2ii
1/))./Université de Montréal
Progrès dans la synthèse de J’analogue [Val6, (6S)-Me12aa45] de
l’auréobasidine B
Par
James Erich Dettwiler
Département de chimie
Faculté des Arts et des Sciences
Mémoire présenté à la faculté des études supérieures
En vue de l’obtention du grade de
Maître ès sciences (M.Sc.)
En chimie
Octobre 2004
o
\T
C
Université
rAi
de Montréal
Direction des bibliothèques
AVIS
L’auteur a autorisé l’Université de Montréal à reproduire et diffuser, en totalité
ou en partie, par quelque moyen que ce soit et sur quelque support que ce
soit, et exclusivement à des fins non lucratives d’enseignement et de
recherche, des copies de ce mémoire ou de cette thèse.
L’auteur et les coauteurs le cas échéant conservent la propriété du droit
d’auteur et des droits moraux qui protègent ce document. Ni la thèse ou le
mémoire, ni des extraits substantiels de ce document, ne doivent être
imprimés ou autrement reproduits sans l’autorisation de l’auteur.
Afin
de
se
conformer
à
la
Loi
canadienne
sur
la
protection
des
renseignements personnels, quelques formulaires secondaires, coordonnées
ou signatures intégrées au texte ont pu être enlevés de ce document. Bien
que cela ait pu affecter la pagination, il n’y a aucun contenu manquant.
NOTICE
The author of this thesis or dissertation has granted a nonexclusive license
allowing Université de Montréal to reproduce and publish the document, in
part or in whole, and in any format, solely for noncommercial educational and
research purposes.
The author and co-authors if applicable retain copyright ownership and moral
rights in this document. Neither the whole thesis or dissertation, nor
substantial extracts from it, may be printed or otherwise reproduced without
the author’s permission.
In compliance with the Canadian Privacy Act some supporting forms, contact
information or signatures may have been removed from the document. While
this may affect the document page count, it does flot represent any loss of
content from the document.
Ce mémoire intitulé:
Progrès dans la synthèse de l’analogue [Val6, (6S)-Mel2aa4..5] de
I’auréobasidine B
présenté par
James Erich Dettwiler
a été évaluée par un jury composé des personnes suivantes:
Professeur Jeffrey R. Keillor
Président rapporteur
Professeur William D. LubelI
Directeur de recherche
Professeur Andreea R. Schmitzer
Membre du jury
SOMMAIRE
Ce mémoire de maîtrise porte sur la synthèse d’acides aminés non-naturels et leur utilisation dans une approche de synthèse d’un analogue de l’auréobasidine sur support solide.
Les auréobasidines (Abs) forment une famille de puissants antibiotiques produits par une variété de levures noires Aureobasidines pullulans R106. Regroupant une vingtaine d’analogues, ce sont des nonadepsipeptides contenant un a-hydroxyacide et 8 acides Œ-aminés dont 3 ou 4 sont N-méthylés. En solution et suivant les conditions, l’isomérisation de l’amide du N-terminal de la proline engendre un équilibre entre la conformation d’un coude de type H’ (isomère trans) et de type VI (isomère cis). Dans le but d’étudier spécifiquement l’activité biologique de l’isomère trans, un acide aminé indolizidinone (stabilisant le coude
p
de type Il’) pourrait être incorporé dans un analogue de I’auréobasidine.Tous les acides aminés non naturels nécessaires à la synthèse d’un analogue de l’auréobasidine ont été préparés suivant les protocoles garantissant des rendements et des puretés énantiomériques optimisés. Tels que décrits dans le chapitre 2, des protocoles de synthèse novateurs ont été développés pour la 3-hydroxyvaline, la 3-hydroxyméthylvaline et un dérivé N méthylé de l’acide aminé azabicyclique (6S)-l2aa. Par ailleurs, des méthodes de synthèse efficaces d’une série de 3,3-dialkylsérines et de cétones homoallyliques sont aussi présentées.
Dans le chapitre 3, les progrès dans la synthèse sur support solide d’un analogue de l’auréobasine B ainsi que d’un peptide modèle sont décrits. Ceci inclut les études sur l’élongation du peptide en incorporant les acides aminés non naturels cités plus haut dans le but d’obtenir un peptide linéaire ainsi que sa cyclisation sur résine d’oxime.
Mots clés
Antifongique, depsipeptide, hydroxyméthylvaline, ,-dialkyIsérine, cétone homoallylique, acide aminé indolizidinone, acides aminés N-méthylés
The synthesis of series cf unnatural amino acids and their subsequent incorporation into aureobasfdin analogs using solid phase peptide synthesis wiII be presented in this thesis.
Aureobasidins are a family of —20 related potent antifungal antibiotic cyclic depsinonapeptides produced by black yeast. They contain 8 cx-amino acids and an a-hydroxyacid. Dependîng on the nature of the analog, 3 to 4 residues are N-methylated. When in solution, aureobasidins can exist eithec as
trans
orcis
prolyl amide isomers that respectively adopt type Il’(trans
isomer) and type VI(cis
isomer) Ç3 turns. Exploring the importance of turn geometry on biological activity, we have pursued the synthesis of aureobasidin analogs possessing an indolizidinon-2-one amino acid (l2aa) as a constrained mimic of the type Il’ 3-turn.AIl unnatural amino acids components of the aureobasidin analog were synthesized with optimized yields and high enantiomeric purity. As described in chapter 2, new protocols were developed for the synthesis cf 3-hydroxyvaline, 3-hydroxymethylvaline and for N-methylated
(6S)-l2aa. This chapter also includes effective methods for the preparation cf Ç3j3-dialkylserines and
homoallylic ketones.
Advances in the synthesis on solid support of an aureobasidin B analog and a model peptide are described in chapter 3. This includes peptide elongation using previously mentioned unnatural amino acids followed by cyclisation using the PCOR (peptide cyclisation on oxime resin) method.
