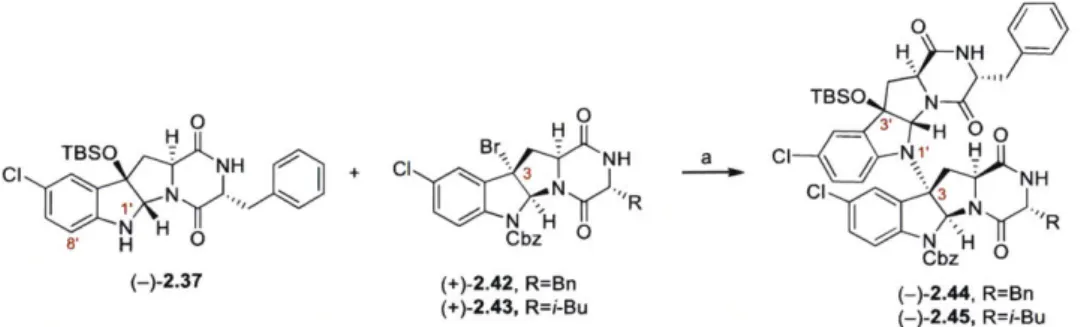
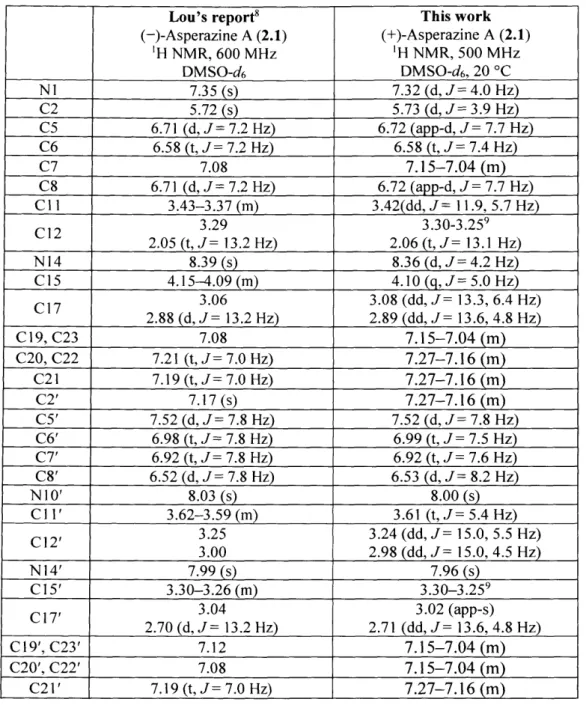
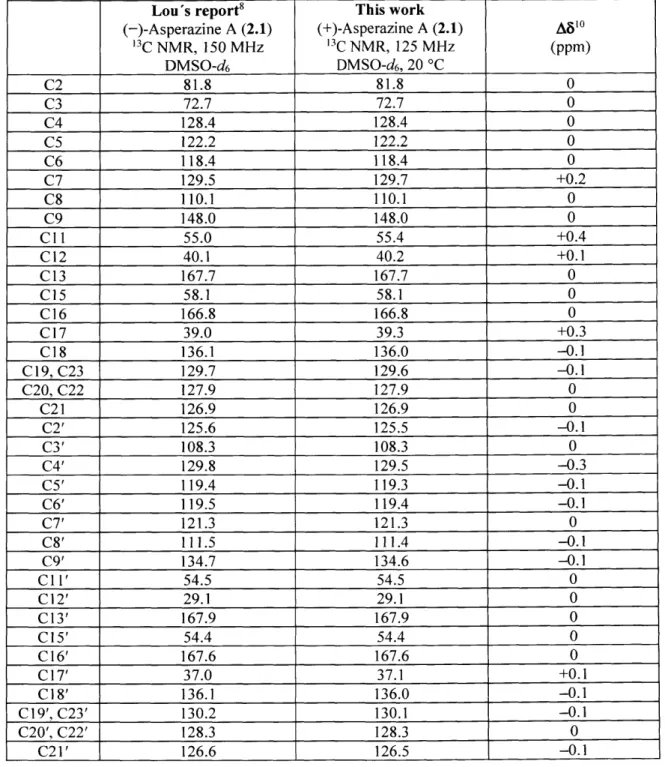
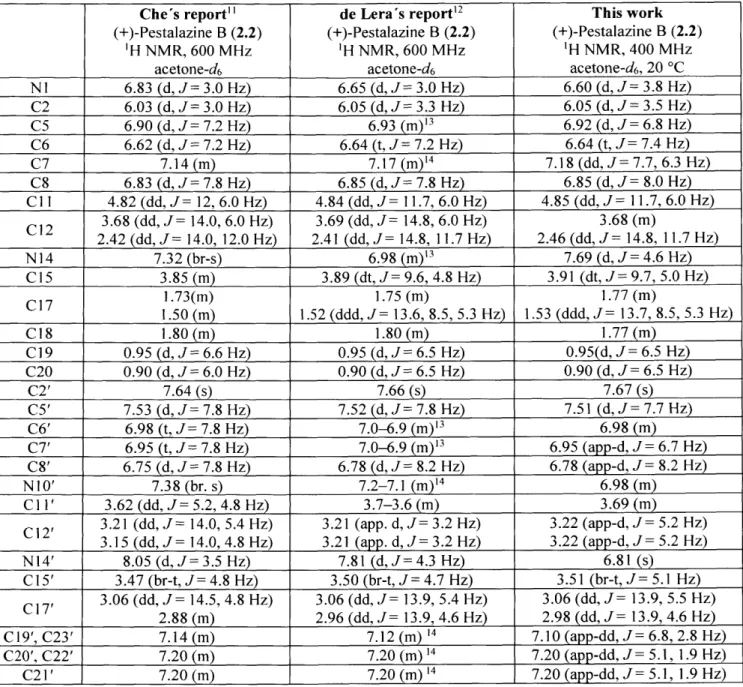
Development and Application of New Strategies for Synthesis of Complex Diketopiperazine Alkaloids
by
Brandon M. Nelson B.S., Chemistry
Illinois State University, 2013 Submitted to the Department of Chemistry
In Partial Fulfillment of the Requirement for the Degree of DOCTOR OF PHILOSOPHY
IN ORGANIC CHEMISTRY at the
Massachusetts Institute of Technology June 2018
0 2018 Massachusetts Institute of Technology All rights reserved
Signature redacted
Signature of Author Certified by_ Department of Chemistry May 1 4th 2018Signature redacted
Pf fesso o ammad Movassaghi ofessor of Chemistry Thesis Supervisor
This doctoral thesis has been examined by a committee in the Department of Chemistry as follows:
Professor Rick L. Danheiser
Professor Mohammad Movass
Signature redacted___
Chairman
aghi
Signature redacted
Thesis Supervisor
Signature redacted
Acknowledgements
First, I would like to thank my advisor, Professor Mohammad Movassaghi, for providing me the opportunity to conduct the research presented in this dissertation. Your enthusiasm for chemistry is unmatched and crucial to the success of the group. I appreciate the hands-on guidance you provided when needed, all of the chemistry discussions through the years, the opportunity to investigate and pursue original ideas, and for continuing to challenge me to improve my chemical intuition to solve difficult problems. The experience I have gained during my time in the group has made me a better chemist than I thought I could become, and has helped train me as a more effective leader and mentor. I would also like to thank Professor Rick Danheiser for serving as my thesis chair during my tenure at MIT and providing guidance and feedback that continue to further my abilities as I move on to the next step in my career.
I would also like to thank my colleagues in the Movassaghi group, both past and present, without whom my experience would have been drastically different. I want to especially thank those group members who helped mentor me as I first joined the group and whose discussions with me helped shape my thought process for addressing significant chemical problems. In particular, I would like to thank Dr. Kolby White for her constant support and leadership and for always demonstrating what a motivated chemist looks like. I would like to thank Dr. Timothy Chang and Dr. Marius Mewald for many great discussions and for making the lab a welcoming and supportive environment. Dr. Petra Lindovska and Alyssa Antropow were my contemporaries the longest and both provided great chemical discussions and support during my tenure at MIT.
I have had the privilege of sharing my experience at MIT with my loving and incredibly supportive fiance Leslie Ramos. Leslie moved to Cambridge with me leaving behind family and friends to be with me during my studies at MIT. She was there for me each night, good and bad, and never ceased to support my goals even when I wavered. It was with her support that I persisted and I believe she deserves as much credit for my successes as I do. I would also like to thank Jack for always being there to greet me when I get home and for always going out of his way to make me smile.
