il t{s0,oS
Rdpublique Alg6rienne Ddmocratique
et
populaire
Ministdre
de
I'Enseignement
Sup6rieur
Universit6
de
Guelma
Facult6 des Math6matiques et de I'Informatique et des Sciences de Ia
Matiire
D6partement des Sciences de laMatiire
Mdmoire
de
fin
d'6tude
26^
annde
master
spdcialitd
:
PHYSIQUE
DE
LA MATIERE
CONDENSEE
Prdsentd
par :
Himoura
fatma
LES
PROPRIETES OPTIQUES DU
coMPoSE
Mgo
Sous
la
Direction
de
:Pr. Dr.S.Djeroud
,
sqq&fr.qEqfr-e.EEEEqqEq-qqq
Eq
qR
Eq
$
lmrorciclntlll
g
-S
-5
1
-:
---
r^^-
T\TETT
,^rrri
acf
q
IJn
trds grand
merci
au
bon
DIEU
qui
est
q
E
.
isent
avec
moi
dans le
bonheur
s
-s
toulours
prgsgnT Avgg
[IUr
\rd.rlD
rw
Lr\,'uu\rr'^
q
-5
dans le
Pire'
q
-\.re
E
qq*
-s...,,1,.{
-S
Je
tiens
d
remercier
vivement
notre
q
-s
\
-\
encadreuse
M*".Dierroud
Professeur
d
$
S
l'universitd
de
Guelma
d'avoir
propos6, q
R
dirigd
et
suivi
ce
travail'
q
E
urrlBg
gL
DI."IIY
l \l\'/
Ul(*Y
f'lllr
5
$R
-\
S
Nous
remercions
les
membres
de
jury
$
EF'
,avoir
acceot6
d,e>
'
t d'dvaluer
R
-q
d'avoir
accePtd
d'examiner
e'
-q
notre
travail.
q
-s
llotrg
LravAil'
R
-58
5
.
, 7 -,,-
{
-S
]rTous
exprimons
dgalement
nos
q
S ,---,.,lll;;;;.; '
'l
rt
q
_R
gratitudes
e
tous
les
enseignants
qul
ol
-$
{-
collabord
}r
notre
formation
aussi
bien
R
-s
vL''oL'::*^;;,;;;;;;.
q
-\
Primaire
qu'universitaire'
-$PIrru4IIvYt't,}lrrYt/l-\,Ir/r.}lrv'ft
E
\q\\\q
\
q\\qE
q
q\ \
\
q
Eq
q
E
q
q
$qqqq\\\q\\\\\q\\ q\\q\
E
\'
-t'\
\-\\\\\\\\\\\\\\\\\qq*
q
.$
Deil-"aGG
$.
-q.
q
$
Avec tout mon
amour,
je
dddie
ce
mdmoire
d
mon
R
$
papasebti
et
ma maman
Darila
qui n,ont
pas
cessd
de
R
$
me
soutenir
durant
toute
ma
scol
afit|,ils
m,ont
6clair6, $
-
$
guidd
et aidd
d
gravir
les dchelons,
pour
cela
je
leur
dddie $
-$
ce
ftavait
on leur
disanr
$
q
E
"eue
dieu
vous
b6nisse,
je
nroubrierai
jamais
ce
que
$
$
-g"=
vous
avez
fait pour
moirr
q
R
A
T'
E
-q
A
mes chdres
s*urs
sihamo
widade
,
zahiraet
rayonne
$.
-$
A
mes
chers frdres
Riad,
billet
q
{5
$
Et
d
toute
ma
famille
et
mes
amies.
$
$
Himou
raratma
*
-qi
q
lq
F\
-qQ.
E
_ER
-gD
t
-E5
Ft
-r:
rftfrnrfthss&'6noar.a,6-a^a-a
E
_\q\q\\\\\qqq\qqqqqqqqqqqqqq
R6sum6
Le
but de cetravail
est d'dtudier les propri6t6s structurelles, 6lectroniques et optiques des oxydes m.talriquesde deuxidme groupe
(alcalinoterreux)
, l,oxyde demagn6sium de structure
rocksalt,
par ram6thode des ondes planes augmentdeslinearis6es
(FP-LAPw)'dans
le"ud"
d"
la th6orie de la fonctionnelle de la densit6 DFT, impl6ment6e dans le code elk.Nous avons
utilisd l'approximation
de ladensit.locale
LDA
et
de L'approximation du gradient gdn6ralis6GGA pour calculer les propri6t6s structurales et les propri6t6s dlectronique (les structure de bandes, nature
du gap et les densit6s d'6tats totales et partielles).
A
l'aide
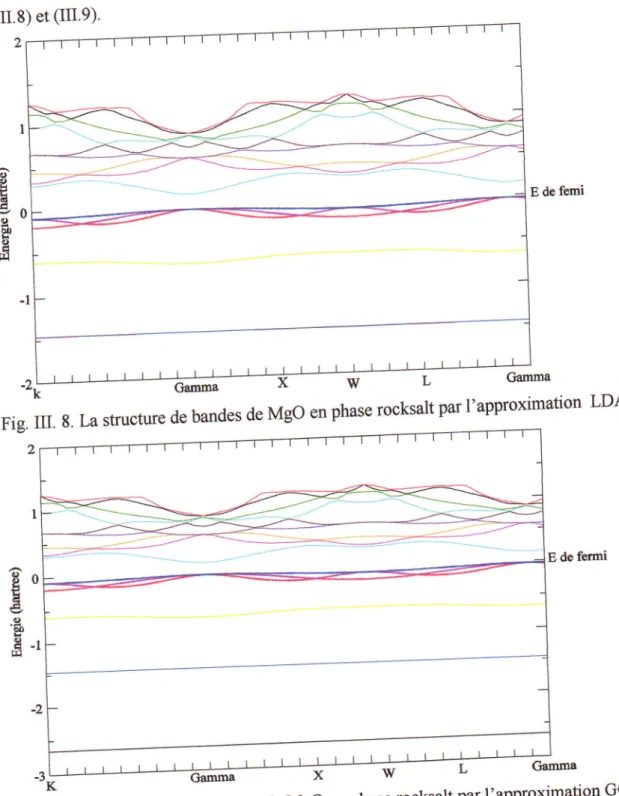
de la m6thode du FP-LAPW,les calculs de la structure de bandes ont montr6 que MgO possdde un gap
direct
au pointf.
Mise d part ces propri6t6s, une grande partie
de
notre
6tude a 6t6 consacr6e l,6tudedes propri6t6s optiques du
Summary
The goal of this work is to
study the structurar, erectronic and opticar properties
of
metallic oxide' magnesium oxideof
structure rocksart, by the methodofincreased full-potentiar rinearised
augmented;;""
wave
(Fp-LApIV).in
the theoryofthe
functional carcurus of density
DFi,
irrior"**rr,"d
in the code elk.we
used the approximation oflocar density
LDA
and
theapproximation of the gradient generarized GGA to
carcurate the sFucturar
*J.rrroonic
properties (the structureof
bands, natureofthe
gap and densities
"rrr"io
r"*r
and partiar).ff:-;T"ff*"i:#?:l,l;lAPw,
structure of bands carcurated showed thatMso
ln
)ommalre
clo
DOmmatre
Introduction gen6raleChapitre
I
Thdorie
de lafonctionnelle
dela
densit6(DFT)
Introduction"'
"'
...3
I. l.Equation de Schrodinger.. ...3 I.2.L'Hamiltonien
total ducristal...
