2006/233
Review
Hypoxia and lipid signaling
Andrea Huwiler1,2and Josef Pfeilschifter1,*
1Pharmazentrum Frankfurt, Klinikum der Johann-Wolfgang-Goethe-Universita¨t, Theodor-Stern-Kai 7, D-60590 Frankfurt am Main, Germany
2Institut fu¨r Pharmakologie, Universita¨t Bern, Friedbu¨hlstrasse 49, CH-3010 Bern, Switzerland
* Corresponding author
e-mail: [email protected]
Abstract
Sufficient oxygen supply is crucial for the development and physiology of mammalian cells and tissues. When simple diffusion of oxygen becomes inadequate to vide the necessary flow of substrate, evolution has pro-vided cells with tools to detect and respond to hypoxia by upregulating the expression of specific genes, which allows an adaptation to hypoxia-induced stress condi-tions. The modulation of cell signaling by hypoxia is an emerging area of research that provides insight into the orchestration of cell adaptation to a changing environ-ment. Cell signaling and adaptation processes are often accompanied by rapid and/or chronic remodeling of membrane lipids by activated lipases. This review high-lights the bi-directional relation between hypoxia and lipid signaling mechanisms.
Keywords: eicosanoids; gene expression; hypoxia;
hypoxia-inducible factor; signal transduction; sphingolipids.
Introduction
To ensure survival in changing environments, mammalian cells have developed mechanisms to respond to reduced oxygen availability (hypoxia) and to maintain energy bal-ance. Hypoxia leads to diminished oxidative phospho-rylation and to a depletion of cellular ATP. Ultimately, chronic hypoxia can result in cell death.
Many physiological responses to hypoxia are molec-ularly controlled by a family of transcription factors denoted as hypoxia-inducible factors (HIF), which have been proposed to be master regulators of oxygen homeostasis. Under normoxia, HIF transcription factors are subjected to a very rapid turnover, in that constitu-tively synthesized HIF protein is rapidly degraded again by the 26S proteasome pathway. Under hypoxic condi-tions, HIF-1 is stabilized instantaneously, resulting in an accumulation of HIF-1 protein that translocates into the nucleus and binds to hypoxia-responsive elements (HREs) in the promoter of hypoxia target genes to acti-vate their expression. A multitude of genes have been
shown to be regulated by hypoxia. These mainly include genes involved in energy metabolism, cell growth and viability, vascular development and remodeling, erythro-poiesis and iron metabolism (for a review, see Semenza, 1999, 2006). However, in pathological processes such as tumor growth, wound healing, retinopathy, and ischemic diseases, hypoxia has also evolved as an important stimulus.
Oxygen-sensing mechanisms
When simple diffusion of oxygen becomes inadequate to provide the necessary flow of substrate, evolution has equipped cells with tools to register and respond to hypoxia by upregulating the expression of specific genes, which allows adaptation to hypoxia-induced stress con-ditions caused by concon-ditions including high altitude, anemia and wound healing, or during development (Semenza, 1999; Pugh and Ratcliffe, 2003). One of the master regulators of these adaptational changes in gene transcription is the HIF family of transcription factors. The functional entity of these transcription factors consists of a/b-heterodimers. HIF-1b, also denoted as the aryl hydrocarbon receptor nuclear translocator (ARNT), is constitutively expressed and, like the a-subunits of HIF, is a member of the basic helix-loop-helix (bHLH)-PAS family (Semenza, 1999, 2006; Pugh and Ratcliffe, 2003). To date, three mammalian HIF a-subunits have been identified, 1a, 2a (also known as EPAS) and HIF-3a, which are all oxygen-sensitive components, but differ in their tissue expressions. Whereas HIF-1a is ubiqui-tously expressed, HIF-2a and -3a show a more restricted expression pattern. HIF-2a is highly expressed in endo-thelial cells (Elvert et al., 1999; Haase, 2006), but is also found in renal interstitial fibroblast-like cells (Haase, 2006) and renal mesangial cells (Petry et al., 2005). HIF-3a exists as at least six splice variants, which can be detect-ed as multiple RNA species in Northern blot analyses with sizes of 1.5, 3.0, 7.0, and 7.5 kb (Maynard et al., 2003). Strong expression of HIF-3a was observed in the heart, placenta, and skeletal muscle, whereas a weak expression profile was found in the lung, liver, and kidney (Maynard et al., 2003). The functions of the different HIF-a proteins in a hypoxic setting are still unclear. How-ever, it has been speculated that HIF-3a, which lacks one of the two transactivation domains present in the other two HIF-a proteins, may serve as a negative regulator of hypoxia-triggered gene transcription by competing for HIF-1b binding (Hara et al., 2001).
Under ambient oxygen conditions, all three HIF-a sub-types are continuously hydoxylated on critical proline residues (Pro402 and/or 564in HIF-1a) by prolyl hydroxylases (PHD)-1, -2 or -3, and subsequently associate with the
von-Hippel-Lindau (VHL) tumor suppressor protein, which is a component of the E3 ubiquitin ligase complex and directs HIF-a subtypes to degradation by the 26S proteasome pathway (Maxwell et al., 1999; Epstein et al., 2001). Importantly, hypoxia slows down the degradation of HIF-1a, because the prolyl hydroxylases are oxygen-dependent and non-hydroxylated HIF-1a is not recog-nized by the VHL protein. As a result HIF-1a accumulates and translocates into the nucleus, where it dimerizes with HIF-1b to bind to HRE, an enhancer element containing the consensus sequence 59-ACGTG-39, located in the promoter regions of many hypoxia target genes, and acti-vates their expression.
Besides proline hydroxylation, HIF-1a is also hydroxy-lated on asparagine (Asp803 in HIF-1a) under normoxic conditions by the factor inhibiting HIF-1 (FIH-1) (Mahon et al., 2001; Peet and Linke, 2006) and this modification prevents the interaction of HIF-1a with its co-activators CREB-binding protein (CBP) and p300 (Dames et al., 2002; Freedman et al., 2002). Under hypoxic conditions, oxygen becomes rate-limiting for FIH-1 activity, resulting in unhydroxylated HIF-1a at Asp803 and consequently allowing association of HIF-1a with CBP and p300. These co-activators act as a link to RNA polymerase II and other factors within the initiation complex.
