HAL Id: tel-01968011
https://tel.archives-ouvertes.fr/tel-01968011
Submitted on 2 Jan 2019HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Yosri Noichri
To cite this version:
Yosri Noichri. Stress oxydant et infarctus du Myocarde. Réseaux moléculaires [q-bio.MN]. Université Paris-Saclay; Université de Monastir (Tunisie), 2016. Français. �NNT : 2016SACLS479�. �tel-01968011�
NNT : 2016SACLS479
THÈSE DE DOCTORAT
DE
L’UNIVERSITÉ DE MONASTIR
ET DE
L’UNIVERSITÉ PARIS-SACLAY
PRÉPARÉE À
L’UNIVERSITÉ PARIS SUD
E
COLED
OCTORALE N°
577
Structure et dynamique des systèmes vivants (SDSV)
Spécialité de doctorat :
Sciences de la vie et de la santé
Par :
Mr Yosri Noichri
Stress oxydant et Infarctus du Myocarde
Thèse présentée et soutenue à Orsay, le 24 Novembre 2016 : Composition du Jury :
Mr CAPY Pierre, Professeur, Université Paris Saclay, Président du Jury Mr ROUIS Mustapha, Professeur, Université Pierre et Marie Curie, Rapporteur Mr MAHDOUANI Kacem, Professeur, Université de Monastir, Rapporteur Mr BAUDIN Bruno, Professeur, Université Paris Saclay, Examinateur
Mr MILED Abdelhédi, Professeur, Université de Monastir, Directeur de thèse
REMERCIEMENTS
Je tiens à exprimer mes vifs remerciements à mes directeurs de thèse : Pr Abdelhédi Miled et Dr Michel Tolédano. Je vous suis très reconnaissant de l’honneur qui vous m’avez fait en acceptant de diriger cette thèse et de m’avoir accueilli dans vos laboratoires, pour vos suggestions et vos conseils judicieux.
Je voudrais témoigner ma reconnaissance au Pr Pierre Capy, Je suis très reconnaissant à l’honneur d’accepter de présider le jury de mon mémoire. Je suis toujours touché par votre gentillesse illimitée, votre modestie et vos compétences scientifiques et je suis très honoré de vous contacter de prés le long de l’élaboration de ce travail.
J’adresse toute ma gratitude à Madame le Pr Salima Ferchichi, pour Vos conseils. Vos encouragements m’ont aidé dans l’élaboration de ce travail.
J'exprime ensuite mon estime et mes remerciements aux membres de mon jury : Le Pr Mustapha Rouis et le Pr Kacem Mahdouani, qui m'ont fait l'honneur de juger ce travail de thèse et de me faire ainsi bénéficier de leurs compétences et de leurs connaissances.
Le Professeur Bruno Baudin pour sa collaboration efficace à ce travail, ses explications pertinentes et sa participation à mon jury de thèse.
Je souhaite également remercier très chaleureusement tous les membres du laboratoire : Stress Oxydant et cancer (au CEA de Saclay) et du laboratoire de biochimie à l’hôpital Hached de Sousse et plus particulièrement:
A Mr Gael Palais, Dr Agnès Delaunay-Moison, Dr Aied Igbaria et Madame Catherine Doreau (Laboratoire stress oxydant et cancer – CEA de Saclay),
A Dr Abdelkader Chalghoum, Madame Rachida Mansour, et Madame Halima Limem (Laboratoire de biochimie – Hôpital Hached) : Toujours prêt à nous rendre service, avec bonne humeur et votre profonde gentillesse. Merci.
INTRODUCTION ... 1 SYNTHESE BIBLIOGRAPHIQUE I. INFARCTUS DU MYOCARDE : ... 3 1. Définition ... 3 2. Données épidémiologiques... 3 A. En Tunisie : ... 4 B Dans le Monde : ... 5 3. Facteurs de risque : ... 5 A. Age : ... 5 B. Sexe et ménopause : ... 5 C. Dyslipidémies : ... 6 D. Diabète : ... 6 E. Hypertension artérielle : ... 6 F Tabac :... 7 G. Obésité : ... 7 H. Hyperhomocystéinémie : ... 8 I. Hyperuricemia : ... 8 J. Stress émotionnel: ... 8 4. Etiologies :... 9 A. Athérosclérose coronaire : ... 9 B. Embolies coronaires : ... 10 C. Traumatismes thoraciques : ... 10 D. Spasme coronaire : ... 10 E. Anomalies hématologiques : ... 10 5. Physiopathologie : ... 10
6. Marqueurs biologiques de Diagnostic :... 12
7. Complications de l’infarctus du myocarde : ... 16
A. Complications précoces : ... 16
A. Prise en charge hospitalière : ... 17
B. Traitements pharmacologiques adjuvants : ... 18
II. STRESS OXYDANT : ... 20
1. Définition : ... 20
2. Principaux éléments prooxydants : ... 20
3. Génération des prooxydants ... 22
4. Paradoxe des espèces réactives d’oxygène :... 24
A. Rôles physiologiques des espèces réactives d’oxygène : ... 25
B. Toxicité des radicaux libres : ... 26
5. Antioxydants ... ….29
A. Systèmes enzymatiques Antioxydants ………...………..……...29
. Peroxyredoxine - Thioredoxine - Sulfiredoxine: ……… 29
. Superoxide Dismutase ……… ... …... . 31
. Catalase ……….. 32
. Système de Glutathion …………..………..……… ………..32
B. Systèmes antioxydants non enzymatiques :……… 33
III. STRESS OXYDANT – INFARCTUS DU MYOCARDE ... 35
1. Sources de génération des espèces réactives d’oxygène sous ischémie-reperfusion: 35 - Dysfonction de la chaine de transport d’électron au niveau mitochondriale : ... 35
- Infiltration des cellules immunitaires : ... 36
- Activation de la Xanthine oxydase : ... 36
- Activation de la NADPH oxydase : ... 36
- Activation du monoxyde d’azote synthase NOS: ... 37
2. Implication des ERO dans la mort des cellules myocytaires ………. ………………...37
3. Rôle des antioxydants enzymatiques dans le contrôle de la voie d'apoptose ...38
4.L'horloge Circadienne ………..………………..39
Partie 1 : Exploration de la variation des paramètres de stress oxydant :
I. Population d’étude : ... 44
II. Méthodes :... 45
1.Prélèvements: ………....45
2. Dosage des apolipoprotéines : ………45 3. Dosage de l’homocystéine : …………………...46
4. Dosage des marqueurs antioxydants :………………46
5. Dosage des TBARS: ……….…...48
Partie 2: Etude des mécanismes de Suroxydation des Peroxyredoxines chez les Pmefs : 1.Culture et synchronisation cellulaire ………..49
2.Immunoblot ………...…………49
3.PCR Quantitative …...………………..50
4. Dosage des ROS par FACS ……………….54
RESULTATS ET DISCUSSION……….……… …..…………………55
DISCUSSION
.