...3 L3
Approximation
de Born Oppenheimer (l,approximationadiabatique)... ... . .. ... ....4
I.4.L'approximation de Hartree- fock..
...5
L5.Th6orie de la fonctionnelle de la
densitd(DFT).
...7
I.5.1 Les thdordmes de Hohenberg et
kohn.
... ...7 I.5.2. Les dquations de
kohn_sfurm...
...g
I.6.L'approximation
de la densitd locale(I_DA)..
.... ...g
1.7.L'approximationdugradientgdndralisd(GGA)...
...10
I.8.Rdsolution des dquations de
Kohn_Sham..
. ... ....
l0
I.9.1'auto-cohdrence dans les calculs... ..
References
"'""'11
Chapitre
II
la m6thodedes ondes planes augment6es lin6aris6es
(rP-LAPW)
Introduction"'"'
...13
II.l.
La mdthode des ondes planesaugmentdes(Apw)...
...14
rr'2'
la mdthode des ondes pranes augmentdes rindarisdes(Fp-LAplv)...
.
... .. 16II.2.l.
Principe de la methodeFp_LApW...
.
. ... . 16
II.3.
Les rdles des 6nergies de lindarisation(81)..
...17
II.4.
construction des fonctionsradiales..
Sommaire
II.4.1. Les fonctions radiales non relativistes. II.4.2. Les fonctions radiales relativistes II.5. Rdsolution de l,dquation de poisson
Il.6.Amdlioration
de la mdthodeFp-LApW
II.6. 1.Les fenCtres d'dnergie multiples Il.6.2.Ddveloppement en orbital local
II.T.Trcitement
des e{fets de spin_orbite18
l9
24 24 RdferencesChapitre
III
III. 1.D6tails de calculs.
Rdsultats et discussion
26
III.
l.l.Test
de convergence.27 III. 2.Propri6tds structurales
III. 3 Propridtds dlectroniques III.3.
L
structure de bandesIIL3.2.
La densitd d,dtars (DOS)32
34
III.4.
Proprietds optiques.III.4.
l.Fonction
didlectrique4l
41
45 R6ferences
Urlgles
figures
Liste
des
figures
figures Titres f)rr:o pages Fig. I.1 Fnfpn,tiol
us 14 rcs{rruuon ces equattons Kohn-Sham
t2
Fig. II.1
urr reseau carre A deux dimensions:
(a) potentiel total, et (b) potentiel
muffin-tin
I
t3
Fig.
IL2
rvwucrvr\\ tYll%
E'va*-l^ .|^ f^
lrrtl-lln
2)(MrJ
T4
Fig.
II.3
ryrw \|v rsueltsilvoc un elat seml-CGur
(t-24
Itig.IILl
,Ar.xr[ (lc rvrguA^ t>!-- - -- '
I 27
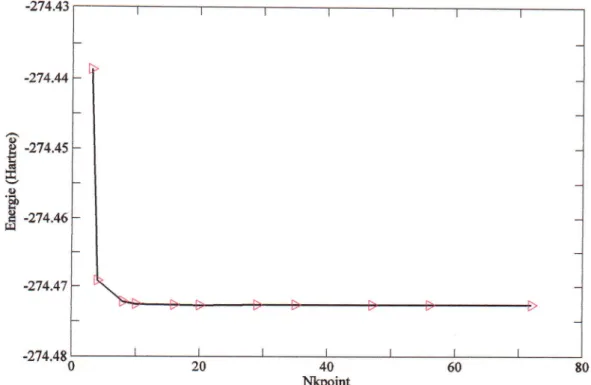
ftig.III.2
v,
ysrrelrv'
uu 1E'.'rgr'
roEile en tonctlon des nombre de points
k
(LDA)
1 a
:;::;; ;: ; ;;;l.
28
Fig.III.3
se vqrra'w'
uE r Elcrglororale en lonction des nombre de
points k
(cGA)
28Fig.III.4
vrgrsrorare en tonctron des valeurs de
param6fre Rvn*K*u,(LDA)
To A I
29
Fig.III.5
rrB,rE roriile en lonctron des valeurs de
parametre
Rrwr*K*u*(GGA)
T
)^
30
Fig.III.6
l'approximation
rniss
torale en ronctlon du volume pourI
3l
Fig.III.7
ue
vs,r@Lrv' tre relcrgle
rotale en tonctlon du volume pour
l'approximation
GGA3l
Fig.III.8
e .'ts uiluscsoe
vlgu
en phase rocksalt(LDA)
I
Fig.III.9
u4uugr ucrvrg\., en pnase rocksalt
(GGA)
f\a*ci+A .t
JJ
Fig.IIl
l0
svuprlv
r.r t'r.rlD uJ\-rDJIolale
cerocksalt par l'approximation
LDA
MgU
dansla
structure 35 Fig.IIL 11vvr'rrv
Lr sr<rrsur\JD/ prmleueOe
Mg
(3S) dans la strUctufe rocksalt par I'approximationLDA
T\^-^:+1,
36
Fig.IIL
l2
vvl,Drreu
{rr4Qr(1^-rDl parTrelle deMg
(2p) dans la structure rocksalt par I'approximationLDA
36Fig.III.13
svuilLv
''| sr.rlD trr\,rD,, paruelle Oe
U
(ZS) danSla
StruCtUfeListe
des
fi
Fig.III.l4
Densitdd'6
rocksgllpgr
L'approxlmauonLDA
Fig.III.15
-Densitd O'Ugrocksalt par I'approximation GGA"
Fig.III.l6
Densitd d'drocksalt par I'approximation
LDA
Fig.III.l7
Densiti
d'dturocksalt par
I'approxmatton
GGAFig.III.18
DensitdO'rot
rocksalt par I'approximation GGA Fig.IIL
lg
Densitd0'eorr(t
rocksalt par I'approximation GGA
Fig.IIL20
La partiet*uttn
lg
structure rocksalt deI'approxim"ri""
fOa
Fig.III.2l
La partiet
rU.
*ructufe
, rocksalt del,approximation-'ipa
Fig.III.22
Lapartie
m
lqstructure rocksalt de l,approximaliJn-
GGA
Fig.III.23
La partiet*
structure
rocksalt de I'approximationListe
des
tableaux
Liste
des
tableaux
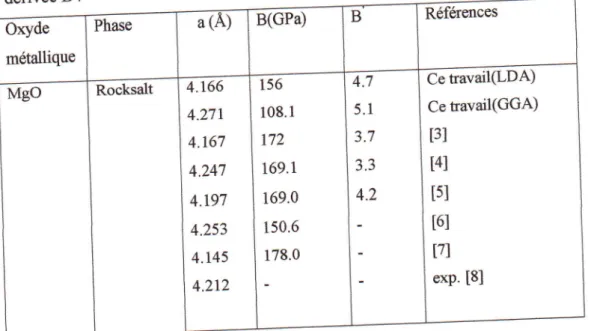
I.
p.u*dttt
B et sa ddrivde B'.
l'erreurret@
I'dnergie Or
*u
fntroduction
Notre but concerne I'etude des
diverses propridtds d savoir les proprietds dlectroniques et optiques du composd binaire
Mgo
cristallisantdans la structure rocksalt.