Key words
Antifungal, depsipeptide, hydroxymethylvaline, 3,3-dialkylserine, homoallylic ketone, indolizidinon-2-one amino acid, N-methylated amino acids
II
TABLE DES MATIÈRES
Page SOMMAIRE
TABLE DES MATIÈRES iii
LISTE DES FIGURES viii
LISTE DES TABLEAUX x
LISTE DES ABRÉVIATIONS ET DES SIGLES xi
REMERCIEMENTS xiv
CHAPITRE 1: INTRODUCTION I
1.1 Les infections fongiques 2
1.2 Les agents antifongiques 3
1.2.1 Les médicaments antifongiques 3
1.2.2 Les polyènes 3
1.2.3 Les azoles 4
1.2.4 La flucytosine 5
1.2.5 Les échinocandines 5
1.3 Structure des peptides et des protéines 6
1.3.1 Interactions 6 1.3.2 Niveaux d’organisation 8 1.3.3 Les repliements y et f3 9 1.4 Peptidomimétisme 10 1.4.1 Mimétisme de coudes
f3
10 1.5 Les auréobasidines 131.5.1 Structure et activité biologique 13
1.5.2 Analogues de l’auréobasidine restreints conformationnellement 18
1.6 Synthèses de I’auréobasidine 19
1.6.1 Synthèses en solution 19
1.6.2 Synthèse mixte sur support solide et solution 27
1.7 Conclusion 29
CHAPITRE 2 SYNTHÈSE DES ACIDES AMINÉS NON NATURELS 33
2.1 Les acides Œ-aminés N-Boc-N-méthylés 34
2.2 L’acide (S)-2-hydroxy-3-méthylbutanoïque (Hmb) 35
2.3 L’acide aminé (3S, 6S, 9S)-2-oxo-3-N-(Boc)amino-1-azabicyclo [4.3.0]nonane-9- 36 carboxylique (Boc-I2aa-OH)
2.4 L’acide aminé (3S, 6S, 9S)-2-oxo-3-N-(Boc)-N-méthyl-amino-1-azabicyclo 38 [4.3.OJnonane-9-carboxyllique (N-Boc-N-Me-I2aa-OH)
2.5 La fi-hydroxyvaline (13-HOVaI) et la N-méthyl-13-hydroxyvaline (13-HOMeVaI) 40 25.1 La 3-hydroxyméthylvaline, les 3,p-dialkylsérines 43
25.2 La N-méthyl-f3-hydroxyméthylvaline 47
2.6 Les depsipeptides : Boc-MeVaI-Hmb-OH (125a) Boc-f3-HOMeVaI-Hmb-OH (125b) 51
2.7 Conclusion 53
2.7 Références du chapitre 2 54
CHAPITRE 3 : SYNTHÈSE D’UN ANALOGUE DE L’AURÉOBASIDINE SUR 57 RÉSINE D’OXIME
3.1 Synthèse de peptide sur résine d’oxime 58
3.2 Synthèse de la [Val6, (6S)-Me12aa45]AbB et de la [Hmb1, Val6, (6S)-MeI2aa5]AbG 59
sur résine d’oxime
3.2.1 Ancrage de la Boc-Phe-OH sur la résine d’oxime 60
3.2.2 Élongation du peptide sur résine d’oxime 61
3.3 Cyclisation de l’analogue [Hmb1, Val6, (6S)-Mel2aa5IAbG (142) sur résine 7f d’oxime
3.4 Conclusion 72
3.5 Références du chapitre 3 74
V
CHAPITRE 5: PARTIE EXPÉRIMENTALE, SYNTHÈSE DES ACIDES AMINÉS NON 79
NATURELS
5.1 Généralités $0
5.2 5.2 N-(Boc)-N-(méthyl)amino acides (59) et (61) 81
5.2.1 Dipeptides hydroxamates Boc-Val-Phe-NHOH(65a) et Boc-Phe-Phe-NHOH 82
(65b)
5.2.2 Pureté énantiomérique de la N-f Boc)-N-(méthyl)-L-valine (59) et de la N-(Boc)- 83 N-(méthyl)-L-phénylalanine (61)
5.3 (S)-2-Hydroxy-3-méthylbutanoate de benzyle (Hmb-OBn, 67) 84
5.3.1 L’acide (S)-2-hydroxy-3-méthylbutanoïque (Hmb, 66) 84 5.3.2 (S)-2-Hydroxy-3-méthylbutanoate de benzyle (Hmb-OBn, 67) 84
5.4 Acide (3S,6S,9S)-2-oxo-3-N-(Boc)amino-1 -azabicyclo[4.3.OJnonane-9-carboxylique 85
(73)
5.5 Acide (3S,6S,9S)-2-oxo-3-N-(Boc)-N-(méthyl)amino-1-azabicyclo[4.3.0]nonane-9 $6
carboxylique (74)
5.5.1 Acide (3S, 6S,9S)-2-oxo-3-N-(Boc)amino-1 -azabicyclo[4.3.0]nonane-9-benzyle 86 ester (75)
5.5.2 Acide (3S, 6S,9S)-2-oxo-3-N-(2-nitrobenzenesulfonyl)amino-1 -azabicyclo[4.3.0] 87 nonane-9-benzyle ester(76)
5.5.3 Acide (3S, 6$,9S)-2-oxo-3-N-f 2-nitrobenzenesulfonyl)-N-f méthyl)amino-1- 88 azabicyclo[4.3.0] nonane-9-benzyle ester (77)
5.5.4 Acide (3S, 6S,9S)-2-oxo-3-N-(Boc)-N-f méthyl)amino-1 -azabicyclo[4.3. 0]nonane-9- 89 benzyle ester (78) 5.5.5 Acide (3S, 6S,9S)-2-oxo-3-N-fBoc)-N-(méthyl)amino-azabicyclo[4.3.0]nonane-9- 90 carboxylique (74) 5.6 (S)-f3-Hydroxy-N-(Boc)-N-(méthyl)valine (99) 91 5.6.1 (4S)-N-(9-phénylfluoren-9-yl)-oxazolidine-4-carboxylate de méthyle (116) 91 5.6.2 f4S)-4-( 1 -hydroxy-méthyléthyl)-3-f9-phénylfluoren-9-yI)-oxazolidine (117) 92 5.6.3 (4R)-O-acétoxyméthyl-5,5-diméthyl-3-(9-phénylfluoren-9-yl)-oxazolidine (123) et 93 (4R)-4-hydroxyméthyl-5, 5-diméthyl-(9-phénylfluoren-9-yl)-oxazolidine (122) 5.6.4 f2R)-2-N-(9-phénylfiuoren-9-yI)amino-3-méthyl-1 ,3-butanediol f118) 94 5.6.5 (2R)-2-N-( Boc)amino-3-méthyl-1 ,3-butanediol (119) 95 5.6.6 (S)-3-Hydroxy-N-fBoc)-N-(méthyl)valine (99) 96
5.7 Synthèse des depsipeptides (125) 99
5.7.1 Procédure générale 99
5.7.2 Boc-MeVal-Hmb-OBn (124a) 100
5.7.3 Boc-HQMeVaI-Hmb-OBn (124b) 100
5.7.4 Boc-HOVaI-Hmb-OBn (124c) 101
5.7.5 Procédure générale déprotection benzyle des dipepsipetides (124) 102
5.7.6 Boc-MeVal-Hmb-OH (1 25a) 103
5.7.7 Boc-HOMeVaI-Hmb-OH (125b) 103
5.7.8 Boc-HOVaI-Hmb-OH (125c) 104
5.8 Références du chapitre 5 105
