Identification in silico du site de fixation à l’ARN de la protéine PTBP1 : découverte de motifs par utilisation de l’outil RSAT via des données CLIP-Seq publiques
25
0
0
Texte intégral
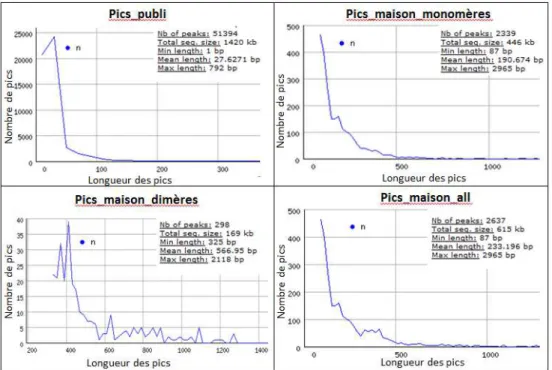
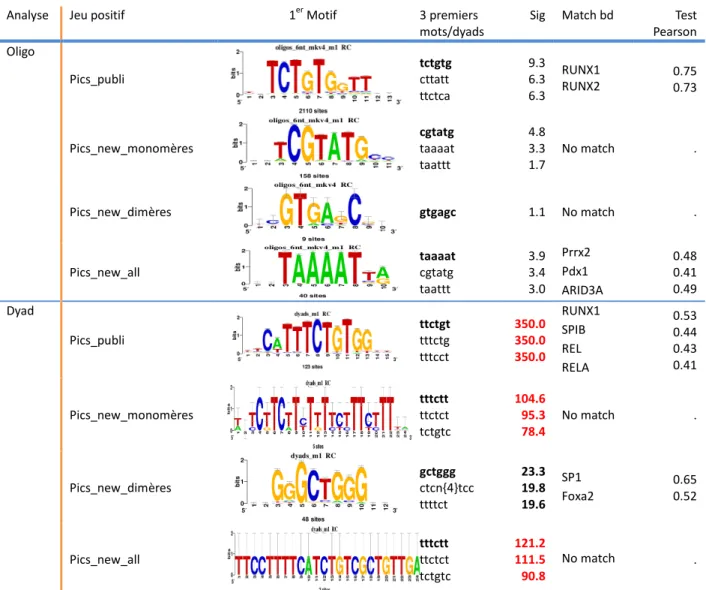
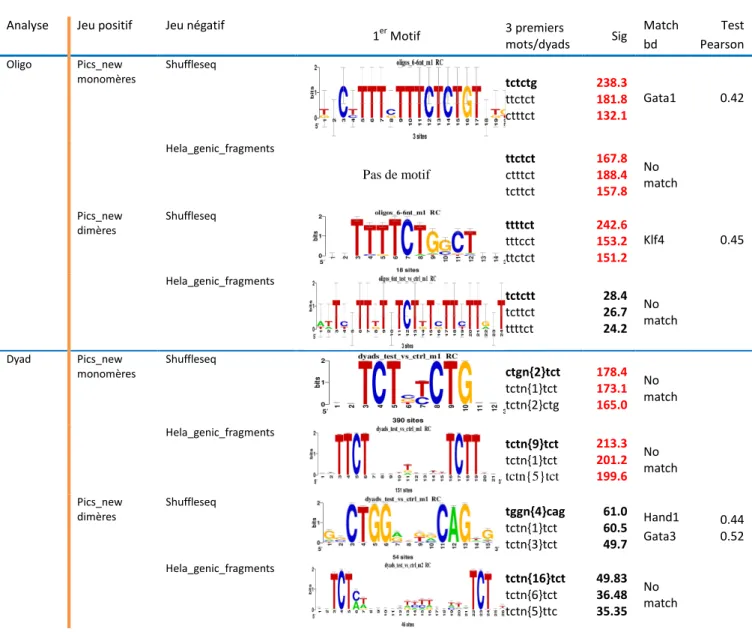
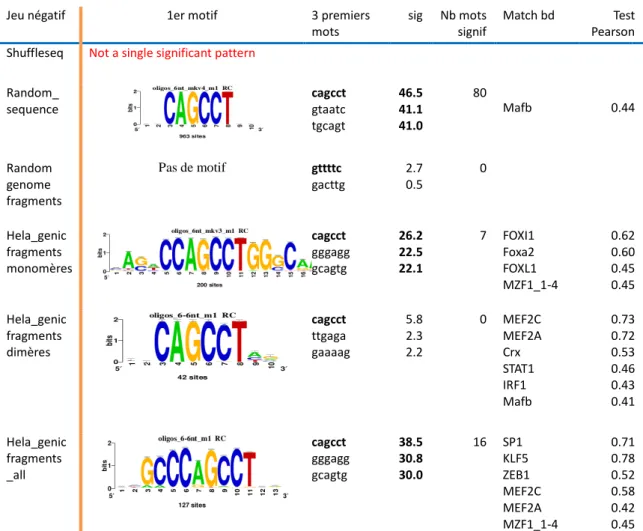
Figure
Documents relatifs