNAQ et GNA11 dans un état constitutivement actif, et mènent ainsi à l’activation constitutive de leurs voies de signalisation en aval. La substitution p.R183C cause seulement une perte
28 partiale de l’activité GTPase. Les mutations Q209 et R183 sont mutuellement exclusives. Les mutations de GNAQ/11 apparaissent chez la moitié des patients et sont présentes dans les tumeurs à toutes les étapes de la progression du MU, et notamment dans les nævi, premier stade du développement tumoral, (Vader et al. 2017) suggérant qu’elles constituent un évènement précoce du MU (Onken et al. 2008). Le knockdown de GNAQ dans des cellules de MU inhibe la viabilité et la migration cellulaires (Liu et al. 2015).
Plusieurs modèles animaux ont été développés afin de mieux comprendre les effets de ces mutations dans le développement du MU et des métastases. Trois principaux modèles de souris génétiquement modifiées pour produire des tumeurs reproduisant le MU ont été générés ces dernières années (Richards et al. 2020). Dans le premier, un système Tet-on est utilisé pour induire l’expression de GNAQQ209L dans des souris déficientes pour
p16Ink4a et p19Ink4b (Feng et al. 2014). Plus de la moitié des souris développent des
mélanomes cutanés, mais pas de MU. Malgré cela, les tumeurs cutanées de ce modèle démontrent une activation de YAP en aval de GNAQ, comme c’est le cas dans les MU, comme nous le verrons dans le chapitre suivant. Dans un autre modèle, l’expression de GNAQQ209L est induite par le système de recombinaison Cre-Lox sur le locus Rosa26, et
entraine un MU chez la souris dans les 3 mois (Huang et al. 2015). Ce modèle utilise un Mitf-cre pour initier l’expression de l’oncogène. Dans ce modèle, les cellules tumorales disséminent par voie sanguine et forment des métastases au poumon. Enfin, le dernier modèle, dans lequel GNA11Q209L est activé par un système similaire Tyrosinase-cre, produit
un phénotype comparable mais un peu plus tardif (Moore et al. 2018).
Le développement de xénogreffes dérivées de patients (Patient Derived Xenografts : PDX) dans la souris a permis d’obtenir des modèles d’étude de la biologie de la tumeur in vivo et de la réponse aux thérapies. Ces modèles ont l’avantage de présenter des phénotype et génotype très similaires à la tumeur d’origine (Laurent et al. 2013). Des modèles de MU ont également été établis dans le zebrafish, utilisant des cellules de MU primaires ou métastatiques exprimant une protéine fluorescente, permettant de suivre l’efficacité de certaines drogues (van der Ent et al. 2014).
GNAQ et GNA11 sont activés par les récepteurs à la sérotonine (5-HT) 2A et 2B (HTR2A et HTR2B) (Benhassine and Guérin 2018; Giulietti et al. 2014). Le rôle de la sérotonine dans les mélanocytes uvéaux est inconnu mais son implication dans la prolifération, la morphologie et la migration des mélanocytes a été décrite dans la peau de la grenouille (Lobikin et al. 2015). De manière intéressante, HTR2B est surexprimé dans les MU à fort risque métastatique, malgré le fait que les mutations de GNAQ et GNA11 activent déjà la voie de signalisation (Onken et al. 2004a)(Figure 8).
Les récepteurs A et B à l’endothéline (EDRNA et EDNRB) sont des récepteurs couplés aux protéines G (RCPG), et signalent également via GNAQ/11 (Ivey et al. 2003). L’endothéline 2 est différentiellement exprimée dans le MU (Zuidervaart et al. 2003) et son récepteur EDNRB est sous exprimé dans le MU métastatique (Smith et al. 2002).
29 La voie GNAQ/11 est d’autant plus importante dans le développement du MU que des mutations en amont et en aval ont été mises en évidence récemment, grâce à ces études à grandes échelles. Ainsi, des analyses de séquençage ont mené à l’identification d’une mutation activatrice dans le gène du récepteur aux cystéinyl-leucotriènes 2 (CYSLTR2). CYSLTR2 est un récepteur à 7 domaines transmembranaires couplés aux protéines G appartenant à la famille des récepteurs « rhodopsin-like » et signalant via l’activation de GNAQ/11 (Sakmar 2002). La mutation p.L129Q de CYSLTR2 survient dans 4% des MU primaires, et est une mutation oncogénique favorisant la transition vers une conformation active, menant à une activation constitutive de la voie GNAQ/11 (Moore et al. 2016).