Finally, I would like to thank my family for all the support and encouragement they have provided throughout the years. To my brother James, all of your amazing ideas for inventions remind me of my curiosity and why I became a scientist to begin with. To my sister Jacqui, while you often thought I was competing with you, I always have wanted to live up to the amazing example you have set for me. To my brother Caleb, we may not
Preface
Portions of this work have been adapted from the following articles that were co-written by the author and are reproduced in part with permission from:
Nelson, B. M.; Loach, R. L.; Scheisser, S.; Movassaghi, M. "Concise Total Synthesis of (+)-Asperazine A and (+)-Pestalazine B" Org. Biomol. Chem. 2018, 16, 202. Bischoff, A.; Nelson, B. M.; Niemeyer, Z. N.; Sigman, M.; Movassaghi, M. "Quantitative Modeling of Bis(pyridine)silver (I) Permanganate Oxidation of Hydantoin Derivatives" J Am. Chem. Soc. 2017, 139, 15539.
Development and Application of New Strategies for Synthesis of Complex Diketopiperazine Alkaloids
by
Brandon M. Nelson
Submitted to the Department of Chemistry On May 31st, 2018 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in
Organic Chemistry Abstract
I. Quantitative Modeling of Bis(pyridine)silver (I) Permanganate Oxidation of Hydantoin Derivatives: Guidelines for Predicting the Site of Oxidation in Complex
Substrates
The bis(pyridine)silver(I) permanganate promoted hydroxylation of diketopiperazines has served as a pivotal transformation in the synthesis of complex epipolythiodiketopiperazine alkaloids. This late-stage C-H oxidation chemistry is strategically critical to access N-acyl iminium ion intermediates necessary for nucleophilic thiolation of advanced diketopiperazines en route to potent epipolythiodiketopiperazine anticancer compounds. In this study, we develop an informative mathematical model using hydantoin derivatives as a training set of substrates by relating the relative rates of oxidation to various calculated molecular descriptors. The model prioritizes Hammett values and percent buried volume as key contributing factors in the hydantoin series while correctly predicting the experimentally observed oxidation sites in various complex diketopiperazine case studies. Thus, a method is presented by which to use simplified training molecules and resulting correlations to explain and predict reaction behavior for more complex substrates.
II. Total Synthesis of (+)-Asperazine A and (+)-Pestalazine B
III. Concise Total Synthesis of (-)-Lasiodiplines E and F and (+)-Desmethyl
Lasiodipline E
The concise, biogenetically inspired total synthesis of (-)-lasiodiplines E and F and
(+)-desmethyl lasiodipline E was achieved. The unique structural architechture of
(-)-lasiodipline F, a previously unknown architecture in diketopiperazine natural products, required an advanced biosynthetic analysis and the acyclic thiolated diketopiperazines provided the opportunity to develop new methods for stereoselective thiolation. The use of a tetracyclic cyclotryptophan core to control sulfidation stereochemistry before revealing the acyclic core proved to be highly efficient and enabling. This total synthesis allowed for the revision of the stereochemistry of the C15 methyl sulfide of (-)-lasiodipline E to the cis stereoisomer.
Thesis Supervisor: Mohammad Movassaghi Title: Professor of Chemistry
Table of Contents
I. Quantitative Modeling of Bis(pyridine)silver(I) Permanganate Oxidation of Hydantoin Derivatives: Guidelines for Predicting the Site of Oxidation in Complex Substrates
Introduction 12
Results and Discussion 16
Conclusion 30
Experimental 32
II. Total Synthesis of (+)-Asperazine A and (+)-Pestalazine B
Introduction 92
Results and Discussion 98
Conclusion 104
Experimental 107
III. Concise Total Synthesis of (-)-Lasiodiplines E and F and (+)-Desmethyl Lasiodipline E
Introduction 134
Results and Discussion 143
Conclusion 149
Experimental 152
Appendix A: Spectra for Chapter I 188
Appendix B: Spectra for Chapter II 354
Appendix C: Spectra for Chapter III 389
Abbrieviations
A
angstrom[a] specific rotation
app apparent aq aqueous atm atmosphere Boc tert-butyloxycarbonyl br broad Bu butyl 0C degree Celsius
CAM ceric ammonium molybdate
Cbz carbooxybenzyl
cm~- wavenumber
d days
d doublet
d deuterium
6 parts per million
DART direct analysis in real time DMAP 4-(dimethylamino)pyridine DMF NN-dimethylformamide DMA NN-dimethylacetamide DMSO dimethylsulfoxide dr diastereomeric ratio ee enantiomeric excess El electronspray ionization Et ethyl FT fourier transform g gram
gCOSY gradient-selected correlation spectroscopy
h hour
HMBC heteronuclear multiple bond correlation HPLC high performance liquid chromatography HRMS high resolution mass spectroscopy HSQC heteronuclear single quantum correlation
Hz Hertz
i iso
p micro Me methyl Mhz megahertz min minute mol mole M.p. melting point MS mass spectrometry m/z mass to charge N normal
NMR nuclear magnetic resonance
nOe nuclear Overhauser effect
NOESY nuclear OVverhauser effect spectroscopy
Nu nucleophile o ortho p para Ph phenyl piv pivaloyl PMP para-methoxyphenyl
ppm parts per million
Pr propyl q quartet Rf retention factor s sec s singlet s strong str stretch t tert t triplet TBS tert-butyldimethylsilyl
TFA trifluoroacetic acid
THF tetrahydrofuran
TLC thin layer chromatography
UV ultraviolet
Chapter I
Quantitative Modeling of Bis(pyridine)silver (I)
Permanganate Oxidation of Hydantoin Derivatives:
Guidelines for Predicting the Site of Oxidation in Complex
Introduction and Background
Potassium permanganate oxidations were first discovered over a century ago and have historically been used for a variety of applications. Oxidation of phenol derivatives and kinetic studies were carried out in the early 1900's by Hinshel."2 Further investigation into the use of permanganate ions as an oxidant led to the discovery of the oxidation of toluene 1.1 to benzoic acid 1.4, shown in Scheme 1.1. The kinetics and mechanism of the oxidation were more thoroughly investigated in the next few decades.3 It was found that toluene was oxidized in a stepwise fashion each step increasing the oxidation state from benzyl alcohol 1.2 to benzaldehyde 1.3 and finally benzoic acid 1.4 (Scheme 1.1).
H
[
OH 0 0 H OHH~O OHI abstraction * rebound;
AcOH/HHO
jbsi
-12J hydrolysis50 *C,4
0
1.1 1.2 1.3 1.4 1.1 1.5 1.2
Scheme 1.1. Potassium permanganate oxidation of toluene.
A more developed understanding of the mechanistic picture was lacking until more recently.4 The prevailing belief is the reaction involves a rate limiting C-H abstraction leading to the benzylic
substituted ammonium ions has increased the scope of permanganate oxidations and has allowed
reactivity that was previously inaccessible.8,9
Permanganate based oxidants were developed, namely bis(pyridine)silver (I) permanganate,0
were introduced for the oxidation of aldehydes via C-H abstraction. This class of oxidants was of
interest to our group as we recognized the unique potential for this oxidant in the oxidation of the
Ca position of diketopiperazines. Application of this permanganate oxidation to complex
diketopiperazines has proven successful in a variety of total synthetic efforts." As illustrated in
Scheme 1.2A, the bis(pyridine)silver (I) permanganate promoted oxidation of dimeric
diketopiperazine (+)-1.6a led to the formation of the corresponding tetrahydroxylated dimer
(+)-1.7a, with hydroxylation at C 1 and C15 (with no partial oxidation products observed), en route
to the synthesis of (+)-11,1 1'-dideoxyverticillin A (1.8a).lIa Similarly, hydroxylation of dimeric
diketopiperazine (+)-1.6b afforded the corresponding tetrahydroxylated dimer (+)-1.7b (Scheme
2A). "' Notably, the desired hydroxylation proceeds even at the more electron-deficient C 15-center
next to the acetoxy group to give tetraol (+)-1.7b without any partial oxidation observed.'2 However, oxidation of the Cl -epimer of dimeric diketopiperazine (+)-1.6a (not shown) under
identical conditions only afforded a diol product where C-H oxidation is limited to the
C15-positions without oxidation at the C II-C15-positions, la highlighting the impact of the diketopiperazine
stereochemistry on the reaction outcome. Ia The hydroxylation of diketopiperazine (+)-1.9 using
substituent, the double oxidation at C15 was surprising given the monohydroxylation of the structurally related diketopiperazine of (+)-1.9. Interestingly, bis(pyridine)silver (I) permanganate promoted hydroxylation of diketopiperazine (+)-1.15,
A) Tetrahydroxylation of a Dimeric Diketopiperazine:
S2Ph SO2Ph RO N Py2AgMnO4 R MeN NH CHC2 MeN HO HO H e 23 *C OH NMe 0 NN N 1 R 0 N R -N HH (+)-1.7a N H OH
(+)-1.6a, R=Me S2Ph 0 63%, R=Me (+)-1.7b SO2Ph 0
(+)-1.6b,R=CH20Ac 55%, R=CH20Ac
B) Dihydroxylation of a Diketopiperazine:
H H
N N
/ 0
H 0 n-Bu4NMnO4 HOO
NMe CHC S2 N
NN Ny~ H 23 'C N, H
&IN HH N NH OH
(+)-1.9 SO2Ph
0 41% (--.O S02Ph 0
C) Triple Oxidation of a Diketopiperazine:
Boc N / B 0 Py2AgMnO4 12 NMe CH2CI2 - )N 15. H 23 C N NH )iH (--.2S02Ph 0 45% Boc N /
K
Boc Ne CP 6 HO 0 NMe (-N N (--.3 SO2Ph0 M H SN 0 MMeN S NMe H 0(+)-1 .8a, R=Me (+)-1 1,11 '-dideoxyverticillin A
(+)-1.8b, R=CH2OAc (i)- Chaetocin A
H N / MeS 0 NMe N H He H A (+)-Gliocladin B (1.11) H N OH 0N~ S" N~ H 0 (+)-bionectin A (1 .14)
D) Mono Oxidation of a Diketopiperazine: E) Double Oxidation of a Diketopiperazine:
H 0 HO 0 AcO 0
Br,. NCHO Py2AgMnO4 Br, NCHO Me NMe Py2AgMnO4
S
AcO 0
hydroxylation of diketopiperazine (+)-1.17 gave the triketopiperazine (+)-1.18 (Scheme 1.2E) with double oxidation at the methylene, consistent with our observations in the oxidation of diketopiperazine (-)-1.12, without C-H oxidation adjacent to the acetylated diketopiperazine nitrogen, consistent with the lack of oxidation at C15 with diketopiperazine (+)-1.15.