………..……………….....86CONCLUSION ET PERSPECTIVES REFERENCES
Liste des Abréviations
IDM: Infarctus du myocarde SCA : Syndrome Coronarien Aigu OMS : Organisation Mondiale de la Santé ST- : Segment T moins
ST+ : Segment T plus
HTA: Hypertension Artérielle ECG : Electrocardiogramme
MONICA: Monotoring of trends and determinants of cardiovascular disease HDL-C: Hight-Density-Lipoprotein-Cholesterol
LDL-C : Low-Density- Lipoprotein-Cholesterol TG : Triglycerides
IMC: Indice de masse corporelle Apo A1: Apolipoprotéine A1 Apo B: Apolipoprotéine B
h-FABP: Heart Fatty acid binding Protein IL : Interleukines
Hcy : Homocystéine Lp(a) : Lipoprotéine a CPR : Protéine C Réactive
BNP : Peptide Natriurétque de type B Tn : Troponine
TnIc : Troponine cardiaque TnT : Troponine T
CK : Créatine Kinase
LDH : Lactate Déhydrogénase ASAT : Aspartate Aminotransférase IEC : Inhibiteur Enzyme de conversion ARNm : Acide Ribo Nucléique messager ADN : Acide Désxiribo Nucléique Prx : Péroxyredoxine
Trx : Thioredoxine Srx : Sulfiredoxine
ASK-1 : Apoptosis Signal Kinase 1
Keap1: Kelch-Like ECH-Associated Protein 1 SOD : Super-Oxyde Dismutase
GPx : Glutathion peroxydase GR : Glutathion réductase SAT : Statut anti-oxydant total ERO : Espèces Réactives d’oxygène
TBARS : Thiobarbituric acid reactive substances PCR: Polymerisation Chain Reaction
EDTA : Acide Ethylenediamine Tétra Acétique ELISA : Enzyme Liked Immunoa-Sorbent Assay SDS : Sodium dodecyl sulfate
GLDH: Glutarate Déshydrogénase TAE : Tris-Acide Acétique-EDTA D.O : Densité Optique
Ac : Anticorps Ag : Antigène
FPIA: Fluorescent Polarisation Immunoassay SAH: S Adénosyl Homocystéine
DTT : Dithiothréitol pH : potentiel Hydrogène
SPSS: Statistical Package for the Social Sciences Kcat : La constante catalytique
UK: United Kingdom
gHb: gramme d’hémoglobine
NADPH: Nicotinamide Adénine Dinucléotide Phosphate reduit. HPLC: Chromatographie Phase Liquide Haute Performance BET : Bromure d’éthidium
TAE : Tris Acide Acétique EDTA. dNTP: Désoxy-nucléoside Tri Phosphate Mefs : Primary mouse embryo fibroblast DMEM : Dulbecco's Modified Eagle Medium SVF : Sérum de veau Foetal
Pmsf :Le fluorure de phénylméthylsulfonyle NEM : N-Ethylmaleimide
EGTA: ethylene glycol tetraacetic acid BSA: Albumine de serum de bovin TBS: Tris Buffered Saline
DTT:Dithiothréitol
DNTP: Deoxynucleoside triphosphates DCF: dichlorodihydrofluorescein
LISTE DES FIGURES
Figure 1 : Mortalité causée pardes maladies coronariennes dans le monde
Figure 2 : Physiopathologie de l’infarctus du myocarde Figure 3 : Structure de la troponine
Figure 4 : Cinétique des marqueurs biologiques de l’infarctus du myocarde Figure 5 : Réactions de peroxydation lipidique
Figure 6 : Mécanisme d’action du système Peroxyredoxine thioredoxine et sulfiredoxine Figure 7: Génération des ERO au cours de l’IDM
Figure 8 : Organisation hiérarchique du système circadien
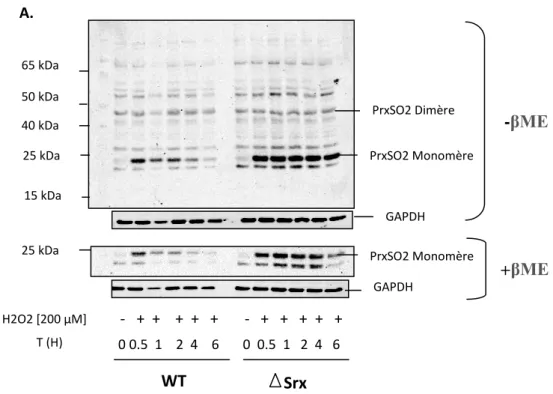
Figure 9 : A. Etude cinétique de la Suroxydation des Prx. B. Etude de la variation de forme
Prx2 chez des Pmefs WT traitées avec 100µM en H2O2.
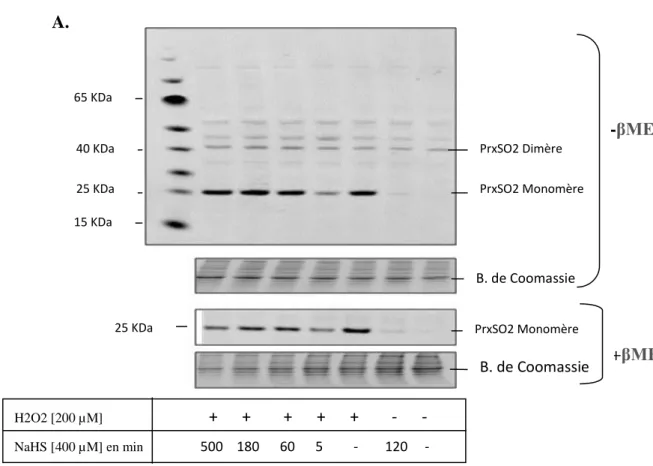
Figure 10. A. Etude de l’implication de H2S dans la suroxydation des Prxs
Figure 11 : A. Etude de l’implication de la H2S dans la variation des interactions
différentielles Prx- Prx.
Figure 12 : Variation du taux des ROS intracellulaires chez des cellules Pmefs WT et Pmefs
Srx-/- synchronisées par le choc sérique et marquées au H2 -DCF-DA
Figure 13: A. B. Etude de rythme de suroxydation des Prxs chez des Pmefs Wt et Srx -/-Figure 14.A.B : Etude de la variation circadienne de la Prx1 chez des Pmefs synchronisés Figure 15.A.B : Etude de la variation circadienne de la Prx2 chez des Pmefs synchronisés Figure 16: Etude de la variation de la Trx chez des Pmefs WT et Pmefs Srx -/-
Figure 17 : Etude de la variation de la Srx chez des Pmefs WT
Figure 18 : A. B : Etude de la suroxydation des Prxs dans les tissus cardiaques des souris
Figure 19.A.B : Etude de la variation de la suroxydation des Prxs dans les tissus du
cérébraux des souris WT et Srx-/- synchronisés par les cycles lumière/ obscurité
Figure 20.A.B : Etude de la variation Prx1 dans les tissus cardiaques des souris WT et
souris Srx-/- synchronisés par les cycles lumière/ obscurité
Figure 21.A.B: Etude de la variation Prx2 dans les tissus cardiaques des souris WT et
souris Srx-/- synchronisés par les cycles lumière/ obscurité
Figure 22.A.B : Etude de la variation de la Prx1 dans les tissus de cérébraux des souris WT
et souris Srx-/- synchronisés par les cycles lumière/ obscurité
Figure 23.A.B : Etude de la variation de la Prx2 dans les tissus de cérébraux des souris WT
et souris Srx-/- synchronisés par les cycles lumière/ obscurité
Figure 24: A. Etude de l’expression de la Prx1 dans le tissu cardiaque chez des souris WT
et Srx -/-. B. Etude de l’expression de la Prx2 dans le tissu cardiaque chez des souris WT et Srx -/-. C. de la Srx dans le tissu cardiaque chez des souris WT
LISTE DES TABLEAUX
Tableau 1 : Taux d’incidence de l’infarctus du myocarde pour 100 000 habitants dans les
L’infarctus du myocarde (IDM) constitue l’une des principales causes de morbi-mortalité
dans les pays développés. On estime que cette pathologie, connue par la nécrose ischémique du muscle cardiaque, peut toucher, chaque année, plus de 3 millions d’habitants dans le monde. Malgré les améliorations thérapeutiques qui ont légèrement diminué les taux alarmants de décès durant les dernières années, l’IDM reste actuellement une urgence médicale et un défi socio économique important [1]
Les facteurs de risque de l’IDM sont multiples (HTA, diabète, obésité, dyslipidémie, tabagisme), traduisant le nombre élevé des acteurs, métaboliques et environnementaux impliqués dans l’étiologie, la physiopathologie et ses diverses complications. Face à la gravité accrue de cette pathologie liée aux complications parfois fatales, des travaux fondamentaux convergent pour souligner l’implication de stress oxydant dans la physiopathologie de l’IDM. [2].
Le stress oxydant se définit par une situation de déséquilibre entre la production des espèces réactives (oxygénées et azotées) et les mécanismes de défense et de détoxification de ces dernières. Ainsi, les acteurs du stress oxydant peuvent également avoir des effets délétères sur de nombreux constituants des cellules (protéines, lipides, acides nucléiques), engendrant des perturbations moléculaires. Celles-ci peuvent évoluer vers l’induction de la mort cellulaire soit par nécrose, soit par apoptose, aboutissant ainsi à l’altération de la fonction contractile de myocarde. Pour faire face à ces effets négatifs, la cellule dispose de différents moyens de lutte dits « antioxydants ». Les systèmes de défense sont très variés car ils permettent soit de capter, soit de neutraliser les molécules réactives ou bien encore d’éliminer et de remplacer les molécules endommagées
Les antioxydants enzymatiques tels que la Superoxide Dismutase, la Catalase, la Glutathion Peroxydase, sont considérés comme la première barrière de la détoxification des pro oxydants sous ischémie reperfusion myocardique.