Pour le calcul de ces propri6t6s on ufilise la mdthode
Fp-LApw
avec
I,utilisation
de la
theorie
de fonctionnelle de la densite.Le
Mgo
possede de nombreuses applications technologiques.L,anatase a un indice de rdfraction 6levd et absorbe dans le
visible
[6].ce
travail est organisd en trois chapikes avec une introductionet une conclusion.
Le
premier chapitre est consacrd d un rappel deprincipe de la fonctionnelle de la densitd
(DFT), et I'approximation
de Iadensitd locale
(LDA)
et l,approximation
dugradient gdneralise (GGA).
Dans
le
second chapitre,nous
rapperonsle
principe de
ra
mdthode des ondesplanes augmentdes lindarisdes (Fp_
LAPIV).
Le
troisidme chapitre, regroupe les rdsultats obtenus avec leurinterprdtation
pour les
propridtds structurales, €lectroniqueset
optiquesd,oxyde
de
magndsiumMgo.
une
conclusion gendrale est donnde d lafin
de cefntroduction
Rdf6rences
[1] H'Mathiew'
physique des semi-conducteurset
des composants dlectronique.Rl.
ldasson (199S).[2J Landolt-B"ornstein, Numerical
daa
andfimctional
relationships.semiconduc,ors: Physics
of
II-vI
andI-vII
compounds, semimagnetic semiconductors science and Technology ,
vol.
17,part
b, ed. byo.
hrladelung
(Berlin,
springer r 9g4).[3J p.Hohenberg and W.Kohn, phys.Rev.B
136(tg64)
564.[
]
W.Kohn and L.J. sham, phys Rev.t40Al
t33(1965). [5]
D.M.
Roessler and W.C.Walker, phys. Rev.159,
733{tg67).
16l
A
schleife, F. Fucrrs, J. Furthm"uller,Chapitre
I
Thdorie
de
la
fonctionnelle
de
la
Itr9orie
de
la
fonctionnelle
de
la
densitd
DF'T
I.
Th6orie
dela fonctionnelle
defntroduction
la densird(DFT)
Les d6veloppements du 20i6-" siecle dans tous les domaines scientifiques surtout ra mecanique quantique ou la reconnaissance presque de toutes pour les propri6tds
des matdriaux et peuvent €tre dtudides par
des outils de calcul convenables. Mais, Ies electrons et les noyaux qui composent les matdriaux constituent un systdme
d plusieurs corps fortement interagissant,
il
est pratiquement impossible de rdsoudreun systeme d'dquations d plusieurs particules.
La solution possible de ce probldme est de remplacer le systdme d'dquation d plusieurs particules par un autre systdme d,dquations
d une seule particule
et de reprdsenter toutes les interactions par un potentiel
effectif
Aprds les efforts de borne Oppenheimer[l]
et Harhee-fock [2.3J.Les proprietds physiques d'un solide sont extr€mement liees au comportement des electrons qui le constituent.
Pour
une comprdhssion fondamentale de la structure dlectronique et des propridtds des
materiaux, le ddveloppement de la thdorie de Ia fonctionnelle de la densitd
DFT
avec les approximations
LDA
et GGA djoud
un r6le trds important dans la matidre condensde
[4].
I.l.Equation
deSchriidinger
un
corpscristallin
est un ensemble de noyaux et desdrectrons en interaction. En
1926'le
physicienAutrichien
schnidingera proposd une dquation qui d6crit
toutes ces interactions la rdsolution de l'dquation de
schrtidinger
caracterisant ce systdme permettrait de ddcrire ses
propridtds quantiques et elre est donnde
par:
[5]HY:EV
(11)
oi
E:est I'dnergie totale du systdmeetv(rii,il1;
est la fonction d,onde, et
H
est I'Hamiltoniende ce systdme, pour un systdme ayantN noyaux et n dlectrons.
L2. LoHamiltonien
total
ducristal
L'dtude des structures de bandes
permet
d'interpreter plusieurs phdnomdnes physiques, qui se ddroulent dans les corps solides. Dans
Chapitre
I
Ihdorie
de
la
fonctionnelle
de fa densit6
::::::"::::*
ro1or.
de routes res interactions exisranr entre res noyaux et res:l'"*t
adopter des modeles simplifids pour
pouvoir obtenir des solutions approch.es
t5l
L'Hamiltonien total est ddfini
par:
H:T"+1o+Vu"*V*+V*
T"
reprdsente I'energie cinetique deselectrons.
T,,
l'dnergie cindtique des noyaux.V""
I'dnergie dlnteraction dlectron-dlecnon.V*
ldnergie d'interaction dlectron -noyau.V*
I'dnergie d'interaction noyau-noyau.L'hamiltonien s€crir
(r 2)
ufiz,tv,iET*o#zr Y#i*trfrv,,,*)x{
p
ZkZtzt
4neRp1Ori : m est la masse de l'6lectron. +
t;
: est la distance entre l'dlectroni
et I'electronj.
M2r: est la masse du noyau.
R11: est la distance entre les centres des noyaux k et I Zx,
Zt:
les nombres atomiques des noyauxk
et I
Les diverses mdthodes de calcul de la structure de bandes dlectroniques
des mat6riaux d I'dtat solide misss au point au cours des
dernidres ddcennies reposent sur un certain nombre d'approximations rdparties sur trois niveaux :
L3
Approximation
deBorn
oppenheimer (r'approximatiou
adiabatique)
Pour
simplifier
l'dquation de schr<idinger en considerant la grande diffdrencede 4
Cha
fndo"i*
0*
tu
fun.tl
ffe
Oqla
densitd
FT
masse entre les €lectrons et les
noyaux' Les noyaux se
ddpracent donc tres rentement par rappolt aux dlectrons probldme du solide peut €tre
divise
en
deux
composants : le mouvement des ions dans un espaceuniformdment
chargd par les dlectrons.ce
ddcouplage entre ces deux mouvements ndcessite
une
justification,
l,approximationde Born- oppenheimer I'offre, elle considdre que les ions sont immobiles
ce qui indique que la foncfion
dbnde
ddpend seulementdes coordonndes des dlectrons. Donc nous pouvons
ddfinir
un nouvel Hamiltonien,H:T"+U._"+U*-n
(r.4) L'dquation de Schrridinger s'6crit alors
:
lilP:
t-z{
fivi
-Li,,ffi+
xo,#
*
EnJy
=
Ey
Le terme d'interaction entre les noyaux que l'on
notera
E est considdrd comme'ne
constante.