Physiological and pathophysiological aspects of hypoxia
Hypoxia triggers both acute and chronic physiological responses. Acute responses may occur as early as sec-onds after exposure to low oxygen and involve the mod-ulation of constitutively expressed proteins, including ion channels. The first ion channels shown to be sensitive to hypoxia were Kq
channels in cells of the carotid body (Lopez-Barneo et al., 1988, 1999). However, it now seems that various Kq
channels are differentially affected by hypoxia (Hool, 2005). For example, in pulmonary artery smooth muscle cells, particularly the Kv1.5 chan-nel is inhibited by hypoxia, leading to membrane depo-larization, influx of Ca2q
through voltage-dependent channels and myocyte contraction (Archer et al., 2004). On the other hand, in systemic arteries and cerebral ves-sels, hypoxia rather increases Kq
currents, which causes vasodilatation and leads to an increase in blood supply to poorly perfused tissue. In renal arteries, hypoxia also stimulates an increase in outward Kq
current associated with dilatation of the arteries (Michelakis et al., 2002).
In addition, various Ca2q
channels are affected by hypoxia. Although Ca2q
channels may be indirectly affected by hypoxia-altered Kq
currents, it has now become evident that Ca2q
channels are also directly influ-enced by hypoxia (for a review see Lopez-Barneo et al., 1999). In voltage-clamped arterial myocytes, hypoxia selectively reduces the influx of Ca2q
through voltage-dependent L-type Ca2q
channels (Franco-Obregon et al., 1995; Fearon et al., 1997), whereas T-type Ca2q
channels were unaltered by hypoxia (Franco-Obregon et al., 1995). Chronic hypoxia-mediated cell responses require hours and mainly involve gene expression leading to de
novo protein synthesis. It has become clear that HIF-1a
regulates a multitude of genes that are involved in phys-iological adaptive processes. More than 70 genes have
been identified as being HIF-regulated, including genes involved in energy metabolism, erythropoiesis, angio-genesis, proliferation, cell survival and apoptosis (for reviews see Semenza, 1999, 2006). However, HIF-medi-ated gene transcription is not the only mechanism by which hypoxia modulates cellular events. Thus, oxygen-consuming enzyme reactions may also be affected by hypoxia (see the section ‘hypoxia-inducible genes’).
Interestingly, HIF-1a is upregulated not only by hypox-ia, but also under certain conditions during normoxia. In this context, pro-inflammatory cytokines such as inter-leukin-1b (IL-1b) and tumor necrosis factor a (TNFa) have been shown to increase HIF-1a activity in normoxic human hepatoma cells (Hellwig-Burgel et al., 1999), human gingival and synovial fibroblasts (Thornton et al., 2000), and even in experimental wounds during acute inflammation (Albina et al., 2001).
Moreover, the gaseous molecule nitric oxide (NO) exerts a dual role in the regulation of HIF-1a expression. Whereas it induces HIF-1a expression under normoxic conditions, it reduces HIF-1a expression under hypoxic conditions (Sogawa et al., 1998). Mechanistically, it was proposed that NO modulates the prolyl hydroxylase activity and thereby alters HIF-1a accumulation (Metzen et al., 2003; Callapina et al., 2005).
Additional factors that have been shown to induce HIF-1a expression include insulin in HepG2cells (Zelzer et al., 1998; Stiehl et al., 2002) and L8 myoblasts (Zelzer et al., 1998), thrombin in vascular smooth muscle cells (Go¨rlach et al., 2001), angiotensin II in renal tubuloepithelial cells (Sanchez-Lopez et al., 2005), and oxidized low-density lipoprotein (oxLDL) in human macrophages (Shatrov et al., 2003). For both angiotensin II and oxLDL, this was shown to mechanistically involve the intermediate gen-eration of reactive oxygen species (ROS) (Shatrov et al., 2003; Sanchez-Lopez et al., 2005).
Finally, prostaglandins have evolved as important reg-ulators of HIF-1 expression. In this context, Liu et al. (2002) and Zhong et al. (2004) demonstrated that pros-taglandin E2(PGE2) specifically stabilizes HIF-1a expres-sion in various tumor cells. Furthermore, overexpresexpres-sion of cyclooxygenase-2 (COX-2) in gastric carcinoma cells resulted in increased VEGF production and increased cell proliferation and tube formation of HUVECs, an effect that was abolished by HIF-1a antisense transfection (Huang et al., 2005). In gastric microvascular endothelial cells, hypoxia-triggered HIF-1a accumulation and angio-genesis were reduced in the presence of non-steroidal anti-inflammatory drugs (NSAIDs), which are classical COX inhibitors. However, this effect was only partially reversed by addition of prostaglandins, suggesting that both prostaglandin-dependent and -independent mech-anisms are involved (Jones et al., 2002). Similarly, the highly selective COX-2 inhibitor NS398 also blocked hypoxia-induced HIF-1a expression, an effect that was detected in COX-2-expressing as well as COX-2-defi-cient colon cancer cell lines (Zhong et al., 2004). More-over, Palayoor et al. (2003) even demonstrated that the NSAID-mediated reduction in HIF-1a occurred in a com-pletely COX-2-independent way. All these partly conflict-ing data show that prostaglandins do have an impact on HIF-1a expression, although the mechanism involved remains uncertain in particular settings.
Remarkably, under disease conditions HIF-1a has also evolved as an important transcription factor. Inflamed tis-sues have lower levels of oxygen than healthy tistis-sues. This causes an accumulation of HIF-1a and helps to establish a rapid and fully developed inflammatory response. In this context, it is known that leukocytes, the key players of the innate immune system, have adapted to hypoxia and exhibit high degrees of lactate formation, even under normoxic conditions. Moreover, tissue hypoxia is a common feature in solid tumors and a driv-ing force for tumor angiogenesis and progression.
The generation of homozygous HIF-1a-/- mice has been attempted. However, the complete deficiency of HIF-1a resulted in developmental arrest and death by day 11 of embryogenesis, with manifestations of neural tube defects, cardiovascular malformations, and marked cell death within the cephalic mesenchyma (Iyer et al., 1998). In a recent study, conditional knockouts of HIF-1a have been generated (Cramer et al., 2003). It was shown that HIF-1a is essential for myeloid cell infiltration and for the regulation of glycolytic capacity of myeloid cells. When HIF-1a was absent, the cellular ATP pool was drastically reduced. The metabolic defect resulted in a severe dysfunction of myeloid cell aggregation, motility, invasiveness, and bacterial killing (Cramer et al., 2003).
