HAL Id: pastel-00578502
https://pastel.archives-ouvertes.fr/pastel-00578502
Submitted on 21 Mar 2011
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
- SILICE OU OXYDE DE TITANE GENERE IN SITU :
SYNTHESE, STRUCTURE ET PROPRIETES
Amadou Lamine Diop
To cite this version:
Amadou Lamine Diop. NANOCOMPOSITES POLY(DIMETHYLSILOXANE) - SILICE OU
OXYDE DE TITANE GENERE IN SITU : SYNTHESE, STRUCTURE ET PROPRIETES. Chimie-Physique [physics.chem-ph]. Université Pierre et Marie Curie - Paris VI, 2010. Français. �pastel-00578502�
THESE DE L’UNIVERSITE PARIS VI - Pierre et Marie Curie UFR de Physique
présenté par Amadou Lamine DIOP
pour obtenir le titre de
DOCTEUR DE L’UNIVERSITE PARIS VI
Ecole doctorale : La Physique de la particule à la matière condensée
NANOCOMPOSITES
POLY(DIMETHYLSILOXANE) - SILICE OU OXYDE DE TITANE
GENERE IN SITU : SYNTHESE, STRUCTURE ET PROPRIETES
soutenue le 15 mars 2010
devant le jury composé de :
Mme. Liliane BOKOBZA ESPCI Paris Directrice de Thèse
Mme. Régine PERZYNSKI Université Paris VI Présidente
Mme. Madeleine DJABOUROV ESPCI
M. Bassel HAIDAR ISM Mulhouse Rapporteur
M. Alain PONTON Université Paris VII Rapporteur
M. Pierre-Antoine ALBOUY LPS Orsay
Remerciements
Ce travail a été réalisé au Laboratoire de Physico-Chimie des Polymères et des Milieux Dispersés (PPMD) de l’Ecole Supérieure de Physique et Chimie Industrielle de la ville de Paris, sous la direction du professeur BOKOBZA Liliane.
J’exprime toute ma reconnaissance au professeur BOKOBZA Liliane pour sa disponibilité et la compréhension dont elle a fait montre pendant ces années. Elle a su me guider au cours de ce travail tout en me laissant souvent une grande autonomie.
Mes plus vifs remerciements vont à M. HAIDAR Bassel de l’Institut de Sciences des Matériaux de Mulhouse et M. PONTON Alain de l’Université Paris VII, pour l’intérêt qu’ils ont porté à ce travail en acceptant d’en être les rapporteurs.
Je remercie aussi Mme. PERZYNSKI Régine, Mme DJABOUROV Madeleine, M. ALBOUY Pierre-Antoine et M. BONN Daniel de m’avoir fait l’honneur de faire partie du jury.
Un grand merci à KOLODZIG Mélanie qui a beaucoup fait pour me mettre à l’aise à mon arrivé au laboratoire. Je la remercie aussi pour sa gentillesse, sa disponibilité et pour toute l’aide précieuse qu’elle m’a apportée.
Mes remerciements vont à MARIOT Sandrine pour son aide et ses conseils expérimentaux lors des manipulations de DSC et d’ATG. Son apport a été très précieux.
Merci à MONTES Hélène pour son aide en rhéologie et pour les solutions expérimentales qu’elle m’a fournies notamment pour les problèmes de collage que j’ai rencontrés.
Je remercie chaleureusement GARNAUD Gilles pour sa gentillesse, sa disponibilité et pour tous les services qu’il a aimablement acceptés de me rendre durant toutes ces années.
Merci à toutes les personnes (Vincent, Leila, Marjolie) avec qui j’ai partagé le bureau par moment pour l’atmosphère conviviale particulièrement à RAHMANI Mostafa.
Au laboratoire LPS d’Orsay, encore merci à Pierre-Antoine ALBOUY d’avoir mis une machine de traction à notre disposition avec beaucoup de gentillesse. Merci aussi à KLEIN Vincent d’avoir pris de son temps et de s’être déplacé jusqu’à l’ESPCI pour configurer la machine de traction.
Merci à tous ceux qui nous ont permis de faire des mesures de DSC quand notre appareil était en panne notamment Severin.
Enfin, je n’oublie pas bien sur mes amis, ma famille : mes parents, mes tantes, mes oncles, ect. Il y a deux personnes sans qui rien n’aurait été possible et pour qui j’ai une pensée toute particulière mes grands-parents maternels : Merci grand-mère et grand-père.
Introduction
Les nanotechnologies sont reconnues comme une direction de recherche multidisciplinaire très prometteuse et en pleine émergence. Dans le domaine des matériaux, l’élaboration de nanocomposites polymère est une activité qui suscite un intérêt sans cesse grandissant et dont les résultats pourraient élargir le champ d’application des polymères. En effet, l’introduction de particules de taille nanomé-trique dans une matrice polymère permet d’obtenir des matériaux aux propriétés améliorées (méca-niques, électriques, optiques) ou nouvelles (retardateur de feu ou effet barrière).
Une grande variété de nanoparticules a été étudiée dans le but d’améliorer les polymères pour leurs applications. Les argiles (silicates lamellaires) sont les nanocharges qui ont reçu les plus vifs intérêts de la part de la communauté scientifique. Un certain nombre de nanocomposites basé sur des silicates ont été commercialisé récemment, composé de polyamide-feuillets nanocomposites (Toyota 1992), polypropylène renforcé d’argile (Général Motors 2002). Un attrait important est également porté sur d’autres charges nanométriques comme les nanotubes de carbone, la silice, le noir de carbone, l’oxyde de titane, etc. qui peuvent être classés en fonction du nombre de dimension à l’échelle du nanomètre. Les activités sur les nanocomposites polymères se concentrent sur les méthodes de synthèse et de caractérisation pour distinguer les différents facteurs influant sur l’effet renforçant. Les méthodes de synthèse les plus souvent explorées vont du mélange direct à des techniques plus élaborées (polyméri-sation in situ, mélange à l’état fondu, technique de modification de la surface de la charge). Les aspects souvent explorés pour l’étude des matériaux nanocomposites sont : la morphologie, l’état de disper-sion, les caractéristiques de surface de la charge qui déterminent l’interaction particule-matrice. Ces facteurs conditionnent le comportement des matériaux (mécanique, électrique, optique, thermique). Certains polymères comme le poly(diméthylsiloxane) ont des propriétés mécaniques très faibles d’où il est nécessaire de les renforcer par des particules. Dans cette optique, les charges utilisées sont la silice, l’oxyde de titane, le noir de carbone et le zirconium pour n’en citer que quelques types les plus couramment utilisées. Ces dernières années, une nouvelle méthodologie de renforcement consistant à générer les particules in situ par le procédé sol-gel a fait l’objet de beaucoup d’études.
Le procédé sol-gel consiste en l’hydrolyse et la condensation d’un alcoxyde M(OR)men présence d’un
catalyseur pour former M-O-M. Il s’utilise sous différentes formes pour maîtriser la composition et la structure du nanocomposite. Une solution consiste à synthétiser les composantes in situ : les charges dans le polymère, le polymère en présence des nanocharges ou encore les deux composantes simulta-nément, pour former des matériaux hybrides organiques-inorganiques.
La méthode de génération de particules de silice par procédé sol-gel au sein d’une matrice poly-mère préformée a été initiée par Mark. Plusieurs études ont été menées avec la même méthode dans différentes matrices (poly(diméthylsiloxane), résines époxy, thermoplastiques). Elle fournit de bons résultats, c’est la raison pour laquelle nous l’avons étendue à la génération de particules d’oxyde de titane dans une matrice préformée de poly(diméthylsiloxane).
L’objet de ce travail a été de synthétiser puis comparer des nanocomposites PDMS −T iO2et PDMS − S iO2pour lesquels les deux types de particules ont été générés in situ par voie sol-gel. Il a fallu adapter
la synthèse in situ de silice (Mark, Ludivine) par voie sol-gel à la synthèse de l’oxyde de titane au sein de la matrice PDMS. En effet, vu les différences (réactivité, viscosité) entre les alcoxydes de titane et de silicium, la transposition de la méthode n’est pas directe. Nous avons essayé de voir comment les paramètres de synthèse (catalyseur, nature de l’alcoxyde) interviennent dans la structure de la phase minérale. Une caractérisation des matériaux a été menée par différentes méthodes pour faire le lien entre synthèse, structure et propriétés. En conséquence, ce manuscrit est organisé de la manière sui-vante :
pelle quelques caractéristiques principales des élastomères et particulièrement du PDMS ainsi que les notions générales de l’élasticité caoutchoutique et du renforcement. Ce chapitre revient aussi sur le principe de synthèse des particules minérales par voie sol-gel et son utilisation pour le renforcement. Les charges utilisées (silice : S iO2, oxyde de titane : T iO2), leurs propriétés de surface et leur structure sont mises avant dans ce chapitre.