Informed by prior biosynthetic studies of sirodesmin by Howlett,13
a and the cysteine feeding
experiments by Kirby,13b and given the presence of various polysulfane congeners in distinct
families of natural ETPs, we posited4 that the introduction of the carbon-sulfur bonds in the biosynthesis of these alkaloids may involve a C-H hydroxylation followed by nucleophilic glutathione thiolation of N-acyl iminium ion intermediates (Scheme 1.3).1 ab
o 0
H HO 0YS 0 0
R' NMe R' NMe R' SkNMe R NMe
RN R" RN R" RN HR" RN S
0 H 0 OHOH
DKP nucleophilic 1 ETP
hydroxylation thiolation synthesis Scheme 1.3. Biosynthesis of epidithiodiketopiperazines.
Importantly, this biogenetically inspired approach to the chemical synthesis of epipolythiodiketopiperazines led to the development of our permanganate promoted hydroxylation of diketopiperazines and laid the foundation for the synthesis of a number of natural and designed complex epipolythiodiketopiperazines." Additionally, consistent with this hypothesis, C-H hydroxylation of a phenylalanine-serine diketopiperazine followed by nucleophilic addition of
epipolythiodiketopiperazine syntheses,6a we have sought to better understand the critical substrate characteristics that govern the reaction outcome.
Results and Discussion
The work presented in this chapter was a collaboration with Professor Matt Sigman, Amanda Bischoff, and Dr. Zach Niemeyer at the University of Utah. The work presented represents a collaborative effort and would not have been possible without contributions from both groups. My
work focused on the synthesis and oxidation of all hydantoin and diketopiperazine derivatives as well as physical organic experiments. The Sigman group provided their expertise in the
mathematical modeling and multivariate analysis presented below and measured the relative rates of oxidation of all hydantoins. In development of the presented linear regression model, discussions between all collaborators was necessary to develop a model that satisfied both mathematical and chemical aspects. All experimental data/spectra presented in this chapter are the work of the thesis author.
Synthesis and Oxidation of Hydantoins. While we have gathered a wide range of experimental observations related to this hydroxylation chemistry (Scheme 1.2), the multiple oxidation
provide excellent substrate variability needed for model development. Notably, these substrates offer control over both the steric and electronic environment of the single activated C-H bond that would be subject to hydroxylation. An array of hydantoins 1.25 was assembled on multi-gram scale from readily available amino acid derivatives 1.23 and phenyl isocyanate.16 Hydantoins 1.24 along with the corresponding N-substituted hydantoins 1.25 provided a diverse training set of substrates for our study.
0
O ~ Ph0Ph
OMe PhNCO OPh PhX
R1 NH2 N
N N
H H H R2
1.23 1.24 1.25
Ph -single activated C-H bond for study
N 0 -rapid access to an array of substrates I -isolation of steric and electronic factors R1 N excellent for comparative analysis
R2
Scheme 1.4. Hydantoin synthesis and diversification.
We focused on the use of bis(pyridine)silver(I) permanganate as the oxidant for hydantoin hydroxylation (Table 1.1) under typical conditions employed in the case studies illustrated in Scheme 1.2. Importantly, a standard set of hydroxylation reaction conditions were used for all hydantoins in this study, unless noted otherwise, to quantify the relative success of each individual case.'7 We began our investigation with the oxidation of the simple alanine derived hydantoin 1.26a, which underwent permanganate promoted oxidation to provide alcohol 1.26b (Table 1.1, entry 1). As we increased the size of the Ni-substituents to methyl and phenyl (1.27a and 1.28a,
yield under the standard conditions. Increasing the size of the NI substituent in the presence of the isopropyl group at C5 caused a significant reduction in the reaction rate and yield (Table 1.1, entries 5-6). Hydroxylation of N-methylated hydantoin 1.30a afforded only 41% yield of alcohol
1.30b with a significant amount of starting material remaining, while the N-phenyl hydantoin
1.31a proved highly recalcitrant toward permanganate oxidation providing 13% yield of alcohol
1.31b and returning 71% of substrate 1.31a.
We next sought to vary the C5-H bond environment. Use of phenyl glycine to form hydantoin 1.32a provided a substrate that has the benefit of a weakened C-H bond due to the adjacent 71
system. Despite increased steric encumbrance, oxidation of hydantoin 1.32a (Table 1.1, entry 7) to alcohol 1.32b proceeded efficiently to give the product in 72% yield. Substitution of the NI position of the phenyl glycine derived hydantoins did not significantly decrease the rate and efficiency of the hydroxylation reaction. Both hydantoins 1.33a and 1.34a were oxidized completely under standard conditions, providing products 1.33b and 1.34b, respectively (Table 1.1, entries 8-9). The complete oxidation of the phenyl glycine derived hydantoins is consistent with a weakened C-H bond due to optimal alignment of the n system of the phenyl group with the C-H orbitals.
We also prepared hydantoins with varied NI steric hindrance using hydantoin 1.26a as starting
material. We prepared NI -iso-butyl hydantoin 1.42a. Hydroxylation of hydantoin 1.42a proceeded
efficiently to alcohol 1.42b (Table 1.1, entry 17), demonstrating that steric hindrance at N 1, slightly
removed from the site of reactivity, has a measurable but reduced impact on the yield of the
product. In an effort towards further electronic variation at N 1, introduction of a
2,2,2-trifluoroethyl group at NI gave hydantoin 1.43a. Hydroxylation of hydantoin 1.43a proceeded to
give alcohol 1.43b in 33% yield along with returning 35% of starting hydantoin 1.43a (Table 1.1,
entry 18). Hydroxylation of NI -benzyl hydantoin 1.44a provided an internal competition between
the C5-H and the benzylic CHs. Interestingly, the preferred site of oxidation for hydantoin 1.44a
remained the C5-H, providing alcohol 1.44b (Table 1.1, entries 19), albeit in diminished yields
compared to the standard substrate 1.26a.
