HAL Id: tel-01433358
https://tel.archives-ouvertes.fr/tel-01433358
Submitted on 12 Jan 2017HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Evaluation d’un variant d’antithrombine dans différentes
indications thérapeutiques
Yasmine Bourti
To cite this version:
Yasmine Bourti. Evaluation d’un variant d’antithrombine dans différentes indications thérapeutiques. Pharmacologie. Université Paris Saclay (COmUE), 2016. Français. �NNT : 2016SACLS404�. �tel-01433358�
NNT: 2016SACLS404
T
HÈSE DE DOCTORAT
DE L’U
NIVERSITÉP
ARIS-S
ACLAYPRÉPARÉE À“UMR-SINSERM1176:HEMOSTASE,INFLAMMATION ET THROMBOSE”
E
COLED
OCTORALE N° 569
Innovation thérapeutique : Du fondamental à l'appliqué
Spécialité: Sciences pharmacologiques
Par
Yasmine BOURTI
Evaluation d'un variant d'antithrombine dans
différentes indications thérapeutiques
Soutenue au « Kremlin-Bicêtre », le « 14 novembre 2016 » : Composition du Jury :
Monsieur le professeur Nicolas LEROLLE, PU- PH CHU d’Angers Président du jury Madame, le professeur Delphine BORGEL, PU-PH INSERM UMR-S1176 Directeur de thèse Madame le docteur Marie-Christine BOUTON, Directeur de recherche INSERM U1148 Rapporteur Monsieur le professeur Laurent MACCHI, PU-PH INSERM U1082 Rapporteur Monsieur le docteur François SALLER MCU, INSERM UMR-S1176 Examinateur
REMERCIEMENTS
Cette thèse de doctorat a commencé par la réponse favorable de Madame le Professeur Delphine BORGEL pour un stage de Master 1. Je tiens à la remercier infiniment de m’avoir confié ce travail de recherche dont le succès n’aurait pas pu être concrétisé sans son encadrement, son optimisme infaillible, sa disponibilité, son sens de l’écoute et ses précieux conseils tout au long de ses années.
Je tiens à remercier Madame le Docteur Elsa BIANCHINI avec qui mon aventure avait commencé au laboratoire et sans qui mon premier projet de recherche n’aurait pas donné de fruits. Une particulière gratitude va à Monsieur le Docteur François SALLER pour ses précieux conseils, sa disponibilité sans faille et sa grande sympathie. Nos discussions scientifiques sans fin vont beaucoup me manquer.
J’exprime ensuite ma profonde gratitude à Madame le Docteur Marie-Christine BOUTON, et Monsieur le Professeur Laurent MACCHI d’avoir accepté d’évaluer, et d’être rapporteurs de cette thèse. Leurs remarques et suggestions précieuses ont permis d’améliorer la qualité de ce travail. Je tiens aussi à remercier Monsieur le Professeur Nicolas LEROLLE d’avoir pris part à ce jury.
Ce travail n’aurait pu aboutir sans l’aide de nombreuses personnes. Que me pardonnent celles que je pourrais oublier, mais j’adresse une pensée particulière au personnel des plateformes à Chatenay-Malabry : Madame Valérie Domergue, Julie et Ayma (Animex), Claudine Déloménie (TRANSPROT). Je vous remercie de m’avoir permis de réaliser ce travail dans de bonnes conditions.
J’ai pu travailler dans un cadre particulièrement agréable, d’abord grâce à l’ensemble des membres de l’ex équipe EA 4531 et ensuite au sein de l’UMRS1176 grâce à l’accueil du Dr Cécile DENIS que je remercie chaleureusement. Une pensée particulière va à, Allan de Quevilly, Christelle Repérant et toutes les personnes que j’ai eu la chance de côtoyer au sein de mon équipe et de toute l’unité UMRS1176.
Une grande pensée à toutes les personnes qui sont passées par le bureau des étudiants à Chatenay : Judicael, Sonia, Maria et le bureau des doctorants à Bicêtre Marion : Awatef, Tiffany, Fatou, Mahita, Ziane, Aleria, Sandra. Merci pour votre présence dans les moments difficiles et de m’avoir toujours permis de travailler dans la bonne humeur.
Mes dernières pensées iront vers mes deux grands-mères, mes parents et ma petite sœur ainsi que toute ma famille, et surtout à mon mari pour leurs soutiens sans faille.
2
RESUME
Notre équipe s’intéresse à la relation structure-fonction d’une protéine, l’antithrombine (AT), un inhibiteur physiologique de la coagulation, en vue d’un développement thérapeutique. Cette protéine anticoagulante, capable de lier un motif pentasaccharidique sur les dérivés hépariniques, possède en outre, à fortes concentrations (500%), des propriétés anti-inflammatoires médiées par sa liaison aux héparan-sulfates cellulaires. Ce profil a mené à l’évaluation de l’AT dans des situations associant un emballement de la coagulation et de l’inflammation, comme c’est le cas au cours du sepsis sévère et d’autres situations d’ischémie-reperfusion (I/R). Cependant, les fortes concentrations utilisées dans les études précliniques nécessiteraient d’administrer des doses d’AT incompatibles avec le profil de sécurité de cette protéine anticoagulante.
Dans ce contexte, nous avons, au cours de ce travail, caractérisé un variant d’AT (AT-N135Q-Pro394) dépourvu d’activité anticoagulante et doué d’une affinité augmentée pour l’héparine. Ce variant est capable de piéger des dérivés hépariniques et apparait comme un candidat idéal pour une utilisation comme antidote en cas de surdosage en héparine non fractionnée (HNF), héparines de bas poids moléculaire (HBPM) ou fondaparinux. Par ailleurs, ce variant pourrait être utilisé à des doses cytoprotectrices, sans risque hémorragique.
Afin de tester cette dernière hypothèse, nous avons développé un modèle d’I/R rénale chez la souris, qui s’accompagne d’une augmentation significative de marqueurs de dysfonction rénale (Urée, Créatinine, Kim-1) et de l’inflammation (expression tissulaire de cxcl-1, il-6). L’AT avait déjà été montrée protectrice (Mizutani et al, Blood 2003) dans un modèle murin comparable. De façon surprenante, nous n’avons observé aucun des effets protecteurs décrits, ni sur l’inflammation ni sur la fonction rénale, et que ce soit avec de l’AT plasmatique, de l’AT recombinante ou encore avec un mélange équimolaire d’AT latente et native. Ce même modèle nous a pourtant permis de mettre en évidence les effets nephroprotecteurs et anti-inflammatoires d’une autre protéine anticoagulante, la protéine C activée. Ces résultats décevants font écho à la rétractation pour fraude de l’article de Mizutani et al. en 2013. Le travail approfondi que nous avons mené nous permet de clarifier la littérature et d’affirmer que l’AT, d’origine plasmatique ou recombinante, ne possèdent pas
3
d’effet protecteur dans l’I/R rénale chez la souris. Dans ces conditions, le variant AT-N135Q-Pro394 n’a pas été testé.
Concernant la seconde indication, l’AT-N135Q-Pro394 avait déjà été évaluée in vivo comme antidote aux dérivés hépariniques, HNF, HBPM et fondaparinux, avec d’excellents résultats. Néanmoins, cet effet antidote a été exploré spécifiquement par mesure de l’activité anti-facteur Xa alors que l’AT inhibe plusieurs enzymes de la cascade de coagulation tel que les facteurs VIIa, IXa et IIa. Nous avons donc exploré cet effet antidote dans un test plus global de la coagulation, le test de génération de thrombine (TGT) pour pouvoir le comparer aux autres stratégies non spécifiques utilisées pour antagoniser les dérivés hépariniques (facteur VII activé recombinant, concentré de complexes prothrombiques activés ou protamine). De façon intéressante, dans un plasma mimant un surdosage, notre variant présente un effet antidote supérieur aux agents hémostatiques et au sulfate de protamine à-vis du fondaparinux et des HBPMs, respectivement, et équivalent au sulfate de protamine à- vis-à-vis de l’HNF. Enfin, dans du plasma en l’absence d’anticoagulant, l’AT-N135Q-Pro394 ne montre aucun effet sur la génération de thrombine contrairement aux agents hémostatiques et au sulfate de protamine qui, ajoutés seuls dans du plasma, modifient significativement le profil des TGT.