Enfin, ce chapitre apporte des informations sur les différentes catégories de nanocharges disper-sées dans un polymère et les méthodes de synthèse des nanocomposites.
+ Chapitre 2 : Synthèse des matériaux
Ce chapitre décrit la méthode de synthèse de particules de silice et d’oxyde de titane in situ au sein d’une matrice polymère préformée, l’objectif étant d’obtenir un protocole expérimental re-productible. Le taux d’avancement de la réaction de synthèse des particules minérales est suivi par spectroscopie d’absorption infrarouge et le taux de charge dans le polymère est déterminé par pesée. La présence d’eau dans les matériaux finaux est mise en évidence par spectroscopie infrarouge. Les doutes liés à la méthode de synthèse ont été levés par des mesures de MET et de RMN.
+ Chapitre 3 : Morphologie des particules in situ
Ce chapitre se concentre sur l’étude de l’état de dispersion, la forme et la taille des charges générées in situ. Deux techniques sont utilisées pour cette étude : la microscopie électronique à transmission et la diffusion de neutrons aux petits angles. Nous avons cherché à voir l’influence de différents paramètres notamment des conditions de synthèse (catalyseur, nature de la charge) sur la morphologie des objets obtenus.
+ Chapitre 4 : Etude de l’interaction matrice-charge in situ
L’analyse calorimétrique et le gonflement sont très souvent utilisés pour mettre en évidence l’interaction polymère-charge et donner la force des liaisons à l’interface. A ces deux méthodes d’analyses, des compléments par RMN du proton et mesure diélectrique sont évoqués sur la mobilité des chaînes et l’existence de couche immobilisée à l’interface polymère-charge. Les résultats obtenus par ces différentes méthodes montrent une différence d’interactions entre les systèmes PDMS − S iO2et ceux PDMS − T iO2. Néanmoins dans les deux systèmes, il apparaît
une forte interaction PDMS-particule qui induit une augmentation de la densité de réticulation et une restriction de la mobilité des chaînes dans les matériaux. Dans tous les systèmes, nous avons noté l’existence d’une couche de polymère immobilisée, mais la manière dont elle est mise en évidence dépend de la charge (S iO2ou T iO2).
+ Chapitre 5 : Stabilité thermique et dégradation
La stabilité thermique des échantillons ainsi que leurs mécanismes de dégradation sont évoqués dans ce chapitre. Il montre notamment un retard à la dégradation obtenu pour les échantillons chargés de S iO2par rapport au PDMS non-chargé. Ce retard est lié au taux de charge et il existe
un seuil en taux de charge au-delà duquel l’ajout de charge devient défavorable à la stabilité thermique.
Pour les échantillons T iO2, un effet contraire est observé, le début de dégradation est anticipé
par rapport au réseau pur. Cette accélération de la dégradation est attribuée à la nature cataly-tique des particules d’oxyde de titane. En fin de dégradation, les réseaux PDMS-T iO2donnent
des résidus beaucoup plus importants par rapport aux réseaux PDMS − S iO2 en raison de la
présence : d’un environnement Ti-OH qui joue le rôle de catalyseur et d’une couche de PDMS plus importante dans les systèmes PDMS − T iO2.
+ Chapitre 6 : Etudes du comportement mécanique
Ce chapitre porte sur une manifestation directe du renforcement c’est-à-dire le comportement mécanique des matériaux. Nous avons cherché à faire le parallèle entre les deux types de charge en essayant de voir comment les différences (nature de la charge, niveau de l’interaction PDMS-charge, morphologie) intervenaient sur la nature du renforcement. Nous avons sollicité nos ma-tériaux de différentes manières :
. Traction uniaxiale ;
. Cycle de déformation (effet Mullins) à différentes températures ; . Sollicitation dynamique (effet Payne).
Bien que les deux types de charge donnent un bon renforcement, nous avons pu constater que le renforcement n’était pas de même nature en regardant les courbes de traction uniaxiale. Des similitudes et des différences sont observées sur le renforcement apporté par les deux charges. Un comportement plastique observé pour les forts taux de charge dans les échantillons S iO2
apparaît même pour les faibles taux de chargement en T iO2. Concernant l’effet Payne, il est
Liste d’abréviations
C ATG : Analyse Thermogravimétrique
C ATR-FTIR Attenuated Total Reflectance-Fourier Transform Infrared C dA : diacétate de dibutyle étain
C DEA : Diétylamine
C dL : Dilaurate dibutyle étain
C DTG : Derivative Thermogravimetric C DSC : Differential Scanning Calorimetry C HCl : Acide chlorhydrique
C IR : Infrarouge
C MET : Microscopie électronique à transmission C MMA : Méthylmétacrylate
C MWNT : Multi-walled nanotube C pce : Pour cent
C PDMS : Poly(diméthylsiloxane) C PMMA : Polyméthylmétacrylate C PMPS : Poly(méthylphénylsilane) C RMN :Résonance Magnétique Nucléaire C S iO2: Silice
C TEOS : Tétraétoxysilane
C TGA : Thermogravimetric Analysis C TIBO : n-butoxyde de titane C T iO2: Oxyde de titane
C TSDC : Thermally Stimulated Depolization Currents C SANS : Small Angle Neutron Scattering
C SAXS : Small Angle X-Ray Scattering C SBR : Styrène-butadière rubber
Table des matières
Introduction v
1 Généralités 5
1.1 Elastomères siliconés : Poly(diméthylsiloxane) . . . 7
1.2 Elasticité caoutchoutique . . . 8
1.2.1 Modèle affine . . . 8
1.2.2 Modèle fantôme . . . 8
1.2.3 Modèle des jonctions contraintes . . . 9
1.2.4 Modèle semi-empirique de Mooney-Rivlin . . . 9
1.2.5 Modèle de Rubinstein et Panyukov . . . 9
1.3 Renforcement . . . 9
1.4 Phénomènes de dissipation d’énergie . . . 11
1.4.1 Effet Payne . . . 11
1.4.2 Effet Mullins . . . 12
1.5 Synthèse des particules minérales par voie sol-gel . . . 13
1.5.1 Mécanisme de formation des gels polymériques . . . 13
1.5.1.1 Mécanisme réactionnel . . . 14
1.5.1.1.1 Hydrolyse . . . 14
1.5.1.1.2 Condensation . . . 14
1.5.1.2 Nature de l’atome métallique . . . 17
1.5.1.3 Nature des groupements alkyles . . . 17
1.5.1.4 Influence du catalyseur et du pH . . . 17
1.5.1.5 Influence de la quantité d’eau . . . 18
1.5.2 Géométrie fractale . . . 19
1.5.3 Processus de croissance . . . 20
1.5.3.1 Processus de croissance à l’équilibre . . . 20
1.5.3.2 Processus cinétique de croissance . . . 20
1.5.3.2.1 Croissance "monomère-cluster" . . . 20
1.5.3.2.2 Croissance "cluster-cluster" . . . 21
1.6 Les charges . . . 22
1.6.1 La silice . . . 22
1.6.1.1 Chimie de surface de la silice . . . 22
1.6.1.2 Structure chimique de la silice . . . 25
1.6.2 L’oxyde de titane . . . 26 1.6.2.1 Chimie de surface de T iO2 . . . 26 1.6.2.2 Applications de T iO2 . . . 28 1.7 Matériaux nanocomposites . . . 28 1.7.1 Définition . . . 28 1.7.2 Différents nanocomposites . . . 29
1.7.2.1 Nanocomposites à base de nanotubes de carbone . . . 29
1.7.2.2 Nanocomposites à base de nano-feuillets . . . 29
1.7.2.3 Nanocomposites à base de nanoparticules sphériques . . . 30
1.7.2.3.2 Le procédé sol-gel . . . 30
1.7.2.3.3 Le greffage in situ et polymérisation . . . . 31
1.7.3 Réseaux interpénétrés . . . 31
2 Synthèse et caractérisation physico-chimique 37 2.1 Formation des réseaux . . . 39
2.2 Réticulation par hydrosilylation . . . 39
2.3 Réticulation par condensation des silanols . . . 39
2.3.1 Protocole expérimental . . . 40
2.3.2 Quantités mises en jeu . . . 40
2.4 Génération des charges . . . 41
2.4.1 Protocoles expérimentaux . . . 41
2.4.1.1 Sans catalyseur . . . 41
2.4.1.2 Catalyse étain et acide . . . 42
2.4.1.3 Catalyse basique . . . 42
2.4.2 Choix des alcoxydes . . . 42
2.4.3 Génération des charges : résultats . . . 43
2.4.3.1 Etape d’absorption des précurseurs . . . 43
2.4.3.2 Etape de génération des charges : résultats . . . 45
2.4.3.2.1 Présentation des spectres FTIR-ATR du PDMS et des pré-curseurs . . . 45
2.4.3.2.2 Catalyse à base d’étain . . . 47
2.4.3.2.3 Catalyse basique . . . 52
2.4.3.2.4 Catalyse acide . . . 54
2.5 Présence d’eau dans les matériaux . . . 56