Le probleme est maintenant purement electronique et ndglige les
vibrations du r6seau
;
ce qui donne d cette approximatioa re
nom adiabatique, cependant le probldme est simple que I'original mais reste toujours
difficile
d rdsoudre.L4.L'approximation
deHartree.
fock
:cette approximation consiste d supposer que chaque dlectron
se d6prace
inddpendamment dans un champ moyen
$eeparres
autre drectrons et noyaux.on
ramene donc Ie probleme
relatif
ri ungrand
nombre d'dlectrons ii un probleme ii un seuldlectron' L'hamitonien peut 6tre 6crit
comme
une sommedes hamiltoniens ddcrivant un seul
dlectron[4,6].
H:f,;Hl
on
ui:frat+ui
(ri) +vi (ri)
Tel
queui(Fi):-IrFft
0.5)
(r.6) {r.7) (r.8)Cha
Th6ori.
de
la
fon.ctimnelle
de
la
densif6
L'dnergie potentiere de
ldlectron
i
dans res champs de tous res noyaux k.
v(ii):
t/z1,#
C'est le champ
effectif
de Hartree. Le potentieleffectif
est la somme deces deux contributions
v\y(i):vh(F)
+
vn(i)
V
hLe
potentiel de Harhee.Y
(fr 1, F2. .. ... . ..,in):yl
(it)Vz,(FZ)...
... ... ...yn(in)
E:E1+8r...
...E,
{r.e)
(r.10)
VnLe
potentiel d,interaction dlectron awres noyaux.En introduisant le potentiel
effectif
dans I'dquation de schnidinger.on
trouve-ll2
vzvi(F)
+ v"rr(i)
yi?):
eiy6(i)
g.l
1) Lafonction
d'onde du systdme dlectronique a la formed'un produit de fonction d'onde des dlectrons, et
l'dnergie
de ce systdme dgale d la somme de l,dnergiede tous les dlectrons.
(r.12)
(r.13)
L'dquation (I'12)
est bien une solution de I'dquation (I.1I)
mais ne respecte pas Ie
principe
DePauli,
I'approximation de (Hartree-Fock)a 6t6 introduite pour prendre en compte le spin des dlectrons pour la rdsolution de l,dquation de schrodinger. La
difference entre l'6nergie du systdme multielectroniques
r6el, et l,dnergie obtenue dans I'approximation de Harfree comme etant celle representant le reste
des interactions dlecffoniques' L'une de ces interactions qui manque dans le moddle de Hartree est I'dchange et la correlation. L'dchange est d'origine purement quantique. C,est cet
effet qui exprime I'antisymdtrie de la fonction d'onde par rapport
a l,echange des coordonn6es de n'importe quels deux dlectrons menant A ddcrire le
systdme d
N
corps (6lectrons) parI'egalitd :
v
1ri,7o,
7b ,.. ...in):y(i1
,ib,ia.
in)
Chanitre
I
Ihilorie
de
la
fonctionnelle
de
la
densit6
(L 15)
L5.Th6orie
dela fonctionnelle
dela densite{DFT)
:
Le concept fondamental de la fonctionnelle
de la densit6 est que ldnergie d,un systdme dlectronique peut €tre exprimde en fonction de sa densitd. c,est en
fait
une idde ancienne datant principalement des travaux de Thomas
[7]
et defermi
[g].l,utilisation dela densite dlectronique comme variable fondamental pour decrire les propri6tes du systdme existe depuis les premidres approches de la
structure 6lectronique de la matidre mais elle n'a obtenu de preuve que par la ddmonstration des deux thdordmes dits de Hohenberg et kohn [9].
L5.l
Lesth6orimes
deHohenberg
etkohn
Le formalisme de la thdorie de la fonctionnelle de la densitd (DFT) est basd sur les deux thdordmes de Hohenberg et Kohn [10f.
Premidrement'
Hohenberg
et
Kohn
ont
montrds
qu'il
existe une
correspondance biunivoque entrele
potentiel
extdrieuret
la
densite dlectroniquep (r)
permettant de representer
le
premier comme
une
fonctionnelle
de
l'6tat
fondamental
de
ladeuxidme'
Par
consdquent,I'dnergie
totale
du
systdmed
l,dtat
fondamental
est dgalement une fonctionnelle unique universelle dela densitd 6lechonique, soit :
E:
E
[p(r)]
G.l6)
ce
thdordme est d la base de la theorie dela
fonctionnelle dela
densitd et expliqueI'appellation
qui
lui
a
dtd
donnde.ceci
difGre de
la
mdthode Hartree-Fock, dans laquelle l'dnergie totale du systdme est fonctionde la fonction d,onde.
une
consdquenceimmddiate
de ce
th6oreme
est
quo
la
densitd
dlectroruque ddtermine defagon unique
l'opdrateur
hamiltonien
du
systdme.
Ainsi,
enconnaissant
la
densite dlectronique, I'opdrateur hamiltonienpeut €tre ddtermin6 et d travers ce hamiltonien, les differentes propri6t6s du matdriau peuvent €tre calculees. Deuxidmement, Hohenberg et kohn ont montrds que : pour un potentiel
v"s
et un nombre d'dlectronsN
donn6s, I'dnergie totale du systdme atteint sa valeur minimaleY
r(,rn)l
Yn(in)J
Y Doit
6tre antisymdtrique. Donc, elle s'dcrit souslorsque la densitd p(r) conespond d la densitd exacte de
|dtat
fondamental p0(r)
Th6o.ie
de Ia
functionnelle
de
la
densitd
s
(pO):
mina
g)
La fonctionnelle de I'dnergie
tokle
de I'dtat fondamental s,ecritcomme suit :
s tb)
r]:
F
Ip(r)l
+lvext e)
pe)
f
r
(r.18)
ori v"'t
Reprdsente le potentielexterne agissant sur les particules et F [p
(r)]
reprdsente Iafonctionnelle universelle de Hohenberg et kohn, avec :
F
tp
(i)l
:
(4r^
+
v)./l4
(r.17)
(r.1e)
La
connaissancede
cette fonctionnelle permet
de
ddterminerl,energie totale
et
la
densitd de charge del'dtai
fondamental pour un potentiel externe donn6, en utilisantle principe
variationner.
Marheureusement,re
thdordme
de
Hohenberg
et
Kohn
nedonne
aucune indication de laforme
de F
tp
(i)l
L5.2. Les6quation
de kohn-shamkohn-sham ont dcrit la densitd dlectronique comme etant la somme des densitds des particules libres sur l'ensemble des orbitares occupdes
p:X[1f
YiG)'l
G.za)
Ils ont utilis6 le principe
variationnell
pour obtenirl'dnergie de
ldtat
fondamental et ladensitd donnant la fonctionnelle E en montrant que la
waie
densite est donn6e par rdsolution auto-compatible (self-consistent) de l'ensemble
des dquations d une particule de type schrcidinger, encore appeldes dquations de
kohn-sham qui sont donn6es
par:
tfiv'n
vion(r)+Yr*vr"(r)ly;:E
yi
(r);i:l
....N
anVH?)