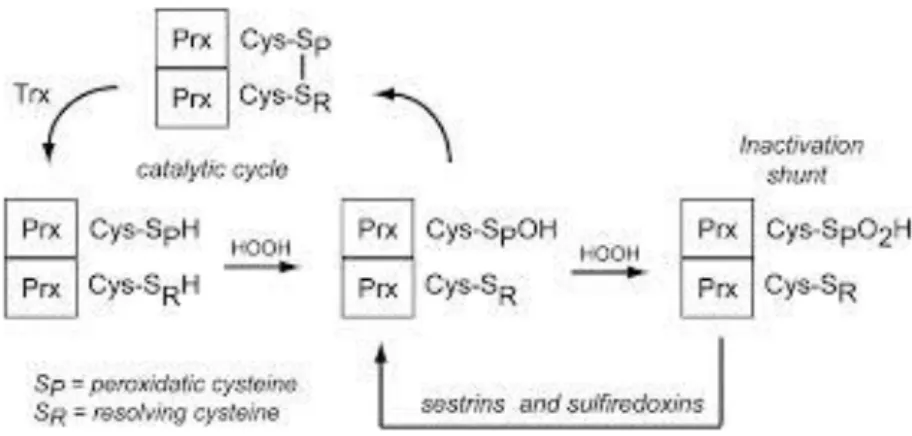
Une famille de peroxydases à thiol, dite peroxyrédoxine (Prx) joue, également, un rôle central dans la régulation des effets cellulaires du peroxyde d’hydrogène, en tant qu’enzymes de détoxification, et qu’effecteurs de signalisation. Elles possèdent en outre une propriété
régulé, par une enzyme : la sulfirédoxine (Srx), possédant une activité sulfinyl réductase unique. La peroxyredoxine et la sulfiredoxine fonctionnent comme un oscillateur. Ce dernier pourrait imposer d’une manière cyclique des variations des teneurs H2O2 et des intéractions entre Prxs et autres protéines régulatrices de la cascade de signalisation pro apoptotique (comme la thioredoxine).
Au niveau cardiaque, la compréhension de rythme d’activité du système Peroxyrédoxine/Sulfirédoxine (Prx/Srx) pourra concevoir le lien entre stress oxydant et ischémie myocardique, avérée survient chez l’homme de façon prédominante au petit matin. La variation des paramètres antioxydants chez des patients hospitalisés pour un IDM, a fait l’objet de la première partie de notre étude. Nous Contribuons, par ce travail, à l’ensemble des analyses portant sur l’implication de la déficience antioxydante dans l’induction de la mort cellulaire, via l’induction de la peroxydation lipidique. Notre exploration a identifié quelques facteurs de risque cardiovasculaire impliqués dans l’aggravation de déséquilibre pro oxydant antioxydant [3].
La deuxième partie de notre travail a été consacrée pour explorer le phénomène d’oxydation et de suroxydation des Peroxyredoxines. Nos travaux visaient à confirmer la variation cyclique de l’état redox des Prxs, et à explorer son impact sur leur conformation et sur leurs interactions différentielles avec autres protéines régulatrices de la cascade de signalisation pro apoptotique. De plus, nous avons cherché les indices qui nous permettront de savoir si la rythmicité de ce système antioxydant est sous le contrôle intrinsèque ou sous le contrôle de l’horloge biologique central.
Nous contribuons par l’ensemble de ce travail aux investigations visant en définitive une meilleure exploration biochimique de l’IDM dans la perspective d’une meilleure prise en charge des personnes atteintes et d’une amélioration des conduites thérapeutiques.
SYNTHESE
I.
INFARCTUS DU MYOCARDE
:L’infarctus du myocarde (IDM) et l’angor instable font partie d’une entité clinique et biologique, appelée : Syndrome coronarien aigu (SCA).
Dans ce contexte, il convient de distinguer selon la présentation électro cardiographique entre un SCA sans décalage persistant du segment ST (angor instable ou IDM sans onde Q) et un SCA avec sus décalage persistant du segment ST (IDM avec onde Q).
La distinction entre un angor instable et un IDM sans onde Q ne peut avoir lieu qu’à l’aide des marqueurs biologiques [6-7].
1. Définition
Selon la troisième définition universelle de l’Infarctus du myocarde, le terme infarctus du myocarde devrait être utilisé lorsqu’il existe des preuves de la nécrose du myocarde dans un contexte clinique d’ischémie myocardique [8].
Cette définition n’exclut pas les symptômes cliniques, aussi les modifications de l’ECG et éventuellement l’imagerie cardiaque.
2. Données épidémiologiques
L’infarctus du myocarde constitue une urgence cardiologique absolue dont l’incidence reste encore élevée dans le monde. Selon les données OMS, les cardiopathies ischémiques demeurent la première cause de décès avec 7,2 millions de décès en 2012.
Ces chiffres nécessitent des investigations statistiques et cliniques importantes avec une bonne maitrise de la variabilité temporelle et spatiale de ces pathologies [9].
Au japon, la mortalité des maladies coronariennes chez l’homme est de 43/100 000 habitants et chez la femme elle est de 22/100 000. Ces valeurs ont été considérées 4 fois moins élevées qu’en Amérique de nord.
En Europe, la mortalité d’origine coronarienne varie également d’un pays à un autre, de 60% à Varsovie, 40% à Belfast et 30% à Newcastle (Figure1)
Figure 1 : Mortalité causée pardes maladies coronariennes dans le monde [10]
A. En Tunisie :
En Tunisie, les maladies cardiovasculaires présentent la première cause de décès (30% des décès) [11].
Les taux d’incidence de l’infarctus du myocarde en Tunisie ont été estimés par Ben Romdhane et al dans une étude qui est limitée aux banlieues de Tunis, inclut les gouvernorats de Tunis, Ariana et Ben Arous. Cette étude a permis de fournir des résultats exprimés par 100000 habitants, représentés dans le tableau 1 [12] :
Tableau 1 : Taux d’incidence de l’infarctus du myocarde pour 100 000 habitants dans les districts de Tunis, 2001 [11]
Tunis Ariana Ben Arous District du tunis
Hommes 114,2 95,5 110,6 107,8
B. Dans le monde
Malgré le progrès enregistré dans la prise en charge des maladies cardiovasculaires, les syndromes coronariens sont responsables de la première cause de mortalité en France. Sur une population de 60 millions, l’incidence annuelle des hospitalisations pour infarctus en France a été estimée entre 60 000 et 65 000 cas. [13]. Selon l’étude MONICA (1990), la fréquence des infarctus du myocarde varie selon les centre d’étude entre 236/100 000 à Lille, 225/100 000 à Strasbourg et 227/100 000 à Toulouse. Le nombre global des patients hospitalisés pour IDM a diminué de 7,4% entre 2002 et 2008. [10,14]
En termes de mortalité par Infarctus du myocarde, la France est classée en première position par rapport aux autres pays industrialisés avec une fréquence de 70/100000 hommes et 16/100000 femmes. A l’échelle géographique, un gradient décroissant nord sud de mortalité coronaire a été montré selon l’étude MONICA. Il pourra être lié aux différences socio-économiques et aux équipements de soins entre les régions. [10, 13]
3. Facteurs de risque :
Les syndromes coronariens ont des origines multifactorielles. La présence des facteurs de risque non modifiables associés à ceux liés au mode de vie permettent de déterminer la progression de cette pathologie. Ces facteurs de risque sont caractérisés par un effet additif, voire synergique [15-16].
Selon des études statistiques, la correction d’un seul facteur de risque permet une réduction significative de l’incidence de complications coronariennes de l’ordre de 20 à 40% [15].
A. Age :
L’âge est considéré comme un facteur dominant des maladies cardiovasculaires. Il a été démontré que le nombre d’IDM exprimé par an et pour 1000 hommes, varie selon l’âge : ce chiffre est de 1,3 entre 35 et 44 ans, de 3,5 entre 45 et 54 ans, de 7,5 entre 55 et 64 ans et de 10,3 après 65 ans [9-10]. Ce facteur de risque est en corrélation avec l’altération de plusieurs propriétés structurales et fonctionnelles des artères de gros calibres incluant le diamètre, l’épaisseur et l’élasticité des parois vasculaires [17].
B. Sexe et ménopause :
Le sexe a été classé parmi les facteurs de risque classiques de l’IDM. Les femmes sont moins sujettes que les hommes aux maladies cardiovasculaires. Selon des études épidémiologiques, l’âge moyen de survenue de l’infarctus est de 10 ans plus élevé chez la
femme : 73 ans en moyenne contre 63 ans chez l’homme [9,18]. Cette différence s’estompe après la ménopause. Celle –ci définit par à une modification du statut hormonal chez la femme ménopausée, incluant la diminution du taux des œstrogènes, associée à un accroissement important de risque cardiovasculaire. [9,19]. L’oestrogénothérapie permettrait de réduire ce risque. [20]
C. Dyslipidémies :
L'élévation du Cholestérol total et du LDL Cholestérol est associée à une augmentation du risque coronarien de façon curvi-linéaire. Une augmentation de 10% de LDL-Cest associée à une augmentation d’environ 20% du risque de cardiopathie ischémique. Un taux plasmatique élevé en LDL peut induire les premiers processus d’athérogénese. Contrairement au LDL-C cette association devienne inverse pour le cas HDL-Cholestérol (HDL-C) dont une élévation de 0,26 mmole par litre réduit de 30% le risque de l’IDM, ce qui confère le rôle protecteur à cette fraction lipidique. Ceci explique la relation linéaire établit entre le rapport Apo A1/Apo B et le risque de l’IDM aigu [21-22].