Hypoxia-inducible genes
A multitude of hypoxia-regulated genes have been iden-tified (for reviews see Semenza, 1999, 2006), including genes involved in various aspects of energy metabolism, such as glucose transporters, glycolytic and gluconeo-genic enzymes, of iron metabolism including transferrin, of catecholamine metabolism, such as tyrosine hydroxy-lase, of vascular control, including nitric oxide synthases and endothelin-1, and of angiogenesis, such as VEGF and PDGF. Moreover, erythropoietin was described as the prototypic HIF-regulated gene, which regulates eryth-ropoiesis and thus, oxygen transport capacity.
Interestingly, there is a partial overlap between hypox-ia-regulated genes and NO- and ROS-targeted genes (Pfeilschifter et al., 2001). This may arise from the fact that, as already outlined above, NO is able to upregulate HIF-1a expression and in this sense mimics hypoxic con-ditions. However, not all cellular events modulated by hypoxia are exclusively explained by HIF-mediated gene transcription. Oxygen-consuming enzyme reactions may also be affected. Such reactions are very prominent in lipid signaling cascades. For example, oxygen is a sub-strate for cyclooxygenases, lipoxygenases and cyto-chrome P450 enzymes, and therefore an effect of hypoxia on these cascades is very obvious.
Lipid signaling
Signal transduction is an important event in cell-cell com-munication and involves the action of second messen-gers to transmit signals from the extracellular to the intracellular space. Cyclic AMP and calcium are the two main second messengers used by hormones for con-trolling physiological cellular processes. However, during the last decades, evidence has accumulated that points
to a key role for lipids in cell signaling. The milestone for a central role of lipids in signal transduction was mani-fested by the discovery of the phosphoinositide (PI) cycle (Berridge, 1984). It was recognized that upon hydrolysis of phosphatidylinositol 4,5-bisphosphate by phospho-lipase C, two important second messengers are gener-ated, the hydrophilic inositol 1,4,5-trisphosphate (IP3) and the lipophilic 1,2-diacylglycerol (DAG). Whereas IP3 inter-acts with an intracellular membrane receptor and leads to the mobilization of Ca2q
, DAG directly binds to and activates protein kinase C (PKC) and thereby initiates important downstream signal transduction cascades (Nishizuka, 1995).
Additional bioactive lipids were identified that can act as second messengers and intracellular modulators (Serhan et al., 1996). These include: arachidonic acid and lysophospholipids, generated by the action of phospho-lipases A2(PLA2); phosphatidic acid (PA), a reaction prod-uct of phospholipase D; 1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine, also known as platelet-activating fac-tor (PAF); phosphatidylinositol 3,4,5-trisphosphate (PIP3) produced by phosphatidylinositol 3-kinases; and the sphingolipids ceramide and sphingosine 1-phosphate (S1P).
Hypoxia and eicosanoids
Eicosanoids largely contribute to the cardinal symptoms of inflammation. The rate-limiting step in the synthesis of eicosanoids (including prostaglandins, leukotrienes and thromboxane) is the availability of free arachidonic acid, generated by the action of a PLA2, which constitutes a superfamily of cytosolic and secreted enzymes (for review see Six and Dennis, 1998; Balsinde et al., 2002). High levels of secreted PLA2(sPLA2) have been found in serum and exudates from patients suffering from inflam-matory diseases, such as rheumatoid arthritis and acute pancreatitis, and trauma (Hara et al., 1989; Nevalainen et al., 2000). In particular, in diseases such as rheumatoid arthritis, atherosclerosis and glomerulonephritis, hypoxia is a prevalent factor that impacts on the recruitment and function of immune cells and other aspects of disease progression (Cramer et al., 2003; Demasi et al., 2003).
For many years it has been known that hypoxia is able to increase PGE2formation, as exemplified in cultures of renal mesangial cells (Jelkmann et al., 1985). It was recently outlined that this hypoxia-triggered PGE2 syn-thesis is due to a potentiation of inflammatory cytokine-induced sPLA2 expression, secretion and activity by hypoxia (Petry et al., 2005). Importantly, hypoxia was unable to induce sPLA2 in the absence of an inflamma-tory stimulus. In mechanistic terms, HIF-2a was identi-fied as the responsible hypoxia-upregulated transcription factor that binds to two identified HRE sites in the sPLA2 promoter sequence of rat mesangial cells (Petry et al., 2005). Such a coordinated action of two signaling path-ways seems to be a typical feature in sPLA2 induction; IL-1b plus NO (Rupprecht et al., 1999) or IL-1b plus per-oxisome proliferator activated receptor (PPAR) a agonists (Scholz-Pedretti et al., 2002) showed similar additive or synergistic effects on sPLA2mRNA expression compared to IL-1b alone.
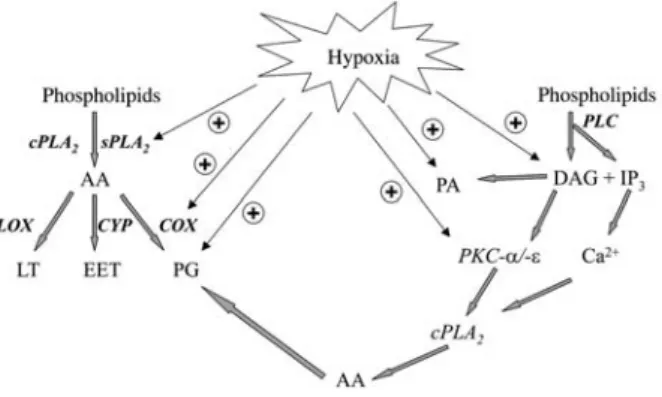
Figure 1 Overview of the effects of hypoxia on eicosanoid-generating enzymes.
Abbreviations: AA, arachidonic acid; COX, cyclooxygenase; cPLA2, cytosolic phospholipase A2; CYP, cytochrome P450;
DAG, 1,2-diacylglycerol; EET, epoxyeicosatrienoic acid; IP3,
ino-sitol trisphosphate; LOX, lipoxygenase; LT, leukotrienes; PA, phosphatidic acid; PG, prostaglandins; PKC, protein kinase C; PLC, phospholipase C; sPLA2, secreted phospholipase A2.