CHAPITRE 6: PARTIE EXPÉRIMENTALE, PROGRES DANS LA SYNTHÈSE DE LA 106 [Val6, (6S)-MeI2aa45JAbB ET DE LA [Hmb1, Val6, (6S)-MeI2aa45lAbG
6.1 Généralités 107
6.2 Résine d’oxime (62) 107
6.3 Résine Boc-Phe-oxime (131) 107
6.3.1 Détermination du degré de la substitution de la résine Boc-L-Phe-oxime (131) 108
6.3.2 Acétylation de la résine Boc-L-Phe-oxime (131) 108
6.4 Résine Boc-MeVal-Phe-oxime (133) 109
6.5 Résine Boc-aa-Hmb-MeVaI-Phe-oxime (135) 109
6.5.1 Résine Boc-MeVal-Hmb-MeVal-Phe-oxime (135a) 109
6.5.2 Boc-HOMeVaI-Hmb-MeVal-Phe-oxime (1 35b) 110
6.6 Résine Boc-Leu-aa-Hmb-MeVaI-Phe-oxime (138) 111
6.6.1 Résine Boc-Leu-MeVal-Hmb-MeVal-Phe-oxime (138a) 111
6.6.2 Résine Boc-Leu-HOMeVaI-Hmb-MeVal-Phe-oxime (1 38b) 112 6.7 Résine Boc-MeVal-Leu-aa-Hmb-MeVal-Phe-oxime (140) 113 6.7.1 Boc-MeVal-Leu-MeVal-Hmb-MeVal-Phe-oxime (140a) 113 6.7.2 Résine Boc-MeVal-Leu-HOMeVal-Hmb-MeVal-Phe-oxime (1 40b) 114 6.8 Boc-VaI-MeVaI-OH (144) 115 6.8.1 Boc-MeVal-OBn (142) 115 6.8.2 Boc-Val-MeVal-OBn (143) 116 6.8.3 Boc-Val-MeVal-OH (144) 117
VII 6.9 Résine Boc-MeI2aa-Val-MeVal-Leu-MeVal-Hmb-MeVal-Phe-oxime (147) 117 6.9.1 Résine Boc-VaI-MeVaI-Leu-MeVaI-Hmb-MeVaI-Phe-oxime (142) + 117 diastéréolsomèce 6.9.2 Résine Boc-MeI2aa-VaI-MeVaI-Leu-MeVaI-Hmb-MeVaI-Phe-oxime (147) + 118 diastéréoisomère
6.10 Synthèse de l’analogue [Hmb1, Val6, (6S)-Me12aa45]AbG (148) sur résine d’oxime 119 et de son diastéréoisomère
6.11 Références du chapitre 6 120
ANNEXE 1: ARTICLES PUBLIÉS xvii
Serine as a Chiral Educt for the Practical Synthesis of Enantiopure N- xviii protected f3-Hydroxyvaline
Diversity-oriented Synthesis of Enantiopure N-protected f3,13- XXi
Diaikylserines
One-pot Synthesis of Homoallylic Ketones from the Addition of Vinyl
Grignard Reagents to Carboxylic Esters xxviii
Advances in a Structure Activity Study cf the Auceobasidin Peptide xxxii Antibiotics: Synthesis of L-N-Boc-methyl-f3-hydroxyvaline
ANNEXE 2 : SPECTRES ET CHROMATOGRAMMES, SYNTHÈSE DES ACIDES xxxiv AMINÉS NON NATURELS
ANNEXE 3 tSPECTRES ET CHROMATOGRAMMES, PROGRES DANS LA cxxxiv
SYNTHÈSE DE LA [Val6, (6S)-MeI2aa45]AbB ET DE LA [Hmb1, Val6, (6S)-Me12aa45]AbG
LISTE DES FIGURES
CHAPITRE 1
Figure 1. Structure de l’amphotéricine B (1) 3
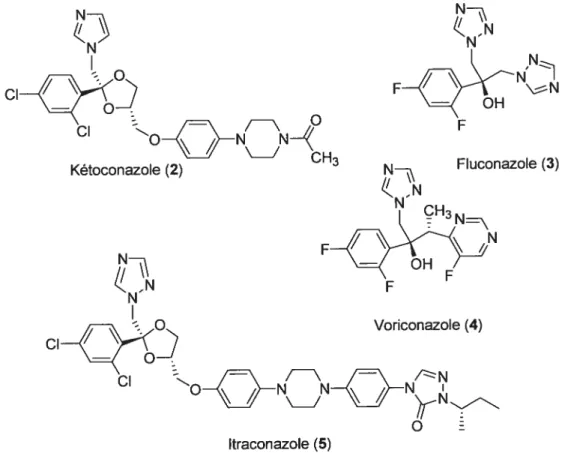
Figure 2. Structures de la kétoconazole (2), la fluconazole (3), la voriconazole (4) 4 et l’itroconazole (5)
Figure 3. Structure de la flucytosine (6) 5
Figure 4. Structure de la caspofungine (7) 6
Figure 5. Formes de résonance du lien amide 6
Figure 6. Isomérisation du lien amide 7
Figure 7. Disposition et angles de torsion de deux liaisons peptidiques rigides par 7 rapport au carbone Ca et définition des trois angles de torsion qui
décrivent la liaison peptidique
Figure 8. Résidus et angles dièdres des tours y et f3
Figure 9. Exemples de mimétismes de tour f3 10
Figure 10. Analogue de LH-RH (11) incorporant un lactame de Freidinger 11 Figure 11. Analogues de la GS [l2aa(6S)445.]GS (12), [Lys2,2’, l2aa(6S)454.5]GS 11
(13) et [l2aa(6R)454’5.]GS (14)
Figure 12. Rotamère trans de l’auréobasidine A trans-(18) 14 Figure 13. Rotamère trans-1$ et cis-18 de l’auréobasidine A 15
Figure 14. Vue stéréoscopique de l’auréobasidine A (19) 16
Figure 15. La réaction rétto-aldol-aldol sur l’auréobasidine A (18) 17 Figure 16. Auréobasidines: Rotamères trans-(21) et cis -(21) et analogues trans 18
(23) et cis (24)
Figure 17. Synthèse des fragments de l’Auréobasidine A (18) en solution par le 20 groupe Takesako
Figure 18. Synthèse totale de l’Auréobasidine A (18) en solution par le groupe 21 Takesako
Figure 19. Synthèse des fragments de l’Auréobasidine A (18) en solution par le 23 groupe Jao
Figure 20. Synthèse totale de l’Auréobasidine A (18) en solution par le groupe Jao 24 Figure 21. Synthèse des fragments de l’Auréobasidine A (18) en solution parle 25
groupe Schmidt
Figure 22. Synthèse totale de l’Auréobasidine A (18) en solution par le groupe 26 Schmidt
ix
Figure 23. Synthèse du fragments Fmoc-Leu8-HOVaI9-D-Hmp1-MeVal-0H2 (42) de 27 l’analogue de l’Auréobasidine A (57) en solution par le groupe Lajoie
Figure 24. Synthèse d’un analogue de l’auréobasidine A (57) utilisant une stratégie 28 solution/support solide (Irt-CI) par le groupe Lajoie
CHAPITRE 2
Figure 25. Synthèse des N-Boc-N-méthyl-amino acides (59) et (61) 34 Figure 26. Evaluation de la pureté énantiomérique des acides aminés Boc-N- 35
méthylés a aa= Val (59) et b aa= Phe (61)
Figure 27. Synthèse de l’acide (R)-2-hydroxy-3-méthylbutanoïque (66) à
partir