To probe the interaction between C5 and NI substituents, we prepared the corresponding
valine derived hydantoins 1.45a and 1.46a. Hydroxylation of hydantoin 1.45a exhibited an even
greater decrease in reaction rate, compared to hydantoin 1.43a, providing only 9% of the
corresponding alcohol 1.45b with 71% recovered hydantoin (Table 1.1, entry 20). Similarly,
hydantoin 1.46a showed greatly diminished reactivity, as compared to hydantoin 1.44a, providing
alcohol 1.46b in a 7% yield with 70% of the starting hydantoin 1.46a recovered (Table 1.1 entry
21). Consistent with the reaction outcome of diketopiperazines (+)-1.15 and (+)-1.17 (Scheme 1.2
0 Ph N R, : N H R2 1.26a-1.47a PY2AgMnO4 CH2CI2, 23 *C 0 Ph R1:IN> HO R2 1.26b-1.46b
Entry Substrate R1 R2 Yield (%) Recovered Krela
Substrate (%) 1 1.26a 2 1.27a 3 1.28a Me H 78 Me Me 60 Me Ph 54 0 1 11 0.21 18 0.14 4 1.29a (CH3)2CH H 79 5 1.30a (CH3)2CH Me 41 6 1.31a (CH3)2CH Ph 13 26 0.04 71 0.12 7 1.32a 8 1.33a 9 1.34a 10 1.35a Ph H 72 Ph Me 85 Ph Ph 64 CH2CH2CH2 16 11 1.36a (CH3)3C H 21 12 1.37a (CH3)2CH2CH 13 1.38a (CH3)2CH2CH H 65 Me 62 14 1.39a Cyclohexyl H 57 15c 1.40a Cyclohexyl Me 16 1.41a Cyclohexyl 72 Ph 30 0 0 Xb Xb 0 0.58 70 0.12 30 0.17 0 0.90 10 0.32 0 0.48 0 0.16 49 0.98 17 1.42a 18 1.43a 19 1.44a Me (CH3)2CHCH2 64 Me CF3CH2 33 Me Bn 30 20 1.45a (CH ) CH CF CH 9 71 0.02 0 0.17 8 35 5 0.26 Xb 0.10
A) Isotope Studies
O N PY2AgMnO4 0 N
Me N solvent, 23 *C Me N
R Me HO Me
1.27a, R=H KlEext (CH2CI2) = 6.97 1.27b
1.27a-dl, R=D KIEint (MeCN) = 2.26
B) Stereochemical Analysis N Py2AgMnO4 0 N Me O -a- Me >o Me - N solvent, 23 'C Me - N Me H H Me HO H 1.36a, 90% ee 1.36b 21%, 90% ee (CH2CI2)
C) Hammett Analysis 49%, 90% ee (MeCN)
R R
PY2AgMnO4
Me Ie N O MeCN, 23 "C Me M :N N 0
H H HO H
R=H, 1.26a R=H, 1.26b
R=Me, 1.48a R=Me, 1.48b
R=N02,1.49a R=N02, 1.49b
Scheme 1.5. Mechanistic investigations of permanganate mediated oxidation of hydantoins
While the mechanism of permanganate oxidation of toluene to benzoic acid has been studied computationally,5,6 supporting a C-H abstraction-rebound pathway, there was need for additional
informative experimental data to assist with a detailed analysis of this reaction in complex settings. We initiated our studies by preparing the C5-D hydantoin 1.27a-di and comparing the rate of oxidation to that of hydantoin 1.27a (Scheme 1.5A). Both competition reactions and independent initial rate measurements confirmed that the rate determining step in the reaction involves C-H
Throughout our synthetic studies, the permanganate mediated diketopiperazine dihydroxylation has been highly diastereoselective, often yielding a single diastereomer. In fact, the data are consistent with a stereospecific hydroxylation except in cases that prevent this outcome due to severe steric blocking leading to a competing reaction manifold or a post-oxidation ionization step. Ild This observation has been both synthetically useful and mechanistically insightful, allowing us to support the hypothesis that the oxygen rebound to the transiently generated radical center is rapid enough to prevent inversion of the radical.' la The hydroxylation
of enantiomerically enriched hydantoin 1.36a provided further support for a stereospecific and stereoretentive hydroxylation under typical conditions (2.0 equiv oxidant, 1 h).'7 While the oxidation proceeded further in acetonitrile as compared to dichloromethane as solvent, in both solvents the product 1.36b was isolated without loss of enantiomeric enrichment (Scheme 1.5B).
We also carried out the hydroxylation reaction with a set of hydantoins that informed a three-point Hammett analysis and found a modest increase in reaction rate for electron withdrawing substituents (krei = 1.16 for 1.49a, Scheme 1.5C). Since C-H abstraction is the rate determining step based on KIE experiments, the observed trend is consistent with the electron withdrawing substituent slightly better facilitating the formation of the short lived radical intermediate, likely due to enhancement of the C4-carbonyl for a captodative stabilization of the radical.
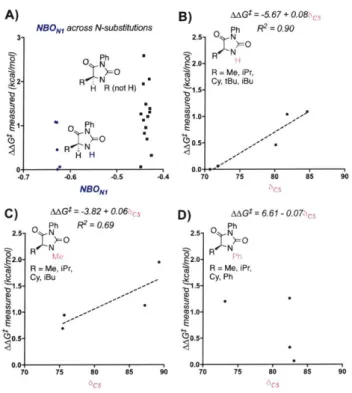
AAGt = 0.74 -0.4 3a(C5) + 0.4 86C5 NBON1 + 0.22NBONI -0. 19%Vbu -2.5- .1 y =0.19 + 0.82x # 2.0- R2 =0.82 Q2 = 0.71 *,/ * .1. . 1. . SC5 0.0. 0.5 1.0 1.5 2.0 2.5 3.0 4
-0.5- Measured AAGt (kca/mol)
.,*G (C5) %Vbur
Figure 1.1. Model comparing predicted AAG: to measured AAG: for the hydantoin library utilizing the Taft parameter Gp, a nitrogen NBO charge, a C NMR shift, and percent buried volume of the abstracted hydrogen.
We next focused on how the oxidation rate of a series of hydantoins could be used to approximate selectivity of more complex diketopiperazine oxidations. To accomplish this, competitive rate measurements were performed between a training set of hydantoins with differing R' and R2 groups (Scheme 1.3) and hydantoin 1.26a (Table 1.1, Entry 1) with the goal of identifying the factors that contribute to the reaction success. A relative rate constant, krei, was obtained for each substrate, and this was converted to a transition state energy difference of AAG+ using the equation AAGI = -RTln(krei). 17 Several molecular descriptors, which have the potential to correlate with AAGI, were then calculated from structures of the substrates computationally
organometallic chemistry,2 3 with spherical radii varying between 1.75 and 3.50 A. (Figure 2). After parameter collection, an optimized correlation between these parameters and AAGI was achieved using linear regression fitting to quantitatively analyze the substituent effects on the oxidation rate.19,20
Previous studies from the Sigman group24 and others2' have identified numerous
multi-dimensional correlations between molecular parameters and reaction outcomes. The computationally derived parameters in this study only revealed relatively complex models to describe the rate measurement, the simplest of which involved four unique terms. These parameters include the Hammett parameter ap of the carbon substituent (Gp(Cs)), the NBO charge of the nitrogen adjacent to the hydrogen to be abstracted (NBONI), the calculated 13C NMR shift of the carbon which is oxidized (8cs), and the %Vbur at a spherical radius of 2.0 A of the abstracted hydrogen (Figure 2). An internal validation employing a leave-one-out analysis (Q2=0.71) suggests a relatively robust model. Considering the apparent complexity of the model likely resulting from the structural variance included in the training set, we selected to deconstruct the terms to facilitate understanding as to how selectivity is imparted by the oxidant.
A) Ph p(C5 B) Ph 0 pC(C5) 2.5 0 N 2.5 SMe N= 2.5 PrN NR R = H, Me, 2.0 R=H,Me, , 2.0 iBu
Isolating the model's parameters revealed notable trends among varying subsets of hydantoins.
Variations in the NI substituent are described by ap(C5) and the NI NBO charge. In considering
Up(C5), it is clear that hydantoins with the same C5 substituent cluster together. However, Ni -phenyl substituents are unique with a much higher ap(c5) in each set (Figure 1.2), corresponding with a
moderately low relative rate for these substrates. Because up describes resonance and electronic
effects, it is reasonable that phenyl would have a greater impact on the models than the remainder of the aliphatic substituents. When NI substituents were compared, the Ni-H substituted
hydantoins were found to have a much lower NI NBO charge than the remainder of the hydantoins, accompanied by their high rate of oxidation (Figure 1.3A). Generation of a model excluding the N 1-H subset resulted in a reasonable model similar to that in Figure 2, with an R2 of 0.75 and a Q2
of 0.56 with the exclusion of the NI NBO charge.'7 Thus, this parameter mainly functions to normalize the NI-H subset.
While ap(Cs) and the NI NBO charge shed light on effects of the NI substituent, variations in
C5 are described by examining the isotropic 3C NMR shift of C5. Holding the Ni-substituent constant, several trends were apparent. For the Ni-H and Ni-Me subsets, as well as a subset describing a variety of other R groups, the energy barrier increases as a function of the 3C NMR chemical shift (Figure 1.313 and 1.3C). Greater electron density thus appears to stabilize radical
formation, in agreement with the KIE studies that suggest rate limiting hydrogen atom abstraction. This trend is not observed in the set of hydantoins with Ni -phenyl substitution (Figure 1.3D). In
subsets with various NI and C5 substituents. This likely occurs because this parameter accounts for integrated steric effects of both substituents.
A)
NBONm across N-substitutions
3. 0 Ph CO 0IN HR (not H) C= (a o) Ph H -0.7 -0.6 -0.5 -0.4 NBON, C) MG$ = -3.82 + 0.06,c,, 2.5 0 Ph R2= 0.69 NO 2.0- MO R = Me, iPr, Cy, iBu C1.0 0.0-70 75 8o 85 90 oC5 B) AAG*=-5.67+0.08S Ph R2= 0.90 2.5. N>=O 2. R N S2.0- H S R = Me, iPr,
Cy, tBu, iBu
o.5 0.5 0.0. 70 75 60 85 90 D) MG* = 6.61 -0.07Hcs 2.5 0 PhNO 2.0 Ph S R= Me, iPr, .5 Cy, Ph 10 0.0 70 75 o 85 9o Hcs5
Figure 1.3. The NBO charge of N, describes the Ni-H subset (A) and the "IC NMR shift describes variation in the C5
substituent (B, C, D).