4
TABLE DES MATIERES
LISTE DES FIGURES ……….. 7
LISTE DES TABLES ……….. 8
LISTE DES ABREVIATIONS………. 9
I. RAPPELS BIBLIOGRAPHIQUES ... 13
CHAPITRE 1: L’ANTITHROMBINE UN ANTICOAGULANT NATUREL MAJEUR ... 14
A. L’hémostase et la coagulation ... 14
B. Régulation de la coagulation ... 16
C. L’antithrombine ... 18
1) Biochimie, biosynthèse et biodistribution ... 19
2) Relation structure fonction ... 20
a) Superfamille des Serpines ... 20
b) Caractéristiques structurales de l’antithrombine native ... 22
c) Différents conformères d’Antithrombine ... 23
c.1 La forme latente et clivée... 23
c.2 Les hétérodimères ... 24
c.3 Les polymères ... 25
d) Mécanisme d’inhibition des serine protéases... 26
e) La potentialisation de l’activité anticoagulante de l’antithrombine par les dérivés hépariniques ... 28
e.1 Activation allostérique de l’AT par le motif pentasaccharidique ... 29
e.2 Potentialisation de l’activité de l’AT matricielle par l’héparine ... 30
3) Déficits congénitaux en antithrombine ... 31
CHAPITRE 2 : LES ANTICOAGULANTS HEPARINIQUES ... 32
A. Physiopathologie et épidémiologie de la maladie thromboembolique ... 32
B. Les anticoagulants hépariniques ... 33
1) L’héparine non fractionnée standard ... 34
a) Structure ... 34
b) Pharmacologie ... 35
2) Les Héparines de Bas Poids Moléculaire (HBPMs) ... 36
a) Structure et préparation ... 36
5
3) Le fondaparinux sodique ... 37
a) Structure ... 37
b) Pharmacologie ... 38
C. Effets secondaires des dérivés hépariniques ... 39
1) Le risque hémorragique ... 39
2) Autres effets secondaires ... 40
3) Stratégies thérapeutiques de neutralisation des dérivés hépariniques ... 40
a) Le sulfate de protamine ... 40
b) Les facteurs procoagulants ... 42
c) Différentes stratégies alternatives proposées pour neutraliser les anticoagulants hépariniques ... 42
CHAPITRE 3 : PROPRIETES CYTOPROTECTRICES ET ANTI-INFLAMMATOIRES DE L’ANTITHROMBINE .. 44
A. Interaction entre l’inflammation et la coagulation ... 44
1) L’ischémie reperfusion rénale ... 45
a) Le rein ... 45
b) Définition et épidémiologie de l’I/R rénale ... 48
c) Physiopathologie ... 49
B. Effets cytoprotecteurs des inhibiteurs physiologiques de la coagulation ... 51
1) Le système de la protéine C ... 51
a) La protéine C activée ... 51
b) La thrombomoduline ... 53
2) Le TFPI ... 55
3) L’antithrombine ... 55
a) Effets cellulaires et anti-inflammatoires de l’AT ... 56
a.1 Effets cellulaires indirects dépendants de la production de prostacycline ... 56
a.2 Effets cellulaires anti-inflammatoires directs ... 57
a.3 Propriétés protectrices de la barrière endothéliale ... 58
a.4 Signalisation cellulaire impliquée dans ses effets anti-inflammatoires ... 59
a.1 Autres propriétés cellulaires de l’AT ... 61
b) Effets protecteurs de l’AT dans l’ischémie/reperfusion et le sepsis ... 62
b.1 Etudes in vivo dans l’ischémie reperfusion et autres défaillances d’organes... 62
b.2 Etudes in vivo dans le sepsis ... 68
b.3 Etudes cliniques ... 70
6
III. Evaluation d’Antithrombines inactivées comme antidotes des dérivés hépariniques dans un test de génération de thrombine (Article 1) ... 75 IV. Evaluation d’Antithrombine inactivée comme agent cytoprotecteur dans l’ischémie reperfusion rénale (Article 2) ... 87 V. DISCUSSION ET PERSPECTIVES ... 102 VI. REFERENCES BIBLIOGRAPHIQUES ... 114
7
LISTE DES FIGURES
Figure 1 : Schéma représentant la cascade de la coagulation……….16
Figure 2 : Représentation schématique des systèmes majeurs de régulation de la coagulation ………..18
Figure 3 : L’archétype de structure tridimensionnelle des Serpines……….. .21
Figure 4 : Structure de l’AT native………...22
Figure 5 : Structures cristallographiques de la forme latente et clivée………...24
Figure 6 :Structure de l’hétérodimère de l’AT………...25
Figure 7 :Mécanisme d’inhibition des Serpines……….26
Figure 8 :Activation allostérique de la forme native de l’AT………....30
Figure 9 : Structure des complexes Michaeliens de l’AT avec le FXa et la thrombine……..31
Figure 10 :Représentation schématique de la structure du pentasaccharide………..38
Figure 11 : Structure primaire et secondaire de la protamine avec l’héparine………....41
Figure 12 :Représentation schématique d’une coupe transversale longitudinale d’un rein...46
Figure 13 : Anatomie du néphron………...47
Figure 14 : Causes d’une réduction généralisée ou localisée du débit sanguin rénal………..48
Figure 15 : représentation schématique de la physiopathologie de l’atteinte rénale aigue…..51
Figure 16 : Représentation schématique des domaines structuraux de la thrombomoduline..54
8
LISTE DES TABLES
Table 1: Mutations au sein de la région P3-P3’ de la boucle réactive obtenues par
9
LISTE DES ABREVIATIONS
ACT Activated coagulation time
AMM Autorisation de mise sur le marché
ANSM Agence nationale de sécurité du médicament et des produits de santé
AOD Anticoagulant oral direct
AVK Anti-vitamine K
AVC Accidents vasculaires cérébraux
AT Antithrombine
CCP Concentré de complexe prothrombique
CCPa Concentré complexe prothrombique activé
CEC Circulation extracorporelle
CIVD Coagulation intravasculaire disséminée
CK18 Cytokeratine 18
COX Cyclo-oxygénase
DDJ Dose définie journalière
EPCR Endothelial Protein C Receptor
FDA U.S Food drug admistration
FT Facteur tissulaire
GAGs Glycosaminoglycanes
HBPMs Héparines de bas poids moléculaire
HBS Heparin binding site
HMGB1 High mobility group box
HNF Héparine non fractionnée
HSPG protéoglycanes à héparans sulfates
HUVECs Human umbilical vein endothelial cells
I/R Ischémie /reperfusion
Kim-1 Kidney injury molecule-1
LPS Lipopolysaccharides
LRP Low density lipoprotein receptor-related protein
MTEV Maladie thromboembolique veineuse
10 PARs Protease activated receptors
PCa Protéine C activée
PF4 Facteur 4 plaquettaire
PGI2 Prostacycline
PNN Polynucléaires neutrophiles
ProS Protéine S
SECR Serpin-enzym complex receptor
Serpin SERine Protease Inhibitors
TAT Complexes thrombine- antithrombine
TCA Temps de céphaline activée
TCP Tube contourné proximal
TFPI Tissue factor pathway inhibitor
TGT Test de génération de thrombine
TIH Thrombopénie induite par l’héparine TLR Toll like receptor
11
La maladie thrombotique est une cause majeure de mortalité et de morbidité dans les pays industrialisés. Parmi les antithrombotiques indiqués dans la prise en charge thérapeutique des thromboses, les anticoagulants hépariniques restent la classe pharmacologique la plus utilisée par voie parentérale. Parmi ces dérivés hépariniques, on retrouve l’héparine non fractionnée (HNF), les héparines de bas poids moléculaires (HBPM) et le fondaparinux. Ils présentent un mécanisme d’action commun qui passe par la potentialisation de l’activité d’un inhibiteur physiologique de la coagulation : L’antithrombine (AT).