de la 36 L-valine (58)Figure 28. Synthèse du (3S,6S,9S)- et du (3S,6R,9S)-2-oxo-3-N-(Boc)amino-1- 37 azabicyclo[4.3.0] nonane-9-carboxylate de méthyle f6S)-72
Figure 29. Synthèse de l’acide (3S,6S,9S)-2-oxo-3-N-(Boc)amino-1- 38 azabicyclo[4.3. OJnonane-9- carboxylique (Boc-l2aa-OH, (6S)-74)
Figure 30. Synthèse de l’acide (3S,6S,9S)-2-oxo-3-N-(Boc)-N-(méthyl)amino-1- 39 azabicyclo[4. 3.0]nonane-9-carboxylique ((6S)-74)
Figure 31. Structure de la luzopeptine E2 (79) 40
Figure 32. Synthèse de la L-N-Boc-f3-hydroxyvaline (83) en 3 étapes par le groupe 41 Shin
Figure 33. Synthèse de la L-ç3-hydroxyvaline (89) en 7 étapes par le groupe Lajoie 42 Figure 34. Synthèse de la L-N-Boc-N-méthyl-3-hydroxyvaline (99) en 8 étapes par 43
le groupe Ciufolini
Figure 35. Synthèse et détermination de la pureté énantiomérique de F3-hydroxyvalines N-protégées (104a,b) en 2 étapes
Figure 36. Synthèse sur une échelle de 7.43 g de la N-Boc-F3-hydroxy-L-valine (83) 45 Figure 37. Addition du bromure de vinyle magnésium sur l’ester méthyilique de la 45
D-N-Boc-sérine (R)-(80)
Figure 38. Mécanisme réactionnel de la formation de la cétone homoallylique (109) 46 Figure 39. Synthèse du L-N-Boc-N-cyclopentyl-f3-hydroxy a-amino acide (113) 47 Figure 40. Synthèse et détermination de la pureté énantiomérique de la L-N-BOc-N- 49
méthyl-f3-hydroxyvaline (99)
Figure 41. Acétylation de l’oxazolidine 117 50
CHAPITRE 3
Figure 43. Synthèse sur résine d’oxime de la [I2aa445]GS 59
Fïgure 44. Structure du peptide modèle [MeI2aa5]AbG (127) ainsi que de l’analogue 60 [Val6, (6S)-MeI2aa5]AbB (23)
Figure 45. Ancrage de la Boc-Phe-OH et détermination de la substitution de la résine 61
12$
Figure 46. Divers agents de couplage pouvant être utilisés en synthèse peptidique 62 Figure 47. Activation d’un acide aminé N-protégé avec du chlorure de bis(2- 63
oxooxazolidin-3-yl) phosphinique (BOP-Cl) par un mécanisme d’activation basique intramoléculaire
Figure 48. Synthèse de la résine Boc-MeVal2-Phe3-Oxime (130) 63 Figure 49. Synthèse des résines Boc-aa9-Hmb1-MeVal2-Phe3-Oxime (132) 64 Figure 50. Synthèse de la résine Boc-Leu8-MeVal9-Hmb1-MeVal2-Phe3-oxime (13$a) 65 Figure 51. Synthèse de la résine Boc-Leu8-HOMeVal9-Hmb1-MeVal2-Phe3-oxime 66
(13$b)
Figure 52. Synthèse des résines Boc-MeVal7-Leu8-aa9-Hmb1-MeVal2-Phe3-oxime (140) 67 Figure 53. Synthèse du dipeptide Boc-Val6-MeVal7-OH (144) 68 Figure 54. Synthèse de la résine Boc-Va16-MeVal7-Leu8-MeVal9-Hmb1-MeVal2-Phe3- 69
oxime+ diastéréoisomère (145)
Figure 55. Synthèse de la résine Boc-MeI2aa-Val6-MeVal7-Leu8-MeVaI9-Hmb1-MeVal2- 70 Phe3-oxime+ diastéréoisomère (147)
Figure 56. Synthèse de l’analogue [Hmb1, Val6, (6S)-Me12aa45]AbG (130) par 72 cyclisation peptidique sur résine d’oxime
LISTE DES TABLEAUX
CHAPITRE I
Tableau I. Valeurs des angles dièdres ‘V2et
p
des acides aminés indolizidinones 12obtenues par rayons X et modélisation moléculaire
xi
LISTE DES ABRÉVIATIONS ET DES SIGLES
[a] Rotation spécifique en [(deg ml) I (g dm)]
A
Angstrom aa Acide aminé Ab Auréobasidine Ac Acétyle Atm Atmosphère Bn Benzyle Boc Tert-butyloxycarbonyle(Boc)20 Dicarbonate de di-tert-butyle
BOP (Benzotriazolyoxy)tris(pyrrolidino)phosphonium hexafluorophosphate
BOP-Cl N,N-bis(2-oxo-3-oxazolidinyl)phosphonium chloride
br Broad (large)
BTSFA N, O-bistriméthylsilyltrifluoroacétamide
c Concentration
°C Degré Celsius
Cl M Concentration inhibitrice minimale
ô Déplacement chimique
d Doublet
dU Doublet de doublet
DBU 1 ,8-diazabicyclo[4. 5.O]undec-7-ène
DCC N,N’-dicyclohexylcarbodiimide
DEC Hydrochlorure de 1 -(3-diméthylaminopropyl)-3-éthylcarbodiimide
DIAD Diisopropyl azodicarboxylate
DIC N,N’-Diisopropylcarbodiimide
DIEA N,N’-Uiisopropyléthylamine
DMAP 4-diméthylaminopyridine
DMF Diméthylformamide
DMP Diméthylphtalate
EAC NOx Ethyl-2-(hydroxyimino)-2-cyanoacétate
EDCI Chlorhydrate de 1 -(3-Uiméthylaminopropyl)-3-éthylcarbodiimide
ee Excès énantiomérique
Et Ethyle
FTIR Infrarouge à transformé de Fourier
h Heure
HATU O-(7-Azabenzotriazol-1 -yl)-1,I ,3,3-tetramethyluronium hexafluorophosphate
H BTU O-(Benzotriazol-1 -yl)-1,1 ,3,3-tetramethyluronium hexafluorophosphate
HIFIP 1,1,1 ,3,3,3-Hexafuoroisopropanol
HMQC Heteronuclear Multiple Quantum Coherence
HOAt 1 -Hydro-7-azabenzotriazole
HOBt Hydroxybenzotriazole
HOOBt 3-Hydroxy-3,4-d ihydro-4-oxo-1 ,2, 3-benzotriazine
HRMS High resolution mass spectrometry
Hz Hertz
I2aa Acide (3S, 6S,9S)-2-oxo-3-N-(Boc)amino-1 -azabicyclo[4.3. O]nonane 9-carboxylique
J Constante de couplage
LRMS Low resolution mass spectrometry
Micro
M Mole par litre