2.6 Questions suscitées par la méthode de synthèse . . . 59
3 Morphologie des particules in situ 65 3.1 Techniques expérimentales . . . 67
3.1.1 Microscopie électronique à transmission (MET) . . . 67
3.1.2 Diffusion des neutrons aux petits angles (SANS : Small Angle Neutron Scat-tering) . . . 67
3.2 Généralités sur la diffusion de rayonnement aux petits angles . . . 67
3.3 Résultats obtenus en MET . . . 70
3.3.1 Echantillons catalysés par un dérivé de l’étain . . . 70
3.3.1.1 Premières constatations pour les échantillons chargés de S iO2 . . . 70
3.3.1.2 Premières constatations pour les échantillons chargés de T iO2 . . . 72
3.3.1.3 Analyse granulométrique . . . 72
3.3.1.3.1 Présentation de la méthode . . . 72
3.3.1.3.2 Application aux échantillons chargés de S iO2 . . . 75
3.3.1.3.3 Application aux échantillons chargés de T iO2 . . . 77
3.3.2 Echantillons basiques . . . 79
3.3.2.1 Premières constatations pour les échantillons chargés de S iO2 . . . 79
3.3.2.2 Analyse granulométrique pour les échantillons chargés de S iO2 . . 80
3.3.2.3 Premières constatations pour les échantillons chargés de T iO2 . . . 81
3.3.2.4 Analyse granulométrique pour les échantillons chargés de T iO2 . . 81
3.4 Résultats SANS . . . 82
3.4.1 Résultats . . . 82
3.4.2 Dimensions fractales . . . 82
3.4.3 Maximum de corrélation . . . 83
TABLE DES MATIÈRES
4 Interaction PDMS-charge in situ 89
4.1 Techniques expérimentales . . . 91
4.1.1 DSC : Analyse calorimétrique différentielle . . . 91
4.1.2 Gonflement des réseaux élastomères chargés . . . 91
4.2 Etude de la mobilité du PDMS par DSC : influence des charges in situ sur la cristalli-sation et la fusion du PDMS . . . 92
4.2.1 Généralités sur la cristallisation, la transition vitreuse et la fusion . . . 92
4.2.2 Etude des différentes transitions : transition vitreuse, cristallisation et fusion . 93 4.2.2.1 Echantillon non-chargé . . . 93
4.2.2.2 Transition vitreuse . . . 94
4.2.3 Influence des charges générées in situ sur la cristallisation du PDMS . . . . . 95
4.2.3.1 Thermogrammes de cristallisation . . . 95
4.2.3.2 Evolution du taux de cristallinité avec la concentration en charge in situ . . . . 96
4.2.3.3 Evolution de la température de cristallisation avec la concentration en charge in situ . . . . 99
4.2.4 Influence des charges in situ sur la fusion du PDMS . . . 100
4.2.4.1 Thermogrammes de fusion . . . 100
4.3 Gonflement des matériaux chargés . . . 102
4.3.1 Généralités et résultats . . . 102
4.3.2 Modèle de Kraus . . . 105
4.3.2.1 Présentation du modèle . . . 105
4.3.2.2 Application du modèle . . . 107
4.3.2.2.1 Application du modèle aux systèmes chargés de S iO2 . . 107
4.3.2.2.2 Application du modèle aux systèmes chargés de T iO2 . . 108
4.3.2.2.3 Récapitulatif . . . 109
4.3.3 Modèle de Lequeux . . . 110
4.3.3.1 Présentation du modèle . . . 110
4.3.3.2 Application du modèle sans couche immobilisée . . . 111
4.3.3.3 Application du modèle avec couche immobilisée . . . 112
4.4 Compléments par RMN1H et TSDC . . . 114
5 Dégradation thermiques des nanocomposites 121 5.1 Technique expérimentale . . . 123
5.2 Dégradation du PDMS non-chargé . . . 124
5.3 Dégradation des réseaux chargés . . . 125
5.3.1 Dégradation du PDMS chargé de silice . . . 125
5.3.2 Dégradation du PDMS chargé à l’oxyde de titane . . . 131
5.4 Comparaison S iO2-T iO2 . . . 136
6 Comportement mécanique des matériaux 139 6.1 Techniques expérimentales . . . 141
6.1.1 Essais de traction simple . . . 141
6.1.2 Essai cyclique à déformation imposée croissante : Effet Mullins . . . 142
6.1.3 Etude du comportement dynamique aux faibles déformations . . . 142
6.2 Etude de traction simple . . . 143
6.2.1 Comportement du réseau non chargé . . . 143
6.2.2 Comportement des réseaux chargés . . . 145
6.2.2.1 Premières constatations : Echantillons catalysés par le dA . . . 145
6.2.2.2 Influence de la nature du catalyseur utilisé pour la génération des charges . . . 147
6.2.2.3 Représentation de Mooney-Rivlin . . . 149
6.2.2.4 Effet du taux de charge sur le module . . . 151
6.3.1 Rappels sur l’interprétation de l’effet Mullins . . . 155
6.3.2 Résultats . . . 157
6.3.2.1 Echantillon non-chargé . . . 157
6.3.2.2 Echantillons chargés de silice . . . 158
6.3.2.3 Echantillons chargés d’oxyde de titane . . . 158
6.3.2.4 Etude comparative de l’endommagement à même taux de charge . 159 6.3.3 Etude de l’effet Mullins à différentes températures . . . 160
6.4 Etude de l’effet Payne . . . 166
6.4.1 Rappels sur l’interprétation de l’effet Payne . . . 166
6.4.2 Résultats et discussions . . . 167 6.4.2.1 Echantillons chargés de S iO2 . . . 167 6.4.2.2 Echantillons chargés de T iO2 . . . 169 Conclusions et perspectives 175 Résumé 179 Abstract 181 Bibliographie : table complète 183 Bibliographie du Chapitre 1 . . . 187 Bibliographie du Chapitre 2 . . . 189 Bibliographie du Chapitre 3 . . . 191 Bibliographie du Chapitre 4 . . . 193 Bibliographie du Chapitre 5 . . . 195 Bibliographie du Chapitre 6 . . . 198
Chapitre 1
Généralités
Sommaire
1.1 Elastomères siliconés : Poly(diméthylsiloxane) . . . 7
1.2 Elasticité caoutchoutique . . . 8
1.2.1 Modèle affine . . . 8
1.2.2 Modèle fantôme . . . 8
1.2.3 Modèle des jonctions contraintes . . . 9
1.2.4 Modèle semi-empirique de Mooney-Rivlin . . . 9
1.2.5 Modèle de Rubinstein et Panyukov . . . 9
1.3 Renforcement . . . 9
1.4 Phénomènes de dissipation d’énergie . . . 11
1.4.1 Effet Payne . . . 11
1.4.2 Effet Mullins . . . 12
1.5 Synthèse des particules minérales par voie sol-gel . . . 13
1.5.1 Mécanisme de formation des gels polymériques . . . 13
1.5.1.1 Mécanisme réactionnel . . . 14
1.5.1.2 Nature de l’atome métallique . . . 17
1.5.1.3 Nature des groupements alkyles . . . 17
1.5.1.4 Influence du catalyseur et du pH . . . 17
1.5.1.5 Influence de la quantité d’eau . . . 18
1.5.2 Géométrie fractale . . . 19
1.5.3 Processus de croissance . . . 20
1.5.3.1 Processus de croissance à l’équilibre . . . 20
1.5.3.2 Processus cinétique de croissance . . . 20
1.6 Les charges . . . 22
1.6.1 La silice . . . 22
1.6.1.1 Chimie de surface de la silice . . . 22
1.6.1.2 Structure chimique de la silice . . . 25
1.6.2 L’oxyde de titane . . . 26 1.6.2.1 Chimie de surface de T iO2 . . . 26 1.6.2.2 Applications de T iO2 . . . 28 1.7 Matériaux nanocomposites . . . 28 1.7.1 Définition . . . 28 1.7.2 Différents nanocomposites . . . 29
1.7.2.1 Nanocomposites à base de nanotubes de carbone . . . 29
1.7.2.2 Nanocomposites à base de nano-feuillets . . . 29
1.7.2.3 Nanocomposites à base de nanoparticules sphériques . . . 30
1.1. ELASTOMÈRES SILICONÉS : POLY(DIMÉTHYLSILOXANE)
1.1 Elastomères siliconés : Poly(diméthylsiloxane)
Les élastomères siliconés sont des systèmes polymères très étudiés. Les silicones ou polyorga-nosiloxanes sont de formule R0(S iOR
2)R0. Le plus étudié d’entre eux est le poly(diméthylsiloxane)
(PDMS). Il est constitué d’unités monomères de masse molaire 74g.mol−1et de formule :
Figure 1.1 –Motif du PDMS
Initialement vers 1940, le PDMS était utilisé uniquement dans l’isolation. Depuis, son utilisation s’est diversifiée. Cette diversification est due aux propriétés suivantes [1] :
+ Biocompatibilité ;
+ Faible évolution des propriétés physiques (viscosité, propriétés diélectriques, capacité ther-mique, etc...) avec la température ;
+ Isolation thermique ;
+ Hydrophobie et perméabilité aux petites molécules O2et CO2;
+ Résistance aux UV ;
+ Inertie chimique à l’oxydation et l’hydrolyse due à la solidité des liaisons S i − O et S i − C. Sa configuration géométrique (le plus grand angle S i − O − S i est de 143°, la distance interatomique S i − O mesure 0.164nm) et l’absence de substituant sur l’atome d’oxygène lui confèrent une chaîne flexible et mobile. Ces caractéristiques de la chaîne entraînent, en plus des propriétés citées plus haut, un comportement élastique lorsque le PDMS est réticulé. Nous rappellerons aussi quelques notions essentielles concernant l’élasticité caoutchoutique.