=
i
f
ffid3r1d3r2
: est le potenriel de Harrree-fock.vr"(r) =
W:
est le potentiel d'6change et de correlation.vien: est le potentiel ionique qui est une fonction locale rdelre de r.
Les dquations de kohen-sham sont probablement les plus importantes de la DFT d travers lesquelles le traitement du probleme d plusieurs dlectrons en interaction. se
Cha
Th6orie
de
la
fonctionneile
de
la
densit6
rdduit d I'dtude d d'un systdme d'dlectrons inddpendants
baignant dans un potentiel
effectif
(V*:vs
*vioorv*.),
quicontient toutes les interactions possibles entre res Electrons et on peut Ie
qualifier
de local caril
ne ddpend que de r, cette mdthode est formellement
exacte mais pratiquement, le calcur de
|dnergie
d,dchange et decorrelation ndcessite
d'introduire
certaines approximations.
L6.L'approximation
dela densit6locale
(LDA)
Dans l'approximation de la
densit€ locale (Locar Density Approximation
LDA),
ir est supposd que la densitd dlectronique peutctre
traitkelocalement sous Ia forme d,un gaz d'dlectrons uniforme.ce
quirevient d poser les deux hypothdses suivantes
:
'
Les effets d'dchange-corrdlation sont dominds parla densite situee au point r.
'
La densitd p (i)
est une fonctionvariant lentement vis_d-vis de
i.
cette approximation consiste donci
consid.rerque la contributio
nde
E,,k
(41
d l'dnergie totale du systdme peut 6tre additionneede fagon cumulee
it
partirde chaque portion du gaznon uniformeconrme
s,il
dtait locarementunifome.
L'dnergie d'dchange-correration(LDA)
peut 6tre dcrite sous Ia forme:
r*!Af,p(i)l:
!
p(i)r?rot
p(i)lor'
$.22)
Dans laquelle
f
e*?Al
p (F)lrepr6sentel'.nergie
d,echange et de condration par dlectron dans un gaz d'llectrons dont la distribution
est supposde uniforme.
Apartir de
t
sllALpG)l,lepotentield'dchange-corrdlation
vigA{ilpeut6treobrenu
d'une fagon variationnelle selon l,6quation
:
VIF A
Gl:
atl p @ ekDcA Lprlll
6p(f)
Pour les systemes magndtiques, le spin
dlecfonique introduit
un degrd de liberte suppldmentaire et la
LDA
doit €tre alors dtendue drApproximation
de la Densitd Locale de
spin (LSDA
:Local
spin Density Approximation),oi
l,energie d,echange et corrdlation est fonctionnene des deux densitds de spin haut et bas
:
E*fooIpt,
pil
=
f
p?)r*!rn
[ptZ),p1(i)J
d3r
$.24)
La
LDA
suppose que la fonctionnelleer"
fp|ff)lest
purement locale. cette energie est divisee en deux termes :(r.23)
€*"
Lp(i)]
=
€,
[p(i)J
+e"
[p(i)]
Cha
Th6o.ie
de ta
foo.fi
lle
de
la
densit6
{DFT
oir
:e'
tp(i)i
est I'dnergie d'dchange et e"[p(i)
| est r,*nergie de corrdration. L'dnergie d'dchange pour un gazd'6lectrons uniforme est donnd e,
en
pllpar
la formulede Dirac-Fermi et ddfinie,
en unitds atomiques comme suit :
s*DAIp(nl:
A.45BUr,
$.26)
Avec
p
:
(4nr"3/3)-l Ie terme de corr6lation a€td estimd en premier par
wigner
par :'*'A[p(nl:-#
(r.27)
Par ailleurs' l'6nergie de corrdlation d'un
gaz d'dlectrons libres a dtd mod6lis6e par Ceperly er
Atder
[il].
LT.Lrapproximation
dugradient
g6n6ralis6(GGA)
:c'est
une amerioration de laLDA
dansle traitement de l,dnergie d,echange_ corrdlation qui consiste d Ia rendre ddpendante non seulement
de la densitd dlectronique mais dgalement de son gradient
IVp(i)l
'
Ainsi
la fonctionnefleE*
to(i)1
rend compte du caractdre non uniform e du gaz d'dlectrons. Dans ce cas, ra contribution
de
Er"
[p(i)ld
l,6nergie totale du systeme peut€tre additionnde de fagon cumurde d partir de chaque portion
du
gaznon uniforme commes'il
etait locarement non uniforme.Elle
s'dcritde Ia forme :
s*foo[p?)]:{
p(F)
s,,
[p?).lvp(fi1]d31
(r.2s)
ou
er"
Ip(i),lvp(i)l]reprdsente
l,energie d'echange-correlation par dlectron dans un systdme d'dlectrons en interaction
mutuelle de densitd non uniforme.
L'utilisation
des fonctionneiles de typeGGA
permetd'accroitre de fagon significative la prdcision des
calculs en comparaison avec la description fournie par Ia
LDA
enparticulier pour l'dnergie de liaison des moldcules.ce
quiest d
l,ongine
deI'utilisation
massive de la DFTpar les chimistes dans res anndes 90.
on
trouve differentes para-m6trisations pourla GGA dont celles de perdew et al (1991) [12] et Perdew et al (1996) [13] et les versions les plus
utilisees sont celles de perdew et Wang [14] et Perdew [15].
f.S.R6solution des 6quations de
Kohn-Sham
: Pour r6soudre res dquations de Kohn-sham,il
fautchoisir une base pour les fonctions d'onde que
I'on
peut prendre comme une combinaison lindaired,orbitales. appelds orbitales de Kohn-Sham
(KS)
:Yi
&,4
:lci$1(k,r)
10
Cha
r@ie
de
la
fonctionnelle
de
la
densitd
(D
oir
les9i(k' r)
sont les fonctionsde base et les
er
les coefficients de ddveloppement,.,,t:1.:::l:o":
o:t
equarions de Kohn et shamrevient d determiner res coefficientsvr9lltJ
cji
potx res orbitaresoccu$es
qui minimisent l,dnergietotale. La rdsolution des dquations de
Ks
pour les pointsde symdtrie dans la premidre zone de
Brigouinpermet
de
simplifier
les calculs' cette rdsolutionse
fait
d'unemanidre
iterat'een
ut'isanr
uncycle d'itdrations auto cohdrent.
ceci
est
rdalisd
en injectant ra densitd de chargeinitiale
pin pour diagonaliser l'dquationsdcuraire :
(H-s;s)
:
o
(I.30) otr
ll
reprdsente Ia matrice hamiltonienneet ,s la matrice de recouwement. Ensuite,
ra nouvelle densitd de charge out
p
estconstruite avec res vecteurs propres de cette
dquation sdculaire en utilisant la densite de charge
totale qui peut €tre obtenue par une sommation sur toutes les orbitales occupdes {II.16).
si
I'on
n'obtient pas la convergence des carcurs, on mdrangeres densitds de charges pin et
poat
dela maniere suivante:
pll':
g-o)pln+aplur
(r.31)
i
represente la idme it6ration et aun parametre de mixage.