Ces anticoagulants, utilisés depuis plus de 60 ans, sont largement utilisés dans le traitement et la prévention dans la maladie thrombo-embolique. Cependant, comme tout anticoagulant, leur utilisation est associée à un risque hémorragique non négligeable. L’HNF possède un antidote utilisé en clinique, le sulfate de protamine. Il neutralise de façon efficace les HNFs, mais seulement partiellement les HBPMs. Par ailleurs, son utilisation est associée à de graves complications cardiovasculaires et allergiques. Le sulfate de protamine étant inefficace vis-à-vis du fondaparinux, des stratégies non spécifiques utilisant des hémostatiques comme le Facteur VII activé (FVIIa) recombinant ou des Concentrés de Complexe Prothombique activés (CCPa), peuvent être administrés en cas d’hémorragies chez des patients présentant un surdosage au fondaparinux. Au sein du laboratoire, la connaissance de la relation structure fonction de l’AT a permis le développement d’ATs inactivées, avec une affinité à l’héparine augmentée, comme antidote au fondaparinux. L’utilisation de ces ATs antidotes obtenues par génie génétique ou modification chimique a déjà fait ses preuves
in vitro et in vivo mais n’a été caractérisée qu’à l’aide d’un test mesurant l’activité inhibitrice
de l’AT vis-à-vis du Facteur X activé (FXa). Dans une première partie de ce travail, nous avons utilisé un test global de la coagulation, le test de génération de thrombine (TGT), pour comparer cet effet antidote aux stratégies actuellement utilisés en thérapeutique pour reverser l’activité anticoagulante des dérivés hépariniques. Ce test nous a également permis d’évaluer l’impact de fortes concentrations de nos ATs inactivées, sur la coagulation.
L’AT, utilisée à des doses supra physiologiques (supérieures à 500%) aurait par ailleurs des propriétés anti-inflammatoires et cytoprotectrices dans différents modèles animaux d’ischémie-reperfusion ou de sepsis sévère. L’administration de telles doses est cependant incompatible avec le profil de sécurité de la protéine. L’AT inactivée recombinante développée comme antidote pourrait permettre de s’affranchir de cette limite. En effet, les propriétés antiinflammatoires de l’AT nécessitent l’intégrité du site de liaison à l’héparine et seraient indépendantes de son activité anticoagulante. L’objectif de la seconde partie de ce
12
travail de thèse était d’évaluer notre AT inactivée comme agent cytoprotecteur au cours de l’ischémie reperfusion (I/R) rénale. Ce type d’I/R entraine une atteinte rénale aigue et peut se rencontrer dans différentes situations pathologiques associées à une réduction du flux sanguin comme le choc septique, un acte chirurgical majeur ou au cours d’une transplantation rénale.
La première partie de ce mémoire est consacrée à des rappels bibliographiques organisés en trois principaux volets. Le premier abordera la place de l’antithrombine dans la coagulation, sa relation structure fonction, et le second volet, les anticoagulants hépariniques. Le troisième volet concernera les propriétés anti-inflammatoires et cytoprotectrices des inhibiteurs de la coagulation et plus particulièrement celles de l’AT. Les résultats des travaux menés au cours de ce travail de thèse, présentés sous forme de deux manuscrits, constitueront la seconde partie de ce mémoire. Le premier article, publié dans la revue « Thrombosis and Haemostasis » en Juillet 2016, compare deux ATs inactivées aux stratégies non spécifiques utilisées en thérapeutique pour reverser l’activité anticoagulante des dérivés hépariniques. Le second manuscrit est une lettre à l’éditor, acceptée dans la revue « Thrombosis and Haemostasis », qui décrit l’utilisation de l’AT comme agent protecteur dans l’ischémie reperfusion rénale.
13
14
CHAPITRE 1: L’ANTITHROMBINE, UN ANTICOAGULANT NATUREL MAJEUR A. L’hémostase et la coagulation
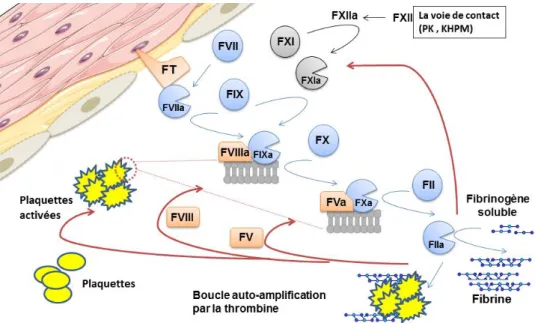
L’hémostase décrit l’ensemble des phénomènes qui participent au maintien de la circulation sanguine et de l’intégrité vasculaire. Elle fait intervenir des protéines plasmatiques, les plaquettes sanguines ainsi que les cellules endothéliales tapissant la lumière vasculaire. Lors d’une lésion vasculaire, l’hémostase permet la formation d’un thrombus pour colmater la brèche vasculaire et stopper l’hémorragie. Ce mécanisme implique, dans un premier temps, l’intervention des plaquettes qui s’activent au contact d’une surface endothéliale endommagée, et qui agrègent pour former un clou plaquettaire plus ou moins stable. Les plaquettes activées changent de morphologie et exposent des phospholipides anioniques à leurs surfaces, cette surface chargée négativement étant nécessaire pour l’interaction de complexes enzymatiques responsables de la production de thrombine localisée. La thrombine générée permettra la production de fibrine à partir du fibrinogène et ce réseau de fibrine ainsi formé va consolider le clou plaquettaire pour former le caillot fibrino-plaquettaire (1).
La génération de thrombine a pour origine une cascade de réactions enzymatiques au cours desquelles des précurseurs inactifs d’enzymes de la coagulation, appelés zymogènes, sont activés pour former des enzymes actives. L’activation de la coagulation est déclenchée par l’exposition du facteur tissulaire (FT) au flux sanguin. Le FT, protéine membranaire exprimée habituellement par les cellules extravasculaires (fibroblastes, cellules musculaires lisses), est capable de recruter les traces de facteur VII activé (FVIIa) circulantes (2). L’association du FVIIa au FT à la surface cellulaire constitue le premier complexe pro-coagulant qui permet l’auto-activation du FVII ainsi que l’activation des facteurs FIX et FX en facteur IX activé (FIXa) et facteur X activé (FXa), respectivement (Fig-1). Une fois activés par clivage protéolytique, ces derniers vont pouvoir à leur tour activer leur substrat. En effet, le FIXa catalyse alors l’activation du FX en FXa, ce dernier généré en quantité suffisante sera responsable de l’activation de la prothrombine en thrombine(2).
Les premières traces de thrombine produites lors de cette phase d’initiation sont capables d’activer les plaquettes ainsi que les cofacteurs de la coagulation, le facteur V (FV) et le facteur VIII (FVIII). En présence d’ions Ca2+
, les FIXa et FXa, ainsi que leurs cofacteurs respectifs, les facteurs FVIIIa et FVa, viennent se fixer aux phospholipides anioniques
15
exposés à la surface des plaquettes activées, pour former deux complexes procoagulants, le complexe tenase et le complexe prothrombinase. Lorsque le FIXa est associé au FVIIIa au sein du complexe tenase, la vitesse d’activation du facteur FX est multipliée d’un facteur 105
à 106 par rapport à celle rapportée pour le FIXa seul (2). Quant au complexe prothrombinase, constitué du FXa et FVa, il permet l’accélération de la conversion de la prothrombine en thrombine d’un facteur 3. 105
comparativement au FXa seul. En plus de sa capacité d’activer les plaquettes, le FV et le FVIII, la thrombine est aussi capable d’amplifier sa propre production via l’activation du facteur XI (FXI). Le FXIa ainsi produit va activer le FIX circulant, et donc indirectement le FX et la prothrombine par la suite. Tous ces mécanismes d’amplification aboutissent à une génération explosive de thrombine qui est alors présente en quantité suffisante pour cliver le fibrinogène soluble en un réseau de fibrine insoluble. De plus, la thrombine active le Facteur XIII (FXIII) en FXIIIa responsable de la stabilisation du caillot préformé en catalysant la formation de liaisons covalentes entre les monomères de fibrine (3). Ce réseau de fibrine n’est dissout grâce au système de la fibrinolyse qu’une fois les tissus vasculaires sous-jacents régénérés.
Le facteur XI activé par la thrombine au cours de la phase d’amplification peut être activé par une autre voie de la coagulation ; la voie de contact. Celle-ci comprend la prékallikréine, le facteur XII (FXII) et le kininogène de haut poids moléculaire comme cofacteur (4). L’activation du système de contact est déclenchée par la liaison du FXII à des surfaces chargées négativement (comme le kaolin) ou des activateurs physiologiques comme les acides nucléiques et le collagène, ou bien encore des agents pathogènes comme les bactéries (5). En physiologie, son rôle reste mal connu car les sujets et les souris déficients en FXII ne présentent pas de risque hémorragique (6, 7). Néanmoins, il a été montré que ces souris présentaient un défaut de stabilisation et de formation du thrombus riche en plaquettes (7). Il a été alors proposé que l’activation de la voie tissulaire par une lésion vasculaire initie la formation du thrombus, celui-ci constituant ensuite une surface propice à l’activation de la voie de contact, cette dernière jouant ainsi un rôle dans le développement et la stabilisation du thrombus (5).