Caractéristiques PDMS
Densité 0,97g.cm−3
Température de transition vitreuse ≈ -123°
Température de fusion ≈ -45°C
Tension superficielle(mN.m−1) 21
Indice de réfraction 1,4
Table 1.1 –Caractéristiques principales du PDMS
Néanmoins le PDMS présente des propriétés mécaniques faibles ainsi qu’en témoigne la courbe force-allongement représentée sur la figure 1.2. Le module élastique et les caractéristiques de rup-ture (déformation et contrainte à la ruprup-ture) peuvent être significativement améliorés par l’ajout de particules minérales. A l’inverse, le caoutchouc naturel qui cristallise sous traction, présente un
auto-renforcement qui se caractérise par une forte remontée de la contrainte aux grandes déformations.
Figure 1.2 –Comparaison des propriétés mécaniques du PDMS pur et du caoutchouc naturel
1.2 Elasticité caoutchoutique
L’élasticité caoutchoutique est essentiellement de nature entropique. Elle peut permettre d’étudier le comportement de matériaux capables de supporter des déformations importantes et réversibles. L’étude du PDMS entre dans ce cadre là. En effet, le PDMS se caractérise par une température de transition vitreuse très basse (Tg= −123°C) qui résulte de la grande flexibilité et de la mobilité de ses chaînes. Quelques notions simples de l’élasticité caouchoutique sont abordées ci-dessous au travers des principaux modèles.
1.2.1 Modèle affine
C’est un modèle développé par Kuhn, Wall et Flory [2]. Il repose sur le fait que le réseau est considéré idéal. Il n’existe pas d’interactions entre les chaînes en dehors des jonctions et le déplace-ment des jonctions par rapport à la déformation macroscopique est affine et se fait à volume constant. L’équation suivante donne la contrainte nominale en fonction de la déformation sous ces conditions.
σnom= SF 0 = NkT V0 (λ − 1 λ2) (1.1)
F est la force exercée et S0la section initiale de l’échantillon.
λ = L
L0 est l’allongement macroscopique de l’échantillon et N le nombre de chaînes du réseau.
1.2.2 Modèle fantôme
Dans un réseau fantôme, les jonctions et les chaînes fluctuent au cours du temps sans être gênées par la présence des chaînes voisines. Les positions moyennes des jonctions varient linéairement avec la déformation macroscopique et l’amplitude des fluctuations ne dépend pas de l’état de déformation [3]. Dans ces conditions, l’équation de la contrainte nominale s’écrit :
σnom= (1 − 2 f) NkT V0 (λ − 1 λ2) (1.2)
1.3. RENFORCEMENT
1.2.3 Modèle des jonctions contraintes
Aussi connu sous le nom de modèle de Flory - Erman [4], ce modèle s’applique aux réseaux réels qui présentent un comportement intermédiaire entre les deux cas extrêmes. Ce modèle prédit une variation du module avec la déformation. Aux faibles taux de déformation, les contraintes sont importantes alors qu’aux taux de déformation élevés, les effets des contraintes sont moindres et le réseau tend vers un comportement fantôme.
1.2.4 Modèle semi-empirique de Mooney-Rivlin
C’est la constatation de la différence entre les données expérimentales et les prévisions des mo-dèles affine et fantôme concernant l’évolution de la contrainte réduite en fonction de la déformation qui justifie son élaboration. Ainsi Mooney et Rivlin [5]ont proposé l’équation semi-empirique :
σreduite= σvraie
λ2− 1 λ
= 2(C1+C2
λ ) (1.3)
2C1et 2C2sont des constantes indépendantes de la déformation et n’ont pas à priori de sens physique. 2(C1+ C2) est une approximation du module aux faibles déformations (modèle affine)
et
2C1une approximation du module aux fortes élongations (réseau fantôme).
1.2.5 Modèle de Rubinstein et Panyukov
C’est un nouveau modèle de comportement élastique non-linéaire [6]. Il prend en compte la dé-pendance en déformation du confinement des chaînes. Ils ont réécrit l’équation de Mooney sous la forme suivante :
σr´eduite= Gc+ Ge
0.74λ + 0.61λ−1/2− 0.35 (1.4)
avec :
Gcest le module fantôme
et
Geest le module du réseau enchevêtré.
1.3 Renforcement
La figure 1.3 présente l’amélioration des propriétés mécaniques induites par l’introduction de particules à une matrice silicone par rapport à ces propriétés intrinsèques. Le module élastique, La résistance à la déchirure et les caractéristiques de rupture (déformation et contrainte à la rupture) sont améliorés [7, 8], ce sont ces effets qui définissent le renforcement.
Figure 1.3 –Influence de l’ajout de charge sur les propriétés mécaniques d’un réseau silicone
Pour prédire le module élastique des matériaux renforcés, plusieurs modèles ont vu le jour. La plupart d’entre eux est inspirée de l’équation d’Einstein qui porte sur la viscosité d’une suspension de particules. Ce modèle relie la viscosité à la fraction volumique de charge (équation 1.5) et est basé sur l’effet hydrodynamique induit par la présence des particules.
η = η0(1 + 2.5φ) (1.5)
φ est la fraction volumique.
Dans l’équation 1.5, la viscosité η est remplacée par le module élastique E pour donner l’équation d’Einstein - Swallwood [9] :
E = E0(1 + 2.5φ) (1.6)
E0est le module élastique de la matrice non-chargée.
Guth et Gold [10] ont modifié l’équation d’Einstein-Swallwood pour prendre en compte les fractions volumiques élevées et ont ajouté un terme d’interactions entre particules.
E = E0(1 + 2.5φ + 14.1φ2) (1.7)
Cette équation ne prend en compte que l’effet hydrodynamique c’est-à-dire les charges sont considé-rées inertes et il n’y a pas d’interactions physico-chimiques. Beaucoup d’études ont montré une grande divergence entre le module élastique estimé par cette équation et les mesures à forts taux de charge [11, 12]. Dès les premières concentrations de silice, des valeurs de module au-dessus de celles calcu-lées par le modèle de Guth et Gold sont observées. Cette divergence avec l’équation de Guth et Gold était prévisible. En effet, cette équation ne prend pas en compte l’existence de liaisons chimiques entre la charge et la matrice qui se traduit par l’augmentation des points d’ancrage c’est-à-dire la densité de réticulation et du module élastique. Une formulation très intéressante de l’effet de renforcement a été donnée par Bokobza et Rapoport [14], qui ont défini le module élastique d’un élastomère chargé comme le produit du module du non-chargé G0et de deux facteurs X et Y :
G = G0XY (1.8)
G0X étant la théorie de Guth et Gold qui n’a trait qu’à l’effet hydrodynamique, et Y représente les interactions charge-matrice.
1.4. PHÉNOMÈNES DE DISSIPATION D’ÉNERGIE
cas où on a affaire à des agrégats structurés, une partie de la matrice peut être piégée en leurs seins : c’est la gomme occluse. Cette partie de polymère ne subirait plus l’effet de sollicitations mécaniques donc ne participerait pas à la déformation mécanique. Ainsi, il est nécessaire de modifier la fraction volumique de charge pour utiliser une fraction volumique de charge dite efficace. A cela s’ajoute le fait que quand le taux de charge augmente, l’arrangement des particules en chemin continu d’agrégats (percolation) apporte une contribution au module élastique.
La complexité du problème due à la diversité des paramètres (dispersion, nature de la charge, matrice, méthode de synthèse,...) fait que beaucoup d’autres modèles ont vu le jour pour se rapprocher des ré-sultats expérimentaux. Parmi ces modèles, on peut citer celui de Kerner qui sera présenté au chapitre 6.