In line with these findings, it was reported that during ischemia, endothelial cells also respond with increased PLA2 activity, which was suggested to belong to the group of secreted PLA2s, but was not further specified (Michiels et al., 2002). Furthermore, an increased amount of arachidonic acid is also liberated from neuroblastoma cells (Petroni et al., 2002) and from cultures of rat hip-pocampal slices (Arai et al., 2001) upon ischemic injury. However, these PLA2 activities were attributed to cyto-solic PLA2 rather than secreted PLA2 (Arai et al., 2001). Especially in the neuronal system, the inflammatory responses occurring upon ischemic injury may contribute to neuronal degeneration and the clinical manifestation of Alzheimer’s disease.
Hypoxia was also identified as a co-stimulus for induc-tion of another pro-inflammatory enzyme, inducible NO synthase (iNOS). Thus, in macrophages, hypoxia poten-tiated interferon-g-induced iNOS expression without showing an effect alone (Melillo et al., 1996).
Furthermore, hypoxia may also affect cyclooxyge-nases, which are important enzymes in the generation of prostaglandins. In this context, Chida and Voelkel (1996) showed that exposure of isolated perfused rat lungs to acute hypoxia was sufficient to induce COX-2 expres-sion. In various other cell types, including endothelial cells (Schmedtje et al., 1997), epithelial cells (Bonazzi et al., 2000) and human neural cells (Bazan and Lukiw, 2002), hypoxia was also able to induce COX-2 expres-sion or potentiate cytokine-induced COX-2 expresexpres-sion.
It is tempting to speculate that hypoxia may also affect the more downstream enzymes such as PGE2synthase and prostacyclin synthase to consistently and coordi-nately upregulate eicosanoid biosynthesis. This concert-ed action of hypoxia on the eicosanoid-generating cascade of enzymes (for an overview see Figure 1) could explain the well-documented increase in prostaglandin synthesis in conditions of limited oxygen availability in brain, kidney and heart, as well as in cell culture of endo-thelial and mesangial cells (Busse et al., 1984; Jelkmann et al., 1985; Kurtz et al., 1985; Michiels et al., 2002).
Hypoxia and diacylglycerol
Diacylglycerol (DAG) is an important lipid second mes-senger directly regulating protein kinase C activity, which, in turn, is involved in the regulation of many cell responses including gene expression (for reviews see Mellor and Parker, 1998; Aschrafi et al., 2003). Recently, it was shown that hypoxia is able to increase cellular DAG levels in HeLa cells (Aragones et al., 2001) and 293 T-cells (Temes et al., 2004). This hypoxia-mediated increase in DAG was abolished by the putative phospha-tidylcholine-specific phospholipase C inhibitor D609, thus suggesting that hypoxia activates this enzyme (Ara-gones et al., 2001). However, since PKC inhibitors had no effect on hypoxia-induced HIF expression, it was con-cluded that DAG targets other than DAG-activated PKC were involved in HIF accumulation. Since phosphatidic acid levels are also increased upon hypoxia due to stim-ulated DAG kinase activity (Aragones et al., 2001), it was proposed that this pathway of DAG/PA generation may rather contribute to HIF activation.
In contrast, other groups could show that hypoxia indeed leads to translocation and activation of the two PKC-a and -´ isoenzymes in neonatal rat ventricular myocytes (Goldberg et al., 1997). The role of PKC in ischemia-reperfusion is currently unclear owing to many conflicting reports. On the one hand PKC may be involved in cell survival signaling, and on the other hand it may be involved in detrimental processes (for a review see Bright and Mochly-Rosen, 2005). In this context, it is worth mentioning that PKC-a and -´ are considered to mediate, in a cell type-specific manner, cPLA2 acti-vation and subsequent eicosanoid formation, which pro-vides an additional amplification loop to the interaction of hypoxia and PLA2/COX-2 isozymes described above (Huwiler et al., 1991; Huwiler and Pfeilschifter, 1993).
Hypoxia and sphingolipids
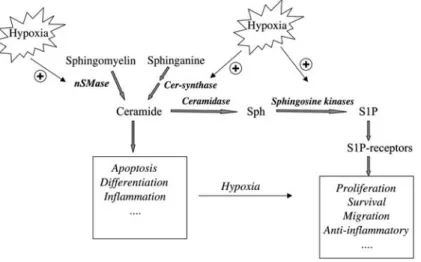
The sphingolipids ceramide and S1P have been attrib-uted central but opposing roles in the regulation of cell growth and death. Whereas ceramide is an inducer of apoptosis in many cell types, S1P rather acts as an anti-apoptotic agent and promotes cell proliferation (for reviews see Huwiler et al., 2000; Spiegel and Milstien, 2003). Since hypoxia is also connected to cell prolifera-tion, it is tempting to speculate that sphingolipids can serve as mediators of hypoxia-induced proliferation, or may themselves be affected by changing oxygen avail-ability (for an overview see Figure 2). Indeed, Yun and Kester (2002) were able to show that in smooth muscle cells, hypoxia-triggered cell proliferation is abolished in the presence of a putative sphingosine kinase inhibitor. In addition, hypoxia led to an increased cellular level of S1P with a concomitant decrease in ceramide as a con-sequence of hypoxia-inhibited sphingomyelinase activity (Yun and Kester, 2002). In line with these findings, Ahmad et al. (2006) reported that in pulmonary smooth muscle cells, hypoxia upregulated sphingosine kinase mRNA expression and was able to potentiate S1P-stimulated mitogen-activated protein kinase activity. In contrast, it was reported that in cultures of rat renal tubular epithelial cells, 1 h of hypoxia with glucose deprivation induced a
Figure 2 Schematic overview on the effects of hypoxia on sphingolipid metabolism.
Abbreviations: Cer-synthase, ceramide synthase; nSMase, neutral sphingomyelinase; sph, sphingosine; S1P, sphingosine 1-phosphate.
significant increase in ceramide, which was further accel-erated after 1 h of reoxygenation (Basnakian et al., 2005). This effect occurred independent of sphingomyelinase activity and was due to increased ceramide synthase activity (Basnakian et al., 2005).