16
Figure 1 : Schéma représentant la cascade de la coagulation déclenchée à la suite de
l’exposition du facteur tissulaire à la surface vasculaire adapté d’après (8)
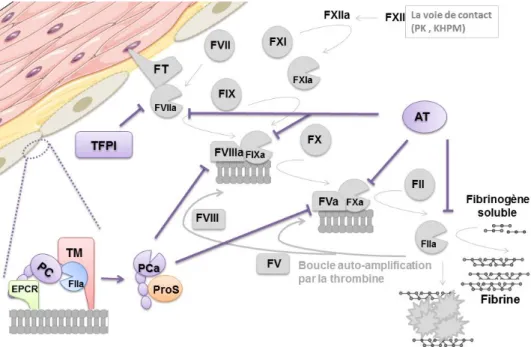
B. Régulation de la coagulation
La cascade de la coagulation reste localisée et ne s’étend pas à distance de la brèche vasculaire, en partie grâce au phénomène de dilution maintenu par le flux sanguin. En effet, la cascade de la coagulation est finement régulée à plusieurs niveaux, principalement par trois systèmes inhibiteurs (Fig-2) : l’inhibiteur de la voie du facteur tissulaire au niveau de la phase d’initiation, le système de la protéine C au niveau de la phase d’amplification ou encore l’antithrombine (AT), inhibiteur de la majorité des enzymes de la coagulation. Il existe d’autres inhibiteurs enzymatiques appartenant à la même famille que celle de l’AT, les Serpines, comme le second cofacteur de l’héparine, l’inhibiteur dépendant de la protéine Z, la protéase nexine-1 et l’inhibiteur de la protéine C1 du complément, ou appartenant à d’autres familles différentes, comme l’α2 macroglobuline.
La phase d’initiation est régulée par l’inhibiteur de la voie du facteur tissulaire (TFPI : Tissue factor pathway inhibitor), un inhibiteur enzymatique à 3 domaines types Kunitz. Le TFPI existe sous deux principales isoformes α et β qui résultent d’un épissage alternatif. Il est synthétisé par les cellules endothéliales majoritairement au niveau de la microcirculation (9). Ces deux isoformes diffèrent par le domaine Kunitz III absent dans l’isoforme β qui présente à la place un groupement C-terminal « glycosylphosphatidyl
17
inositol » nécessaire pour s’ancrer à la surface des cellules endothéliales. Le TFPIα est aussi capable de se lier à l’endothélium sur les glycosaminoglycanes (GAGs) grâce à une extrémité C-terminale très basique (10) Le TFPI est capable d’inhiber le complexe FVIIa-TF. Ainsi, le domaine Kunitz I interagit avec le FXa et le domaine Kunitz II avec le site actif du facteur VIIa, formant ainsi un complexe quaternaire inactif FXa-TFPI-FVIIa-TF (11). Récemment, le rôle du TFPIα a été mis en évidence dans l’inhibition du complexe prothrombinase au niveau de la phase d’initiation grâce à son extrémité C-terminale basique (12). Cette partie du TFPIα est capable d’interagir avec le FV et certaines formes de FVa qui possèdent une région acide retrouvée lors d’un clivage partiel par d’autres protéases que la thrombine.
Le second inhibiteur physiologique de la coagulation est le système de la protéine
C qui comprend la protéine C, son cofacteur la protéine S (ProS), et deux récepteurs
membranaires, le récepteur endothélial de la protéine C (EPCR) et la thrombomoduline (TM).
La protéine C est une protéine sous forme zymogène dont l’activation implique principalement la thrombine et la TM. Une fois liée à la TM, la thrombine perd ses propriétés procoagulantes et devient capable d’activer la PC. Ainsi, la présence de TM accélère la génération de protéine C activée (PCa) d’un facteur 1000 par rapport à la thrombine libre (13). L’EPCR, un second récepteur capable de fixer la protéine C peut également potentialiser son activation par le complexe thrombine-TM, mais dans une moindre mesure (environ 20 fois seulement) (14). L’EPCR est présent en plus grande proportion au niveau des gros vaisseaux, au niveau desquels l’EPCR aurait pour rôle de concentrer la protéine C (15).
La PCa est une sérine protéase vitamine K-dépendante qui dégrade par protéolyse limitée les FVa et FVIIIa. Cette activité protéolytique est modestement (facteur 20 au plus) potentialisée par la ProS et le FV (16). La dégradation par la PCa des FVa et FVIIIa ne permet plus aux complexes tenase et prothrombinase de fonctionner et la cinétique de production de la thrombine devient très lente. L’activité catalytique de la PCa est elle-même régulée par deux inhibiteurs de la famille des Serpines majoritairement par l’inhibiteur de la protéine C (PCI : Protein C inhibitor) et d’un effet moindre par l’α1 antitrypsine.
La protéine S, cofacteur de la PC est un autre facteur vitamine K dépendant, circulant à 60-70% sous forme inactive liée à la protéine de transport du système du complément C4bBP (C4b-binding protein) (17). La ProS est aussi capable d’interagir comme cofacteur du TFPI via son domaine Kunitz III et accélérer son inhibition du FXa, mais cette propriété n’a été mise en évidence qu’in vitro (18).
18
Figure 2 : Représentation schématique des systèmes majeurs de régulation de la coagulation
(8).
Le troisième inhibiteur majeur de la coagulation est l’antithrombine (AT) capable d’inhiber différentes serine protéases de la coagulation: la thrombine, le FXa, le FIXa, le FXIa, le FXIIa et le FVIIa (19). L’AT appartient à la superfamille des Serpines (pour SERine
Protease INhibitors), et possède à son extrémité C-terminale une boucle réactive avec
certaines protéases de la coagulation qui lui permet de former un complexe enzyme-inhibiteur covalent irréversible. L’AT est un inhibiteur enzymatique dont l’activité anticoagulante peut être accélérée après liaison à l’héparine.
C. L’antithrombine
Contrairement à son nom laissant penser qu’elle n’inhibe que la thrombine, l’AT est un inhibiteur physiologique majeur capable d’inhiber de nombreuses autres sérine-protéases de la cascade de coagulation (comme le FXa), ainsi que la majorité des enzymes de la voie du FT (comme le FIXa, FVIIa) et de la voie de contact (comme le FXIIa, le FXIa). De ce fait, les déficits congénitaux en AT sont associés à des manifestations thromboemboliques et les souris dont le gène codant pour l’AT a été inactivé ne sont pas viables (20). Par ailleurs, l’AT possède des propriétés cellulaires, notamment en termes d’inflammation et d’angiogenèse.
19 1) Biochimie, biosynthèse et biodistribution
L’AT est une glycoprotéine monocaténaire de 432 acides aminés présentant 3 ponts disulfures (Cys8-Cys128 ; Cys21-Cys95 ; Cys247-Cys430) et 4 sites des N-glycosylation sur les asparagines (N96, N135, N155, N192). La position 135 n’est glycosylée que partiellement en raison d’une séquence consensus de type N-X-S moins favorable que la séquence optimale N-X-T pour l’attachement de la chaine glycane (21). Ainsi, dans le plasma, deux isoformes d’AT circulantes sont retrouvées: l’isoforme α avec 4 chaines N-glycanes (58 kDa) qui représente 90 % de l’AT circulante, et l’isoforme β avec 3 chaines N-glycane (56 kDa) qui compte pour 10% de l’AT plasmatique (22). L’absence de la chaine N-glycane en position 135, région proche du site de liaison à l’héparine, augmente significativement l’affinité de l’AT pour l’héparine. L’AT est synthétisée au niveau hépatique, et circule à une concentration plasmatique moyenne comprise entre 2 et 3 μM (23). Dès lors, les taux d’AT plasmatiques sont diminués chez les patients qui présentent une insuffisance hépatique (24).