1.4 Phénomènes de dissipation d’énergie
1.4.1 Effet PaynePhénomène caractéristique des élastomères chargés, l’effet Payne est la non-linéarité du compor-tement viscoélastique qui apparaît à faible déformation (ε < 50%). Aux faibles déformations, on observe le plateau G0
0. Lorsque la déformation augmente, le module de conservation G’ chute jusqu’à
une valeur G0
∞où elle se stabilise. La figure 1.4 montre l’évolution de G’ (module de conservation),
G"(module de perte) et tanδ (déphasage entre contrainte et déformation) au cours de ce phénomène. tanδ est défini par :
tanδ = G”
G0 (1.9)
La chute du module de conservation coïncide avec un maximum du module de perte G" traduisant une importante dissipation d’énergie.
Figure 1.4 –Effet Payne [13]
L’effet Payne dépend de paramètres tels que la température, le taux de charge, l’état de dispersion et la nature de la charge. La figure 1.5 montre l’influence du taux de silice dans le PDMS sur l’effet Payne.
Figure 1.5 –Influence du taux de silice sur l’effet Payne [13]
Il est interprété comme un processus de désagglomération, réagglomération du réseau de charge ou des mécanismes mis en jeu à l’interface charge-matrice (adsorption et désorption de l’élastomère, libération de la gomme occluse inter-agrégats). Nous reviendrons plus largement sur l’interprétation de l’effet et discuterons nos résultats sur l’effet Payne au chapitre 6.
1.4.2 Effet Mullins
Aux grandes déformations, le comportement mécanique des élastomères chargés est modifié après l’application d’une première extension. Lors de la seconde traction, l’élastomère chargé présente un ``ramollissement de contrainte´´appelé effet Mullins (figure 1.6).
Figure 1.6 –Effet Mullins
Plusieurs théories ont été avancées pour tenter d’expliquer ce phénomène. Une de ces théories considère que lors de la première traction, il y a rupture ou glissement de chaînes liées à la charge [15, 16] du fait de la distribution en longueur des chaînes inter-particulaires. A chaque déformation, un certain nombre atteint leur extensibilité limite (figure 1.7). Ainsi, le nombre de points de réticula-tion efficace diminue, expliquant cet abaissement de la contrainte lors de la seconde tracréticula-tion.
1.5. SYNTHÈSE DES PARTICULES MINÉRALES PAR VOIE SOL-GEL
Figure 1.7 –Modèle de Bueche
Une autre théorie relie l’effet Mullins aux phénomènes à l’interface charge-matrice [17]. Cette théorie est basée sur un mécanisme de glissements des chaînes adsorbées à l’interface sur la surface de la charge (figure 1.8).
Figure 1.8 –Modèle de Dannenberg
Nous reviendrons plus en détail sur les interprétations de l’effet Mullins au chapitre 6.
1.5 Synthèse des particules minérales par voie sol-gel
Le procédé sol-gel a été mis en œuvre en 1864 par Thomas Graham [18] en étudiant des gels de silicium. Il consiste à transformer un mélange de précurseurs liquides en solide par des réactions ayant lieu à température ambiante. Ces précurseurs sont nommés sol et peuvent être de deux natures :
+ Des particules colloïdales dispersées dans un liquide ; + Des précurseurs organométalliques.
C’est l’agrégation ou la polymérisation de ces précurseurs qui aboutit à la formation d’un solide stable appelé gel. En fonction du type de précurseur utilisé, on obtient des gels colloïdaux ou des gels poly-mériques.
Notre travail porte essentiellement sur les gels polymériques.
1.5.1 Mécanisme de formation des gels polymériques
La synthèse de ces gels est basée sur l’hydrolyse et la condensation d’un alcoxyde métallique M(OR)men présence d’un catalyseur neutre, acide ou basique. Le processus sol-gel est décrit par les
Figure 1.9 –Mécanismes procédé sol-gel
Ce sont les taux relatifs d’hydrolyse KH et de condensation KC qui déterminent la structure et la
morphologie des produits finaux. La contribution de ces différentes réactions dépend de paramètres tels que la nature de l’alcoxyde (l’atome de métal, la nature du groupement alkyl et la complexité moléculaire), du taux d’hydrolyse r = [H2O]
[M] , du choix du catalyseur et sa concentration (pH), du
solvant si nécessaire et de la température. Dans notre travail, nous utiliserons deux alcoxydes : un à base de métal de transition Ti et un à base de métal du groupe IV Si.
1.5.1.1 Mécanisme réactionnel 1.5.1.1.1 Hydrolyse
L’hydrolyse de l’alcoxyde métallique commence dès l’apport d’eau au système. Elle est matérialisée par la réaction suivante :
M(OR)4+ 4H2O - M(OH) + 4ROH (1.10)
En l’absence de catalyseur, cette réaction est une substitution nucléophile qui commence par la fixation d’une molécule d’eau sur le métal (1) avec un transfert de proton (2) et au final un départ d’un groupe ROH (3) comme indiqué sur la figure 1.10.
Figure 1.10 –Mécanisme d’hydrolyse des alcoxydes M(OR)4
La vitesse d’hydrolyse est très dépendante du catalyseur (acide, basique, neutre), de la quantité d’eau et du métal central.
1.5.1.1.2 Condensation
La condensation peut se faire selon plusieurs processus : alcoxolation, oxolation, alcoolation et ola-tion. Les conditions expérimentales déterminent la réaction qui est favorisée parmi ces processus. C’est aussi une substitution nucléophile mais cette fois-ci, c’est un intermédiaire réactionnel (un li-gand hydroxo) qui joue le rôle de molécule attaquante. Nous présentons les mécanismes des réactions d’alcoxolation, d’oxolation, alcoolation et d’olation.
+ L’alcoxolation
L’alcoxolation se produit entre deux alcoxydes dont l’un seulement est partiellement hydrolysé. Son mécanisme réactionnel est le suivant :
1.5. SYNTHÈSE DES PARTICULES MINÉRALES PAR VOIE SOL-GEL
Figure 1.11 –Mécanisme d’alcoxolation des alocoxydes M(OR)m
+ L’oxolation
L’oxolation se produit entre deux alcoxydes partiellement hydrolysés avec départ d’une molé-cule d’eau. Le mécanisme réactionnel de l’oxolation est le suivant :
Figure 1.12 –Mécanisme d’oxolation des alocoxydes M(OR)m
L’alcoolation et l’olation sont des réactions de polymérisation par coordination. Elles se pro-duisent lorsque la coordination du métal (N-z est différent de 0, N étant le nombre de coordina-tion du métal central et z son état d’oxydacoordina-tion) n’est pas satisfaite sous sa forme alcoxyde. + Alcoolation
Figure 1.13 –Mécanisme d’alcoolation des alocoxydes M(OR)m
+ Olation
L’olation se produit lorsque l’insaturation de coordination N − Z du centre métallique est supé-rieur à 0. La figure 1.14 donne le mécanisme de l’olation.
Figure 1.14 –Mécanisme d’olation des alocoxydes M(OR)m
Les réactions d’hydrolyse et de condensation sont en concurrence dans la transformation des précur-seurs alcoxydes métalliques en oxydes métalliques. La prédominance de l’une ou de l’autre réaction influence fortement la structure et la morphologie de l’oxyde final. Le fait qu’une de ces réactions soit favorisée dépend des conditions expérimentales notamment de paramètres internes (nature de l’atome métallique, des groupements alkyles et la structure des précurseurs moléculaires) et externes (nature du catalyseur, taux d’hydrolyse etc...).
1.5. SYNTHÈSE DES PARTICULES MINÉRALES PAR VOIE SOL-GEL
1.5.1.2 Nature de l’atome métallique
Le titane appartient au groupe des éléments de transition du tableau de classification de Mendelev alors que le silicium Si est un élément du groupe IV. Plusieurs éléments différencient les alcoxydes métalliques du groupe des éléments de transition de ceux siliconés du groupe IV.
(i) La plus faible électronégativité des métaux de transition et leur caractère plus électrophile par rapport au silicium (Tableau 1.2) rendent les alcoxydes à base de métaux de transition plus sensibles à l’hydrolyse, la condensation et aux autres réactions nucléophiles.
Eléments EN δM r (nm) N
Si 1.74 +0.32 0.26 4
Ti 1.32 +0.60 0.61 6
Table 1.2 –Caractéristiques physiques des centres métalliques avec EN : électronégativité du métal, δM : charge
partielle, r : rayon ionique, N : nombre de coordination maximum de métaux tétravalents (Z=4) [19]
(ii) Les métaux de transition présentent souvent plusieurs états de coordination stables, et quand un état est instable, ils sont capables d’étendre leur coordination via une association nucléophile. (iii) Un contrôle accru de l’humidité et de l’état d’hydrolyse est nécessaire pour les alcoxydes à base de métaux de transition en vue de la préparation de gels homogènes plutôt que de précipi-tés.
(iiii) La rapidité des réactions nucléophiles rend fondamentalement plus difficile l’étude des réac-tions d’hydrolyse des alcoxydes à métaux de transition par rapport aux alcoxydes métalliques du silicium.