Les récepteurs CYSLTR sont activés par les leucotriènes cystéinés (LTC4, LTD4 et LTE4) qui sont des médiateurs lipidiques inflammatoires. Les leucotriènes jouent un rôle dans de multiples maladies, incluant le cancer. Ainsi, il a été montré que LTD4, agoniste de CYSLTR2, régule la survie et la migration des cellules cancéreuses du colon chez l’homme (Massoumi et al. 2003), et facilite la survie et la prolifération cellulaires des cellules épithéliales via l’activation de la β-caténine (Mezhybovska et al. 2005).
Une autre mutation récurrente a été identifiée, survenant quant à elle en aval de GNAQ/11, dans le gène de la phospholipase C β4 (PLCβ4). La mutation p.D630Y est localisée dans le domaine catalytique de la PLC et mène à l’activation constitutive de la voie de signalisation en aval de GNAQ/11 (Johansson et al. 2016).
Les mutations des gènes GNAQ, GNA11, CYSLTR2 et PLCβ4 sont mutuellement exclusives et constituent les mutations initiatrices du mélanome uvéal. Elles sont non significatives du pronostic.
b) Mutations secondaires associées au pronostic
D’autre mutations surviennent plus tardivement dans le développement du mélanome uvéal et sont associées au développement des métastases et au pronostic.
Les MU métastatiques sont associés à des mutations somatiques de BAP1 dans 80% des cas, entrainant généralement la production d’une protéine tronquée. L’inactivation de BAP1 est associée à un haut risque métastatique et un mauvais pronostic (Harbour et al. 2010). BAP1 présente une activité de gène suppresseur de tumeur dans les cellules cancéreuses, et des mutations BAP1 sont retrouvées dans des cancers du sein et du poumon (Jensen et al. 1998; Wood et al. 2007) Son inactivation suit la perte classique d’un suppresseur de tumeur ; mutation, puis perte de l’hétérozygotie (LOH). BAP1 est localisé sur le chromosome 3 et code pour une déubiquitinase (Jensen et al. 1998). Il joue un rôle clé dans le remodelage de la chromatine en interagissant avec ASXL1 pour former le complexe déubiquitinase répresseur Polycomb PR-DUB (polycomb-repressive deubiquitylase) qui est impliqué dans la pluripotence des cellules souches (Scheuermann et al. 2010). La séquence de BAP1 contient des domaines de liaison pour BRCA1 (Breast Cancer 1) et BARD1 (BRCA1- associated RING domain 1). BAP1, BRCA1 et BARD1 peuvent alors former un complexe
30 hétérodimérique suppresseur de tumeur. Il a été montré, dans des cellules HeLA, que BAP1 module l’activité de la ligase E3 du complexe formé par BRCA1 et BARD1, qui est requis pour la réponse aux dommages à l’ADN (Nishikawa et al. 2009). La mutation de BAP1 altère donc sa fonction dans la réparation des cassures double brin de l’ADN (Ismail et al. 2014), et dans le maintien de l’intégrité du génome, et peut contribuer à l’apparition d’altérations génétiques plus tardives.
BAP1 joue également un rôle dans la régulation du cycle cellulaire en agissant sur HCF1 (host cell factor 1) et E2F1 (Pan et al. 2015).
Enfin, BAP1 régule la transcription et la structure de la chromatine via la déubiquitination de l’histone H2A (Scheuermann et al. 2010; He et al. 2019), et à travers son interaction avec HCF1 et le facteur de transcription YY1 (Yin Yang 1), affectant ainsi la transcription de plusieurs gènes (Yu et al. 2010). BAP1 régule ainsi les programmes de transcription impliqués dans la différentiation cellulaire et bloque la transition des cellules vers un phénotype de cellule souche (Matatall et al. 2013).
L’ensemble des fonctions de BAP1 sont attribuées à sa localisation nucléaire.
Cependant, Bononi et al. ont récemment découvert que BAP1 est également localisé au réticulum endoplasmique (RE). Il se lie, déubiquitine, et stabilise alors le récepteur IP3R3 (récepteur de l'inositol 1,4,5-trisphosphate de type 3) qui contrôle la libération du calcium, du RE vers le cytoplasme et les mitochondries, menant à l’apoptose. L’étude montre que des niveaux réduits de BAP1 dans les tumeurs mutées, causent une diminution des niveaux d’IP3R3 et de la signalisation calcique, empêchant les cellules mutées de rentrer en apoptose (Bononi et al. 2017).
Ainsi, la perte de BAP1 mène à l’augmentation des dommages à l’ADN (due à son activité nucléaire), à une diminution de l’apoptose (due à son activité cytoplasmique) et donc à une augmentation des conditions favorables à la transformation cellulaire.