L’hypertriglycéridémie, constitue également un facteur de risque des cardiopathies ischémiques indépendant de l’élévation de cholestérol [21].
D. Diabète :
Des études épidémiologiques prospectives portant sur une large cohorte de diabétiques ont pu déceler une association entre le niveau de la glycémie et le risque coronarien. Le risque de développer un syndrome coronarien est 2 à 4 fois plus élevé chez les sujets diabétiques [21,23]. Dans le diabète de type II, les perturbations de la glycorégulation ne sont le plus souvent qu’un élément parmi d’autres perturbations (dyslipidémie, HTA…) entrant dans le cadre du syndrome polymétabolique.
Selon l’étude UKPDS, une baisse de 0,9% du taux d’hémoglobine glyquée entraine une réduction de 16% de risque de survenue d’IDM [10].
E. Hypertension artérielle :
Il s’agit d’un facteur de risque modifiable dont la hausse du niveau d’hypertension augmente le risque des complications cardiovasculaires. Le lien entre niveau tensionnel et risque cardiovasculaire est continu, ce qui signifie qu’il n’y a pas de seuil individualisé en dessous duquel le risque peut être considéré comme nul. La pression artérielle systolique (PAS) a la signification pronostique la plus forte. Le rôle délétère de la pression pulsée (ou
pression différentielle = PAS – PAD) a été mis en évidence : son augmentation traduit une altération de la compliance (ou fonction d’amortissement) des gros vaisseaux. Une méta analyse a montré que le risque relatif d’un accident coronarien est de 5 chez les hypertendus par rapport aux sujets normaux.
La relation linéaire entre le niveau de la pression artérielle diastolique et le risque de développer un IDM a été mise en évidence dans plusieurs études épidémiologiques. Une élévation de 30 mmHg de pression diastolique multiplie par 4 le risque d’IDM [10,21].
F. Tabac :
Ce facteur de risque à été classé parmi les facteurs de risque comportementaux. Selon l’OMS, le tabac est responsable de 5 millions de décès par an dans les pays occidentaux. Pour évaluer ce risque, plusieurs renseignements doivent être nécessairement fournis tels que la quantité journalière du tabac consommé, la durée de tabagisme ainsi que l’âge de début [15,21].
Plusieurs études ont exploré la relation entre le nombre de cigarettes consommés quotidiennement et le taux de risque relatif de présenter un IDM. Une corrélation linéaire a été prouvée pour un nombre de cigarettes compris entre 0 et 25, voire être plafonnée pour un nombre supérieur à 25 cigarettes par jour [15].
De nombreux composants du tabac jouent un rôle délétère favorisant les complications de l’athérosclérose :
- L’oxyde de Carbone (CO) favorise l’athérogénèse par hypoxie de l’intima des artères. - La fumée de tabac a un effet toxique sur la vasomotricité endothélium-dépendante par
inactivation du NO et par l’oxydation des LDL.
- Le tabac est un facteur thrombogène favorisant l’activation plaquettaire
- La nicotine favorise la libération des catécholamines ce qui majore la fréquence cardiaque et la pression artérielle [24]
G. Obésité :
L’obésité est définie par l’excès généralisé de masse adipeuse au niveau de l’organisme [25]. Les études épidémiologiques ayant recours, le plus souvent, à l’index de la masse corporelle (IMC) qui est calculé en divisant le poids de la personne par le carré de sa taille. Si cet indice est supérieure à 30 kg/m2, le sujet est considéré obèse [25].
Il existe une véritable relation entre le risque cardiovasculaire et la surcharge pondérale. Un excès de poids de 10 à 30% augmente le risque relatif de développer une cardiopathie ischémique à 1,4. Le risque est élevé à 2 pour un excès de poids dépassant 30% [21].
H. Hyperhomocystéinémie :
L’hyperhomocystéinémie se définit par l’excès du taux plasmatique en homocystéine (Hcy). L’homocystéine est un acide aminé qui joue un rôle important dans le métabolisme de la méthionine. Elle peut être métabolisée de deux façons : soit par trans-sulfuration, conduisant à la formation de cystéine, soit par reméthylation en méthionine. Le métabolisme de l’homocystéine est dépendant de l’apport en plusieurs facteurs vitaminiques : vitamines B6 et B12, acide folique. Les valeurs normales d’homocystéinémie sont comprises entre 5 et 15 μmol/l. [26].
L’hyperhomocystéinémie peut être classée en fonction de sa sévérité et de sa cause. Elle peut résulter d’un défaut génétique touchant l’une des enzymes impliquées dans le métabolisme de l’Hcy, ou d’une déficience en une ou plusieurs vitamines participants au cours de ce cycle métabolique [9,26].
Au niveau vasculaire, plusieurs effets délétères sont engendrés par l’Hcy, dont une partie est liée aux propriétés prooxydantes qui sont dues à son groupement thiol réducteur. L’homocysteine peut diminuer la production de la NO par la NO Synthase endotheliale, induisant l’altération des mécanismes de contrôle de la vasomotricité au niveau endothéliale [27]. Ceci rend de l’hyperhomocyctéinémie un des facteurs athérogènes et un facteur de risque indépendant de la survenue d’IDM [9,26].
I . Hyperuricemie :
L’acide urique est le produit final de métabolisme des purines. L’acide urique induit la prolifération des cellules musculaires lisses et l’induction de la synthèses des substances pro inflammatoires et des substances à propriétés vasoconstrictives. Des taux élevés en acide urique ont été détectés dans les plaques athéromateuses provoquant l’adhesion plaquettaire et la formation du thrombus. [28]
J. Stress émotionnel :
Ce facteur inclut le stress professionnel, domestique, financier ou lié à des événements pénibles [15]. Le stress émotionnel est associé avec l’infarctus du myocarde dans 4 à 18% des cas. La stimulation du système sympathique, qui en résulte, peut expliquer cette relation [9].
4. Etiologies :
A. Athérosclérose coronaire :
C’est l’étiologie de la quasi-totalité des cas d’IDM (95% des cas) [9]. La définition de l’athérosclérose selon l’OMS est la suivante « c’est une association variable des remaniements de l’intima des artères de gros et de moyen calibre, consistant à une accumulation focale de lipides, de glucides complexes de sang, de produits sanguins, de tissus fibreux et de dépôts calcaires. Le tout s’accompagnant d’une modification du média » [29]. Il s’agit d’une pathologie inflammatoire liée à l’infiltration majeure des cellules immunitaires (macrophages dérivés des monocytes et lymphocytes) et des lipides, notamment les LDL-C au niveau des lésions artérielles, conduisant à l’apparition des cellules spumeuses. Ceci est précédé par la migration des cellules musculaires lisses qui vont synthétiser à leur tour une matrice extracellulaire plus ou moins abondante.
Schématiquement, cette matrice évolue en 5 stades :
Le stade I se caractérise par la présence au niveau intimal de cellules spumeuses isolées : des
macrophages contenant dans leur cytoplasme des lipides en quantité abondante.
Le stade II se caractérise par des stries lipidiques constituées par un grand nombre de cellules
spumeuses d’origine macrophagiques, aussi de cellules musculaires lisses (CML) gorgées de lipides. Il n’existe pas à ce stade de lipides extracellulaires. Ces cellules spumeuses forment de petits amas, ou couches cellulaires visibles à l’œil nu, et qui font légèrement saillie dans la lumière artérielle. Les stries lipidiques peuvent régresser ou évoluer vers les deux stades suivants (III et IV) correspondant à la progression de la lésion d’athérosclérose.
Le stade III ou pré-athérome, qui peut être présent chez « le jeune adulte », correspond au
stade précédent associé à une accumulation abondante de dépôts lipidiques extracellulaires.
Le stade IV se caractérise par le regroupement des lipides extra et intracellulaires en amas
appelé coeur (ou noyau) lipidique constituant l’athérome ou plaque simple.
Le stade V correspond à la plaque d’athérosclérose mature. La fibrose ou chape fibreuse
entoure le noyau lipidique l’isolant de la lumière artérielle. Ce tissu fibreux est produit par les CML. Il est constitué de collagène, de mucopolysaccharides, d’élastine et de fibrine. [9,29-30].
B. Embolies coronaires :
Les infarctus du myocarde par embolies coronaires sont des cas rares. Ces embolies prennent naissance principalement à partir des tumeurs endocardiques, des thrombus muraux et des valvulopathies du cœur gauche. Les embolies gazeuses peuvent être également signalées au cours d’une chirurgie cardiaque [9,31].