1.5.1.3 Nature des groupements alkyles
Il a été montré que la vitesse d’hydrolyse décroît lorsque la taille des substituants augmente aussi bien pour les alcoxydes de silicium [20, 23, 24, 25] que pour les alcoxydes de titane [26]. Livage et al. [19] déterminent la distribution de charge partielle dans l’alcoxyde et déduisent que l’augmentation de la taille des substituants entraîne une diminution de la charge partielle positive sur les centres métalliques et sur l’atome d’hydrogène (tableau 1.3). Ils relient cette diminution de la charge partielle à la sensibilité à l’hydrolyse. R δ(T i) δ(OR) δ(H) δ(S i) δ(OR) δ(H) kh102M−1s−1[H+]−1 CH3 +0.66 -0.16 +0.12 +0.36 -0.09 +0.14 -C2H5 +0.63 -0.16 +0.10 +0.32 -0.08 +0.11 5.1 n − C4H9 +0.61 -0.15 +0.09 +0.30 -0.08 +0.09 1.9 n − C6H13 +0.60 -0.15 +0.08 +0.29 -0.07 +0.08 0.83 n − C9H19 +0.59 -0.15 +0.07 +0.28 -0.07 +0.08 0.3 T i(OR)4 S i(OR)4
Table 1.3 –Distribution de charges dans les alcoxydes T i(OR)4et S i(OR)4[19]
Donc lorsque la taille du groupement alkyl augmente, la charge partielle sur le métal diminue et la vitesse d’hydrolyse s’en trouve ralentir.
1.5.1.4 Influence du catalyseur et du pH
Une façon d’affecter les vitesses relatives d’hydrolyse et de condensation est de jouer sur le pH en utilisant un catalyseur acide (HCl) ou basique (NH4OH).
Pour une catalyse acide, un groupe alcoxyde portant une charge partielle négative est facilement pro-toné. La densité électronique du centre métallique est diminuée ce qui le rend encore plus électrophile et la protonation augmente le caractère nucléofuge d’un groupe partant. Ce type de catalyse améliore la cinétique de production du groupe partant. Dans ces conditions, tous les groupes OR sont hydrolysés
si une quantité d’eau suffisante est ajoutée. Pour Livage et al. [19] en présence de H3O+la
condensa-tion se produit sur les intermédiaires M(OH)x(OR)(z − x) qui se sont formés rapidement.
La catalyse basique n’a pas le même effet sur les alcoxydes de silicium et de titane. L’utilisation de NH4OH comme catalyseur pour les alcoxydes de silicium active l’hydrolyse. C’est dû à l’activation
nucléophile du silicium par la paire libre de l’azote [20, 21]. Inversement Bradley [22] trouve que l’utilisation de NaOH comme catalyseur rend plus difficile l’hydrolyse de T i(OBu)4par rapport à une catalyse neutre ou acide. Mais Wang et Mark [72] considèrent que NH4OH est le meilleur catalyseur
pour générer in situ T iO2 dans une matrice PDMS. Ils obtiennent des forts taux de charge pour des
temps de gonflement assez courts. 1.5.1.5 Influence de la quantité d’eau
Le paramètre qui matérialise l’influence de la quantité d’eau est le taux d’hydrolyse h défini par : h = [H2O]
[M(OR)z]
(1.11) Cette quantité intervient dans le déroulement et la compétition entre les réactions d’hydrolyse et de condensation et donc sur la structure et la morphologie des produits finaux. Généralement un rapport r important favorise l’étape d’hydrolyse [27, 28].
Pour les alcoxydes de silicium, dans des proportions sous stœchiométriques en eau (r << 2), le mé-canisme de condensation par départ d’alcool est favorisé, tandis que le départ d’eau est largement favorisé pour r > 2 [28].
Pour les alcoxydes à base de métaux de transition (Ti, Zr), trois principaux domaines sont observés [19] :
h < 1 Dans ce domaine, la condensation est principalement gouvernée par les réactions d’alco-lation et l’alcoxod’alco-lation. La fonctionnalité du précurseur envers l’alcoxod’alco-lation est toujours infé-rieure à 1, alors que pour l’alcoolation la fonctionnalité peut aller jusqu’à z-1 (soit trois molé-cules d’eau pour un métal tétravalent). L’hydrolyse est précisément contrôlée, il n’y a aucune précipitation, ni gélation (il n’y a pas d’excès local d’eau).
1 < h < z Livage et al. [19] pour analyser l’effet du taux d’hydrolyse ont donné la valeur des gradients de charge dans l’iso-propoxyde de titane comme exemple. Ils remarquent à partir de ces valeurs que la première étape d’hydrolyse se produit pour δ(OR) > 0 et δ(T i) > 0. En raison de la charge positive suriPrOH, ce sont les mécanismes d’oxolation et d’alcoxolation
qui sont en concurrence. Dans ces conditions, les chaînes de polymères s’accordent avec le modèle linéaire ci-dessous :
Figure 1.15 –
De telles structures ont été obtenues par Boyd [29] et Winter [30] avec T i(OBu)4.
Avec une hydrolyse plus poussée, la charge partielle des groupements alcoxy devient de plus en plus positive. Ainsi, le transfert de proton peut devenir l’étape limitante. L’hydrolyse ne peut alors devenir complète même pour h=4. C’est pour cette raison que l’oxolation devient très compétitive quand le nombre maximum de coordination du métal est satisfait (TMOS, TEOS). Dans le cas des alcoxydes avec métaux de transition, l’olation peut agir préférentiellement car les conditions de charges requises (δ(OH) << 0, δ(M) >> 0, N − z >> 0) sont satisfaites.
1.5. SYNTHÈSE DES PARTICULES MINÉRALES PAR VOIE SOL-GEL
h > z Quand un excès d’eau est ajouté à l’alcoxyde, des polymères ramifiés, gels ou précipités peuvent être obtenus. Il a été observé que le taux d’hydrolyse [31, 32] influe fortement sur la taille et la masse des macromolécules obtenues. Cette observation est valable aussi bien pour les alcoxydes de silicium que pour ceux de titane. Par utilisation d’un excès d’eau, des poudres mono-disperses de T iO2 sont obtenues par précipitation contrôlée de T i(OEt)4 [33]. Comme
la précipitation est un processus lent, c’est probablement l’olation et non l’oxolation qui est le mécanisme prépondérant pour la condensation.
1.5.2 Géométrie fractale
Au cours des dernières années, la notion de fractale a ouvert la voie pour obtenir des réponses à la plupart des questions concernant la géométrie des particules et la relation entre la forme, la structure et les processus qui créent ces systèmes. Ces systèmes conservent leur forme géométrique lorsque l’on change d’échelle, ils sont dits auto-similaires.
Les fractales sont des systèmes géométriques de structures complexes dont la création ou la forme met en jeu des règles utilisant le fractionnement. Elles sont à la base d’un nouveau système géométrique permettant de représenter des objets très irréguliers.
Dans les premières approches de ces systèmes désordonnés, le désordre était considéré comme une perturbation d’un système idéal. L’hypothèse, dans cette approche, est que ces différents systèmes héritent des propriétés physiques et chimiques des systèmes idéaux, si les écarts avec les systèmes sont petits. Or la notion de fractale a prouvé son efficacité en traitant le problème comme intrinsèque plutôt qu’une perturbation.
Les particules fractales peuvent être caractérisées par un seul paramètre D, la dimension fractale, qui est l’exposant qui permet de relier la masse M d’un objet à sa taille R (équation 1.14). La figure 1.16 montre la cinétique de croissance et les structures désordonnées trouvées pour diverses dimensions fractales par simulation.
Figure 1.16 –Exemple de structures de clusters de silice avec à gauche représentation des sites des siliciums et à droite représentation des périmètres [35]
Les fractales les plus souvent évoquées sont la masse et la surface fractale. La masse fractale est donnée par l’équation suivante :
M ∝ RD (1.12) D peut varier de 1-3, avec 1 affecté à des structures linéaires et 3 pour les structures les plus denses. La surface fractale se caractérise par une dimension fractale Ds qui relie la surface d’un objet à son rayon :
S = aDs (1.13)
Ce type de fractale peut prendre n’importe quelle valeur entre 2 et 3. Pour Ds= 2, la surface de l’objet
est uniformément lisse alors que Ds= 3 correspond à une surface rugueuse.
1.5.3 Processus de croissance
1.5.3.1 Processus de croissance à l’équilibre
Flory [36, 37] et Stockmayer [38] furent les premiers à proposer un modèle pour décrire la poly-mérisation des polymères organiques. Ce modèle est basé sur la formation au hasard de liaisons entre fonctions réactives d’espèces voisines sur un réseau infini. Ce modèle a comme avantage de pouvoir décrire certains résultats de diffusion de rayonnement aux petits angles pour des systèmes fractales et ouverts, le point gel mais ne permet pas de rendre compte de la formation de structures cycliques. Pour pallier aux faiblesses du modèle à l’équilibre, Stauffer [39] et De Gennes [40] ont proposé le modèle de percolation. Dans ce modèle, les liaisons sont formées de manière aléatoire entre nœuds voisins sur un réseau d-dimensionnel. Cette approche permet de prédire la formation de cycles et les volumes exclus sont pris en compte [41].