Ainsi
Ia procddure iterative peut €tre poursuiviejusqu'd
ce que la convergence soit realisde.f.9.L'aufo-coh6rence
dans les calculsPour
simplifier
les carcurs, enrdsorvant les equations de KS pour les points de symetrie dans la premidre zone de brouillon. ces solutions s,obtiendront d,une manidre itdrative en utilisant un cycle d'itdrations auto-cohdrent
illustre par l,organigramme de la Figure I.1.
on
commence par une densitd d'essai p;npourla premidre itdration. Typiquement on utilise une superposition des
densitds atomiques puis on calcul la matrice de
Kohn-sham' et en rdsolvant les dquations pour les
coefficients d'expansion pour obtenir les orbitales de kohn-sham, d cette dtape, en calculant la nouvelle densitd
pout.sila
densit6 ou I'dnergie a beaucoup change (criterede convergence), on retoume d ra premidre *tape, et en mdrangeant res deux densites de charge
_pinet
poutde
la manidre suivante:p'iit:
(1+s)
pln+ spiu,
tr.32)
I : represente le nombre de I'it6ration.
s
: Un parametre de mdlange (mixage).Ainsi
Ia procedure iterative peut 6tre poursuivie jusqu'dIa convergence soit rdalisde
Chapitre
J
Iltrlgligje
la
fonctionnelle
de
la
densit6
Fig.
r.l:organigramme
de ra resolutiondes dquations Kohn-sham [12]
on
peut representer cette procddure parre sch.ma ci-aprds.
Ftn
Calcule I"(r/
Rfsoudrc
les6rluations
^E_f
Ddteluiner.Er
CeIcuIepr*
nSon,/roo****;\
\/
Oni
pj;t
=
{t
* c)pil
+
spl*
Cakuler
L2Chapitre
I
Theorie
de
la fonctionnelre
de ra
densit6(DFT
R6fdrences
[ 1 ]M.Born.R. oppenheimer, Ann,
phys g7,45 7
(1927)
[2] D.R.Harhee, proc. Camp.phil. soc.24, 39( 1 92 g)[3]
V.FOCK,Z,phys,
61,t26(tg30) ;62,200)
[4] Mdmoire de magister universitd de
m'sila
par kouriche
athmane,2006[5]
m6moire universit6 dem'sila
de magister parme*agfadila ,2003
[6]S'J.lee, les,T.s kwon, k.nahm, c.k.kim,J.phus. condens. Mayyer2(1gg0)
3253. [7]Mdmoire de magistdre, universite Mohamed Boudiafde
m'sila,
Baraka fatiha-2AA4t8l E.
fermi,Z.phys,48,73(t928).
[9] M6moire universitd de
m'sila
de magister
par zerargafures .200g[0]
P. Hohenberg and W. Kohn,phys.
Rev.136,8g64
(1964).[11]
D' M.
ceperly
and B. J. Arder,phys.
Rev.Lett.B
23, 504s (19g0). [12] J. P. Perdew and
y.
Wang, phys. Rev.B
45,13244(1992).1[13] J. P. Perdew, S. Burke and
M.
Emzerhof, phys. Rev. Lett.77,3g65
(1ee6).[14] J. P. Perdew and
y.
Wang,phys.
Rev.B
33, g800 (19g6).[15] J. P. Perdew
in'Electronic
structure ofsolids,,Academie
Verlag,Berlin,
1 1(1eet).
[16] K.A.TheseMajister, Universit6 de M,sila,(2009 ).
:
A1
.
t-h
antfre
TT
:
\.-/
rr14f, r LI v II
T
/t7
",
aaugmentdes linearisdes
(Fp_LApv/\
I
,,,
l-^:
-^
--Chapitre
II
la mdthode
des ondes planes augment6es lin6arisSes
(rP-LAPW)
tr
la
mdthode des ondes planes augmentdes tin6aris6es(Fp-LAplv)
Introduction
La mdthode
LAPW
(Linearized Augmented Plane Wave) developpde par Andersen [1], est fondamentalement une amdlioration de la mdthode dite des ondes planesaugmentdes
(APW)
5labaree par Slater, en 1937 [2, 3] {les ddtails de cette mdthode peuvent 0tre trouvds dans lelivre
de Loucks[4]
cette mdthode devienne la methode des ondes planes augmentees lin6airement (FP-LAPW).Une nouvelle technique pour resoudre l'dquation de Poisson
[5]
a 6td ajoutdei
la mdthodeLAPW
pour que nous puissions traiter I'absorption moldculaire sur les surfaces.Pour d6crire le potentiel
cristallin,
Slaterintroduit
I'approximation du potentiel lVluffin-tin, ce potentiel est reprdsent€ dans les figures(II.1)-(IL2).
Selon cette approximation, le potentiel est sphdriquement sym6trique dI'intdrieur
des spheres atomiques du rayonr,
et assume constanti
l'ext6rieur
le potentiel developpe sousla
forme suivante :
..,-,,_f
Xr-
v6(r)
\*
a I'int€rieurde
ta
sph|re
"t'/-tXr
vrrrik'
h{ext*rieurde
la sph*re.
(rL1)
Ainsi, avant
d6crire la m6thode FP-LAPW, nous rappellerons les bases de la m6thode APW.Fig.
Ill:Potentiel
cristallin
d'un rdseau carrd d deux dimensions: (a) potentiel total, et (b) poteiltielmuffin-tin
[6].chapitre
rr
la mdthode
des ondes pranes
augmentfes
rin6aris6es
ILl.
La
mdthode des oudes planesaugment6es(Aply)
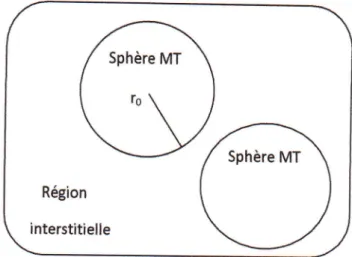
slater considdre que I'espace est
divisd en deux types de rdgions
(figure
Ir.2):rdgion
de cmur et region interstitielle ; la rdgion pr6s du noyau a un potentiel
et une foncrion d'onde similaire d ceux
d'un
atome isold alors, le potentiel varie fortement).Cette rdgion est limitde par une sphdre atomique
(s)
de rayonre et le potentiel possede la symdtrie sphdrique. Dans la region interstitielle les fonctions d,ondes sont planes et le potentiel est constant. Donc la fonction d,onde
s'dcrit
sous la forme :@
(d:{h''s
"i@+fii
lE,*AmIh
(r)ym
T)
ToT
lfa
(rr.2)O: Volume de la maille unitaire. Y6n: les harmoniques sphdriques.