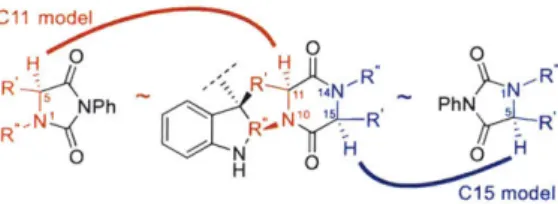
Using the model developed above (Figure 1.1) and the mechanistic insight regarding influential
- 'A
undergo oxidation, to afford the same parameters that were extracted in our hydantoin analysis (Figure 1.4).17 C11 m O O 0H0 R ~ ~ ~ -.-: - II1 .R ~, 4N -R PhN I'N R5 NPh R R"' R-( R,.N '1 H R 0 H R' C15 model
Figure 1.4. Structural correlation used to calculate parameters for hydantoins and the corresponding diketopiperazines.
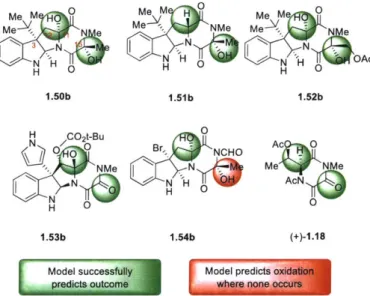
Using the model generated from the hydantoin derivatives, the predicted AAGI was calculated for each of the diketopiperazine substrates. This required that the parameters for hydantoin and diketopiperazine reactive sites be normalized together, but the intention of this analysis was to use the information gained from the hydantoin series to explain diketopiperazine trends and not to directly compare the diketopiperazine reactivity to the hydantoin reactivity. It therefore must be emphasized that the calculated AAG: does not represent an actual energy barrier between the diketopiperazines and substrate 1.26a, but rather an abstract, but nevertheless descriptive, numerical output by which the model generated by the hydantoin derivatives can categorize the diketopiperazine derivatives. These calculations divided the diketopiperazines into two subsets: one with a calculated energy barrier of >0.15 kcal/mol corresponding to oxidized diketopiperazines, and another with a calculated energy barrier of <0.15 kcal/mol corresponding
the successful oxidation of both CiI- and C15-positions to give diol 1.50b (Figure 1.5).26
Furthermore, when the corresponding Cl -epimeric diketopiperazine was used as substrate for this analysis the model correctly predicts the mono oxidation at C 15-position to give alcohol 1.51b consistent with prior experimental data.' Ia The model also predicted the formation of dihydroxylated C3-t-butyl diketopiperazine 1.52b, an outcome reminiscent of the oxidation of diketopiperazine (+)-1.6b to tetraol (+)-1.7b (Scheme 1.2A). Interestingly, application of the model to the C3-pyrrole diketopiperazine as a surrogate for diketopiperazine (-)-1.12 (Scheme 1.2C) led to the correct prediction of oxidation at both C11- and C15-positions to afford triketopiperazine 1.53b (Figure 1.5), corresponding to triketopiperazine (-)-1.13 (Scheme 1.2C). While the model successfully predicts hydroxylation at C 1-position of diketopiperazine (+)-1.15 (Scheme 1.2D), it fails to predict the lack of reactivity at the C 15-position and offers diol 1.54b (Figure 1.5) as the oxidation product instead of the alcohol (+)-1.16 (Scheme 1.2D). However, application of the model to the analysis of the simpler diketopiperazine (+)-1.17 (Scheme 1.2E) led to the correct prediction for the formation of triketopiperazine (+)-1.18 (Figure 1.5) as the oxidation product in agreement with our experimental findings. Importantly, the model not only predicts double oxidation at the methylene of diketopiperazine (+)-1.17, it also predicts the lack of oxidation at carbon adjacent to the acetylated nitrogen. Indeed, this model demonstrates high
several complex diketopiperazines. The %Vbur for a spherical radius of 2.00 A was found to be higher than 75% for substrates that did not undergo oxidation, and lower than 75% for those which were oxidized. This single parameter approximation proved slightly less descriptive than the full model prediction, accurately predicting 12 of the 14 diketopiperazine potential sites of oxidation. The %Vbu parameter incorrectly predicts oxidation at the C15 site to yield 1.54b (Figure 1.5), the same site incorrectly predicted by the model. This parameter also incorrectly predicts that oxidation will not occur at one CII position, which is oxidized, thus yielding a triketopiperazine derivative of 1.53b with no oxidation at the CII site.
M!VMeV V M e M Mee Me e '_1 N'H
YO
105 OAc H 0 H 0 1.50b 1.51b 1.52b H CO2t-Bu 0 N 0/ 0.0 Br,, NOHO Ac 0 1.53b 1.5b (+)-1.18Model successfully Model predicts oxidation
me e Nur e
Figure 1.5. Relation of complex diketopiperazine oxidation outcomes predicted by model to experimental outcomes.
Conclusions
In summary, the hydroxylation of complex diketopiperazines promoted by bis(pyridine)silver (I) permanganate has been a critical transformation for the total synthesis of a variety of
provided excellent substrate variability with control over both the steric and electronic environment of the activated C-H bond subject to hydroxylation. The model prioritizes Hammett values and %Vbur as key contributing factors in the hydantoin series. Importantly, the model may be applied to more complex substrates as illustrated in Figure 1.5 and it can correctly predict the hydroxylation outcome with a high level of agreement with experimental results. This will provide a roadmap to synthetic design and application of this late-stage oxidation reaction in complex synthesis.
(1) Hinshelwood, C. N. J Chem. Soc., Trans., 1919,115, 1180-1188.
(2) Hinshelwood, C. N.; Winkler, C. A. J. Chem. Soc., 1936, 368-370.
(3) Cullis, C. F.; Ladbury, J. W. J Chem. Soc., 1955, 5 55-560.
(4) Brauman, J. I.; Pandell, A. J. J. Am. Chem. Soc.1970, 92, 329-335. (5) Strassner, T; Houk, K. N. J. Am. Chem. Soc. 2000, 122, 7821-7822.
(6) K. A. Gardner, J. M. Mayer, Science 1995,269, 1849-1851.
(7) Firouzabadi, H.; Sardarian A. R.; Naderi, M.; Vessal, B. Tetrahedron 1984, 40, 5001-5004. (8) Firouzabadi, H.; Vessal, B.; Naderi, M. Tetrahedron Lett. 1982, 23, 1847-1850.
(9) Sala, T.; Sargent, M. V. J. Chem. Soc., Chem. Commun., 1978, 253-254.
(10) Firouzabadi, H.; Vessal, B.; Naderi, M. Tetrahedron Lett. 1982, 23, 1847.
(1 )(a) Kim, J.; Ashenhurst, J. A.; Movassaghi, M. Science 2009, 324, 238. (b) Kim, J.; Movassaghi, M. J Am. Chem. Soc. 2010, 132, 14376. (c) Boyer, N.; Movassaghi, M. Chem. Sci. 2012, 3, 1798. (d) Coste, A.; Kim, J.;
Adams, T. C.; Movassaghi, M. Chem. Sci. 2013, 4, 3191. (e) Adams, T. C.; Payette, J. N.; Cheah, J. H.; Movassaghi, M. Org. Lett. 2015, 17, 4268.
(12) For discussion on impact of electron withdrawing groups on C-H oxidation, see: (a) Ishihara, Y.; Baran, P. S.
Synlett 2010, 12, 1733. (b) Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem. Int. Ed. 2012, 51, 8960. (c)
Cernak, T.; Dykstra, K. D.; Tyagarajan, S.; Vachal, P.; Krska, S. W. Chem. Soc. Rev. 2016, 45, 546. (d) Engle, K. M.; Yu, J.-Q. J. Org. Chem. 2013, 78, 8927. (e) Neufeldt, S. R.; Jim~nez-Os~s, G.; Huckins, J. R.; Thiel, 0. R.;
(19) Milo, A.; Bess, E. N.; Sigman, M. S. Nature 2014, 507, 210-214.
(20) Santiago, C. B.; Milo, A.; Sigman, M. S. J Am. Chem. Soc. 2016, 138, 13424. (21) Hammett, L. P. Chem. Rev. 1935, 17, 125.
(22) (a) Taft, R. W. J. Am. Chem. Soc. 1952, 74, 2729. (b) Taft, R. W. J Am. Chem. Soc. 1952, 74, 3120. (c) Taft, R.
W. J. Am. Chem. Soc. 1953, 75, 4538. (d) Hansch, C.; Leo, A.; Hoekman D. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology; American Chemical Society, Washington DC, 1995.
(22) (a) Clavier, H.; Nolan, S.P. Chem. Commun. 2010, 46, 841. (b) Falivene, L.; Credendino, R.; Poater, A.; Petta,
A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. Organometallics, 2016, 35 (13), 2286.