La biodistribution de l’AT a été étudiée chez le lapin où l’administration d’AT plasmatique radiomarquée a mis en évidence la répartition de la protéine dans un troisième compartiment extravasculaire en plus de la partie plasmatique et de celle liée à la surface vasculaire. Dans une étude pharmacocinétique chez l’homme, l’AT présente la même distribution : dans le plasma (39,3 %), liée à la surface vasculaire (10,9 %) et extravasculaire (49,6 %) (25). L’AT semble suivre deux étapes de distribution : une première étape de liaison rapide à la surface vasculaire grâce à sa capacité de lier aux héparans sulfates cellulaires ; puis un passage plus lent au niveau du compartiment extravasculaire (26). L’AT possède une demi-vie d’élimination d’environ 2,8 jours (27), et la forme β semble avoir une demi-vie 2,5 fois plus courte que celle de la forme α (28), ceci pouvant s’expliquer par son affinité augmentée pour les héparans sulfates.
Dans la circulation, la clairance de l’AT semble être consécutive à sa complexation avec ses enzymes cibles, en particulier la thrombine. En effet, les complexes thrombine- antithrombine (TAT) sont éliminés très rapidement, en 5 min environ, comparativement à la forme native (29). De plus, la clairance des complexes TAT peut entrer en compétition avec d’autres complexes Serpine-enzyme montrant une voie commune d’élimination. Ces complexes sont clairés au niveau hépatique grâce à la présence de récepteurs Serpine-enzyme
20
comme les Serpin-enzym complex receptor (SEC-R), ou encore, des récepteurs récepteurs de la famille des lipoprotéines (LRP : Low density lipoprotein receptor-related protein). La cytokeratine 18 (CK18) de la famille des filaments intermédiaires, la vitronectine et les protéoglycanes joueraient aussi un rôle dans la clairance des complexes TAT (30). Un modèle intégrant l’ensemble de ces acteurs a été proposé : les complexes TAT se lient aux protéoglycanes à héparans sulfates (HSPGs) par l’intermédiaire de la vitronectine, et ces HSPGs vont permettre le transfert du complexe TAT-vitronectine à la CK18 et au récepteur LRP responsable de leur internalisation (31).
2) Relation structure fonction
L’AT appartient à la superfamille des Serpines dont les membres présentent une très grande analogie structurale.
a) Superfamille des Serpines
En 1980, Hunt and Dayhoff ont été les premiers à montrer des similitudes entre l’AT humaine et l’ovalbumine chez le poulet (32). Plus tard, Carrell a utilisé pour la première fois l’acronyme de SERPIN (pour SERine Protease INhibitors) (33) car la majorité des membres de cette famille étaient des inhibiteurs des serine protéases. Par la suite, plus de 500 protéines de type Serpine ont été identifiées dans le génome des procaryotes et des eucaryotes, et ont été impliquées dans des fonctions biologiques très variées (34). Chez l’homme, 37 Serpines ont été clonées et représentent à elles seules 10% des protéines circulantes totales (19). Elles possèdent très peu d’homologie de séquence (≈25%) les unes par rapport aux autres, mais gardent toutefois une grande homologie de structure dont l’archétype est la structure cristallographique de l’α1-antitrypsine résolue dans les années 80 (35). La structure des serpines se caractérise par la présence de 3 feuillets β (A–C) et de 7 à 9 hélices α (A-I)
(Fig-3). En outre, elles possèdent toutes un feuillet β A central à 5 brins et une boucle réactive
flexible, au sommet de la protéine, capable d’interagir avec les protéases (Fig-3). Chez les eucaryotes, elles sont classées en 16 familles (A-P) et la protéine est désignée de la façon suivante: SERPINXy, « X » indiquant la famille d’appartenance et « y » le numéro de la Serpine dans la famille). L’AT est ainsi nommée SERPINC1 (36).
21
Comme leur acronyme l’indique, la majorité des Serpines sont des inhibiteurs des serine protéases mais certaines participent en outre à d’autres fonctions, comme le transport d’hormones ou la signalisation cellulaire (37). Leur structure caractéristique leur permet de fonctionner comme des inhibiteurs suicides. En effet, la boucle réactive de la Serpine est clivée par les protéases cibles et forme un complexe acyl-enzyme covalent, ceci entrainant un réarrangement structural par l’insertion de la boucle réactive pour former un brin supplémentaire au sein du feuillet β A, tout en entrainant l’enzyme liée vers le bas de la Serpine, comme dans un mécanisme de piège à souris (38).
Chez l’homme, les Serpines possèdent un rôle important dans des phénomènes physiologiques nécessitant une étroite régulation, comme l’inflammation et le système du complément (α-antitrypsine, l’inhibiteur de la protéine C1 du complément), la fibrinolyse (α2
-antiplasmine) et la coagulation (antithrombine, protéase nexine-1, second cofacteur de l’héparine)(39).
Figure 3 : L’archétype de structure tridimensionnelle des Serpines
: «
-1 antitrypsine » lefeuillet A coloré en violet ; feuillet B coloré en orange; feuillet C coloré en vert. 9 hélices (A à I) colorées en gris. Boucle réactive (RCL) colorée en bleu ciel (code PDB 1QLP) (40).
Acylation Translocation
22
La flexibilité de la structure des Serpines est nécessaire à son activité inhibitrice, et des mutations affectant une région charnière (Hinge region dans la Fig-4), responsable de la mobilité de la boucle réactive, peuvent entrainer une rétention intracellulaire, une perte de fonction, ou la génération d’autres conformères (39). Ces dysfonctions et/ou déficits de différentes Serpines peuvent causer de graves pathologies appelées « Serpinopathies » allant de la démence concernant la Neuroserpine, à des emphysèmes pulmonaires pour l’α1-antitrypsine, ou à des thromboses veineuses dans le cas de l’AT (41).
b) Caracteristiques structurales de l’antithrombine native
La première structure cristallographique de l’AT native a été résolue par Carrell R.et al. en 1994 (42). Elle adopte l’arrangement tridimensionnel des Serpines avec 3 feuillets
centraux (A, B, C) et 9 hélices (A à I). Ainsi, les feuillets A, B, et Cde l’AT sont organisés respectivement en 5, 6, et4 brins, respectivement (Fig-4). De plus, comme toutes les Serpines, l’AT présente une séquence de 24 résidus (377-400) organisée en une boucle très flexible exposée au solvant qui est responsable de sa fonction inhibitrice.
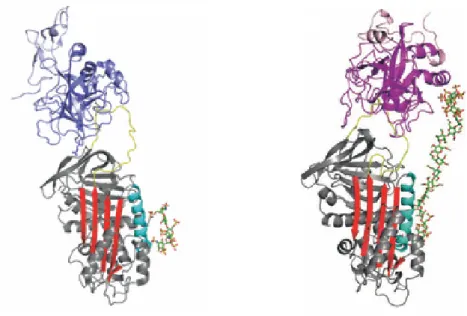
Figure 4 : Structure de l’AT native avec le feuillet β A (bleu), l’hélice D (cyan) constituant le
site de liaison à l’héparine, la boucle réactive (rouge) avec le résidu P1 (arg 393) critique pour l’activité anticoagulante de la Serpine, la région charnière (Hinge, rouge) partiellement insérée dans le feuillet β A. (Code PDB 1T1F) (44)
23
Cette boucle réactive de l’AT possède une arginine en position 393 critique à l’activité de l’AT (43), désignée P1 selon la nomenclature de Schechter et Berger. Les acides aminés situés à l’extrémité N-terminale du résidu P1 sont numérotés P2 jusqu’à P17 alors que les résidus situés en C-terminal du résidu P1 sont désignés P1’ jusqu’à la position P7’.
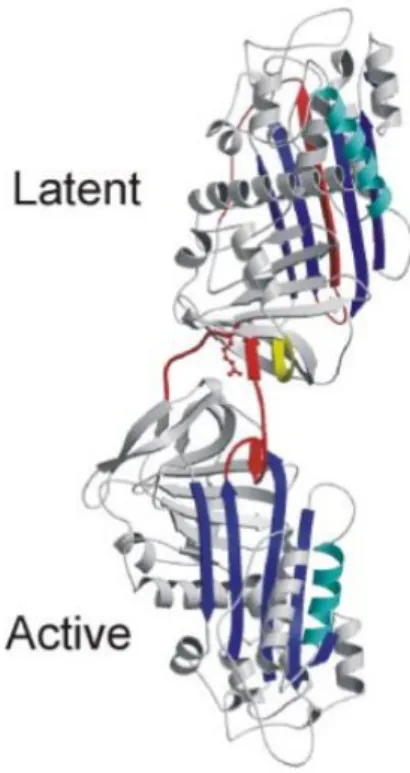
c) Différents conformères d’Antithrombine c.1 La forme latente et clivée
Contrairement à la majorité des protéines, la structure de la forme native de l’AT ne représente pas sa conformation la plus stable. En effet, la boucle réactive est capable de s’insérer dans le feuillet β A constituant le 4ème
brin d’un feuillet qui devient à 6 brins pour adopter une structure plus stable. En absence de clivage de la boucle réactive, cette transition entraine l’apparition d’une forme dite « Latente » qui participerait à la sénescence de la protéine (45).