C. Traumatismes thoraciques :
Il s’agit de contusion myocardique ou déchirure coronaire. La majorité des cas d’infarctus du myocarde, ayant lieu lors d’un traumatisme thoracique, sont dus à un accident de la voie publique [32].
D. Spasme coronaire :
Le spasme coronaire est l’une des causes de l’ischémie myocardique. Ceci peut être prouvé par le fait qu’un spasme coronaire peut déclencher la rupture de la plaque athéromateuse, aggraver la réduction du calibre coronaire sur une plaque athéromateuse rompue et partiellement thrombosée [9,33].
E. Anomalies hématologiques :
Elles sont en rapport avec des états d’hypercoagulabilité, et de thromboses sur des vaisseaux coronaires sains. Ces anomalies surviennent chez des sujets jeunes le plus souvent présentant des déficits en protéines C, ou en protéines S ou en antithrombine [34]. D’autres étiologies peuvent également être mentionnées, telles que les anomalies congénitales des artères coronaires, l’insuffisance aortique, la contusion myocardique, ainsi que la toxicomanie [9,34].
5. Physiopathologie :
L’infarctus du myocarde est un syndrome coronarien aigu, lié à l’occlusion du vaisseau coronaire, et responsable d’un état d’ischémie sévère qui évolue par la suite vers une nécrose myocardique.
Dans la quasi-totalité des cas, l’occlusion coronaire prend naissance de la fissuration d’une plaque athéromateuse, mettant à nu la matrice sous endothéliale. Il s’agit d’une surface très thrombogène qui entraine l’adhésion, l’activation et l’agrégation plaquettaire, aboutissant à la formation d’un thrombus coronaire occlusif. A partir de ce thrombus, des emboles peuvent être libérés aggravant la vasoconstriction en aval (figure 2)[9,35-36].
Figure 2 : Physiopathologie de l’infarctus du myocarde [9] 1 : thrombus, 2 : embolie distale, 3 : vasoconstriction
La réduction des apports de sang au myocytes, qui en résulte, provoque un état de déséquilibre entre l’apport d’oxygène et sa consommation par ces cellules, désigné par le nom d’ischémie myocardique [9].
Au niveau des cellules myocytaires, l’ischémie induit une altération de la pompe ionique, une fuite d’ions potassiques vers le milieu extracellulaire, une surcharge intracellulaire en calcium activant en conséquence les voies de signalisation pro apoptotiques. L’ischémie myocardique est associée avec une ischémie cellulaire. Une fois prolongée, elle peut évoluer vers une nécrose myocardique irréversible. Celle-Ci s’étend du centre de la zone ischémique vers sa périphérie et de l’endocarde vers l’épicarde aboutissant à l’altération de la fonction contractile du myocarde [1,36-37].
La restauration du flux coronaire après quelques dizaines de minutes d’ischémie entraine une réduction de la taille de la nécrose par rapport à la zone initialement à risque. Si la restauration du flux coronaire est tardive, celle-ci s’accompagne des lésions dites : des lésions de reperfusion [9].
6. Marqueurs biologiques de diagnostic:
Le diagnostic doit être établi d’une manière précoce. L’infarctus du myocarde est une maladie de nécrose myocardique. Le diagnostic, doit s’appuyer sur des signes cliniques (douleurs thoraciques, spontanées, retro sternales, constrictives, angoissantes), sur des signes éléctrocardiographiques (sus décalage du segment ST, avec ou sans onde Q). Le diagnostic doit être confirmé par la suite par la détection des marqueurs biologiques spécifiques. Il s’agit des protéines présentes dans les cellules cardiomyocytaires et qui sont libérées dans la circulation sanguine lors d’une nécrose myocardique [35,38].
Ces marqueurs sont capables de nous renseigner concernant la taille de la zone nécrosée, et de suspecter en plus une extension ou une récidive [35].
De part leur sensibilité et de leur spécificité moyennes, le dosage de certains marqueurs cardiaques classiques n’est plus pertinent. Ceci a mené vers la mise en place des nouveaux marqueurs plus spécifiques avec des recommandations précises sur leur utilisation [39].
Troponine :
Grâce à sa grande cardiospécificité, la troponine est devenue le marqueur biologique de choix de l’infarctus du myocarde. La troponine constitue avec l’actine et la tropomyosine l’un des composants protéiques des filaments fins du muscle strié, intervenant dans la régulation de la contraction musculaire. Il s’agit d’un trimère formé de 3 sous unités :
- la troponine C : il s’agit d’un polypeptide de petit poids moléculaire (18 kDa), qui existe sous forme identique dans le myocarde et dans le muscle strié. La troponine C est le site de fixation du calcium.
- la troponine T : son poids moléculaire est important (37 kDa). Elle existe sous de nombreuses formes moléculaires distinctes avec deux isoformes cardiaques et 12 isoformes musculaires.
- la troponine I : fixe le filament d’actine à la troponine C et empêche la contraction en absence de calcium. Elle existe sous trois formes différentes : deux formes spécifiques des muscles striés et une forme spécifique du myocarde (TnIc) (Figure 3) [35-36,38,40-41].
Plusieurs méthodes immunoenzymatiques et d’électrochimiluminescence ont été développées en faveur d’un dosage spécifique, sensible et rapide de deux isoformes cardiaques de la troponine I et T, permettant ainsi d’exclure toute interférence avec les
isoformes squelettiques [35,38]. Un seuil de 0,1 ng/ml est avéré décisionnel pour le diagnosctic [35].
Le progrès réalisé dans les dosages des troponines était concrétisé par l’apparition des dosages dits « ultrasensibles » qui concernent les dosages de la TnIc et de la TnTc. Ce type des dosages se caractérise par une bonne précision analytique avec une limite de détection qui correspond à 2,06 ng/l pour le dosage de la TnIc. Ce type de dosage permet, également, de détecter toute élévation du taux de la TnIc dans un intervalle de temps compris entre 1 à 3 heures [39,42].
Figure 3 : Structure de la troponine [41]
h- FABP :
Les « fatty acid binding protein » (FABP) sont des protéines intracellulaires qui lient de manière réversible et non covalente les acides gras non estérifiés (AGNE). Les FABP regoupent des isoformes « h-FABP ». Il s’agit des petites protéines cytosoliques de 132 acides aminés (14,5 kDa), très abondantes dans les cellules cardiomyocytaires [39-43].
Le dosage de la h-FABP repose sur des méthodes de type ELISA quantitatives. Ce test est positif pour des concentrations supérieures à 6 µg/l. Les applications cliniques du dosage de ce marqueur sont en relation avec ses caractéristiques de marqueur de nécrose précoce, sensible et spécifique du myocarde [39].
Myoglobine :
Il s’agit d’une hémoprotéine de faible poids moléculaire (17500 Da), présente dans toutes les cellules du muscle strié et cardiaque et intervenant dans le métabolisme aérobie en stockant de l’oxygène [25,38].
Vu son faible poids moléculaire, la myoglobine est libérée rapidement dans le sang en cas d’une nécrose myocardique. Elle est rapidement éliminée par voie rénale sauf dans le cas d’insuffisance rénale. Des taux plasmatiques élevés de Myoglobine peuvent être observés en cas d’atteinte musculaire ou d’insuffisance rénale [35,39-40].
Le dosage de la myoglobine a un intérêt lié à sa cinétique rapide permettant de suivre les récidives précoces d’un IDM et l’évaluation des techniques de reperfusion coronaire. La plus grande limite de son dosage est l’absence de cardiospécificité [39-40].
Des méthodes, adoptées à l’urgence médicale, permettent le dosage rapide et automatisé de la myoglobine. Elles incluent les méthodes d’immunonéphélométrie, d’immunoturbidimétrie d’immunoenzymofluorimétrie et d’immunochimiluminescence [39-40].
Les valeurs usuelles du dosage de la myoglobine sont comprises entre 10 et 85 µg/l, au cours d’un infarctus [35].
.
Créatine kinase :
Il s’agit d’une enzyme dimérique de PM 87 kDa, présente dans le cytoplasme des cellules myocytaires et constituée de l’assemblage de 2 sous unités M et B, de 360 acides aminés pour chacune. Cette association 2 à 2 détermine 3 isoenzymes différentes : la CK-MM, l’isoforme squelettique et myocardique, la CK-MB présente essentiellement dans le tissu myocardique et la CK-BB qui prédomine dans le cerveau. Elle est présente aussi au niveau des autres tissus tels que la prostate, le foie, l’utérus et d’autres tissus [35-38].