D’autres gènes sont associés au risque métastatique, notamment ; EIF1AX (Eukaryotic Translation Initiation Factor 1A X-Linked) et SF3B1 (Splicing Factor 3b Subunit 1).
EIF1AX code pour un facteur d’initiation de la traduction essentiel. C’est un composant du complexe de pré-initiation 43S qui induit le recrutement de la petite sous unités ribosomique 40S sur la coiffe 5’ des ARN messagers. Les mutations de EIF1AX sont principalement retrouvées dans les tumeurs de bon pronostic à faible risque métastatique (Furney et al. 2013; Martin et al. 2013)(Figure 4).
SF3B1 code pour un composant du complexe de la petite ribonucléoprotéine nucléaire U2 (snRNP) du spliceosome, impliquée dans la reconnaissance d’un point de branchement en amont du site d’épissage 3’, qui est requis pour un épissage correct de l’ARN (Furney et al. 2013; Alsafadi et al. 2016). Les mutations de SF3B1 mènent à la reconnaissance de points de branchements différents, perturbant l’épissage normal. Ces mutations provoquent l’utilisation de mauvais sites d’épissage alternatif et mènent à la rétention de courtes séquences introniques dans le transcrit mature. Ces aberrations d’épissage peuvent provoquer la dégradation de l’ARNm, ou des changements dans la
31 fonction des protéines traduites. Dans une étude incluant plusieurs types de cancer, dont le MU, il a été montré que la mutation SF3B1 entraine la perte de BRD9, qui est un composant clé du complexe BAF non canonique (ncBAF), impliqué dans le remodelage de la chromatine. Les mutants SF3B1 reconnaissent un point de branchement aberrant dans la séquence de BRD9, induisant l’inclusion d’un exon « poison », menant à la dégradation de l’ARNm de BRD9. La déplétion de BRD9 cause la perte du ncBAF et provoque la mélanomagénèse. Corriger le mauvais épissage de BRD9 dans les cellules mutées SF3B1 en utilisant des oligonucléotides antisens supprime la croissance tumorale, et suggère une nouvelle approche thérapeutique pour les tumeurs présentant ces mutations (Inoue et al. 2019). Les mutations de SF3B1 sont retrouvées dans 10% à 20% des cas de MU (Harbour et al. 2013) et sont associées à un risque métastatique faible à intermédiaire (Martin et al. 2013; Harbour et al. 2013)(Figure 4). Le groupe de Harbour a récemment montré que les mutations de SF3B1 surviennent plutôt dans les tumeurs exprimant l’oncogène PRAME (Preferentially expressed antigen in melanoma) (Field et al. 2016c).
Les MU mutés SF3B1 sont caractérisés par des anomalies chromosomiques que nous décrirons plus tard, touchant notamment le chromosome 8. Cependant, il été montré que des MU non mutés SF3B1 présentent les mêmes altérations chromosomiques, suggérant la présence de mutations dans d’autres gènes du spliceosome. Des mutations ont en effet été observées dans les gènes SRSF2 (Serine And Arginine Rich Splicing Factor 2) et U2AF1 (U2 Small Nuclear RNA Auxiliary Factor 1) (van Poppelen et al. 2019; Robertson et al. 2017). Tout comme SF3B1, il semblerait que ces gènes soient associés à un risque métastatique intermédiaire.
Les mutations de BAP1, EIF1AX, SF3B1, SRSF2 et U2AF1 ne coexistent pas dans le MU. Des études récentes ont analysé l’évolution génétique entre les MU primaires et métastatiques, et certaines nouvelles mutations ont été associées à la progression tumorale. De manière intéressante, dans certains cas, les tumeurs non mutées BAP1 développent des métastases, indiquant que la perte de BAP1 n’est pas obligatoire pour la dissémination métastatique. Cette observation suggère que dans les tumeurs mutées BAP1, la perte de BAP1 n’est pas acquise pendant la progression métastatique mais est un évènement antérieur à la formation de métastases. Des mutations de facteurs de remodelage de la chromatine tels que PRMB1 et EZH2, semblent apparaitre tardivement dans l’évolution de la tumeur, mettant en évidence une suite d’évènements dans le MU, commençant par la mutation de BAP1, puis PRMB1, EZH2 et d’autres gènes, plus tardivement (Shain et al. 2019; Piaggio et al. 2019).
La présence de certaines de ces mutations dans certaines tumeurs et pas dans d’autres, met en évidence une hétérogénéité intertumorale moléculaire, et définit des grandes classes de patients au sein de la même maladie, ne présentant pas la même réponse aux traitements, ni le même pronostic.