In various models of myocardial ischemia-reperfusion, changes in ceramide levels have been reported. In this context, hypoxia-reoxygenation of cardiac myocytes induced rapid activation of a neutral sphingomyelinase activity (Hernandez et al., 2000). Similarly, in vitro ische-mia (oxygen/serum/glucose deprivation) led to a pro-gressive accumulation of ceramide in cardiomyocytes (Bielawska et al., 1997).
Mice that were subjected to 45 min of unilateral renal ischemia showed an approximately 50% decrease in cor-tical ceramide and sphingosine levels (Zager et al., 1997). After reperfusion, sphingosine returned to normal levels, whereas ceramide further increased to supranormal lev-els (Zager et al., 1997).
In a rat heart model, application of ischemia with sub-sequent reperfusion induced apoptosis of myocardial cells, an event that was mimicked in vitro by C2 ceramide (Bielawska et al., 1997). In vivo, the content of ceramide in the ischemic area was significantly elevated, thus sug-gesting that ceramide signaling may be involved in ische-mia/reperfusion death of myocardial cells (Bielawska et al., 1997; Cordis et al., 1998). Furthermore, chronic expo-sure of rats for 8 weeks to 10% oxygen, which equals the oxygen conditions at an altitude of 4000 m, led to a marked increase in cardiac mass in both the left and right ventricles. In the right ventricle, this was associated with a reduction in ceramide content and a slight increase in DAG content (El Alwani et al., 2005).
As ceramide is degraded to sphingosine and may be further converted to S1P, an involvement of S1P in the ischemic reaction could also be envisioned. Indeed, it was recently shown that deficiency of the S1P1receptor subtype resulted in induced expression of HIF-1a and VEGF in deficient mice limbs (Chae et al., 2004).
Another coupling of hypoxia to sphingolipids was shown in skin-specific ARNT/HIF-1b-deficient mice. These mice died shortly after birth due to severe
dehy-dration caused by water loss caused by a defective epi-dermal barrier function (Takagi et al., 2003). Lipid analysis of the skin revealed significant changes in the composi-tion of ceramides. Whereas normal murine epidermis ceramides contain 2-sphingenine and 4-hydroxysphin-ganine, the deficient epidermis contained sphinganine instead of 4-sphingenine and the amount of 4-hydroxy-sphinganine was greatly decreased, thus suggesting a deficiency of dihydroceramide desaturase activity in ARNT/HIF-1b-deficient cells (Takagi et al., 2003).
Perspectives
The observations cited above provide the framework to begin to understand the cross-communication between two important signaling cascades that have become fashionable again. The identification of HIF-1 as the mas-ter regulator of oxygen homeostasis and more recently of the PHDs as the critical oxygen-sensing devices has promoted the return of this research area to the scientific center stage. Similarly, the discovery of lipid-regulated key signaling elements including cell membrane recep-tors (e.g., the S1P receptor family, formerly denoted as Edg receptors) and transcription factors (e.g., PPARs) has shifted the scientific spotlight back to membrane lipids. The vital elegance and elaborate mechanisms that have evolved to regulate fundamental cell responses are attracting the attention of basic scientists, as well as cli-nicians, and promise to provide novel pharmacological approaches for the treatment of human diseases. Further research progress in this area will address the interface between signal transduction and translational medicine; these results are likely to be very exciting.
Acknowledgments
We gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft (PF361/2-2, GRK757/2, FOG784), the Swiss National Science Foundation, the Wilhelm Sander-Stiftung and the European Community (FP6: LSHM-CT-2004-005033).
We apologize to our colleagues for being unable to provide pri-mary citations for much of the work described in this review owing to space limitations.
References
Ahmad, M., Long, J.S., Pyne, N.J., and Pyne, S. (2006). The effect of hypoxia on lipid phosphate receptor and sphingo-sine kinase expression and mitogen-activated protein kinase signaling in human pulmonary smooth muscle cells. Prosta-glandins Other Lipid Mediat. 79, 278–286.
Albina, J.E., Mastrofrancesco, B., Vessella, J.A., Louis, C.A., Henry, W.L. Jr., and Reichner, J.S. (2001). HIF-1 expression in healing wounds: HIF-1a induction in primary inflammatory cells by TNF-a. Am. J. Physiol. Cell. Physiol. 281, C1971–C1877.
Aragones, J., Jones, D.R., Martin, S., San Juan, M.A., Alfranca, A., Vidal, F., Vara, A., Merida, I., and Landazuri, M.O. (2001). Evidence for the involvement of diacylglycerol kinase in the activation of hypoxia-inducible transcription factor 1 by low oxygen tension. J. Biol. Chem. 276, 10548–10555. Arai, K., Ikegaya, Y., Nakatani, Y., Kudo, I., Nishiyama, N., and
Matsuki, N. (2001). Phospholipase A2mediates ischemic
inju-ry in the hippocampus: a regional difference of neuronal vul-nerability. Eur. J. Neurosci. 13, 2319–2323.
Archer, S.L., Wu, X.C., Thebaud, B., Nsair, A., Bonnet, S., Tyrrell, B., McMurtry, M.S., Hashimoto, K., Harry, G., and Michelakis, E.D. (2004). Preferential expression and function of voltage-gated, O2-sensitive K
q
channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: ionic diversity in smooth muscle cells. Circ. Res. 95, 308–318.
Aschrafi, A., Shabahang, S., Pfeilschifter, J., and Huwiler, A. (2003). Regulatory functions of protein kinase C isoenzymes in the kidney. Curr. Top. Biochem. Res. 5, 27–41.
Balsinde, J., Winstead, M.V., and Dennis, E.A. (2002). Phospho-lipase A2regulation of arachidonic acid mobilization. FEBS
Lett. 531, 2–6.
Basnakian, A.G., Ueda, N., Hong, X., Galitovsky, V.E., Yin, X., and Shah, S.V. (2005). Ceramide synthase is essential for endonuclease-mediated death of renal tubular epithelial cells induced by hypoxia-reoxygenation. Am. J. Physiol. Renal Physiol. 288, F308–F314.
