Multi-elemental analysis of solidified mineral melt samples by laser-induced breakdown spectroscopy coupled with a linear multivariate calibration
Texte intégral
Figure
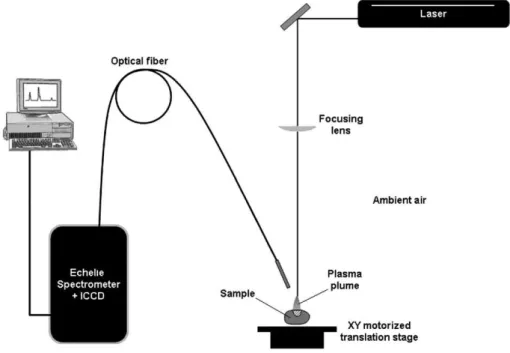
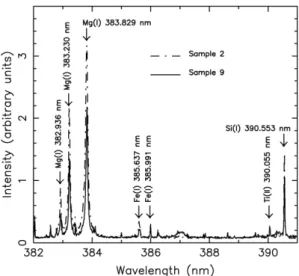
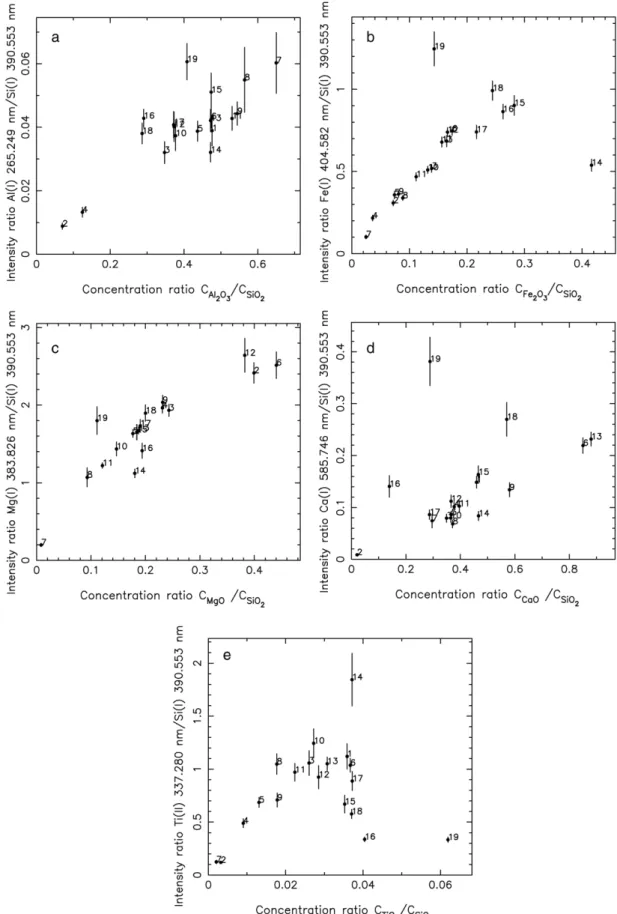
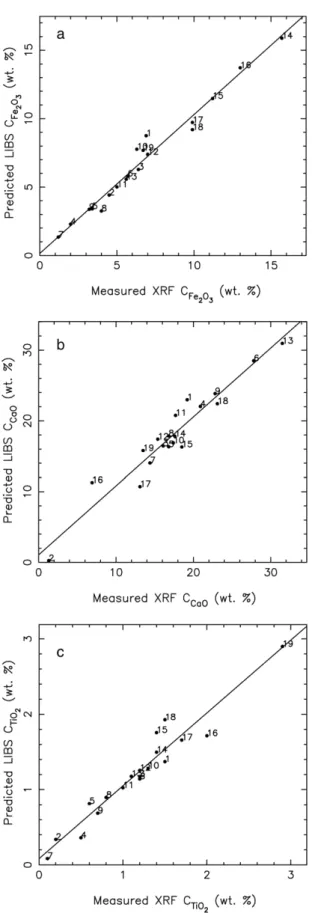
Documents relatifs
Belov, A., Eroshenko, E., Heber, B., Yanke, V., Raviart, A., R¨ohrs, K., M¨uller-Mellin, R., Kunow, H., Wibberenz, G., and Paizis, C.: Latitudinal and radial variation of >2
This paper proposes a domain-specific language (DSL), QIRAL, for the description of Lattice QCD simulations, and its compiler to generate parallel code.. A domain-specific language
Strongly and densely punctuate, punctures as large as or larger than areas between them, elongated, sometimes almost confluent and forming short striae, subrounded on head sides
The study area included a control site in Imdan at about 30 km, eastern Béjaia department and 9 sampling sites located in Bejaia city (Sidi Ahmed (SA), Targa Ouzemour (TO),
ةفرعم ىلع دعاست لا ةيئاهن ميوقت ةيلمع وهف ،ةيساردلا ةنسلا ةياهن يف ايرولاكبلا ناحتما متي و رخآ هنأ ىلع ةنسلا رخآ ناحتما ىلإ نورظني ذيملاتلا حبصأ ثيح
These include the PA700 (19S complex), which assembles with the 20S and forms the 26S proteasome and allows the efficient degradation of proteins usually labeled by ubiquitin
Yet another embodiment of the invention is shown in Fig. This embodiment allows energy to be recovered during both compression and extension of the piston. As the piston 12,
A pooled genome-wide screening strategy to identify and rank influenza host restriction factors in cell-based vaccine production platforms..