32
4) Voies de signalisation
La découverte des acteurs moléculaires clés opérant essentiellement dans la voie des RCPG (GNAQ, GNA11, PLCB4, et CYSLTR2), dont les mutations sont mutuellement exclusives, a mené à l’identification des voies de signalisation impliquées dans le MU.
La liaison d’un ligand aux RCPG, comme CYSLTR2, induit un changement conformationnel du récepteur menant à la liaison de GNAQ/11 au GTP et à la libération des sous-unités β-γ qui transfèrent alors le signal du récepteur aux effecteurs en aval tels que le facteur 6 d’ADP-ribosylation (ARF6). Ces effecteurs vont stimuler plusieurs voies de signalisation incluant la phospholipase C β (PLCβ)/PKC, la cascade des MAPK (mitogen-activated protein kinase) qui contrôle la prolifération cellulaire, la voie PI3K/AKT, la voie des Rho GTPases impliquée dans la réorganisation du cytosquelette et YAP (yes-associated protein), un acteur clé de la voie Hippo (Figure 5).
Figure 4 : Principales altérations génétiques associées au mélanome uvéal
Les mutations activatrices du MU font partie de la voie des récepteurs couplés aux protéines G (RCPG) et des mutations secondaires déterminent le risque métastatique.
Nomenclature acides aminés : Q : Glutamine, R : Arginine, D : Aspartate, L : Leucine.
(Van Raamsdonk et al. 2009b, 2010; Robertson et al. 2017; Johansson et al. 2016; Harbour et al. 2010, 2013; Martin et al. 2013)
33 - ARF6
La petite GTPase ARF6 est un effecteur direct de la signalisation oncogénique GNAQ et il a été montré récemment que ARF6 agit comme l’embranchement central de la signalisation GNAQ en activant toutes les voies en aval (Yoo et al. 2016). Les auteurs montrent que GNAQQ209L forme un complexe avec GEP100 (ARF-guanine Nucleotide
exchange) activant ARF6 qui redirige GNAQ de la surface de la cellule aux vésicules cytoplasmiques, et induit l’activation des voies PLC/PKC et Rho/Rac, menant à l’expression des facteurs de transcription AP-1 et YAP/TEAD. L’activation d’ARF6 mène également à la libération de la β-caténine et à sa translocation dans le noyau où elle induit la transcription et participe à la prolifération cellulaire du MU. Yoo et al. ont également montré que l’inhibition d’ARF6 diminue la prolifération et la tumorigénèse dans un modèle de xénogreffe de MU.
ARF6 est un acteur important de la prolifération, la migration, l’invasion et le développement de métastases dans de nombreux cancers (Morishige et al. 2008; Grossmann et al. 2013; Hu et al. 2009)
- La voie des MAPK/ERK
La cascade des MAPK est suractivée dans 45% à 86% des MU primaires, présentant une augmentation de la phosphorylation des protéines effectrices ERK1/2 (Pópulo et al. 2011; Zuidervaart et al. 2005). L’expression ectopique de l’oncogène GNAQQ209Lstimule la
phosphorylation de ERK1/2 dans les mélanocytes, et les mutants GNAQ/11 présentent une stimulation de l’activité MAPK/ERK via l’induction de la PLCβ et l’activation de PKC (Van Raamsdonk et al. 2009b). Il a été montré que l’inhibition pharmacologique de PKC inhibe la prolifération des cellules de MU en ciblant la voie PKC/ERK1/2 (Wu et al. 2012). La PLCβ, effecteur de GNAQ/11, hydrolyse PIP2 (phosphatidylinositol(1,4,)bis-phosphate) pour générer les messagers secondaires IP3 (inositol trisphosphate) et diacylglycerol (DAG). Le DAG active la voie PKC, qui à son tour active RasGRP3 (guanyl-releasing protein-3) qui active RAS et la voie MAPK (Chen et al. 2017b). L’activation de la voie PKC-ERK par GNAQ/11 survient généralement dans des tumeurs de mauvais pronostic ayant perdu BAP1 (Robertson et al. 2017). L’IP3 est libérée dans le cytoplasme et se lie aux récepteurs IP3R, ce qui déclenche la libération du calcium du réticulum endoplasmique vers le cytoplasme, les mitochondries et les lysosomes. Le flux de calcium entre les différents compartiments cellulaires entraine alors des cascades cellulaires impliquées dans de multiples processus, incluant la mort et la survie cellulaires. Une augmentation de calcium dans les mitochondries entraine l’apoptose. La régulation de l’IP3 et de l’activité des IP3R est donc essentielle au bon déroulement de ces processus cellulaires.