C6:
coefficients de ddveloppement.U;(r)
: la solution rdguliere de I'dquation suivant :[3]
{-ifffiv(r}&}ru,(r):o
Ot
Er : parametre d'dnergie.V(r)
: le composant sphdrique du potentiel dans la sphere(rr.3)
Sphdre MT
SphAre MT
Fig.
tr
2
:porentiel((
Muffin-Tin
>>(MT)
Chapitre
II
la mdthode
des
ondes planes augment6es
lin6aris6es
(rP-rAPW)
L'orthogonalisation de
U'(r)
et deU(r)
d'aprds [26] est donne :ff"
r,
(r)dr =
L
glt2)
Avec le choix de la norme
llui
ll permetI'indication
de rang pour le quel la lin6arisation de I'energie sera une bonne approximation. En particulier, les erreurs sur l'dnergie de linearisation sont acceptables selon Anderson.lluilllEFsl <
1
(rr.13)On E1 est le paramdtre d'dnergie et E l'€nergie des bandes.
Si un
tel
choix n'est pas possible, plusieurs options sont disponibles :1-
On divise les range d'dnergie dans les fen€tres, et chacune de ces fen6tres est traitee sdpardment.2-
Onutilise
un ddveloppement sous la forme d'orbitales locales (ceci est effectivem ent la methode quadratique).3-
On rdduit lataille
de la sphdre. Donc, on rdduit la norme de la ddrivde. Donc la suit, on va exposer les deux param6tr6s m6thodes. La troisidme option a 6td appliqude par goedeker [9].4-
3.4.2. Les fonctionsradiales relativistes
Dans le cas des 6l6ments lourds qui ont un nombre atomique 6lev6, on tient compte de
I'effet
relativiste,Les effets relativistes concernent seulement les fonctions radiales dans les sphdres
MT.
Pour introduire cet effet,il
faut remplacer les dquations (2.9)etQ.$
par les 6quations de Dirac et leurs ddrivdes par rapport d l'6nergie. Dans le but de rdsoudre ces dquations, koelling et Harman [10] trouvaient une technique qui n6gligeI'effet
spin-orbite Roskey, Wood et boring Takeda, Macdonald etAl).
u/.
:l
gkJCkv
1*ur:l--i.f1s67JC6,l
(II'14)
K
: le nombre quantique relativiste..flgy i ost le spin-orbit d deux composants et les coordonn€s radiales a 6td supprim€.
Chapitre
II
la m6thode
des ondes
planes
augment6es
lin6aris6es
(FP-LAPW)
Koelling
et harmon[9] utilisent une nouvellefonction
:.1
arm*'
M:m+#@-v)
g'
: est la derivde radiale de g :m : est la masse.
C : est la vitesse de la lumidre dans le vide.
Ot
.f,"
: est le spinor non relativiste.Ddfinissant et p1rg1 et Q1:rCO1 l'dquation sdculaire relativiste devient P'r:2MCQ+12p1
Q'r:-p+
t#+
(v-Er)l
prI
ins
-o:c1$t
tron r >* t-(T)'
/'
-tl
pr:zgfiqr+tfiq)
+|pr
a;:
-:
a;W
+
u
-
nl]o"\ffi+
r]a
(r.15)
(rr.16)
La solution en fonction des nombres quantiques habittrelle
lmLSls'dcrit
commesuit:
I
SIYImxs
1o'*:l#o,(-9't*
i
*
sp)Tmx,f
(rr.17)
Cette dernidre equation peut 6tre rdsolue numdriquement comme dans le cas de l'dquation de Schnidinger non relativiste en utilisant la methode prddicateur-correcteur par exemple, en donnant les conditions aux limites.
(rr.18)
(rr.le)
(rI.20)
Le terme spin-orbite peut 6tre inclus en additionnant le terme
-(#)f*1)
p.(Au
membre droit de l'dquation(II.16).
La ddriv6e est similaire au cas non relativiste.0r.21)
{rr.22}
Chapitre
II
la mdthode
des ondes planes augmentdes lin6arisSes
(FP-LAPW)
Les composantes
gi
et f1 peuvent €tre ddterminees en utilisant les definitions p1 etQl et (01 les deux composantes sont utilisdes dans la construction de la densitd de charge ou I'dvaluation des 6l6ments de matrice (pour les composantes non spheriques deI'hamiltonien, par exemple).
Ainsi
IJzrlaquantitd est remplac6e dans l'equation(II.l3)
de normalisation par le terme*
* f
.IL5.
R6solution
del'6quation
de poisson :Le potentiel utilise dans les 6quations de KS comprend le terme d'echange et de Corrdlation, et le terme coulombien
VC(r).
Le terme coulombien est la somme du potentiel de Hartree(VH(r))
et du potentiel nucldaire.VC(r)
est ddtermin6 par 1'6quation de Poisson d partir de la densit6 de charge (electronique et nucleaire) :Le potentiel
utilise
dans les dquations deKS
comprendle
terme d'dchangeet
decondlation,
et le
terme
coulombienVC(r). Le
terme coulombien est
la
somme du potentiel de Hartree(Vf(r))
et du potentiel nucl6aire.VCG) est
ddtermindpar l'6quation de
Poisson
d
partir de
la
densit6
de
charge (dlecfronique et nucldaire) :v2%(rF
np(r)
0I.23)
L'intdgration
de cette equation est seulement possible dans l'espace reciproque.La mdthode de resolution dite de
la
<<pseudo-charge r> duei
Hamann [1 1] et Weinert[12] est bas€e sur deux observations :
r
La
densitdde
charge est continueet
varie
lentement dansla
rdgioninterstitielle
et beaucoup plus rapidement dans les spheres.r
Le
potentiel
coulombien dansla
rdgion interstitielle
ddpendd
la
fois
de
la charge interstitielle et du multipOle de la chargei
I'interieur
de la sphdre. Dans la r6gion interstitielle, la densite de charge est ddveloppee en serie de Fourierp{rf
;o(G)giG'r
(rr.24)
Chapifre
II
Ia
mdthode
des ondcs planes augmenf6es
lindaris6es
(Fp-LAPW
G+0
G=0
(r1.2s) (rr.30)K"(r):
IG*
Ylm
{r) Doncuf*(r)lmcr,*vrff
(r)
0r.2e)On ddtermine le potentiel
i
I'intdrieur
dela
sphdreMT
parI'utilisation
de la fonction de Green.Yy(r):v1ffG)HJ.#L-f
dr'r't*Zpu(r')
+
,t
1R6r',"-t
pu{r')
-W
l:.
dr'r't+z
p,(r)\
(II.31)
et les ondes
planes.iG'r
,ont
calculdes d partir de la fonction de Besseljl
Tu,,"
tiG.r
-
4nuiG.raZwtl
j{lellr
-
r))y1i*@)\*(r
_
r,}
(rr.26\
ori
r est la coordonnde radiale,r
la position de la sphdre o etRo
son rayon.%(c)#
(rr.27)
Le potentiel interstitiel
vpw
a€tetrouv|
directement par int€gration de :vp*:
Er*vf{
(r)ym?)