Cette conformation condamnant la boucle réactive à rester insérée au sein du feuillet β A entraine l’inactivité irréversible de l’AT vis-à-vis de la coagulation (Fig- 5A). De plus, la forme latente de l’AT possède une affinité diminuée à l’héparine et une résistance aux agents dénaturants comme le chauffage jusqu’à 127°C ou l’urée à une concentration de 7 M (46). L’AT latente peut être obtenue in vitro par chauffage à 60°C de l’AT native en présence de citrate ou de sulfate d’ammonium (47) et possède une migration électrophorétique plus rapide que celle de la forme native. In vivo, environ 3% de l’AT totale circulante se convertit en forme latente chaque jour (46). Il existe cependant des variants naturels d’AT (N187K/D, T85M/K) thermolabiles qui se convertissent facilement en forme latente in vitro et conduisent à la présence d’environ 10% de forme latente dans la circulation (48,49). Cette instabilité participe fortement au risque thrombotique observé chez les sujets présentant ces mutations.
Cette conformation hyper stable de la Serpine sous sa forme latente est aussi appelée « relachée » alors que l’AT native adopte une conformation dite « stressée ». En plus de la forme latente, l’AT peut se trouver sous forme clivée. En effet, le clivage au niveau de la boucle réactive par des protéases cibles, ou à différents niveaux dans la boucle réactive, entraine l’insertion de celle-ci au sein du feuillet β A (Fig-5B). L’AT latente et clivée, toutes
24
deux inactives vis-à-vis de la coagulation, possèdent en outre des propriétés antiprolifératives (50,51).
Figure 5 : Structures cristallographiques de la forme latente (A) de l’-1 antitrypsine avec la boucle réactive (jaune) insérée dans le feuillet β (rouge) formant le 4ème brin. (B) Forme d’AT clivée avec la boucle clivée (jaune) insérée au sein du feuillet β (rouge) (52)
c.2 Les hétérodimères
La structure tridimensionnelle de l’AT a été résolue par deux groupes différents en 1994 (54,45). Tous deux ont réussi à obtenir la cristallisation de l’AT, mais sous forme dimérique et non monomérique. En effet, le dimère cristallisé est constitué de l’association non covalente d’une molécule d’AT native et d’une seconde molécule d’AT latente (Fig-6). L’hétérodimère est facilement obtenu dans le plasma après une incubation à 37°C pendant 72 h ou simplement par un mélange équimolaire des deux espèces d’AT, native et latente (55). Cet hétérodimère est caractérisé par une mobilité électrophorétique légèrement plus lente que celle de chacune des deux formes d’AT, latente et native. Deux types de dimères ont été décrits selon qu’il résulte de l’association d’une AT latente avec soit une AT native de type α soit avec l’isoforme β de l’AT, ce dernier hétérodimère apparait le plus stabilisé en présence d’héparine (55). In vivo, l’existence et le rôle potentiel de ces hétérodimères restent cependant peu explorés.
25
Figure 6 : Structure de l’hétérodimère de l’AT formé de deux molécules d’AT montrant
l’interaction étroite de la boucle réactive (rouge) de l’AT native (en bas) avec le brin 2C du feuillet β C (jaune) de l’AT latente (en haut), l’hélice D des deux AT représentée en couleur
Cyan et le feuillet β A en magenta (44).
c.3 Les polymères
Les polymères sont les derniers conformères décrits dans la littérature pour l’AT. En effet, l’AT et certaines Serpines sont capables de s’auto-assembler dans certaines conditions dénaturantes (températures élevées, pH acide…) (56) in vitro ou lors de la présence de mutations particulières. Lors du repliement protéique, les Serpines adoptent un état intermédiaire « polymérogène » transitoire à demi-vie très courte. Très peu de polymères peuvent alors être formés mais dans certains cas, la présence de mutations ralentit la renaturation de cet état transitoire en forme native et favorise cet état intermédiaire polymérisé (52). Deux modèles de polymérisation ont été proposés : Le premier par insertion de la boucle réactive d’un monomère dans le feuillet β d’un second monomère, le deuxième modèle a été mis en évidence par étude de la structure cristallographique d’un dimère d’AT stable après incubation à pH 5.7 à 37°C (57,58). Dans ce modèle, les monomères d’AT adoptent une structure particulière avec l’externalisation d’un ensemble constitué de la boucle (brin 4a) et
26
du brin 5a du feuillet β A, qui viennent s’insérer dans un monomère adoptant la même structure (52).
d) Mécanisme d’inhibition des serine protéases
L’AT suit le mécanisme d’inhibition des membres de la superfamille des Serpines en agissant comme un substrat suicide. En effet, les serine protéases reconnaissent une séquence peptidique particulière et clive la cible après formation d’un complexe acyl-enzyme avec le substrat. Dans le cas d’un substrat classique, il y a une déacylation et libération du substrat clivé. Lors de l’interaction de la protéase avec l’AT, il y a d’abord la formation d’un complexe Michaelis-like non covalent grâce aux chaines latérales des résidus P1-P1’ au niveau de la boucle réactive (37). Cette première étape aboutit à la formation d’une liaison covalente de type ester entre le résidu de la protéase (Ser195) avec le groupement carboxyle du résidu P1 (Arg393) issu du clivage de la liaison P1-P1’ de l’AT. Les résidus P1-P1’ définissent la spécificité de la Serpine envers une protéase cible. Enfin, la boucle réactive de l’AT s’insère au niveau du feuillet β A tout entrainant la protéase liée avec elle comme dans un piège à souris (Fig-7) (37).
Figure 7 : Mécanisme d’inhibition des Serpines en plusieurs étapes, la formation du
complexe Michaelien entre la protéase (bleu) et la Serpine qui aboutit à la formation d’un intermédiaire acyl-enzyme irréversible par l’incorporation de la boucle (jaune) au sein du feuillet β A (rouge), entrainant la protéase (60).
27
La dislocation de la protéase entraine une distorsion de son site actif et permet d’éviter une dissociation du complexe Serpine-enzyme, jouant ainsi le rôle d’un substrat suicide
(Fig-7). Enfin, le changement conformationnel du complexe protéase-Serpine permet alors sa
reconnaissance par des récepteurs responsables de sa clairance rapide de la circulation (59).
La séquence P4-P4′ au sein de la boucle réactive joue un rôle important dans l’activité des Serpines présentant une fonction d’inhibiteur enzymatique. Ainsi, les mutations affectant cette région entrainent une perte d’activité anticoagulante comme pour l’AT. De plus, une seconde région appelée « région charnière » (Fig-4 et Fig-8) possède des résidus à chaines latérales courtes qui permettent la flexibilité de la boucle nécessaire à la formation du complexe Serpine-enzyme (61). En dépit du rôle de l’arginine 393 en P1 qui est critique pour l’activité anticoagulante de l’antithrombine, d’autres résidus au sein de la boucle possède un rôle important, comme le résidu P1’ à chaine latérale courte plutôt polaire qui détermine sa sélectivité. En effet, ce résidu interagit avec la poche S1’ de l’enzyme (adjacente au résidu S1 du site actif), cette poche est étroite pour le FXa et plus large pour la thrombine par l’occupation d’une lysine. Ainsi, la substitution du résidu P1’ de l’AT par une isoleucine conduit à une perte de l’activité inhibitrice vis-à-vis de la thrombine (anti-FIIa) et la conservation de l’activité vis-à-vis du FXa (anti-FXa) (62). Des mutations combinées des résidus P3 et P3’ permettent aussi de dissocier ces deux activités. Enfin, la glycine en P2 semble jouer un rôle dans la présentation du site actif dans une conformation optimale afin de faciliter l’interaction avec la sérine 195 de la poche catalytique de l’enzyme. Elle interagirait avec le résidu S2 apolaire de l’enzyme cible, et des mutations l’affectant entrainent une perte de l’activité anticoagulante (63) (Tab-1).