(24) (a) Harper, K. C.; Sigman, M. S. Science 2011, 333 (6051), 1875. (b) Harper, K. C.; Bess, E. N.; Sigman, M. S.
Nat Chem 2012, 4, 366. (c) Milo, A.; Neel, A. J.; Toste, F. D.; Sigman, M. S. Science 2015, 347, 737. (d) Mougel,
V.; Santiago, C. B.; Zhizhko, P. A.; Bess, E. N.; Varga, J.; Frater, G.; Sigman, M. S.; Coperet, C. J. Am. Chem. Soc.
2015, 137, 6699. (e) Neel, A. J.; Milo, A.; Sigman, M. S.; Toste, F. D. J. Am. Chem. Soc. 2016, 138, 3863. (f)
Niemeyer, Z. L.; Milo, A.; Hickey, D. P.; Sigman, M. S. Nat Chem 2016, 8, 610. (g) Santiago, C. B.; Milo, A.; Sigman, M. S. J. Am. Chem. Soc. 2016, 138, 13424. (h) Sigman, M. S.; Harper, K. C.; Bess, E. N.; Milo, A. Acc.
Chem. Res. 2016, 49, 1292. (i) Orlandi, M.; Coelho, J. A. S.; Hilton, M. J.; Toste, F. D.; Sigman, M. S. J. Am. Chem. Soc. 2017, 139, 6803.
(25) (a) Piou, T.; Romanov-Michailidis, F.; Romanova-Michaelides, M.; Jackson, K. E.; Semakul, N.; Taggart, T. D.; Newell, B. S.; Rithner, C. D.; Paton, R. S.; Rovis, T. J. Am. Chem. Soc. 2017, 139, 1296-13 10. (b) Wu, K.;
Doyle, A. G. Nat. Chem. 2017, doi:10.1038/nchem.2741.
(26) The model determines the potential for oxidation at each position and does not provide stereochemical
information. For illustrative purposes, the alcohol stereochemistry of the predicted products is depicted consistent with prior experimental observations, see reference 11.
(27) For substrates (-)-1.12 and (+)-1.15, each diketopiperazine-methylene C-H bond was considered
independently, and parameters were collected from the starting material. Additionally, parameters were collected for partially oxidized hemiaminal intermediates in which the C15 position was oxidized cis or trans with respect to the CII methine. These structures were used to probe reactivity of the methylene C-H bonds and did not display a significant difference in modeling or prediction.
Experimental Section
General Procedures. Unless otherwise stated, all reactions were performed under an argon atmosphere, either in oven-dried or flame-dried round bottom flasks, or modified Schlenk (Kjeldahl shape) flasks. The flasks were fitted with rubber septa and reactions were conducted under a positive pressure of argon. Stainless steel syringes or cannulae were used to transfer air- and moisture-sensitive liquids. Flash column chromatography was performed as described by Still et al. using silica gel (60 A pore size, 40-63 pm, 4-6% H20 content).1 Analytical thin-layer chromatography was
performed using glass plates pre-coated with 0.25 mm 230-400 mesh silica gel impregnated with a fluorescent indicator (254 nm). Thin layer chromatography plates were visualized by exposure to ultraviolet light and/or an aqueous solution of ceric ammonium molybdate (CAM) followed by heating (<1 min) on a hot plate (~250 'C). Organic solutions were concentrated on rotary evaporators at -2 torr (house vacuum) at 25-35 'C, then at -0.5 torr (vacuum pump) unless otherwise indicated.
Materials. Commercial reagents and solvents were used as received with the following exceptions: dichloromethane, acetonitrile, tetrahydrofuran, methanol, NN-dimethylformamide and triethylamine were purchased from J. T. Baker (Cycletainer TM) and were purified by the method of Grubbs et al. under positive argon pressure.2 Permanganate oxidant was stored at 0 0C and remained in good condition for a month. All amino acid derivatives were purchased from Chem-Impex International. All other solvents and chemicals were purchased from either Sigma-Aldrich, Strem Chemicals, or Alfa Aesar-Johnson Matthey.
Instrumentation. Nuclear magnetic resonance (H and 13C NMR) spectra were recorded with Varian
inverse probe INOVA-500, Varian INOVA-500, and Bruker AVANCE III 400 spectrometers, and are reported in parts per million on the 6 scale. 'H NMR are referenced from the residual protium in the NMR solvent (Chloroform-d: 6 7.26 (CHCl3), Acetone-d: 6 2.05 (Acetone-d)). Data are
reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet, q = quadruplet, m
= multiplet), coupling constant(s) in Hertz (Hz), integration, assignment]. 13C NMR are referenced
from the carbon resonances of the solvent (CDCl 3: 6 77.23, Acetone-d: 6 29.92). Data are
reported as follows: chemical shift (assignment). Infrared data (IR) were obtained with a Perkin-Elmer 2000 FTIR, and are reported as follows: frequency of absorption (cm-'), intensity of absorption (s = strong, m = medium, w = weak, br = broad). Optical rotations were measured on a Jasco- 1010 polarimeter. We are grateful to Dr. Li Li for obtaining the mass spectrometric data at the Department of Chemistry's Instrumentation Facility, Massachusetts Institute of Technology. High-resolution mass spectra (HRMS) were recorded on a Bruker Daltonics APEXIV 4.7 Tesla FTICR-MS using an electrospray (ESI) ionization source.
Positional Numbering System. Numbering of the hydantoin and diketopiperazine substrates is shown below. 9 78 67 R 4 _:NN 2 R 5 %R hydantoin 0 5 R. 12 113 NR" 6 3 2 N 5
I
10 16 R' 7 N H 0 8 Ri' tetracyclic diketopiperazine EEE - W.-Diketopiperazine Hydroxylation Case Studies 0 Br, NH N -IMe SN HI SOPh 0 1.15a 1 -formyl benzotriazole-DMAP, DCM 23 'C 53% 0 H Br, NCHO N Me SOPh 0 (+)-1.15
N-formyl Tetracyclic Diketopiperazine (+)-1.15:
A sample of 1-formylbenzotriazole (242 mg, 1.98 mmol, 3.00 equiv) was added in one portion as a solid to a solution of diketopiperazine 1.15a3 (315 mg, 661 ptmol, 1 equiv) and
4-dimethylaminopyridine (242 mg, 1.98 mmol, 3.00 equiv) in dichloromethane (10 mL) under an argon atmosphere. After 24 h, the reaction mixture was diluted in dichloromethane (15 mL) and washed with a saturated aqueous solution of ammonium chloride (1 x 30 mL). The aqueous phase was extracted with dichloromethane (2 x 20 mL) and the combined organic layers were dried over
anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The crude yellow oil was purified via flash column chromatography (eluent: 5% acetone in dichloromethane) to yield N-formyl diketopiperazine (+)-1.15 (176 mg, 349 ptmol, 52.8%) as a white solid.
'H NMR (500 MHz, CDCl3, 25 'C):
13C NMR (125 MHz, CDC 3, 25 'C):
FTIR (thin film) cm-1:
6 9.27 (s, lH, N14CHO), 7.88 (d, J= 7.9 Hz, 2H, SO2Ph-o-H), 7.63 (d, J= 8.2 Hz, 1H, C8H), 7.54 (t, J= 7.4 Hz, I H, SO2Ph-p-H), 7.42 (t, J= 7.8 Hz, 2H, SO2Ph-m-H), 7.40 - 7.35 (m, 2H, C5H, C7H), 7.22 (t, J = 7.6 Hz, I H, C6H), 6.51 (s, 1H, C2H), 4.87 (q, J= 7.1 Hz, 1H, C1sH), 4.10 (dd, J= 10.6, 6.0 Hz, IH, C iH), 3.30 (dd, J= 13.2, 6.0 Hz, 1H, C12Ha), 2.99 (dd, J= 13.2, 10.7 Hz, IH, C12Hh), 1.38 (d, J= 7.1 Hz, 3H, Ci5Me). 6 167.4 (C13), 164.1 (C16), 160.0 (N14CHO), 141.0 (C9), 138.1 (SO2Ph-ipso-C), 133.9 (SO2Ph-p-CH), 132.5 (C4), 131.5 (Cs), 129.2 (SO2Ph-m-CH), 128.0 (SO2Ph-o-CH), 126.6 (C6), 124.7 (C7), 117.9 (Cs), 85.7 (C2), 58.5 (C11), 57.7 (C3), 52.3 (Cis), 46.3 (C12), 18.3 (Ci5Me).