=Euvf*(r)kr(r)
(IL2s)
Soit
Chapitre
II
Ia
m6thode
des
ondes planes
augmentfes
lin€aris€es
(FP-LAPW)
Oir les
pr$')
sont les parties radiales de la densite de charge.Il6.Am6lioration
dela
mdthodeFP-LAPW
:Le but de la methode FP-LAPW est d'obtenir des 6nergies de bande prdcises au voisinage des 6nergies de lindarisation ElU31. Dans la plupart des matdriaux,
il
suffit
de choisir les dnergies E au voisinage du centre des bandes. Cependant, ce n'est pas toujours possible etil
existe de nombreux mat€riaux pour lesquels le choix d'une seule valeur deEI
n'estpas suffisant pour calculer toutes les bandes d'energie: Par exemple, les matdriaux avec des orbitales4f
U4,l5]
et les 6l6ments des m€taux de transition [16, 17,t8].
C'est le probldme fondamental de I'etat desemi-ccur
qui est un 6tat intermddiaire entre I'dtat de yalence et 1'6tat decaur.
Il
existe deux moyens pour traiter cette situation:r
L'usage des fen6tres d'6nergie multiple.o
L'utilisation
d'un d6veloppement en orbitales locales.tr.6.1.Les
fenOtresd'6nergie multiples
:La technique la plus utilisde pour
fiaiter
le probleme du semi-ocow est celle qui consistei
diviser le spectre energetique en fenOtres dont chacune correspond d une {nergieEI
115,191. Cette procddure de traitement est illustree dans la figure(rr.3).
Dans ce traitement par le moyen des fenOtres, une sdparation est faite entre f€tat de valence et celui de semi-ceur ou un ensemble de EI est choisi pour chaque fen6tre pour
fiaiter
les 6tats correspondants. Ceci revient d effectuer deux calculs par lamdthodeLAPW,
ind€pendants, mais toujours avec le m0mepotentiel.
La mdthode FP-LAPW est basde sur le
fait
que les fonctions(fte101
sont orthogonalesi
n'importe quel dtat propre duceur
et, en particulier, d ceux situdsi
la surface de la sphdre.Cependant,les 6tats de semi-ceur satisfont souvent d cette condition, sauf
s'il
y
alapr6sence de bandes < fant0mes>> entfe l'6tat de semi-ceur et celui de valence.Chapitre
II
la m6thode
des ondcs planes augmentdes
lin6arisfes
(FP-LAPw)
I
I
fendtre
E[')+
sl')
2
fen€tres
Yaleuce
Seuri-cew'
Et
t
Fig.
tr.3:
Exemple defen0te
avec un dtatsemi-caur
tr.6.2.D6veloppement enorbital
local
:Le ddveloppement de la m6thode
LAPW
consiste en une modification des orbitales locales de sa base afin{viter
I'utilisation de plusieurs fen6tres.L'idde
principale est de traiter toutes les bandes avec une seule fen€tre d'dnergie en particularisant I'dtat de semi-ccenr. Plusieurs propositions ont etd faites par Takeda[20],
Smrcka [21], Petrul22let
Schanghnessy [23]. R€cemment Singhf2fi
aproposd une combinaison lin6aire de deux fonctions radiales correspondant d deux dnergies differentes et de la d€rivde par rapporti
I'energie de I'une de ces fonctions.Qt*:
IA6IJ{r,81,)
+BwUt$,
Eut) +Cr*{Jr(r,
E2,}lY6$)
(II'32)
Ori les coefficients C1- sont de la m6me nature que les coefficientS
At*
et81*
prdcddemment definis. Par ailleurs, cette modification diminue I'erreur commise dans Ie Calcul des bandes de conduction et de valence.
tr.7.
Traitement
des effets despin-orbite
:Le terme de spin-orbite est important pour le calcul de la structure de bandes et des propri6tds dlectroniques des matdriaux qui contiennent des eldments lourds ou les substances magn€tiques.
Les 6l6ments de la matrice de spin-orbite d I'intdrieur d'une sphdre peuvent €tre calcules,i
priori,
comme suit:, | | ,t
{oEla""
lo;;1:
chapitre
II
la
methode
des
ondes planes augmentdes lin6aris6es
(FP-LAPW)
:
tt*t,*,le;*$)er,*'(G')lui*lu*luii^')]+r;1c)Bv*'(G')\uy*ln'"luy'*'\
+ni*r;lly
*,
{G'}
lui*ln'"lui'*,}+rfo(c)
Bv*' (G')
lui*la*lui'*'\
(II'33)
Avec
\uf*ln*lud*,1
=+not(xlYm*o.Lvv*'lxo'
!
drplo''(r;)'
iX
gI'34)
ori
plest
la partie la plus importante de la fonction radialeul
etv
la partie sphdrique du potentiel.Chapitre
II
la methode
des
ondes planes augmentces lin6aris6es
(rP-LAPW
R6f6rences
tll
O.K.
Andersen, Phys. Rev.812
(1975).[2]
J. C.Slater, Phys. Rev.,81,385(1951).[3]
J.C.Slater,Advences in Quantum Chemistry 1, 35(1964)'[4] T.L.Loucks,((The
Augmented Plane Wave Method)),BenjaminNew YorK1967)'
[5] E.Wimmer,H.Krakauer,M.weinert and A.J.Freemen,phys .Rev 824,864(1981)'
t6]
F'Elhaj-Hassan, Thbse de doctorar,Universite deMetz,(2000).
[7]
J.C.slater, phys' Rev.51,846 (1937)'t8l M.
B Kanoun.Thdse de doctorat. First-principles studyof
structural, elastic andelectronic properties of
AIN
and GaI'{ semiconductors under pressurEffect
and Magentism inAIN
:Mn and GaN :Mn systems, universitd de Tlemcen' (2004)'[9]
S.goedecker, phys Rev .847,9881(1993)'I I 0]
D.Koelling
and B.N Harmonj'phys' C I 0,3 107 (1'97 7)[11] D.R. Hamann, Phys. Rev.
Lett' 42,662
(1979)'t12l M.
Weinert, J.Math' Phys.22,2433
(1981)'tl3l
O.K.
Andersen, Phys. Rev' B12 (1975)' [1a] D. Singh,phys'Rev.B 44, 7 451(1991)'[15]
S.Goedecker andK
.Maschke, Phys'Rev'B42'
8858 (1990)'t16l
D.J. Singh and H. Krakauer, Phys' Rev'B
43' 1441 (1991)'l17l
P.Blaha,D.J Singh, P.L SorantinandK.
Schwarz, Phys'Rev.B 46,1321(1992)'
[ 1 8] D. J. Singh,K. Schwar
z
andP' Blaha, Phys'Rev'B 46'5849
(t992)'
[19] L.F.Mattheiss and D'R.Hamann, Phys'Rev 'F.33' 823 (1986)'
t20l
T. Takeda and J. Kubler, J' Phys' F 5, 661 (1979)'[21] L.Smrcka,Czech.J' Phys.B34 ,694 (1984)'
[22] J.Petru and L.Smrcka,Czech'J' Phys'
B
35' 62 (1985)'[23] D. J. Shaughnessy,G.R.Evans and

![Fig. r.l:organigramme de ra resolution des dquations Kohn-sham [12]](https://thumb-eu.123doks.com/thumbv2/123doknet/14915849.660596/24.892.179.781.169.889/fig-l-organigramme-ra-resolution-dquations-kohn-sham.webp)