La structure de l’AT native possède la particularité par rapport aux autres Serpines de présenter une région charnière (P13-P15) G379-S380-E381 partiellement incorporée dans le feuillet A grâce à des liaisons ioniques avec le domaine d’interaction avec l’héparine (64,65). L’expulsion de cette région charnière suffit à l’activation allostérique de l’AT en favorisant son interaction avec les protéases (66). De plus, certaines structures cristallographiques montrent que le résidu P1 est dirigé vers l’intérieur de la protéine (44) grâce à une liaison ionique entre l’arginine 393 et l’acide glutamique 255. Cette indisponibilité du résidu P1 aux protéases permet de placer l’AT dans une conformation moins active mais il semblerait que l’AT native soit en équilibre sous plusieurs conformations dans lesquelles le résidu P1 est plus ou moins exposé (52).
28
Table 1: Mutations au sein de la région P3-P3’ de la boucle réactive obtenues par
recombinaison génétique ou présentes dans des variants naturels chez des patients
e) La potentialisation de l’activité anticoagulante de l’antithrombine par les dérivés hépariniques
Bien que l’AT soit un inhibiteur physiologique lent dans la circulation, son activité est accélérée grâce à sa liaison aux héparans sulfates ou à l’héparine. Ce mécanisme est à la base de l’utilisation des dérivés hépariniques comme médicaments anticoagulants. In vivo, des GAGs de type héparans sulfates tapissent la paroi vasculaire et semblent potentialiser physiologiquement l’activité anticoagulante de l’AT (69). Dès lors, les patients présentant certaines mutations à l’état homozygote touchant la région de l’AT impliquée dans la liaison aux héparans sulfates présentent une augmentation du risque de thrombose (70). Cette région riche en arginines et lysines de l’AT impliquée dans l’accélération de son activité par l’héparine, a été décrite par Rosenberg et Damus en 1973 (71). Par la suite, la caractérisation de variants naturels d’AT présentant une affinité réduite pour l’héparine a permis d’identifier les résidus basiques impliqués dans le site de liaison à l’héparine (72,73). Cette région appelée « heparin binding site » (HBS) est constituée par une partie de l’hélice A, l’extrémité N-terminale, et la totalité de l’hélice D.
L’héparine ou les héparans sulfates présentent une séquence pentasaccharidique spécifique (représentant 30% de l’héparine) responsable de l’activation allostérique de l’AT.
Position Résidu
Mutations recombinantes /variants naturels
Conséquence sur l’activité de l’AT P1 : Arg R393H « Glasgow », R393P
« Pescara »
- Perte de l’activité anticoagulante (67)
P1’ : Ser S394L « Denver », S394V, C - Baisse modérée de l’activité anticoagulante (68) S394G, A, T -Légère baisse de l’activité anticoagulante (68) S394P, M - Perte totale de l’activité anticoagulante
S394I -Conservation de l’activité anti-Xa et perte de l’activité anti-IIa (62)
P2 : Gly G392D « Stockholm », G392P, V, Q, D
- Perte de l’activité anticoagulante (63) P3 : Ala
P3’ : Asn
Double mutant : A391D N396E - Conservation de l’activité anti-Xa et perte de l’activité anti-IIa (62)
29
L’identification de la structure de ce motif, désigné par « DEFGH » où chaque lettre représente un sucre, a permis de mieux comprendre le changement conformationnel de l’AT nécessaire à son activité anticoagulante, et de mettre en évidence le rôle des différents résidus de l’AT impliqués dans la liaison au pentasaccharide : Lys11, Arg13 (extrémité N-terminale) Arg46, Arg47 (hélice A) Lys114, Lys125 et Arg129 (Hélice D)(74). Ces résidus interagissent principalement par l’intermédiaire de liaisons ioniques avec les groupements sulfates et carboxyles du pentasaccharide, et de liaisons hydrogène (59).
L’activation allostérique de l’AT ne participe qu’à l’accélération de son activité inhibitrice vis-à-vis des facteurs FIXa et FXa. En effet, son activité inhibitrice vis-à-vis de la thrombine nécessite la présence de chaines assez longues d’héparine capables de former un complexe ternaire thrombine/héparine/AT.
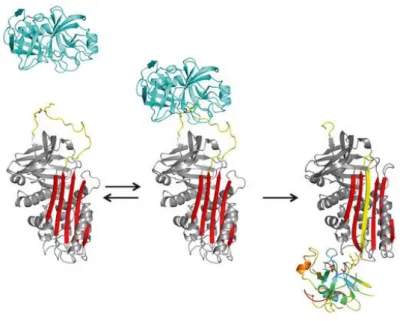
e.1 Activation allostérique de l’AT par le motif pentasaccharidique
La comparaison des structures cristallographiques de l’AT libre et liée à l’héparine montre des changements conformationnels au niveau de l’HBS et de la région charnière. Dans un premier temps, l’extrémité N-terminale de l’hélice A subit une réorientation. Ensuite, l’hélice D subit une rotation avec une élongation de l’extrémité supérieure (située en C-terminal de l’hélice D) et formation d’une nouvelle hélice α P (P pour Pentasaccharide) de l’autre extrémité (en N-terminal de l’hélice D), entrainant finalement un réarrangement des différents résidus basiques impliqués (75). Enfin, la région charnière, enfouie à proximité du feuillet A, est expulsée pour permettre l’exposition de la boucle réactive. En effet, l’activation allostérique de l’AT avec l’élongation de l’hélice D entraine le resserrement du feuillet A et l’expulsion de la boucle réactive qui devient ainsi plus exposée aux enzymes cibles (76)
(Fig-8). En plus de la boucle réactive, une deuxième surface d’interaction de l’AT avec les
protéases a été mise en évidence à partir des années 2000. Située au niveau du feuillet β C au sommet de la protéine, cette région proche de la boucle réactive est appelée « Exosite ». Cet exosite joue un rôle dans l’interaction de l’AT avec les protéases FXa et FIXa, en absence ou en présence d’héparine. L’équipe d’Olson a localisé l’exosite grâce à des mutations de la région du brin 3 du feuillet B (246-258) et mis en évidence deux résidus particulièrement importants (Tyr 253 et Glu 255) et conservés dans les séquences d’AT chez les vertébrés (77).
30
Récemment, un modèle a été proposé pour expliquer le rôle de différents résidus de l’exosite au niveau de la forme native ou activée par l’héparine de l’AT. Sous forme native, l’exosite participe à un équilibre entre des forces d’attraction et de répulsion avec la protéase. Après activation allostérique de l’AT par de l’héparine, les forces de répulsions sont diminuées grâce au changement conformationnel de la protéine (78). Finalement, des interactions directes de l’exosite de l’AT avec le FXa et le FIXa, en plus de l’exposition de la boucle réactive, participent à l’activation allostérique de l’AT vis-à-vis de ces facteurs.
Figure 8 : Activation allostérique de la forme native de l’AT présentant la région charnière
(encadrée) partiellement insérée dans le feuillet β A (rouge), après liaison du pentasaccharide (66).
e.2 Potentialisation de l’activité de l’AT matricielle par l’héparine
L’activation allostérique de l’AT par sa liaison au pentasaccharide n’a qu’un rôle mineur dans l’activité inhibitrice de l’AT vis-à-vis de la thrombine puisqu’elle n’augmente que d’un facteur 2 la vitesse de la réaction d’inhibition alors qu’elle accélère l’inhibition du FXa d’un facteur 300 (59). Cette faible accélération contraste avec l’effet de rapprochement de l’AT et de la thrombine engendré par la présence des chaines hépariniques d’au moins 18 unités saccharidiques, qui entraine une accélération de l’activité anti-IIa d’un facteur 4500 (79). En 2004, la résolution de la structure cristallographique du complexe ternaire «
AT-+ pentasaccharide
Native Activé
31
thrombine-héparine » a permis de confirmer ce mécanisme dit « matriciel » (80). La liaison entre l’AT et l’héparine implique le pentasaccharide et l’HBS de l’AT.