Figure 1.17 –Schématisation des théories de gélification (a) modèle de Flory (f=3) (b) percolation sur réseau carré
1.5.3.2 Processus cinétique de croissance
Les mécanismes cinétiques de croissance se déclinent en deux catégories de processus : croissance "monomère-cluster" et "cluster-cluster". Plusieurs études par simulation numérique ont été effectuées en se basant sur ces mécanismes de croissance.
1.5.3.2.1 Croissance "monomère-cluster"
Ce modèle est limité par la diffusion des espèces ou par la réaction et nécessite une source constante de monomère dans le milieu réactionnel.
+ Modèle d’Eden [42]
Il est utilisé assez souvent pour la modélisation de la croissance cellulaire. Il est basé sur ce que l’on appelle le périmètre qui correspond à l’ensemble des sites inactifs plus proches voisins
1.5. SYNTHÈSE DES PARTICULES MINÉRALES PAR VOIE SOL-GEL
d’au moins un site actif. La règle consiste à choisir un site de façon aléatoire parmi les sites du périmètre et le rendre actif.
Si on modélise le réseau par un ensemble de carrés, à l’étape initiale un carré est choisi au milieu. Son périmètre constitue quatre sites inactifs. Ensuite un de ces sites est sélectionné au hasard et rendu actif. Ce processus est limité par la taille du réseau ou le nombre d’itérations. Le périmètre n’étant jamais vide, la croissance ne peut s’inhiber d’elle-même.
+ Modèle Diffusion Limited Monomer Cluster Aggregation (DLMCA)
Dans le modèle DLMCA ou "Witten-Sander", du nom des auteurs qui l’ont proposé en 1981, les monomères sont relâchés un par un à partir de sites arbitrairement loin d’un cluster central. Les monomères ont un déplacement browmien et se lient au cluster au premier choc entre monomère - cluster en formation.
+ Modèle Reaction Limited Monomer-Cluster Aggregation (RLMCA)
Ce modèle se différencie du modèle DLMCA par l’existence d’une barrière pour la formation des liaisons entre monomère et cluster. Ainsi, pour qu’un monomère se lie de manière efficace et irréversible à un cluster en formation il est nécessaire d’avoir plusieurs chocs.
Figure 1.18 –Simulations de structures résultantes des différents modèles de croissance [43]
1.5.3.2.2 Croissance "cluster-cluster"
Le modèle de croissance cluster-cluster est le résultat d’une marche aléatoire des monomères qui forment une collection de clusters qui vont croître en s’agrégeant entre eux. Ce modèle est limité par la diffusion ou la réaction :
+ Modèle Diffusion Limited Cluster Aggregation (DLCA)
Les clusters s’accrochent de manière irréversible dès le premier choc. La dimension fractale trouvée en simulation numérique est de 1.80.
+ Modèle Reaction Limited Cluster Aggregation (RLCA)
La probabilité d’accrochage est inférieure à un, il faut donc plusieurs chocs pour que les clusters s’accrochent. Une dimension fractale de 2.09 est trouvée avec ce modèle.
Contrairement au modèle d’agrégation "monomère-cluster", les structures fractales formées sont très ouvertes et ne possèdent pas de centre évident. Le modèle de croissance cluster-cluster est attendu lorsqu’il n’existe pas de source de monomères et lorsque la condensation entre espèces faiblement condensées et fortement condensées n’est pas favorisée.
1.6 Les charges
La charge consiste en une phase rigide introduite de différentes manières (mélangeage direct, gé-nération in situ) pour améliorer les propriétés intrinsèques des élastomères. Les charges peuvent être de plusieurs natures selon leur géométrie : sphériques (S iO2, T iO2, noir de carbone...), filaments
(na-notubes de carbone), feuillets (argile). Elles sont caractérisées par :
+ Leur structure : C’est la manière dont les particules sont organisées. On peut distinguer trois échelles de structure : partant des particules élémentaires qui se regroupent sous l’effet des forces colloïdales lors de la synthèse pour donner naissance aux agrégats. Sous l’effet d’attrac-tions plus faibles, les agrégats conduisent à la formation d’agglomérats.
+ Leur surface spécifique : C’est le rapport entre la surface de la charge et sa masse. Elle peut aller jusqu’à 400m2/g pour certaines silices.
+ Leur activité chimique : Elle rend compte de la compatibilité entre la charge et la matrice. Elle influe fortement sur la dispersion et la formation d’interactions entre la matrice et la charge. Elle est exprimée par l’énergie de surface qui détermine les interactions charge-charge et polymère-charge par la formule :
γ = γds+ γssp (1.14)
Le premier terme γd
s est la composante dispersive représentant les interactions entre la charge et
la matrice.
Le second terme γsps est la composante spécifique ou polaire dont dépendent souvent les inter-actions charge-charge.
Nous détaillerons, dans ce qui suit, quelques caractéristiques de la silice et de l’oxyde de titane qui sont les deux charges utilisées dans ce travail.
1.6.1 La silice
La silice ou dioxyde de silicium, de formule générale S iO2, est composée de tétraèdres S iO4
liés par leurs sommets mettant leurs atomes d’oxygène en commun. Elle peut être d’origine naturelle (dans les minéraux tels que le quartz), synthétique, amorphe (verre de silicium) ou cristalline (quartz, tridymite, silices fibreuses ou lamellaires etc.).
1.6.1.1 Chimie de surface de la silice
La nature chimique de la surface d’une particule est un paramètre fondamental dans le renforce-ment des polymères. Dans le cas de la silice, les grouperenforce-ments chimiques prépondérants en surface sont les hydroxyles et l’eau physisorbée ou chimisorbée. Selon la manière dont les hydroxyles sont ar-rangés, on aboutit à plusieurs types de silanols en surface. Il existe trois classifications des différentes structures :
+ Une classification en Qn: Ce modèle est basé sur la chimie du silicium. Les groupements sont
classés par le nombre n d’atomes d’oxygène liés à un atome central. La figure 1.19 récapitule les différents groupements de surface de ce classement.
1.6. LES CHARGES
Figure 1.19 –Différents types de silanols (Qn)
La RMN du silicium permet de mettre en évidence les différents sites de la silice. La figure 1.20 présente les massifs de silice en RMN29S i pour des PDMS chargés de 20 pce de silice
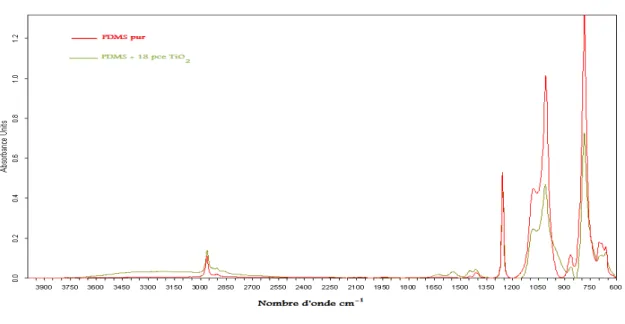
générée in situ en présence de deux types de catalyseurs [12].
Sur la figure 1.20, on peut distinguer trois résonances : vers -90 ppm pour les silanols germinés Q2, vers -100 ppm pour les silanols simples Q3et -110 pour les Q4. La RMN29S i permet donc
de différencier les silanols Qnet même fournir les concentrations relatives.
Par exemple pour de la silice générée in situ au sein d’une matrice PDMS, le tableau 1.4 [12] donne les concentrations relatives trouvées pour différents taux de silice avec des catalyseurs à base "d’étain" (diacétate de dibutyle étain : dA et dilaurate dibutyle étain : dL) .
Taux de silice (pce) catalyseur utilisé Q2(-94 ppm) Q3(-102 ppm) Q4(110 ppm)
60 dA 13% 44% 43%
40 dL 14% 42% 44%
45 dL 10% 47% 43%
Table 1.4 –Décomposition du massif de la silice pour plusieurs échantillons ``étain´´[12]
Fyfe et al. [49] trouvent un ratio entre les Q2,Q3 et Q4 pour des gels commerciaux
respec-tivement de 58%, 37% et 6%. Toutes ces valeurs de concentrations montrent la diversité de répartition des différents types de silanols selon leur mode d’obtention.
+ Une classification basée sur le type d’interactions entre hydroxyles de surface : Les hydroxyles de surface peuvent s’associer par l’intermédiaire de liaisons hydrogènes, entre eux ou avec l’eau de surface, d’où l’existence de silanols libres très disponibles et de silanols vicinaux moins disponibles et moins réactifs (figure 1.21).