Me Méthyle MHz Mégahertz m Multiplet MeOH Méthanol min. Minute NMM N-méthylmorpholine 0m Ornithine p Para Pac Phénylacyle
PCOR Peptide Cyclization on Oxim Resin
PF Point de fusion
Pfp 2, 3,4,5,6-Pentaflurophénol
Ph Phényle
PhF 9-(phénylfluorényle)
ppm Partie par million
PyBrop Bromotris(pyrrolydino)phosphonium hexafluorophosphate
4-pyrpyr 4-pyrrolidinopyridine
q Quadruplet
XIII
RMN Résonance magnétique nucléaire
S Substitution d’une résine (mmollg)
s Singulet
SPPS Synthèse de peptide sur phase solide
t Triplet
TBTU N-oxyde de N-[( 1 H-benzotriazol-1 -yl)(diméthylamino)méthylène]-N méthylméthanaminium tétrafluoroborate
TEMPO 2,2,6, 6-tetramethyl- 1-piperidinyloxy, ftee radical
TES Triethylsilyl
TFA Acide trifluoroacétique
THF Tétrahydrofurane
THP Tétrahydropyrane
TIC Total Ion Current
TLC Thin Layer Chromatography
Tmac2O Anhydride triméthylacétique
TMS Triméthylsilyle
TP Température de la pièce
TR Temps de rétention
Ts p-toluène sulfonyle
p-T5OH Acide para-toluène sulfonique
UV Ultraviolet
REMERCIEMENTS
Ce travail de maîtrise a été réalisé dans les laboratoires de chimie organique de l’Université de Montréal sous la direction du Professeur William D. Lubeli. Je tiens à le remercier très sincèrement pour l’attention constante, sa disponibilité, le dynamisme communicatif, la compétence avec lesquels il a suivi ce travail et de l’excellente formation scientifique qu’il m’a permis d’acquérir. Durant ces études, j’ai eu l’occasion de présenter mes travaux dans cinq forums de recherche: le
3ème symposium international en synthèse organique et organométallique de l’Université de
Montréal SISOUM 2002, le 18ème symposium de peptide APS 2003 à Boston, le 39ème congrès IUPAC et la 86ème conférence de la société de chimie du Canada 2003 à Ottawa et le 14ème Québec-Ontario mini symposium â l’Université du Québec â Montréal (UQAM) 2003 MONTREAL et le let symposium des étudiants graduées en chimie de l’Université de Montréal 2004. De plus, j’ai pu rédiger quatre publications faisant référence à mes travaux et qui ont permis d’ouvrir la voie à de nouveaux sujets de recherche. Une cinquième publication est actuellement en cours de rédaction.
Je remercie également tous les membres du département de Chimie Organique et particulièrement ceux du groupe Lubell, qui grâce à leur amitié, leur bonne humeur, leur aide et leur connaissance, m’ont permis d’effectuer ce travail dans une ambiance dynamique et conviviale. Merci Catherine Girard pour toute l’aide que tu m’as apporté au long de mes études, de m’avoir supporté dans mes délires, de m’avoir soutenu et de m’avoir aidé pour la mise en page de ce mémoire. Merci Jerome Cluzeau pour tes précieux renseignements. Merci Eryk Thouin pour tes suggestions judicieuses. Merci Fabrice Galaud pour ta compagnie et ta bonne humeur dans notre laboratoire. Merci à Dalbir Singh Sekhon pour sa grande aide dans les analyses LC-MS et mes purifications RP-HPLC. Merci encore à : Liliane Halab, Laurent Bélec, Kenza Dairi, Rosa Melendez, Guillame Jeannotte, Silvia Van Cauwenberghe, Priscille Laborde, John et Erin Blankenship, Ramesh Kaul, Karine Gingras, Frederik Rombouts, Yann Brouilelle, Simon Surprenant, Karl Hansford, Aurélie Dôrr, Gil Fridkin, Benoît Jolicœur, Zohreh Sajjadi Hashemi, Damien Boeglin, Félix Marcotte, Mallem Ramana et Mildred Bien-Aimé. Finalement, merci à Guybrush Treepwood pour ses commentaires pertinents et son aide précieuse dans la correction du français.
xv
Je n’oublie évidemment pas ma mère Denise Alice Dettwiler pour son aide, son soutien moral et financier durant la période de ce travail. Mme Marie-France Thomas pour son accueil et m’avoir hébergé à Montréal. Merci Solange Zurfluh pour tes encouragements lots de mes études d’ingénieries. Merci Sébastien Vodoz mon plus vieil ami qui m’a soutenu durant mes études par ses conseils. Merci à tous mes amis: Grégoire Vodoz, Greg Medwed, Dan Tatut, Davide Peduzzi, Lisa Roulet, Luzolo Soba Carlos, Olivier Delessert, Steve Guex, Manuel Junod, Benjamin Zumstein, Rémy Tissot, Christian Zilocchi, Aurélie Auroy, Florian Labarre, Andy Strong, Stéphane Pinaud, Vincent Brossard, Toni Verdini, Willy Schild et Lucienne Lannaz. Ainsi que mes anciens professeurs qui m’ont permis d’acquérir une bonne formation : Dr. Christiane Miéville (Ecole professionnelle de Lausanne EPSIC), Dr. Edmond Goy (Ecole d’Ingénieurs de Genève EIG), Dr. Eva Winter (A sa mémoire, décédée en 2003, Ecole d’ingénieurs de Genève EIG), Pr. Antoni Verdini (Institut de biochimie d’Epalinges).