La créatine kinase joue un rôle important dans l’apport énergétique nécessaire à la contraction du muscle, en catalysant le transfert réversible du phosphate entre l’ADP et l’ATP [38]. L’analyse de ce marqueur enzymatique peut être réalisée soit par la mesure de l’activité catalytique de la créatine kinase totale grâce à une méthode cinétique spectrophotométrique, soit par la mesure de l’activité de la fraction MB grâce à des méthodes électrophorétiques ou par immunoinhibition [35,38,40]. Toute augmentation du taux de la créatine kinase totale est associée à une élévation du taux du CK-MB. Il est nécessaire de déterminer le rapport CK-MB/CK totale multiplié par 100, afin de distinguer entre un IDM et autre situation impliquée dans l’élévation non spécifique de la CK-MB (chirurgie, effort musculaire intense…) [35,38,40].
Les valeurs normales de la CK totale sont comprises entre 30 et 200 U/l à 37°. Pour la CK-MB, les valeurs de référence sont comprises entre 0 et 10 U/l [35].
Lactate deshydrogénase (LDH) :
Il s’agit d’une enzyme de masse moléculaire 134 kDa. Elle catalyse l’oxydation de L-lactate en pyruvate. Cinq isoenzymes ont été définies, résultantes de l’association quatre à quatre de deux chaines peptidiques M et H, dont la proportion de ces enzymes varie d’un tissu à un autre.
La mesure de l’activité de LDH totale est possible grâce à une méthode spectrophotométrique. Les valeurs usuelles sont comprises entre 200 et 400 U/l à 37° [35].
Aspartate aminotransférase (ASAT) :
Il s’agit d’une enzyme dimérique, de masse moléculaire 90 kDa, présente principalement dans les cellules du muscle strié. Elle catalyse en présence du phosphate de pyridoxal le transfert du groupement aminé du L-aspartate sur le 2-oxoglutarate.
L’activité catalytique de l’ASAT est mesurée par une méthode spectrophotométrique, dont les valeurs usuelles sont comprises entre 5 et 45 U/l à 37° [35].
Cinétique des marqueurs biologiques :
La cinétique d’apparition reflète la taille et la demi-vie de chaque marqueur [35,38]. (Figure 4)
En cas d’une nécrose myocardique, la myoglobine est libérée rapidement dans le sang. Ceci justifie son utilisation comme marqueur précoce de l’IDM. Le taux de la myoglobine s’accroit dans les quatre premières heures de début des douleurs thoraciques, atteint le pic entre 6 et 7 heures et retourne à sa valeur normale après 24 heures de début de l’IDM.
L’activité de la CK-MB augmente entre 3 et 12 premières heures, atteint le pic vers 24 heures et revienne à la valeur normale après 48 heures de début de l’IDM.
Le taux de la troponine Ic augmente, également, de 3 à 12 heures, atteint le pic vers 24 heures et retourne à la normale après 5 jours de début des douleurs thoraciques (Figure 4) [39-40].
7. Complications de l’infarctus du myocarde :
La gravité de l’infarctus du myocarde réside dans la survenue des complications. Celles ci sont directement dépendantes de la masse myocardique nécrosée, à l’exception des arythmies ventriculaires initiales.
A. Complications précoces : elles surviennent au cours de la première semaine lors d’un
IDM, elles sont variées, regroupant :
Complications mécaniques
- Rupture de la paroi cardiaque : La forme classique est une rupture aiguë complète de la paroi libre du VG avec effusion de sang dans la cavité péricardique conduisant à un état de collapsus par tamponnade. Parfois, la rupture est incomplète avec suffusion de sang à partir du muscle nécrosé et installation progressive des symptômes.
- Insuffisance mitrale : Celle-ci peut exister soit en forme sur-aigue résultant d’une rupture ischémique plus ou moins complète d’un pilier, soit sous forme aigue résultante d’une dysfonction ventriculaire gauche aigue. L’incidence de l’IM est de 1 à 3% des cas d’IDM. - Communication interventriculaire : Il s’agit de la complication mécanique la plus fréquente des infarctus (1 à 5% des cas).
Troubles rythmiques
- Des troubles de rythme supraventriculaire : incluant la tachycardie sinusale, la tachycardie atriale et l’extrasystole auriculaire.
- Des troubles de rythme ventriculaire : incluant l’extrasystolie ventriculaire, la tachycardie ventriculaire, le rythme idioventriculaire accéléré et la fibrillation ventriculaire.
Troubles de conduction
- Blocs auriculo-ventriculaires : la gravité de cette complication est déterminée selon sa localisation inférieure ou antérieure.
- Dysfonction sinusale : elle est souvent en rapport avec une occlusion très proximale de l’artère coronaire droite, et plus rarement de l’artère circonflexe.
B. Complications tardives : Certaines de ces complications ont été mentionnées au dessus
(l’insuffisance cardiaque et les arythmies ventriculaires). La seule complication avérée spécifique de cette période est le syndrome de Dressler, probablement lié à des phénomènes auto immuns [9,35,44].
8. Traitement de l’infarctus du myocarde: A. Prise en charge hospitalière :
Une fois, le diagnostic est établi, une thérapie de reperfusion sera recommandée, soit par thrombolyse soit par angioplastie, dont le but est d’assurer la restauration rapide et complète de la circulation sanguine coronarienne [45].
Thrombolyse :
Elle est appliquée d’une façon pharmacologique par l’administration d’un agent thrombolytique. L’alteplase administrée en protocole accéléré, est considérée comme étant l’élément thrombolytique de référence [36-46].
Néanmoins, le succès de cette stratégie de reperfusion, se confronte parfois avec un taux faible d’éligibilité au traitement thrombolytique, inclut la contre indication à la fibrinolyse rapportée aux sujets de plus de 70 ans [46].
Angioplastie :
L’angioplastie primaire permet la recanalisation chirurgicale de l’artère obstruée plutôt que la lyse de thrombus. Elle permet d’obtenir un taux de perméabilité coronaire proche de 100% [36-46].
Cette procédure associée à un taux de mortalité faible, permet de traiter simultanément l’occlusion coronaire et la sténose sous-jacente. Elle est classée, la meilleure, en termes d’efficacité, en comparaison avec la thrombolyse coronaire [36-46].
L’angioplastie est utilisée comme traitement de sauvetage dans le cas ou une échéance du traitement thrombolytique sera avérée [36].
B. Traitements pharmacologiques adjuvants :
Le traitement pharmacologique de l’IDM repose essentiellement sur l’utilisation des antiagrégants, des anticoagulants, des béta bloquants, des dérivés nitrés et des inhibiteurs calciques [36].
Antiagrégants plaquettaires :
La justification physiopathologique de leur prescription est de prévenir la survenue des nouveaux accidents coronaires de nature thrombotique [47]
L’aspirine : elle inhibe l’agrégation plaquettaire par son acétylation de la cyclooxygénase 1 [48]. L’utilisation de l’aspirine a été associée avec une réduction du taux de mortalité cardiovasculaire de plus de 50% à un mois par rapport au placebo. Cet agent antiagrégant a montré un effet synergique, une fois associé avec le traitement thrombolytique [46-47]. Le clopidogrel : c’est un agent antiagrégant plaquettaire de la classe des thiénopyridines. Il permet de limiter l’amplification de l’activation plaquettaire. [47].
Anticoagulants :
Une héparinothérapie est recommandée au stade aigue de l’infarctus du myocarde, liée à son rôle dans la réduction des risques de réocclusion coronaire et de thrombose inter ventriculaire.
Les anticoagulants permettent également de prévenir les accidents thromboemboliques veineux liés à l’alitement [35-36].
Bétabloquants :
Leur administration intraveineuse dès les premières heures de l’infarctus sans traitement de revascularisation permet de réduire le taux de mortalité. L’incidence de fibrillation ventriculaire et la taille de l’infarctus [49]. La prescription des bétabloquants par voie intraveineuse est actuellement recommandée. Ils sont contre indiqués en présence d’une décompensation cardiaque aigue [46-49].
Dérivés nitrés :
Les dérivés nitrés sont prescris lors d’un IDM, grâce à leur action anti angineuse d’une part et d’autre part à leur effet vasodilatateur coronaire. Ils ont le double avantage de favoriser la circulation collatérale et de pouvoir lever un éventuel facteur spastique à l’endroit de l’occlusion coronaire [36].
Inhibiteurs de l’enzyme de conversion (IEC) :
La prescription des inhibiteurs de l’enzyme de conversion lors d’un IDM avec sus-décalage du segment ST, peut être liée à leur impact favorable sur le remodelage ventriculaire, l’amélioration des paramètres hémodynamiques et la diminution de l’incidence de l’insuffisance cardiaque [50].