Figure 1.21 –Différents types de silanols selon le type d’associations
+ Une classification basée sur le rapport à l’eau : L’eau s’adsorbe par liaisons hydrogènes sur les silanols isolés et vicinaux (figure 1.22) avec une adsorption préférentielle sur les silanols vicinaux.
1.6. LES CHARGES
1.6.1.2 Structure chimique de la silice
En raison de son affinité avec les silicones du fait de la présence de la liaison Si-O, la silice est la charge la plus couramment utilisée pour renforcer les silicones. Les atomes de silicium en surface maintiennent leur coordination tétraédrique et les atomes d’oxygènes complètent leur valence libre en se liant à un atome d’hydrogène pour former un silanol. Les structures obtenues sont les suivantes :
Figure 1.23 –Différentes espèces de siliciums dans la silice
Les silanols libres sont des groupements hydoxyles OH situés sur des sites silicium Q3. Ils peuvent être détectés par spectroscopie infrarouge avec un nombre d’onde autour de 3750cm−1.
Les silanols vicinaux sont des hydroxyles situés sur des sites Q3contigus et peuvent interagir par des
liaisons hydrogènes.
Les silanols géminés sont deux groupes OH liés à un même atome de silicium.
La densité surfacique de silanols est un paramètre important, des études théoriques ont permis de dé-terminer la densité de OH par nm2. La plus simple de ces théories élaborée par Iller [44] est basée sur des considérations géométriques et la densité de S iO2amorphe. Sa théorie aboutit à la conclusion
qu’il devrait y avoir 7.8 atomes de silicium par nm2en surface ou très proche de la surface. Il suppose
que ça devrait être 7.8 groupes SiOH par nm2de surface. Cependant, Boehm [45] fait remarquer que les atomes de silicium ne peuvent pas être tous exactement en surface, certains doivent être surfaciques et d’autres internes, et, par conséquent seule la moitié des atomes de silicium porte des groupes OH ce qui donne 3.9OH/nm2de surface.
Iller [44] a également suggéré en se basant sur une densité et un indice de réfraction de la silice amorphe proches de ceux de la cristobalite et de la tridymite que la concentration d’hydroxyles de surface peut être estimée à partir des structures cristallines. En choisissant la surface 1.0.0 de la cris-tobalite cette méthode aboutit à 8OH/nm2et en appliquant la même méthode à la tridymite elle donne
4.6OH/nm2.
En plus de ces méthodes théoriques, des méthodes expérimentales sont aussi utilisées (infrarouge, résonance magnétique nucléaire RMN, thermogravimétrie, méthodes chimiques). Par exemple, Zhu-ravlev [46, 47] pour déterminer la quantité de OH par nm2et distinguer la surface et les OH utilise une
méthode d’échange deutéré [48]. Pour des essais sur une centaine d’échantillons, il trouve des valeurs comprises entre 4 et 6 OH/nm2. Plus récemment, il [48] a montré que ce nombre pouvait atteindre 30
Figure 1.24 –Nombre d’hydroxyles par nm2en fonction de la surface pour plus de 170 silices rapportées dans la
littérature [48]
1.6.2 L’oxyde de titane
L’oxyde de titane existe sous plusieurs formes dont quatre naturelles : rutile, anatase, brookite et T iO2(B). Quelques formes ont été synthétisées à haute pression dont T iO2(II), T iO2(H) hollandite.
Parmi toutes ces formes, le rutile et l’anatase sont les plus étudiés dans la recherche et dans l’industrie chimique.
Ces deux structures (anatase, rutile) sont basées sur l’interconnexion d’octaèdre T iO6. Dans ces deux
cas, l’atome de titane est entouré de six atomes d’oxygène. Ces deux structures se distinguent par la manière dont sont connectés les octaèdres. Ils sont liés par les arêtes et les sommets dans le cas du rutile et uniquement par les sommets pour l’anatase. La figure 1.25 donne la structure du rutile et de l’anatase selon Linsebigler et al. [50] et les caractéristiques de la maille.
Figure 1.25 –Structure d’anatase et rutile [50](# O, Ti)
Plusieurs études ont été menées pour voir comment les différentes faces des octaèdres réagissent en présence d’une autre molécule surtout en présence d’eau. Il sera fait état dans le paragraphe suivant de la manière dont l’eau et les OH s’arrangent à la surface de l’oxyde de titane.
1.6.2.1 Chimie de surface de T iO2
La surface de T iO2 est capable d’adsorber, de dissocier ou de réagir (oxydant, réducteur) avec
plusieurs molécules organiques ou inorganiques sous certaines conditions. La plus importante d’entre elles est la molécule d’eau qui peut être adsorbée à la surface de T iO2 de manière moléculaire ou dissociative, ce qui détermine sa dispersibilité dans des solutions aqueuses ou non. Les résultats ex-périmentaux et théoriques obtenus jusqu’à présent sur les détails de l’adsorption sur T iO2ne font pas
1.6. LES CHARGES
l’unanimité et sont parfois contradictoires. Les résultats obtenus à température ambiante sur l’adsorp-tion sur T iO2:
+ la surface de T iO2est complètement inerte ;
+ H2O s’adsorbe seulement dissociativement ; + H2O s’adsorbe de manière moléculaire seulement ;
+ pas de chimisorption pour H2O, ni pour OH.
Ces contradictions peuvent être imputables aux différentes morphologies de T iO2obtenues selon la
méthode de synthèse.
Plusieurs études ont porté sur l’adsorption d’eau, l’organisation des groupements hydroxyles et des molécules d’eau à la surface de T iO2. Nous en récapitulons quelques-unes ici.
Vittadini et al. [51] se sont intéressés à l’adsorption d’eau sur les surfaces (101) et (001) en fonction du taux de couverture θ. Ils définissent θ comme le ratio entre le nombre de molécules de H2O
adsor-bées et le nombre de surface de Ti coordonnée par cinq sites. Pour la surface (101), l’adsorption est moléculaire quel que soit θ. En revanche pour la surface (001), le type d’adsorption moléculaire ou dissociative dépend de la valeur de θ. Pour θ ≤ 0.5, les molécules d’eau se dissocient spontanément sur la surface alors que pour θ ≥ 0.5, les molécules d’eau sont adsorbées de manière dissociative jus-qu’à la moitié d’une monocouche et les molécules restantes forment des liaisons hydrogènes avec les groupes OH de surface et les oxygènes pontés.
Des études expérimentales et théoriques [52, 53, 54] sur la surface (100) du rutile ont conclu à deux types d’adsorption de l’eau : dissociative et moléculaire et ceux indépendamment des mesures, des défauts et des sites T i3+présents. Mais Langel et al. [55] n’arrivent pas à la même conclusion pour la même surface. Arrouvel et al. [56] ont montré que l’eau peut être, en premier, chimisorbée dans un état non dissocié sur un atome de titane, qui se comporte comme un acide de Lewis. L’eau peut ensuite se dissocier sur un atome de titane et d’oxygène, site basique de Lewis, conduisant à la formation de deux groupes hydroxyles à la surface. Il peut arriver qu’il y ait une physisorption des molécules d’eau sur les groupes OH précédemment formés.
Après cette adsorption, Nosaka et al. [57] ont proposé un arrangement des molécules d’eau à la sur-face. Par des mesures de RMN du proton, ils suggèrent une séparation des molécules adsorbées en trois couches (figure 1.26). La couche I serait constituée de molécules d’eau ou de groupes hydroxyles avec mobilité réduite prés de la surface, la couche II de molécules d’eau plus mobiles et la couche III contiendrait les molécules d’eau les plus mobiles.
Figure 1.26 –Structure des couches d’eau sur une surface idéale de T iO2déduite des observations en RMN du


![Figure 1.16 – Exemple de structures de clusters de silice avec à gauche représentation des sites des siliciums et à droite représentation des périmètres [35]](https://thumb-eu.123doks.com/thumbv2/123doknet/2905066.75161/30.892.91.771.609.1031/figure-exemple-structures-clusters-représentation-siliciums-représentation-périmètres.webp)
![Figure 1.18 – Simulations de structures résultantes des différents modèles de croissance [43]](https://thumb-eu.123doks.com/thumbv2/123doknet/2905066.75161/32.892.191.627.403.752/figure-simulations-structures-résultantes-modèles-croissance.webp)
![Figure 1.27 – Structure de l’eau sur la surface de T iO 2 d’après [58]](https://thumb-eu.123doks.com/thumbv2/123doknet/2905066.75161/39.892.318.636.254.580/figure-structure-l-eau-surface-io.webp)



![Figure 3.1 – Schéma de la diffusion aux petits angles par des agrégats, des particules élémentaires et des structures atomiques [1]](https://thumb-eu.123doks.com/thumbv2/123doknet/2905066.75161/78.892.267.569.679.998/figure-schéma-diffusion-agrégats-particules-élémentaires-structures-atomiques.webp)