Finalement, je remercie le Conseil de recherche en sciences naturelles et en génie du Canada pour le support financier.
A ma Mère, Denise Alice Dettwiler et à la mémoire de mon Père Erich Alfred Dettwiler
(1939-1981) grâce à qui j’ai pu faire mes études.
Le savoir ne réside pas seulement dans les livres, les laboratoires, les fiches, mais dans et
par l’amitié, celle des hommes, celles des bêtes et des étoiles.
Chapitre I
Introduction
tf Les infections fongiques
Les défis particuliers présentés par les infections fongiques ont stimulé de nombreux efforts tant au niveau diagnostique que thérapeutique durant les deux dernières décénnies.1 En effet, l’incidence des infections pulmonaires, urinaires et des septicémies d’origine fongique a augmenté considérablement depuis les années 1990.1 Des données statistiques montrent clairement une hausse de la mortalité due aux infections fongiques aux Etats-Unis durant les deux dernières décennies.2 Cette augmentation peut principalement être attribuée à la croissance de la population immunosupprimée que ce soit suite à des traitements contre les leucémies, en prévention des rejets d’organes ou encore suite à des infections au VIH.3 Les difficultés à traiter les infections fongiques viennent en partie du fait qu’elles sont encore difficiles à diagnostiquer.4 De plus, le nombre d’espèces différentes responsables d’infections est en croissance.4 Finalement, le spectre d’agents antifongiques disponibles est relativement restreint.4
Les infections fongiques peuvent être divisées selon le tissu impliqué et l’étendue de l’infection les infections cutanées ou sous-cutanées sont généralement traitées efficacement avec des agents topiques.5 Par contre, les infections systémiques, souvent issues de la dissémination d’une infection pulmonaire,6 engendrent des taux de mortalité pouvant excéder 9Q%4 Bien que certaines espèces aient un potentiel pathogénique plus élevé que d’autres, le statut immunologique de l’hôte est d’une importance primordiale pour déterminer la sévérité d’une infection fongique.6
La pneumonie à Aspergillus est une complication courante sérieuse chez les greffés et les sidatiques.7 L’Aspergilus peut aussi se disséminer dans le sang d’un patient immunosupprimé via les poumons, les voies aériennes ou un traumatisme mineur de la peau. La colonisation du cerveau ou de d’autres organes devient alors possible.6
Les manifestations les plus courantes de l’infection à Cîyptococcus neoformans sont la pneumonie et la méningoencéphalite.6 Ces infections sont le lot des patients dont l’immunité cellulaire est profondément supprimée, par exemple les sidatiques.3
Toutes les espèces de Candida qui sont pathogènes pour l’humain sont aussi rencontrées comme commensales de la flore normale.6 Le Candida peut être la cause d’infections cutanées ou des muqueuses relativement bénignes chez les hôtes en bonne santé. La prévalence de la candidose vaginale est très élevée chez la femme et elle est chronique chez plusieurs personnes infectées.6 Chez les patients dont l’immunité est déficiente comme les nouveaux nés, les diabétiques ou les patients traités avec des glucocorticoïdes, l’infection à Candida peut dégénérer en arthrite septique ou en oesophagite.6 Chez les patients immunosupprimés la dissémination hématologique peut engendrer des méningites et des abcès cérébraux.6
Chapitre 1 3
1.2 Les agents antifongiques
La cible principale des agents antifongiques utilisés pour traiter les mycoses graves est l’enveloppe cellulaire qui comprend la membrane et la paroi.5 L’intégrité de ces structures est essentielle à la survie cellulaire puisque sa perturbation entraîne une lyse osmotique.8 La paroi cellulaire présente un intérêt pharmaceutique particulier parce qu’elle n’a pas d’équivalent chez les mammifères. En conséquence, un agent spécifique contre la paroi cellulaire devrait être peu toxique pour les humains.9 L’accroissement de l’incidence des infections fongiques graves, ainsi que les problèmes de résistance aux médicaments actuels ont stimulé la recherche sur les agents antifongiques.9
1.2.1 Les médicaments antifongiques
Un grand nombre de composés a montré une activité antifongique in vitro. Par contre, peu d’entre eux ont pu franchir toutes les étapes nécessaires à la commercialisation. Présentement, les médicaments disponibles contre les infections fongiques se limitent aux polyènes, aux composés azole, la flucytosine et les échinocandines.9
1.2.2 Les polyènes
Les polyénes ont été les premiers médicaments antifongiques développés. Ils agissent en se combinant à l’ergostérol de la membrane cytoplasmique entraînant une augmentation de la perméabilité et, éventuellement, une lyse cellulaire.56 Actuellement, l’amphotéricine B (1) est le traitement de choix contre les infections fongiques systémiques. Les indications les plus courantes sont l’histoplasmose, la blastomycose, la candidose et la cryptococcose.6 Environ 80% des patients traités avec ce médicament présenteront des complications rénales possiblement fatales.1 Divers complexes colloïdaux d’amphotéricine B (1) qui entraînent moins de complications sont disponibles actuellement, mais leur prix prohibitif empêche leur usage de routine.10
OH
1.2.3 Les azoles
Les azotes forment la famille d’agents antifongiques la plus utilisée en clinique, ils incluent les imidazoles (kétoconazole (2)) et les triazoles (fluconazole (3), voriconazole (4), itraconazole (5) et ravuconazole) (Figure 2). Ils inhibent la synthèse d’ergostérol, nécessaire à l’intégrité de la paroi fongique.5 Ils peuvent aussi causer des dommages directs à la membrane cytoplasmique.5 Un très grand nombre d’interactions avec d’autres médicaments a été documenté de plus, ils sont tératogènes et sont sujets au développement de résistance.1° Une variété d’imidazoles (kétoconazole, sulconazole, oxiconazole,
...)
est disponible pour les infections fongiques superficielles. La kétoconazole,la première imidazole utilisée pour les infections systémiques, a été maintenant remplacée
par les triazoles. Cette dernière classe montre moins d’hépatotoxicité et a moins d’impact sur la synthèse hormonale. De plus, le spectre d’activité est plus large. La fiuconazole (3) est utilisée pour les candidoses superficielles et systémiques, ainsi que les coccidioses. Les effets secondaires principaux sont des allergies et des nausées.6 Le voriconazole (4), un dérivé de la fluconazole (3) a un spectre beaucoup plus large.5 L’itraconazole (5) est le seul triazole pouvant être utilisé contre I’aspergillose et la tolérance est grandement améliorée par rapport à l’amphotéricine B (1).