1731 (m), 1699 (s), 1398 (m), 1367 (m), 1265 (w), 1221
0 H Br,, NCHO N Me N "IHN SO2Ph 0 (+)-1.15 0 HO PY2AgMnO4 Br, - NCHO DCM, 23 *C - y N Me 11% SO2Ph 0 (+)-1.16
N-formyl Tetracyclic alcohol (+)-1.16:
Bis(pyridine)silver(I) permanganate (137 mg, 360 pmol, 3.00 equiv) was added in one portion as a solid to a solution of N-formyl diketopiperazine (+)-1.15 (50.0 mg, 99.1 jimol, 1 equiv) in dichloromethane (1.2 mL) under an argon atmosphere resulting in a dark purple solution. After 2 h, the brown/purple reaction mixture was diluted in ethyl acetate (20 mL) and washed with a saturated aqueous solution of sodium bisulfite (25 mL) and the aqueous layer was extracted with ethyl acetate
(2 x 15 mL). The organic layers were combined and washed with a saturated aqueous solution of
ammonium chloride (1 x 20 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure to yield a crude white solid. The crude solid was purified via flash column chromatography on silica gel (eluent: 3-*5% acetone in dichloromethane) to yield N-formyl-tetracyclic alcohol (+)-1.16 (5.6 mg, 10.8 ptmol, 10.9%) as a white solid, and separately the recovered diketopiperazine (+)-1.15 (20.5 mg, 40.6 ptmol, 41.1%).
'H NMR (500 MHz, CDCl3, 25 'C):
13C NMR (125 MHz, CDCl3, 25 'C):
FTIR (thin film) cm-':
HRMS (DART) (m/z): 6 9.29 (s, 1H, N14CHO), 7.94-7.85 (m, 2H, SO2Ph-o-H), 7.66 (d, J= 8.3 Hz, I H, CsSO2Ph-o-H), 7.55 (t, J= 7.5 Hz, I H, SO2Ph-p-H), 7.50-7.37 (m, 4H, SO2Ph-m-H, C5H, C7H), 7.26 (d, J= 14.9 Hz, 1H, C6H), 6.60 (s, I H, C2H), 4.86 (q, J= 7.1 Hz, I H, C15H), 3.35 (d, J= 14.5 Hz, I H, C12Ha), 3.28 (d, J= 14.5 Hz, 1H, C12Hb), 2.72 (s, I H, C I OH), 1.51 (d, J = 7.1 Hz, 3H,C CiMe). 6 166.2 (C13), 165.5 (C16), 160.4 (N14CHO), 140.3 (C9), 138.6 (SO2Ph-ipso-C), 134.8 (C4), 134.0 (SO2Ph-p-CH), 131.6 (C), 129.2 (SO2Ph-m-CH), 128.1 (SO2 Ph-o-CH), 126.8 (C6), 124.4 (C7), 118.8 (C8), 88.1 (Cii), 87.0 (C2), 56.5 (C3), 53.4 (C12), 53.2 (Cis), 19.1 (CI5Me). 3247 (br-w), 1730 (s), 1717 (s), 1675 (s), 1409 (m), 1368 (s), 1242 (m),, 1171 (s), 913 (w), 758 (w), 727 (m). calc'd for C2H22BrN406S, [M+NH4]': 537.0438, found: 537.0449.
HO 0 H MeH NMe HN 0 1.17a AcO H0 Ac20, DMAP M Ne DCM, 23*C AcN 0 S070 (+)-1.17 Diketopiverzine (+)-1.17:
Acetic anhydride (343 ptL, 4.36 mmol, 3.00 equiv) was added dropwise over 30 seconds to a fine suspension of diketopiperazine 1.17a (250 mg, 1.45 mmol, 1 equiv) and 4-dimethylaminopyridine (371 mg, 3.63 mmol, 2.50 equiv) in dichloromethane (15 mL). After 2.5 h, the reaction mixture was diluted in dichloromethane (20 mL) and washed with a saturated aqueous solution of sodium bicarbonate (1 x 50 mL). The aqueous phase was extracted with ethyl acetate (2 x
25 mL) and the combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The crude yellow oil was purified via flash column chromatography (eluent: 20% acetone in dichloromethane) to afford diketopiperazine (+)-1.17 (289 mg, 1.13 mmol, 77.8%) as a white solid.
'H NMR (500 MHz, CDCl3, 25 'C):
3C NMR (125 MHz, CDCl
3, 25 'C):
FTIR (thin film) cm-1 :
HRMS (DART) (m/z):
[X]D 23 :
6 5.27 (qd, J= 6.4, 4.2 Hz, 1H, CHOAc), 5.19 (d, J
4.3 Hz, 1H, CHC=O), 4.26 (d, J= 18.4 Hz, 1H, CH2C=O), 3.92 (d, J= 18.4 Hz, IH, COCH2), 2.90 (s,
3H, NCH3), 2.48 (s, 3H, NCOCH3), 1.93 (s, 3H, CHOCOCH3), 1.18 (d, J = 6.5 Hz, 3H, CHCH3). 6 170.8 (NCOMe), 169.1 (CHOCOMe), 167.4
(AcNC=O), 164.9 (MeNC=O), 72.1 (CHOCOMe), 58.9 (CHC=O), 53.5 (CH2), 33.4 (NCH3), 26.5 (NCOCH3), 21.1 (CHOCOCH3), 17.4 (H3CCHOAc). 2989 (w), 1742 (s), 1713 (s), 1674 (s), 1369 (s), 1218 (s), 1084 (w), 1041 (w), 911 (m), 784 (w), 726 (s). calc'd for C1 H,7N205, [M+H]*: 257.1132, found: 257.1114. +191 (c = 0.70, CHCl3).
AcO 0 M H Py2AgMnO4 Me W~e AcN DCM, 23 *C 0 44% (+)-1.17 AcO 0 H Me NMe AcN 0 0 (+)-1.18 Triketopiperzine (+)-1.18:
Bis(pyridine)silver(I) permanganate (449 mg, 1.17 mmol, 3.00 equiv) was added in one portion as a solid to a solution of diketopiperazine (+)-1.17 (100 mg, 390 Imol, I equiv) in dichloromethane (5.0 mL) under an argon atmosphere resulting in a dark purple solution. After 2 h, the brown/purple reaction mixture was diluted in ethyl acetate (20 mL) and washed with a saturated aqueous solution of sodium bisulfite (50 mL). The aqueous layer was extracted with ethyl acetate (2
x 25 mL). The organic layers were combined and washed with a saturated aqueous solution of ammonium chloride (1 x 50 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure to yield a crude white solid. The crude solid was purified via flash column chromatography on silica gel (eluent: 5% acetone in dichloromethane) to yield triketopiperazine (+)-1.18 (46.4 mg, 172 pmol, 44.0%) as a white solid.
'H NMR (500 MHz, CDC13, 25 'C):
3C NMR (125 MHz, CDCl3, 25 'C):
FTIR (thin film) cm-1:
HRMS (DART) (m/z): [a]D2 3: TLC (5% acetone in dichloromethane), Rf. 6 5.58 (d, J= 1.7 Hz, IH, CHC=O), 5.31 (qd, J= 6.5, 1.8 Hz, IH, CHOAc), 3.21 (s, 3H, NCH3), 2.64 (s, 3H, NCOCH3), 1.91 (s, 3H, CHOCOCH3), 1.23 (d, J= 6.6 Hz, 3H, CHCH3). 5 171.0 (NCOMe), 168.8 (CHOCOMe), 166.4 (MeNC=O), 157.4 (MeNC=O(C=O)), 154.4
(AcNC=O(C=O)), 72.0 (CHOCOMe), 59.2 (CHC=O), 27.8 (NCH3), 26.4 (NCOCH3), 20.7 (CHOCOCH3), 17.8 (H3CCHOAc). 1741 (m), 1693 (s), 1356 (m), 1325 (m), 1211 (s), 1041 (w), 910 (m), 802 (w), 729 (s). calc'd for C1 H18N306, [M+NH4]': 288.1196, found: 288.1173. +56 (c = 0.39, CHCI3). 0.62 (UV, CAM).
Hydantoin Synthesis
1. PhNCO 0 NEt3, MeCN, 23 *C
Me OMe 2. HCI (aq), MeOH M
o
NH2 *HCI 100OIC Me HN==
S1.26 94% 1.26a
Representative procedure-A for synthesis of hydantoin substrates. Hydantoin 1.26a:
Triethylamine (5.50 mL, 39.4 mmol, 1.10 equiv) was added to a solution of L-alanine methyl ester hydrogen chloride S1.26 (5.00 g, 35.8 mmol, 1 equiv) in acetonitrile (300 mL) under an argon atmosphere. Phenyl isocyanate (4.10 mL, 37.6 mmol, 1.05 equiv) was added dropwise via syringe over 3 min to the resulting solution. After 3 h, saturated aqueous sodium chloride solution (150 mL) was added, the layers were separated, and the aqueous layer was extracted with ethyl acetate (3 x 100
mL). The combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting light yellow foam was dissolved in methanol (100 mL) and aqueous hydrogen chloride solution (6 N, 100 mL) and heated to 100 'C. After 3 h, the reaction mixture was allowed to cool to ambient temperature and diluted with deionized water (100 mL). The reaction mixture was extracted with ethyl acetate (4 x 200 mL) and the combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure to yield a white solid. The crude product after workup is of high enough purity for use in the subsequent step. For analytical purposes, the crude solid was purified via flash column chromatography on silica gel (eluent: 10% acetone in dichloromethane) to yield hydantoin 1.26a (6.40 g, 33.6 mmol, 94.0% over two steps) as a white solid.