Bazan, N.G. and Lukiw, W.J. (2002). Cyclooxygenase-2 and pre-senilin-1 gene expression induced by interleukin-1b and amyloid b 42 peptide is potentiated by hypoxia in primary human neural cells. J. Biol. Chem. 277, 30359–30367. Berridge, M.J. (1984). Inositol trisphosphate and diacylglycerol
as second messengers. Biochem. J. 220, 345–360. Bielawska, A.E., Shapiro, J.P., Jiang, L., Melkonyan, H.S., Piot,
C., Wolfe, C.L., Tomei, L.D., Hannun, Y.A., and Umansky, S.R. (1997). Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am. J. Pathol. 151, 1257–1263.
Bonazzi, A., Mastyugin, V., Mieyal, P.A., Dunn, M.W., and Lania-do-Schwartzman, M. (2000). Regulation of cyclooxygenase-2 by hypoxia and peroxisome proliferators in the corneal epithelium. J. Biol. Chem. 275, 2837–2844.
Bright, R. and Mochly-Rosen, D. (2005). The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke
36, 2781–2790.
Busse, R., Fo¨rstermann, U., Matsuda, H., and Pohl, U. (1984). The role of prostaglandins in the endothelium-mediated vasodilatory response to hypoxia. Pflu¨ger’s Arch. 401, 77–83. Callapina, M., Zhou, J., Schnitzer, S., Metzen, E., Lohr, C., Deit-mer, J.W., and Bru¨ne, B. (2005). Nitric oxide reverses des-ferrioxamine- and hypoxia-evoked HIF-1a accumulation –
implications for prolyl hydroxylase activity and iron. Exp. Cell Res. 306, 274–284.
Chae, S.S., Paik, J.H., Allende, M.L., Proia, R.L., and Hla, T. (2004). Regulation of limb development by the sphingosine 1-phosphate receptor S1P1/EDG-1 occurs via the hypoxia/ VEGF axis. Dev. Biol. 268, 441–447.
Chida, M. and Voelkel, N.F. (1996). Effects of acute and chronic hypoxia on rat lung cyclooxygenase. Am. J. Physiol. 270, L872–L878.
Cordis, G.A., Yoshida, T., and Das, D.K. (1998). HPTLC analysis of sphingomylein, ceramide and sphingosine in ischemic/ reperfused rat heart. J. Pharm. Biomed. Anal. 16, 1189– 1193.
Cramer, T., Yamanishi, Y., Clausen, B.E., Foster, I., Pawlinski, R., Mackman, N., Haase, V.H., Jaenisch, R., Corr, M., Nizet, V., et al. (2003). HIF-1a is essential for myeloid cell-mediated inflammation. Cell 112, 645–657.
Dames, S.A., Martinez-Yamout, M., De Guzman, R.N., Dyson, H.J., and Wright, P.E. (2002). Structural basis for HIF-1a/CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. USA 99, 5271–5276.
Demasi, M., Cleland, L.G., Cook-Johnson, R.J., Caughey, G.E., and James, M.J. (2003). Effects of hypoxia on monocyte inflammatory mediator production: Dissociation between changes in cyclooxygenase-2 expression and eicosanoid synthesis. J. Biol. Chem. 278, 38607–38616.
Elvert, G., Lanz, S., Kappel, A., and Flamme, I. (1999). mRNA cloning and expression studies of the quail homologue of HIF-2a. Mech. Dev. 87, 193–197.
El Alwani, M., Usta, J., Nemer, G., Sabban, M.E., Nasser, M., Bitar, H., Souki, R., Dbaibo, G.S., and Bitar, F.F. (2005). Reg-ulation of the sphingolipid signaling pathways in the growing and hypoxic rat heart. Prostaglandins Other Lipid Mediat. 78, 249–263.
Epstein, A.C., Gleadle, J.M., McNeill, L.A., Hewitson, K.S., O’Rourke, J., Mole, D.R., Mukherji, M., Metzen, E., Wilson, M.I., Dhanda, A., et al. (2001). C. elegans EGL-9 and mam-malian homologs define a family of dioxygenases that regu-late HIF by prolyl hydroxylation. Cell 107, 43–54.
Fearon, I.M., Palmer, A.C., Balmforth, A.J., Ball, S.G., Mikala, G., Schwartz, A., and Peers, C. (1997). Hypoxia inhibits the recombinant a 1C subunit of the human cardiac L-type Ca2q
channel. J. Physiol. 500, 551–556.
Franco-Obregon, A., Urena, J., and Lopez-Barneo, J. (1995). Oxygen-sensitive calcium channels in vascular smooth mus-cle and their possible role in hypoxic arterial relaxation. Proc. Natl. Acad. Sci. USA 92, 4715–4719.
Freedman, S.J., Sun, Z.Y., Poy, F., Kung, A.L., Livingston, D.M., Wagner, G., and Eck, M.J. (2002). Structural basis for recruit-ment of CBP/p300 by hypoxia-inducible factor-1a. Proc. Natl. Acad. Sci. USA 99, 5367–5372.
Goldberg, M., Zhang, H.L., and Steinberg, S.F. (1997). Hypoxia alters the subcellular distribution of protein kinase C isoforms in neonatal rat ventricular myocytes. J. Clin. Invest. 99, 55–61.
Go¨rlach, A., Diebold, I., Schini-Kerth, V.B., Berchner-Pfannsch-midt, U., Roth, U., Brandes, R.P., Kietzmann, T., and Busse, R. (2001). Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22(phox)-containing NADPH oxidase. Circ. Res. 89, 47–54.
Haase, V.H. (2006). Hypoxia-inducible factors in the kidney. Am. J. Physiol. Renal Physiol. 291, F271–F281.
Hara, S., Kudo, I., Chang, H.W., Matsuda, K., Miyamoto, T., and Inoue, K. (1989). Purification and characterization of extra-cellular phospholipase A2from human synovial fluid in
rheu-matoid arthritis. J. Biochem. 105, 395–399.
Hara, S., Hamada, J., Kobayashi, C., Kondo, Y., and Imura, N. (2001). Expression and characterization of hypoxia-inducible factor (HIF)-3a in human kidney: suppression of
HIF-medi-ated gene expression by HIF-3a. Biochem. Biophys. Res. Commun. 287, 808–813.