Des antagonistes aux récepteurs de l’angiotensine sont utilisés comme alternative recommandable aux IEC dans le cas d’une intolérance à ce traitement [49].
II. LE STRESS OXYDANT
:1. Définition :
Le stress oxydant se définit par un déséquilibre profond entre les antioxydants et les prooxydants en faveur de ces derniers. Cette situation peut résulter d’une déficience ou défaillance en systèmes antioxydants, des troubles de production, de distribution ou d’une abondance accrue des prooxydants [51-52]. Des études ont mis en évidence l’implication des facteurs alimentaires, notamment les micronutriments dans la modulation de ce phénomène [53].
Le mot prooxydant désigne toute espèce réactive de l’oxygène et de l’azote, radicalaire ou non radicalaire [54]. Un radical libre est une espèce chimique, molécule ou atome, capable d’avoir une existence indépendante en contenant un ou plusieurs électrons célibataires. Le radical libre a la propriété d’être instable, très réactif avec une durée de vie très courte.
Pour devenir stable, il aura toujours tendance à remplir son orbitale en captant un électron, par l’oxydation d’un autre composé [55]. Cette propriété rend les radicaux libres aptes à réagir avec différentes molécules et composantes cellulaires telles que l’acide nucléique, les lipides, les protéines et les composés glucidiques.
2. Principaux éléments pro oxydants :
Les espèces dérivées de l’oxygène comme le peroxyde d’hydrogène, ne sont pas des radicaux libres mais elles sont aussi réactives et peuvent jouer le rôle des précurseurs des radicaux. L’ensemble de ces radicaux libres et leurs précurseurs sont appelés : des espèces réactives d’oxygène (ERO) [56].
Anion superoxyde O°-2 :
L’anion superoxyde provient de l’ajout d’un seul électron à la molécule d’oxygène selon la réaction suivante : O2 + électron O2-°
L’anion superoxyde est un radical stable, peu réactif avec un pouvoir oxydant faible [57-58]. Il est produit dans toutes les cellules aérobies (particulièrement dans les mitochondries). De même, l’anion superoxide est généré par la NADPH oxydase présente au niveau des cellules vasculaires endothéliales. L’apparition de radicaux superoxydes peut résulter de l'auto-oxydation (l'auto-oxydation par l'oxygène) des composés tels que des neuromédiateurs (adrénaline, noradrénaline, dopamine…), des thiols (cystéine), des coenzymes réduits (FMNH2, FADH2).
L’anion superoxyde peut être généré lors de la détoxification des xénobiotiques (toxiques, médicaments) par le système des cytochromes P450 présents au niveau du réticulum endoplasmique [59].
Peroxyde d’hydrogène H2O2
La dismutation de l’anion superoxyde constitue la source majeure de peroxyde d’hydrogène [60]:
2H+ + O2°- + O2°- SOD H2O2 + O2
La production de peroxyde d’hydrogène peut résulter de la réduction biélectronique de l'oxygène en présence d'oxydases (aminoacides oxydases, glycolate oxydase, urate oxydase…) qui se trouvent principalement dans des organites cellulaires bien individualisés comme les peroxysomes. La membrane mitochondriale externe renferme une monoamine oxydase capable de catalyser la désamination oxydative de certaines amines, avec production simultanée de H2O2 [59]
Le peroxyde d’hydrogène est un oxydant puissant, liposoluble, capable de diffuser facilement à travers les membranes cellulaires [55,61-62]
Radical hydroxyl OH°
Le radical hydroxyl est extrêmement réactif avec une durée de vie très courte (10-9 S) et une action très délétère. Les cibles biologiques des radicaux hydroxyls sont les molécules organiques et inorganiques des cellules, en particulier, l’ADN, les protéines, les lipides, les acide-aminés, les sucres et les métaux. Le radical •OH est produit au sein des cellules, suite soit à la réaction de Haber-Weiss, soit à la réaction de Fenton qui nécessite des catalyseurs métalliques [55, 61].
Réaction de Fenton : Métal n+
+ H2O2 métal n+1 +OH° + OH-
Réaction de Haber-Weiss : H2O2 + O2° O2 +OH° + OH-
Monoxyde d’azote NO°
Le monoxyde d’azote posséde un seul électron célibataire. Il est généré au niveau des tissus biologiques par une flavoenzyme calcium dépendante : l’oxyde nitrique synthase [63]. La NOS produit le monoxyde d’azote NO par oxydation de la L-arginine par l’oxygène [62] Trois isoformes distinctes de cette enzyme ont été identifiées : l’oxyde nitrique synthase neuronale (NOSn), endothéliale (NOSe) et celle inductible (NOSi) [64]. La NOSn et la NOSe
dont l’expression est constitutive, sont activées physiologiquement par des hormones stéroïdes et des neurotransmetteurs. Au contraire, la NOSi est exprimée dans toutes les cellules nuclées [64-65].
Le monoxyde d’azote est soluble en phase aqueuse et lipidique, ce qui lui permet de diffuser facilement à travers les membranes cellulaires, et jouer le rôle d’un médiateur paracrine avec un large spectre d’actions physiologiques : le contrôl du tonus vasculaire, l’inhibition de l’agrégation plaquettaire, et la régulation des voies de transduction du signal [63-64]. Le monoxyde d’azote est doué des propriétés pro apoptiques et anti apoptiques à la fois. L’action pro apoptique fait intervenir l’anion superoxide O2, en activant indirectement les caspases et en conduisant à la fragmentation de l’ADN. L’action anti apoptotique de Monoxyde d’azote est liée à l’inihibition de l’activité catalytique des caspases. [66]
Peroxynitrite ONOO°
Le peroxynitrite est un agent oxydant et nitrosant à la fois. Il est liposoluble. Il provient de l’interaction entre l’anion superoxyde et l’oxyde d’azote générés lors d’un processus inflammatoire [61,63]:
NO° + O2-° ONOO°
Le peroxynitrite est un oxydant toxique, capable de diffuser facilement à travers les membranes cellulaires. Le peroxynitrite est impliqué dans l’activation des voies pro apoptotiques, chez les érythrocytes, via l’activation des caspases 8 et 3.
Radical peroxyl ROO°
Il résulte de l’action d’un radical libre sur un acide organique tel que les lipides membranaires. Le radical peroxyl est très réactif, réagissant avec la molécule d’hydrogène la plus proche ou avec les composés contenant des doubles liaisons s dans son environnement [54,67].
3. Génération des prooxydants
Au niveau de la mitochondrie :
La respiration mitochondriale conduit essentiellement à la réduction de l’oxygène en eau par transfert de quatre électrons via le cytochrome oxydase du complexe IV. Le bilan énergétique permet la phosphorylation de l’ADP en ATP [58-68] :
Une réduction incomplète peut concerner une fraction de 2 à 5% de l’oxygène consommé par la mitochondrie. Celle-ci aboutit à l’apparition d’anion superoxyde au niveau de plusieurs sites potentiels dont la NADPH déshydrogénase du complexe I et le site ubisemiquinone du complexe III [58-68].
NADPH oxydase :
La NADPH oxydase est une enzyme cellulaire connue dans les cellules phagocytaires, à l’origine de la production extracellulaire d’anions superoxydes nécessaires à la destruction d’éléments étrangers. Cette enzyme a été mise en évidence, après, dans les cellules endothéliales vasculaires, dans les cellules musculaires et dans les fibroblastes de l’adventice. La réaction catalysée par la NADPH oxydase est la suivante [58,69-70]:
NADPH + 2O2 NADP+ + H+ + 2O2-°
Les NADPH oxydases sont caractérisées par des structures similaires, cependant distinctes de celles des neutrophiles, ce qui suggère des mécanismes d’activation et d’action différente : la NADPH vasculaire est constitutivement active, alors que la NADPH oxydase des neutrophiles peut être induite par des produits microbiens tels que les liposaccharides bactériens, des lipoprotéines et des cytokines [64, 68, 70].
Myeloperoxydases :
La myeloperoxydase (MPO) est une enzyme qui est présente dans les neutrophiles. Elle est considérée parmi les systèmes antimicrobiens formés dans les phagosomes. Le produit initial du système myeloperoxydase – peroxyde d’hydrogène – ion chlore est l’acide hypochloreux :
H2O2 + Cl- MPO HOCl + OH-
Ces produits peuvent eux même donner naissance aux autres composés toxiques tels que le chlore sous forme de gaz (Cl2) et l’oxygène singulet [69,71].
Xanthine oxydase :
Sous conditions physiologiques, la xanthine oxydase est localisée en particulier dans le foie et dans la muqueuse de l’intestin grêle [72].