ClÔ Kétoconazole (2) Fluconazole (3) F(4(/N Voriconazole (4) Cl Itraconazole (5)
Chapitre 1 5
1.2.4 La flucytosine
La flucytosine (6) est convertie dans la cellule fongique en fluorouracile, un composé qui empêche la transcription de l’ARN et la synthèse de l’ADN.5 La flucytosine (6) est rarement utilisée en monothérapie à cause du développement rapide d’une résistance.6 Par contre, elle peut être utilisée en combinaison avec l’amphotéricine B (1) contre les infections à cryptococcus et à candida.
À
haute dose, elle interfère avec la fonction de la moelle osseuse et prédispose aux colites.6N H2
NkfF
Figure 3 Structure de la fiucytosine (6)
1.2.5 Les échinocandines
Les échinocandines sont des hexapeptides cycliques liés à une chaîne lipidique qui sont isolés de la souche Aspergilus ruglosus.5 Les membres de cette famille de peptides sont des inhibiteurs de la (1 ,3)-3-D-glucane synthase, une enzyme essentielle à la synthèse de la composante majeure de la paroi cellulaire fongique.8 Depuis les années 70, diverses échinocandines sont entrées en phase clinique, de ces recherches, seule la caspofungine (7) a montré une toxicité suffisamment faible pour être utilisée chez les humains.5 Actuellement, la caspofungine (7) est approuvée dans le traitement des aspergilloses invasives, ainsi que des candidoses oropharyngées.8 L’incidence des effets indésirables de 7 est comparable à celle du fluconazole (3) et nettement inférieure à celle de l’amphotéricine B (f)•8 De plus, les interactions avec d’autres médicaments sont nettement plus rares que dans le cas des triazoles.8 L’absence d’antagonisme avec les autres classes d’antifongiques en fait un candidat intéressant pour les polythérapies.8 La biodisponibilité orale de la caspofungine (6) est très limitée et elle ne peut être administrée que par voie intraveineuse.8
rn
H2NFigure4 : Structure de la caspofungine (7)
1.3 Structure des peptides et des protéines
Les peptides et les protéines sont constitués d’acides aminés condensés les uns à
la suite des autres par des liaisons amides.11 Les acides aminés naturels constituant les
protéines sont au nombre de vingt et la plupart de configuration L. Ceci souligne le nombre extraordinaire de peptides ou de protéines possibles. Par définition, les chaînes d’acides aminés de masse moléculaire inférieure à 10,000 Daltons sont des peptides.12
1.3.1 Interactions
Les structures protéiques peuvent être stabilisées par deux classes de liaisons fortes (liaisons peptidiques et ponts disulfures) et/ou trois classes de liaisons faibles (liaisons hydrogènes, interactions hydrophobes et interactions électrostatiques).
La liaison peptidique possède une forme de résonance qui donne le caractère d’une
double liaison partielle entre le carbone et l’azote (Figure 5). C’est-à-dire que la rotation autour de la liaison amide CŒ-C(O)-N-Ca (angle w) est singulièrement inhibée et, de ce fait, les six atomes (carbone du carbonyle, l’oxygène, l’azote et l’hydrogène) impliqués se retrouvent dans un même plan (Figure 5).
HO 7
Chapitre 1 7
Le lien amide peut exister sous la forme de deux isomères: l’isomère trans (w =
1800) et l’isomère cis (w = 0°) (Figure 6). Du point de vue énergétique, la rotation du N,N
diméthylacétamide vaut 19,3 kcal/mol (cis —trans).13 L’isomère trans a la plus basse énergie et il est retrouvé dans tous les liens peptidiques sauf pour les acides N,N disubstitués. En comparaison, il existe une grande liberté de rotation autour des autres liaisons dans la chaîne peptidique.14 De plus, la forme de résonance du lien amide offre à la structure peptidique une caractéristique importante qui est la polarité, essentielle à la formation de liaison hydrogène.14
O w=1$00 O w=0°
Trans Cis
Figure 6: Isomérisation du lien amide
Les quatre angles de torsion qui décrivent l’arrangement de la chaîne peptidique sont l’angle (p qui décrit C(O)-N-CŒ-C(O), l’angle i qui décrit N-CŒ-C(O)-N,
l’angle w qui décrit l’angle de torsion du lien amide et l’angle
x
qui définit l’angle des chaînes latérales (Figure 7)15 Des limites de degré de liberté de rotation des plans de liaison peptidique selon les deux angles (p et u,i sont généralement imposées par des contraintes stériques. Ces restrictions angulaires s’expriment aisément dans le diagramme de Ramachandran qui donne en fonction de (p. Cette méthode graphique permet de schématiser la conformation de l’ensemble des atomes de carbone CŒ dans une protéine.16OXR1 O
Figure7: Disposition et angles de torsion de deux liaisons peptidiques rigides par rapport au carbone Ca et définition des trois angles de torsion qui décrivent la liaison peptidique
Mis à part le lien peptidique, l’autre interaction forte qui stabilise la conformation des protéines est le pont disulfure (liaison covalente). La liaison disulfure est formée normalement par l’oxydation des chaînes latérales de deux résidus de cystéine et est relativement stable et résistante aux conditions de dénaturation des protéines.12
Parmi les interactions faibles (non covalentes), on retrouve les liaisons hydrogènes
qui sont formées entre les chaînes latérales des acides aminés composant les peptides ou
entre les atomes d’hydrogène (donneur de lien hydrogène) et d’oxygène ou de l’azote (accepteur du pont hydrogène) des liaisons peptidiques. Les ponts hydrogènes dans les chaînes peptidiques sont un facteur stabilisant très important dans les structures secondaires.17
Le caractère hydrophobe des résidus apolaires joue un rôle très important dans les
repliements protéiques. En général, le centre des protéines naturelles ne contient pas d’eau et regroupe surtout des acides aminés apolaires et hydrophobes. Inversement, les acides aminés possédant des charges et autres acides aminés polaires se retrouvent essentiellement à la surface protéique en contact avec l’eau.14
Les chaînes latérales des acides aminés de charges opposées ont la possibilité d’interagir entre elles pour former des interactions électrostatiques (ponts salins) qui peuvent à leur tour stabiliser une structure protéique.14
1.3.2 Niveaux d’organisation
La structure des protéines est organisée selon plusieurs niveaux.
La structure primaire18 d’un peptide ou d’une protéine représente la séquence
linéaire des résidus des acides aminés composant la chaîne peptidique. Par convention, le premier acide aminé de la structure primaire est celui dont la fonction aminée n’est pas engagée dans une liaison peptidique (extrémité NH2-terminale), et le dernier acide aminé est situé à l’extrémité COOH terminale.