Hellwig-Burgel, T., Rutkowski, K., Metzen, E., Fandrey, J., and Jelkmann, W. (1999). Interleukin-1b and tumor necrosis fac-tor-a stimulate DNA binding of hypoxia-inducible factor-1. Blood 94, 1561–1567.
Hernandez, O.M., Discher, D.J., Bishopric, N.H., and Webster, W.A. (2000). Rapid activation of neutral sphingomyelinase by hypoxia-reoxygenation of cardiac myocytes. Circ. Res. 86, 198–204.
Hool, L.C. (2005). Acute hypoxia differentially regulates Kq
chan-nels. Implications with respect to cardiac arrhythmia. Eur. Biophys. J. 34, 369–376.
Huang, S.P., Wu, M.S., Shun, C.T., Wang, H.P., Hsieh, C.Y., Kuo, M.L., and Lin, J.T. (2005). Cyclooxygenase-2 increases hypoxia-inducible factor-1 and vascular endothelial growth factor to promote angiogenesis in gastric carcinoma. J. Bio-med. Sci. 12, 229–241.
Huwiler, A. and Pfeilschifter, J. (1993). A role for protein kinase C-a in zymosan-stimulated eicosanoid synthesis in mouse peritoneal macrophages. Eur. J. Biochem. 217, 69–75. Huwiler, A., Fabbro, D., and Pfeilschifter, J. (1991). Possible
reg-ulatory functions of protein kinase C-a and -´ isoenzymes in rat renal mesangial cells. Stimulation of prostaglandin syn-thesis and feedback inhibition of angiotensin II-stimulated phosphoinositide hydrolysis. Biochem. J. 279, 441–445. Huwiler, A., Kolter, T., Pfeilschifter, J., and Sandhoff, K. (2000).
Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim. Biophys. Acta 1485, 63–99. Iyer, N.V., Kotch, L.E., Agani, F., Leung, S.W., Laughner, E.,
Wen-ger, R.H., Gassmann, M., Gearhart, J.D., Lawler, A.M., Yu, A.Y., and Semenza, G.L. (1998). Cellular and developmental control of O2homeostasis by hypoxia-inducible factor 1a.
Genes Dev. 12, 149–162.
Jelkmann, W., Kurtz, A., Fo¨rstermann, U., Pfeilschifter, J., and Bauer, C. (1985). Hypoxia enhances prostaglandin synthesis in renal mesangial cell cultures. Prostaglandins 30, 109–118. Jones, M.K., Szabo, I.L., Kawanaka, H., Husain, S.S., and Tar-nawski, A.S. (2002). von Hippel Lindau tumor suppressor and HIF-1a: new targets of NSAIDs inhibition of hypoxia-induced angiogenesis. FASEB J. 16, 264–266.
Kurtz, A., Jelkmann, W., Pfeilschifter, J., and Bauer, C. (1985). Role of prostaglandins in hypoxia-stimulated erythropoietin production. Am. J. Physiol. 249, C3–C8.
Liu, X.H., Kirschenbaum, A., Lu, M., Yao, S., Dosoretz, A., Hol-land, J.F., and Levine, A.C. (2002). Prostaglandin E2induces
hypoxia-inducible factor-1a stabilization and nuclear locali-zation in a human prostate cancer cell line. J. Biol. Chem.
277, 50081–50006.
Lopez-Barneo, J., Lopez-Lopez, J.R., Urena, J., and Gonzales, C. (1988). Chemotransduction in the carotid body: Kq
current modulated by PO2 in type I chemoreceptor cells. Science
241, 580–582.
Lopez-Barneo, J., Pardal, R., Montoro, R.J., Smani, T., Garcia-Hirschfeld, J., and Urena, J. (1999). Kqand Ca2qchannel
activity and cytosolic wCa2q
xin oxygen-sensing tissues. Res-pir. Physiol. 115, 215–227.
Mahon, P.C., Hirota, K., and Semenza, G.L. (2001). FIH-1: a nov-el protein that interacts with HIF-1a and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15, 2675–2686.
Maxwell, P.H., Wiesener, M.S., Chang, G.W., Clifford, S.C., Vaux, E.C., Cockman, M.E., Wykoff, C.C., Pugh, C.W., Maher, E.R., and Ratcliffe, P.J. (1999). The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent pro-teolysis. Nature 399, 271–275.
Maynard, M.A., Qi, H., Chung, J., Lee, E.H., Kondo, Y., Hara, S., Conaway, R.C., Conaway, J.W., and Ohh, M. (2003). Multiple splice variants of the human HIF-3a locus are targets of the
von Hippel-Lindau E3 ubiquitin ligase complex. J. Biol. Chem. 278, 11032–11040.
Melillo, G., Taylor, L.S., Brooks, A., Cox, G.W., and Varesio, L. (1996). Regulation of inducible nitric oxide synthase expres-sion in IFN-g-treated murine macrophages cultured under hypoxic conditions. J. Immunol. 157, 2638–2644.
Mellor, H. and Parker, P.J. (1998). The extended protein kinase C superfamily. Biochem. J. 332, 281–292.
Metzen, E., Zhou, J., Jelkmann, W., Fandrey, J., and Bru¨ne, B. (2003). Nitric oxide impairs normoxic degradation of HIF-1a by inhibition of prolyl hydroxylases. Mol. Biol. Cell 14, 3470–3481.
Michelakis, E.D., Rebeyka, I., Wu, X., Nsair, A., Thebaud, B., Hashimoto, K., Dyck, J.R., Haromy, A., Harry, G., Barr, A., and Archer, S.L. (2002). O2sensing in the human ductus
arte-riosus: regulation of voltage-gated Kqchannels in smooth
muscle cells by a mitochondrial redox sensor. Circ. Res. 91, 478–486.
Michiels, C., Renard, P., Bouaziz, N., Heck, N., Eliaers, F., Ninane, N., Quarck, R., Holvoet, P., and Raes, M. (2002). Identification of the phospholipase A2isoforms that
contrib-ute to arachidonic acid release in hypoxic endothelial cells: limits of phospholipase A2inhibitors. Biochem. Pharmacol.
63, 321–332.
Nevalainen, T.J., Haapamaki, M.M., and Gronroos, J.M. (2000). Roles of secretory phospholipases A2in inflammatory
dis-eases and trauma. Biochim. Biophys. Acta 1488, 83–90. Nishizuka, Y. (1995). Protein kinase C and lipid signaling for
sus-tained cellular responses. FASEB J. 9, 484–496.