Toutefois, la xanthine oxydase existe initialement sous la forme d’une déhydrogénase intracellulaire. A l’issu d’une réaction inflammatoire, cette déhydrogénase sera convertie suite à une oxydation réversible des résidus cystéine ou suite à une protéolyse limitée en xanthine oxydase. Celle-ci est impliquée dans la catalyse de l’oxydation de l’xanthine générant de l’acide urique et d’anions superoxydes [72-73].
Cytochromes P450 :
Les isoenzymes P450 réduisent l’oxygène moléculaire en anion superoxyde au cours de la détoxification des doses aigues en toxines. Cette production pro oxydante est considérée comme étant la contre partie du maintien de la survie cellulaire en cas d’une intoxication [64].
Oxyde nitrique synthase (NOS) :
Les oxydes nitriques synthases constituent une famille d’enzymes impliquées dans la neurotransmission, la vasodilatation et dans les processus immunitaires. Cette implication est liée à la synthèse de l’oxyde nitrique (NO°) et de L-citrulline à partir de L-arginine en présence d’oxygène et de NADPH [74].
Trois isoformes distinctes de cette enzyme ont été identifiées : l’oxyde nitrique synthase neuronale (NOSn), endothéliale (NOSe) et celle inductible (NOSi) [64].
La NOSn et la NOSe dont l’expression est constitutive, sont activées physiologiquement par des hormones stéroïdes et des neurotransmetteurs. Au contraire, la NOSi est exprimée dans toutes les cellules nucléées, générant des taux élevés en oxyde nitrique en réponse aux cytokines, aux produits des bactéries, aux stimulus immunologiques et autres [64,74].
Enzymes peroxysomales :
De par leur implication dans le métabolisme oxydatif, les enzymes peroxysomales constituent une source importante des ERO notamment le peroxyde d’hydrogène, assurant le transfert d’hydrogène de leurs substrats respectifs à une molécule d’oxygène. Parmi ces enzymes, on peut citer : l’acyl-CoA oxydase, l’urate oxydase et autres [75].
4. Paradoxe des espèces réactives d’oxygène :
Ce paradoxe provient de deux rôles contradictoires de ces espèces réactives envers l’organisme humain. Dans le temps ou ces éléments sont toxiques, susceptibles d’engendrer de nombreuses pathologies, elles constituent également des espèces indispensables à la vie, remplissant ainsi plusieurs fonctions physiologiques [56,76].
A. Rôles physiologiques des radicaux libres :
Régulation des voies de signalisation cellulaire :
Au niveau vasculaire, les espèces réactives d’oxygène (ERO) sont impliquées dans la régulation des différentes fonctions physiologiques. Parmi les EROs, le monoxyde d’azote (NO°), synthétisé par la NOS endothéliale, intervient dans le contrôle du tonus vasculaire [70]. Divers mécanismes ont été explorés, via lequels, le NO° est impliqué dans la transduction de signal tel que la S-nitrosylation de plusieurs protéines récepteurs (RTK receptor tyrosine kinase, TNF …). [77] Le NO° exerce d’autres effets importants en inhibant l’agrégation plaquettaire, l’adhérence des leucocytes et la prolifération des cellules musculaires lisses [64,70].
Autre que le monoxyde d’azote, le peroxyde d’hydrogène, intervient également dans la croissance et la prolifération des cellules endothéliales via différents mécanismes tels que l’oxydation réversible des acides aminés, incluant les résidus cysteine, methionine, tryptophane, et les résidus thyrosines [77-78].
Le Peroxyde d’hydrogène est impliqué dans la régulation de l’activité des tyrosines phosphatases, des protéases telles que les caspases et les métalloproteinases via l’oxydation de leurs cysteines catalytiques. Chez les cellules des mammifères, le peroxyde d’hydrogène est impliqué dans la régulation des activités des protéines chaperones, incluant les Hsp 25, 60 et 70 via le mécanisme d’oxydation des cysteines actives. |77]. Ces espèces oxydantes sont considérées comme des modulateurs de l’expression de plusieurs gènes participant à l’homéostasie vasculaire, et à la croissance cellulaire par l’intermédiaire de facteurs de transcription redox sensibles [56, 62, 68]. La S-nitrosylation et la S-glutathionylation des différents facteurs de transcription tels que la Nf-KB et le C-Jun membre de la famille AP-1 peuvent diminuer leurs intéractions avec l’ADN entrainant la diminution de niveau d’expression des gènes.
Au cours des processus inflammatoires :
La production massive des espèces réactives d’oxygène joue un rôle clé dans l’activation des processus inflammatoires. Les radicaux libres activent un facteur de transcription le Nf-KB induisant la transcription des gènes codants pour des protéines impliquées dans la réponse inflammatoire, incluant les cytokines. [79]
La production d’interleukine-2 par les cellules lymphocytaires T est induite par des taux physiologiques en anions superoxydes et en peroxydes d’hydrogène [63-64].
Les radicaux libres, générés en circulation sanguine, sont capables d’activer l’expression des molécules d’adhésion et des récepteurs des cellules endotheliales, rendant ces cellules beaucoup sensibles aux effets proinflammatoires des cytokines, et augmentant le potentiel de recrutement des cellules leucocytaires [79]. De plus, les radicaux libres jouent un rôle clé dans la défense contre les agents pathogènes phagocytés par les macrophages et neutrophiles. Ce mécanisme de défense est induit suite à l’activation de certaines enzymes prooxydantes au sein des phagosomes telles que la NADPH oxydase et la myeloperoxydase [56, 64].
Les espèces réactives d’oxygène peuvent intervenir dans différents processus physiologiques : elles peuvent servir de relais entre les cellules, activer certaines enzymes, des canaux ioniques, et des récepteurs membranaires. Elles peuvent réguler aussi la synthèse des prostaglandines par les cyclooxygénases [56,64].
B. Toxicité des radicaux libres :
La production excessive des radicaux libres provoque des lésions directes des composantes cellulaires par oxydation d’ADN, des protéines, des lipides, et des glucides. Les conséquences biologiques qui en résultent sont extrêmement variables selon la dose et le type cellulaire soumis à cette action toxique [56,63].
Oxydation des acides désoxyribonucléiques :
Les acides désoxyribonucléiques sont très sensibles à l’attaque par les radicaux libres. Le radical hydroxyl peut réagir avec tous les composés de la molécule d’ADN, endommageant ses bases azotés (plus que 60 lésions ont été identifiées).
La guanine est la plus facile à être oxydée, engendrant ainsi deux produits majeurs : le 8-hydroxyguanine et le formamidopyrimidine [56,63, 80].
Ces espèces radicalaires sont capables d’attaquer la liaison entre la base et le désoxyribose ou attaquer le sucre lui même créant une coupure au niveau de la chaine simple brin [56].
Plusieurs dérivés issus de l’action radicalaire au niveau des acide nucleiques peuvent être dosés, tels que le glycol de thymine, le 8-hydroxyguanine, le 8-hydroxy-adénine, soit par chromatographie gazeuse couplée à une spectrométrie de masse, soit par une méthode HPLC assez sophistiquée [81-82].
![Figure 1 : Mortalité causée par des maladies coronariennes dans le monde [10]](https://thumb-eu.123doks.com/thumbv2/123doknet/14521054.722485/17.892.211.720.147.524/figure-mortalité-causée-maladies-coronariennes-monde.webp)
![Figure 2 : Physiopathologie de l’infarctus du myocarde [9]](https://thumb-eu.123doks.com/thumbv2/123doknet/14521054.722485/24.892.274.618.173.541/figure-physiopathologie-de-l-infarctus-du-myocarde.webp)
![Figure 3 : Structure de la troponine [41]](https://thumb-eu.123doks.com/thumbv2/123doknet/14521054.722485/26.892.233.737.413.687/figure-structure-troponine.webp)
![Figure 4 : Cinétique des marqueurs biologiques de l’infarctus du myocarde [40]](https://thumb-eu.123doks.com/thumbv2/123doknet/14521054.722485/28.892.217.677.789.1066/figure-cinétique-marqueurs-biologiques-l-infarctus-myocarde.webp)
![Figure 5 : Réactions de peroxydation lipidique [85]](https://thumb-eu.123doks.com/thumbv2/123doknet/14521054.722485/41.892.129.775.109.434/figure-réactions-peroxydation-lipidique.webp)
![Figure 7: Génération des ERO au cours de l’IDM [102]](https://thumb-eu.123doks.com/thumbv2/123doknet/14521054.722485/48.892.187.738.573.965/figure-génération-ero-cours-l-idm.webp)
![Figure 8: Organisation hiérarchique du système circadien [127]](https://thumb-eu.123doks.com/thumbv2/123doknet/14521054.722485/53.892.265.733.688.1071/figure-organisation-hiérarchique-système-circadien.webp)