La thrombine, elle, se lie à une autre partie de l’héparine par l’intermédiaire de son « exosite II » riche en résidus basiques (80). Cet effet matriciel semble être aussi retrouvé pour d’autres protéases comme les FXa et FIXa en présence d’ions calcium (81,82)
Figure 9 : Structure des complexes Michaeliens entre l’AT et le FXa en présence du
pentasaccharide (gauche) et la thrombine en présence de la chaine d’héparine (droite) (52)
3) Déficits congénitaux en antithrombine
Les déficits congénitaux en AT sont associés à une augmentation du risque de thrombo-embolie veineuse d’environ 20 fois (70). La plupart des mutations touchant le gène de l’AT « SERPINC1 » sont ponctuelles à transmission autosomale dominante. La très grande majorité des déficits sont hétérozygotes et les rares déficits homozygotes ont été identifiés causant une perte de liaison à l’héparine (AT Budapest) ou une perte de l’inhibition de la thrombine (AT Cambridge II) (83,84). La prévalence des déficits d’AT est comprise entre 1/500 et 1/3000 et ils sont retrouvés chez 2 à 5% des patients souffrant de thrombose veineuse (85). En se basant sur les dosages antigéniques et fonctionnels de l’AT plasmatique utilisés en biologie clinique, les déficits héréditaires en AT ont été classés en 2 groupes (déficits de type I et type II) (86). Une base de données regroupant les mutations du gène de l’AT chez l’homme est accessible en ligne (87). Les déficits de type I sont quantitatifs et par conséquent définis par une diminution parallèle de l’antigène et de l’activité de l’AT,
32
généralement à des taux de l’ordre de 50%. Les déficits de type II sont eux définis par la présence d’une AT présentant une activité anticoagulante réduite.
Ces déficits qualitatifs sont liés à la présence de mutations situées principalement dans 3 régions de l’AT: la région du site actif (Type IIa : positions proches du résidu 394), le site de liaison à l’héparine (Type IIb, Positions 41 ; 47 ; 99 ; 129), et la partie C-terminale (Type IIc : Positions 402 ; 404-407 ; 429). Les variants de la partie C-terminale sont appelés « pléiotropiques » puisqu’ils sont associés non seulement à un taux de sécrétion diminué, mais aussi à une activité abaissée et une affinité à l’héparine diminuée (72). Par ailleurs, les mutations hétérozygotes touchant le site de liaison à l’héparine sont moins thrombogènes (88).
CHAPITRE 2 : LES ANTICOAGULANTS HEPARINIQUES
A. Physiopathologie et épidémiologie de la maladie thromboembolique
Le thrombus, résultant de l’activation de la coagulation et de l’agrégation plaquettaire, peut dans certaines situations pathologiques obturer complètement ou partiellement un vaisseau sanguin. On parle alors de thrombose. Lorsque le thrombus se détache et se retrouve entrainé dans la circulation, en risquant d’obturer un autre vaisseau en aval, on parle d’embolie (89). La thrombose peut prendre différentes formes comme la thrombose veineuse profonde, l’embolie pulmonaire, les accidents vasculaires cérébraux (AVC) ou encore l’infarctus du myocarde.
Les thromboses veineuses profondes sont le plus souvent localisées dans les membres inférieurs et favorisées par trois facteurs principaux regroupés sous le nom de triade de Virchow: (i) la stase ou ralentissement du flux sanguin ; (ii) les lésions de la paroi vasculaire et (iii) la modification de l’hémostase (90). Un déséquilibre de la coagulation, héréditaire ou résultant de différentes situations physiopathologiques (grossesse, cancer, chirurgie…), peut ainsi favoriser l’apparition d’épisodes thrombotiques. La maladie thromboembolique veineuse (MTEV) regroupant les thromboses veineuses profondes et les embolies pulmonaires présente une incidence de 1/1000 (91). L’incidence annuelle de la MTEV est d’environ 120 pour 100 000 en France et entre 60 et 100 pour 100 000 dans le monde. Le taux de mortalité du à la MTEV déclaré en France est de 7.2 pour 100 000, cependant des estimations issues
33
d’autopsies dans le monde montrent entre 0.8 et 1% d’embolies pulmonaires chez les patients hospitalisés (92).
Les thromboses artérielles se manifestent principalement sous forme d’infarctus du myocarde ou d’accidents vasculaires cérébraux (AVC) avec comme étiologie la plus fréquente une défaillance de la paroi vasculaire, comme c’est par exemple le cas dans une maladie chronique, l’athérosclérose.
Le thrombus peut se former dans les artères, les veines, le cœur, ou les capillaires, et il
est composé de cellules sanguines et de fibrine. Cependant, sa composition en proportion de ces différents constituants varie en fonction des conditions hémodynamiques rencontrées lors de sa formation (93). En effet, le flux sanguin élevé présent dans les artères entraine la formation d’un thrombus composé majoritairement d’agrégats plaquettaires et peu de fibrine, alors que dans le système veineux, le thrombus sera formé de globules rouges, très peu de plaquettes et présentera une grande quantité de fibrine. Ainsi, les antiagrégants plaquettaires et les anticoagulants sont tous deux utilisés dans le traitement de la thrombose artérielle (93), alors que dans les thromboses veineuses, seuls les anticoagulants sont indiqués, aussi bien dans la prévention que le traitement de la MTEV (93).
B. Les anticoagulants hépariniques
Les anticoagulants sont des médicaments indispensables et incontournables dans de nombreuses situations cliniques associées à un état procoagulant. Ils sont utilisés aussi bien dans le traitement que dans la prévention des événements thrombo-emboliques. En fonction de leur mode d’administration, on retrouve deux types d’anticoagulants : les anticoagulants oraux et les anticoagulants injectables. Les anticoagulants oraux ont été dominés durant des décennies par une seule classe pharmacologique : « les anti-vitamine K » (AVK) qui agissent en bloquant le cycle de régénération de la vitamine K, et par conséquent la synthèse des facteurs vitamine K-dépendants (II, VII, IX et X). Cependant, une nouvelle classe a vu le jour depuis 2008, les anticoagulants oraux directs (AOD) qui agissent par inhibition directe du FXa (apixaban, rivarixaban, endoxaban) ou de la thrombine (dabigatran).
Les anticoagulants administrés par voie parentérale sont représentés majoritairement par la classe des anticoagulants hépariniques : l’héparine standard ou non fractionnée (HNF),
34
les héparines de bas poids moléculaire (HBPMs) et le fondaparinux. L’héparine standard, le plus ancien anticoagulant utilisé, a été découverte en 1916 par Jay McLean (94). Extraite et purifiée à partir de tissus animaux, elle a été utilisée la première fois en clinique en 1937. En 1982, des préparations de bas poids moléculaire obtenus par fractionnement de l’héparine standard ont été utilisées la première fois en clinique et ont donné naissance aux HBPMs (95). Le fondaparinux, dernier né des dérivés hépariniques, produit par synthèse chimique, a obtenu son autorisation de mise sur le marché (AMM) en 2004 (96). Avant l’arrivée sur le marché des anticoagulants oraux directs, les dérivés hépariniques étaient utilisés en première intention du fait de leur action immédiate. En cas de traitement prolongé, un relai par les AVKs, qui présente un délai d’action de quelques jours, était réalisé. En France, les HBPMs sont largement en tête des utilisations thérapeutiques des dérivés hépariniques avec une augmentation globale des ventes entre 2001 (146 millions de dose définie journalière : DDJ) et 2013 (188 millions de DDJ). Les ventes des HNF sont relativement stables au cours du temps entre 20 et 24 millions de DDJ alors que celles du fondaparinux, ont augmenté progressivement jusqu’en 2010 (16 millions de DDJ) et semblent s’être stabilisées depuis (97). Dans la suite de ce travail, nous nous intéresserons aux anticoagulants hépariniques qui agissent comme cofacteurs de l’AT.
1) L’héparine non fractionnée standard a) Structure
L’héparine est un polymère osidique linéaire de type glycosaminoglycane. Elle est constituée d’une répétition disaccharidique de base associant un acide uronique et une glucosamine. Les unités osidiques sont substituées par des groupements sulfates à différentes positions (N-sulfatation, 6-O-sulfatation, 3-O-sulfatation, 2-O-sulfatation) (102). Trois types de séquence sont retrouvés dans une chaîne d’héparine : une région majoritaire (l’unité disaccharidique régulière trisulfatée), une séquence variable, minoritaire et intercalaire (l’unité disaccharidique disulfatée), et enfin une séquence pentasaccharidique représentant le tiers de la séquence d’héparine. Cette région particulière très sulfatée est caractérisée par une unité centrale de glucosamine 3-O-sulfaté, très importante dans l’activité anticoagulante de l’héparine (98). Les osides composant le pentasaccharide sont nommés, de gauche à droite, DEFGH (Fig-10), et la substitution du groupement sulfate en position 3 du motif F entraine l’abolition de l’activité anticoagulante du motif pentasaccharidique (99).