Palayoor, S.T., Tofilon, P.J., and Coleman, C.N. (2003). Ibuprofen-mediated reduction of hypoxia-inducible factors HIF-1a and HIF-2a in prostate cancer cells. Clin. Cancer Res. 9, 3150–3157.
Peet, D. and Linke, S. (2006). Regulation of HIF: asparaginyl hydroxylation. Novartis Found. Symp. 272, 37–49.
Petroni, A., Papini, N., Blasevich, M., Rise, P., and Galli, C. (2002). Arachidonate release and c-fos expression in various models of hypoxia and hypoxia-hypoglycemia in retinoic acid differentiated neuroblastoma cells. Neurochem. Int. 40, 255–260.
Petry, C., Huwiler, A., Eberhardt, W., Kaszkin, M., and Pfeil-schifter, J. (2005). Hypoxia increases group IIA phospholi-pase A2expression under inflammatory conditions in rat renal
mesangial cells. J. Am. Soc. Nephrol. 16, 2897–2905. Pfeilschifter, J., Eberhardt, W., and Beck, K.F. (2001). Regulation
of gene expression by nitric oxide. Pflu¨ger’s Arch. 442, 479–486.
Pugh, C.W. and Ratcliffe, P.J. (2003). Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, 677–684. Rupprecht, G., Scholz, K., Beck, K.F., Geiger, H., Pfeilschifter,
J., and Kaszkin, M. (1999). Cross-talk between group IIA-phospholipase A2 and inducible NO-synthase in rat renal
mesangial cells. Br. J. Pharmacol. 127, 51–56.
Sanchez-Lopez, E., Lopez, A.F., Esteban, V., Yague, S., Egido, J., Ruiz-Ortega, M., and Alvarez-Arroyo, M.V. (2005). Angio-tensin II regulates vascular endothelial growth factor via hypoxia-inducible factor-1a induction and redox mecha-nisms in the kidney. Antioxid. Redox Signal. 7, 1275–1284. Schmedtje, J.F. Jr., Ji, Y.S., Liu, W.L., DuBois, R.N., and Runge,
M.S. (1997). Hypoxia induces cyclooxygenase-2 via the NF-kB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 272, 601–608.
Scholz-Pedretti, K., Gans, A., Beck, K.F., Pfeilschifter, J., and Kaszkin, M. (2002). Potentiation of TNF-a-stimulated group IIA phospholipase A2expression by peroxisome
proliferator-activated receptor a activators in rat mesangial cells. J. Am. Soc. Nephrol. 13, 611–620.
Semenza, G.L. (1999). Regulation of mammalian O2homeostasis
by hypoxia-inducible factor. Annu. Rev. Cell Dev. Biol. 15, 551–578.
Semenza, G.L. (2006). HIF-1 and human disease: one highly involved factor. Genes Dev. 14, 1983–1991.
Serhan, C.N., Haeggstrom, J.Z., and Leslie, C.C. (1996). Lipid mediator networks in cell signaling: update and impact of cytokines. FASEB J. 10, 1147–1158.
Shatrov, V.A., Sumbayev, V.V., Zhou, J., and Bru¨ne, B. (2003). Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1a (HIF-1a). accumulation via redox-dependent mechanisms. Blood 101, 4847–4849.
Six, D.A. and Dennis, E.A. (1998). The expanding superfamily of phospholipase A2enzymes: classification and
characteriza-tion. Biochim. Biophys. Acta 1488, 1–19.
Spiegel, S. and Milstien, S. (2003). Sphingosine-1-phosphate: an enigmatic signaling lipid. Nat. Rev. Mol. Cell Biol. 4, 397–407. Stiehl, D.P., Jelkmann, W., Wenger, R.H., and Hellwig-Burgel, T. (2002). Normoxic induction of the hypoxia-inducible factor 1a by insulin and interleukin-1b involves the phosphatidylinosi-tol 3-kinase pathway. FEBS Lett. 512, 157–162.
Sogawa, K., Numayama-Tsuruta, K., Ema, M., Abe, M., Abe, H., and Fujii-Kuriyama, Y. (1998). Inhibition of hypoxia-inducible factor 1 activity by nitric oxide donors in hypoxia. Proc. Natl. Acad. Sci. USA 95, 7368–7373.
Takagi, S., Toio, H., Tomita, S., Sano, S., Itami, S., Hara, M., Inoue, S., Horie, K., Kondoh, G., Hosokawa, K., et al. (2003). Alteration of the 4-sphingenine scaffolds of ceramides in
keratinocyte-specific Arnt-deficient mice affects skin barrier function. J. Clin. Invest. 112, 1372–1382.
Temes, E., Martin-Puig, S., Aragones, J., Jones, D.R., Olmos, G., merida, I., and Landazuri, M.O. (2004). Role of diacylgly-cerol induced by hypoxia in the regulation of HIF-1a activity. Biochem. Biophys. Res. Commun. 315, 44–50.
Thornton, R.D., Lane, P., Borghaei, R.C., Pease, E.A., Caro, J., and Mochan, E. (2000). Interleukin 1 induces hypoxia-induc-ible factor 1 in human gingival and synovial fibroblasts. Bio-chem. J. 350, 307–312.
Yun, J.K. and Kester, M. (2002). Regulatory role of sphingomyelin metabolites in hypoxia-induced vascular smooth muscle cell proliferation. Arch. Biochem. Biophys. 408, 78–86.
Zager, R.A., Iwata, M., Conrad, D.S., Burkhart, K.M., and Iga-rashi, Y. (1997). Altered ceramide and sphingosine expres-sion during the induction phase of ischemic acute renal failure. Kidney Int. 52, 60–70.
Zelzer, E., Levy, Y., Kahana, C., Shilo, B.Z., Rubinstein, M., and Cohen, B. (1998). Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1a/ARNT. EMBO J. 17, 5085–5094.
Zhong, H., Willard, M., and Simons, J. (2004). NS398 reduces hypoxia-inducible factor (HIF)-1a and HIF-1 activity: multiple-level effects involving cyclooxygenase-2 dependent and independent mechanisms. Int. J. Cancer 112, 585–595.