'H NMR (400 MHz, CDCl3, 25 'C): 6 7.53-7.44 (m, 2H, C8H), 7.43-7.35 (m, 3H, C7H, C9H), 7.01 (s, 1H, NIH), 4.19 (qd, J= 6.9, 1.3 Hz, I H, CsH), 1.51 (d, J= 7.0 Hz, 3H, C5Me).
13C NMR (100 MHz, CDCl3, 25 *C): 6 173.8 (C4), 157.0 (C2), 131.6 (C6), 129.3 (C7), 128.5 (C9), 126.4 (C8), 53.1 (C5), 17.9 (C5Me).
FTIR (thin film) cm-1: 3212 (br-m), 3108 (w), 1769 (w), 1704 (s), 1501 (m), 1419 (m), 1333 (m), 1182 (m).
Me 0 Me OMe NH2 oHCI S1.29 1. PhNCO NEt3, MeCN, 23 *C
2. HCI (aq), MeOH
100 C 94% ON Me N H Me 1.29a Hydantoin 1.29a:
The hydantoin 1.29a was prepared following the representative procedure-A using L-valine methyl ester hydrogen chloride S1.29 (3.00 g, 17.9 mmol, 1 equiv). The crude product after workup was purified via flash column chromatography on silica gel (eluent: 10% acetone in dichloromethane) to yield hydantoin 1.29a (3.68 g, 16.9 mmol, 94.2% over two steps) as a white solid.
'H NMR (400 MHz, CDCl3, 25 'C):
"C NMR (100 MHz, CDCL3, 25 'C):
FTIR (thin film) cm-1:
HRMS (DART) (m/z): TLC (5% acetone in dichloromethane), Rf: 5 7.53-7.44 (m, 2H, C8H), 7.40-7.33 (m, 3H, C7H, C9H), 6.19 (s, 1H, NiH), 4.08 (dd, J= 3.7, 1.3 Hz, I H, C5H), 2.33 (hd, J= 6.9, 3.7 Hz, 1H, C5CH), 1.10 (d, J 7.0 Hz, 3H, C5CHMea), 1.02 (d, J= 6.8 Hz, 3H, C5CHMeb). 6 172.5 (C4), 157.1 (C2), 131.6 (C6), 129.3 (C7), 128.5 (C9), 126.4 (Cs), 62.3 (C5), 30.8 (CsCH), 18.9 (C5CHCaH3), 16.2 (C5CHCbH3). 3295 (br-m), 1776 (w), 1709 (s), 1500 (w), 1412 (m), 1172 (w), 804 (w). calc'd for C12H15N202, [M+H]+: 219.1128, found: 219.1130. 0.41 (UV, CAM).
O e NH2 *HCI
S1.32
1. PhNCO
NEt3, MeCN, 23 'C, N
2. HCI (aq), MeOH N = O
100C Ic N
H
90% 1.32a
Hydantoin 1.32a:
The hydantoin 1.32a was prepared following the representative procedure-A using L-phenylglycine methyl ester hydrogen chloride S1.32 (2.00 g, 9.92 mmol, 1 equiv). The crude product after workup was purified via flash column chromatography on silica gel (eluent: 10% acetone in dichloromethane) to yield hydantoin 1.32a (2.25 g, 8.92 mmol, 89.9% over two steps) as a white solid. 'H NMR (400 MHz, CDCl3, 25 'C):
13C NMR (100 MHz, CDC
3, 25 *C):
FTIR (thin film) cm-1:
HRMS (DART) (m/z):
TLC (40% ethyl acetate in hexanes), Rf:
8 7.63-7.32 (m, 10H, ArH), 6.53 (s, 1H, N1H), 5.19 (d, J= 1.3 Hz, 1H, CsH). 6 170.8 (C4), 156.6 (C2), 134.2, 131.4, 129.3, 129.3, 129.2, 128.4, 126.6, 126.2, 60.6 (C5). 3304 (br-w), 1779 (w), 1714 (s), 1496 (w), 1411 (m), 1173 (w), 704 (m). calc'd for C15H13N202, [M+H]+: 253.0972, found: 253.0969. 0.30 (UV, CAM).
0 OH NH SOC12 MeOH reflux SI.35 0 OMe NH eHCI SI.35a 1.PhNCO NEt3, MeCN, 23 *C
2. HCI (aq), MeOH
100 *C 60% 0 H N> N 1.35a Hydantoin 1.35a:
L-Proline methyl ester hydrogen chloride S1.35a was prepared from L-proline S1.35 (2.00 g, 17.4 mmol, I equiv) following literature procedure.4 The hydantoin 1.35a was prepared following the representative procedure-A using L-proline methyl ester hydrochloride S1.35a. The crude product after workup was purified via flash column chromatography on silica gel (eluent: 50% ethyl acetate in hexanes) to yield hydantoin 1.35a (2.25 g, 10.4 mmol, 59.8%, over three steps) as a white solid. 'H NMR (400 MHz, CDCl3, 25 "C):
3C NMR (100 MHz, CDCl
3, 25 'C):
FTIR (thin film) cm-1:
HRMS (DART) (m/z):
TLC (50% ethyl acetate in hexanes), Rf:
6 7.49-7.42 (m, 2H, C8H), 7.42-7.37 (m, 3H, C7H, C9H), 4.24 (dd, J= 9.3, 7.5 Hz, I H, CsH), 3.79 (dt, J= 11.3, 7.7 Hz, 1H, Ni CHa), 3.34 (ddd, J= 11.3, 8.3, 4.6 Hz, 1H, NiCHb), 2.35 (dtd, J= 12.4, 7.2, 3.6 Hz, 1H, NICH2CHa), 2.27-2.04 (m, 2H, Ni CH2CHb, C5CHa),
1.85 (dtd, J= 12.6, 9.4, 8.3 Hz, 1H, C5CHb). 6 172.8 (C4), 159.5 (C2), 132.0 (C6), 129.3 (C7), 128.3 (C8), 126.1 (C9), 63.4 (C), 46.0 (N1CH2), 28.0 (N1CH2CH2), 27.1 (CsCH2). 2905 (w), 1775 (w), 1702 (s), 1598 (w), 1495 (w), 1402 (m), 1123 (m), 744 (w). calc'd for C12H,3N202, [M+H]*: 217.0972, found: 217.0976. 0.35 (UV, CAM).
0 Me OH Me NHBoc SOC12 MeOH reflux S1.37 0 Me Y- OMe Me NH2.HCI SI.37a 1. PhNCO NEt3, MeCN, 23 *C
2. HCI (aq), MeOH
100 *C 89% Me >2NNO N Me H 1.37a Hydantoin 1.37a:
L-Leucine methyl ester hydrogen chloride S1.37a was prepared from N-Boc-L-leucine S1.37
(8.00 g, 32.1 mmol, 1 equiv) following literature procedure.4 The hydantoin 1.37a was prepared
following the representative procedure-A using L-leucine methyl ester hydrogen chloride S1.37a. The crude product after workup was purified via flash column chromatography on silica gel (eluent: 35% ethyl acetate in hexanes) to yield hydantoin 1.37a (6.61 g, 28.5 mmol, 88.7%, over three steps) as a white solid.
'H NMR (400 MHz, CDCL3, 25 0C):
13C NMR (100 MHz, CDCl3, 25 *C):
FTIR (thin film) cm-1:
HRMS (DART) (m/z):
TLC (35% ethyl acetate in hexanes), Rf:
8 7.53-7.44 (m, 2H, C8H), 7.42-7.32 (m, 3H, C7H, C9H), 6.47 (s, 1H, N1H), 4.19 (ddd, J= 9.3, 4.0, 1.4 Hz, 1H, CsH), 2.04-1.77 (m, 2H, C5CH2), 1.77-1.54 (m, 1H, CSCH2CH), 1.00 (d, J= 6.4 Hz, 3H, CsCH2CHMea), 0.98 (d, J= 6.3 Hz, 3H, C5CH2CHMeb). 6 173.5 (C4), 156.8 (C2), 131.7 (C6), 129.3 (C), 128.4 (C9), 126.3 (C7), 55.9 (C5), 41.2 (C5CH2), 25.3 (C5CH2CH), 23.2 (C5CH2CHCaH3), 21.9 (C5CH2CHCbH3). 3263 (br-w), 3112 (w), 1774 (w), 1727 (s), 1494 (w), 1430 (m), 1397 (m), 765 (w), 749 (w). calc'd for C13H17N202, [M+H]*: 233.1285, found: 233.1286. 0.34 (UV, CAM).