La structure secondaire19 correspond à un arrangement spatial localisé d’une
portion de la chaîne principale. Les trois structures secondaires les plus courantes sont l’hélice a, le feuillet f3 et les coudes (13 ou y). L’hélice a fait tourner la chaîne peptidique par rapport à elle-même d’un tout tous les 3,7 acides aminés. Elle est stabilisée pat des liaisons hydrogènes entre le carbonyle de la liaison peptidique qui suit l’acide aminé N°1 avec l’amine de la liaison peptidique qui précède l’acide aminé N°5. Le feuillet
13
quant à lui est formé lorsque plusieurs portions de chaînes se joignent bord à bord pour former un plan où les carbonyles de chaque acide aminé d’une portion de chaîne se lient avec les amines de l’autre portion. Actuellement, il est encore difficile de prédire la structure secondaire des peptides sur la base de leur structure primaire puisque les structures secondaires dépendent du milieu.Chapitre 1 9
La structure tertiaire19 correspond à l’arrangement dans l’espace des structures secondaires. Elle est le résultat des interactions faibles entre des acides aminés de la même chaîne peptidiques non voisins dans la structure primaire.
La structure quaternaire19 décrit les arrangements entre des acides aminés de molécules différentes mais qui sont unies en un seul complexe. Les interactions de la structure quaternaire sont les mêmes que celles de la structure tertiaire.
1.3.3 Les repliements y et
13
L’étude des structures secondaires des peptides permet de comprendre les relations existant entre leurs conformations et leurs activités biologiques. Parmi les structures secondaires, les repliements jouent un rôle primordial dans la structure et les fonctions des peptides.
Les structures en coude assurent un renversement rapide du sens de la propagation de la chaîne peptidique. C’est ce qui permet à une protéine de se replier et de maintenir sa structure compacte.19 Les repliements sont classés suivant le nombre de résidus qui sont impliqués dans le tour. Le coude y est constitué de trois résidus d’acide aminé, ces derniers stabilisent la structure par un lien hydrogène intramoléculaire entre le proton de l’amide du résidu 1+2 et le carbonyle du résidu I (C=O’ H-N’2).2° Le coude f3 est constitué de quatre résidus d’acides aminés, ces derniers stabilisent la structure par un pont hydrogène intramoléculaire entre le proton de l’amide du résidu i+3 et le carbonyle du résidu I (C=O’ H-N’3), la structure résultante est un cycle à 10 membres.21 Les coude f3 sont classés en plusieurs types selon les valeurs de l’angle de torsion des résidus 1+1 (92, 412) et
1+2 (93, 413) (Figure 8).21 22
RHI Résidu i+1
Ri
R’ d
NH >-Ri+2 esi u I
o
Partie N-terminalePartie C-terminale Partie N-terminale
Coudey Coude f3 Type Il
Résidu i+ 2 Résidus i+1 et i+2
42= -80° 412 -120° , -80° Ri+1 HN R NH d <7
//
Q: Résidu +2 Résidu i+3 Partie1.4 Peptidomimétisme
Plusieurs peptides naturels présentent une activité biologique intéressante. Cependant, au niveau pharmacologique, ils possèdent des limitations intrinsèques à cause de leur faible stabilité métabolique in vivo, de leur immunogénéicité et de leur faible absorption par voie orale.2324 Actuellement, de nombreuses recherches sont effectuées dans le but de contourner les désavantages du peptide natif cible en élaborant des produits dit peptidomimétiques qui contiennent des structures non-naturelles. Les avantages pharmacologiques recherchés par rapport au produit cible sont: une stabilité métabolique, une bonne biodisponibilité, de hautes affinité et spécificité.25
Le développement de produits peptidomimétiques implique l’élaboration et l’étude d’analogues par l’incorporation de contraintes structurelles. Il est donc important d’identifier les résidus impliqués dans la reconnaissance moléculaire.26 Dans la plupart des études, le peptide natif sert de conduite directrice pour le développement de peptidomimétiques. Ces derniers peuvent être basés sur des oligomères qui miment des structures primaires par l’utilisation d’isostères de lien amides et/ou l’utilisation de modifications du squelette peptidique du peptide natif, incluant des extensions de chaîne peptidique ou l’incorporation d’hétéroatomes.226
1.4.1 Mimétisme de coudes
F
Certaines molécules27’28 ont été synthétisées dans l’objectif d’être des structures qui remplacent les résidus intervenant dans les coudes J3. Ces composés synthétiques peuvent être donc directement incorporés au sein de structures peptidomimétiques.
8
Figure 9: Exemples de mimétismes de coude 13
Un des premiers peptidomimétiques synthétisé et incorporé dans une séquence peptidique est le lactame de Freidinger (8). Une fois incorporé dans la LH-RH (Luteinizing Hormone-Releasing Hormone), il est obtenu un analogue dont l’activité a été augmentée environ neuf fois par rapport au peptide parent.29 Toutefois, ce composé ne contraint que
9 (6S)-1O (6R)-1O
Chapitre 1 11
Arg-Pro-GIy-NH2
p-Glu : pyroglutamate 11
Figure 10 Analogue de LH-RH (Il)incorporant un lactame de Freidinger
Nagai et Sato ont synthétisé le dérivé Uipeptidique bicyclique BlD (9) imitant les positions i+1 et i+2 du coude
p.3°
Celui-ci fixe les angles ‘Y2et à des valeurs proches d’un coude 3 de type li’. Sa versatilité est démontrée par son incorporation dans certains composés biologiquement actifs (Leu-enképhaline, gramicidine S (GS), et un analogue cyclique de la somatostatine).31Le (6S)-l2aa (65)-(1O) et le (6R)-l2aa (6R)-(1O) ont été utilisés dans le groupe Lubeil
pour préparer trois analogues contraints de la gramicidine S (12, 13 et 14), un antibiotique naturel (Chapitre 31), dans le but d’étudier les relations entre la conformation du peptide et son activité biologique (Figure 11)32
Figure 11: Analogues de la GS [l2aa(6S)44’5’]GS (12), [Lys2’2’, l2aa(6S)454’5’]GS (13) et [l2aa(6R) ‘354’5’]GS (14)
L’interprétation des résultats de l’analyse conformationnelle des analogues 12, 13 et 14 indique clairement que l’analogue 12 contenant le diastéréoisomère de l’acide aminé indolizidinone (6S)-l2aa (6S)-(1O) donne un analogue de la GS qui maintient un feuillet 3 et présente une structure générale très semblable. Le replacement de l’ornithine du composé
12 par la lysine engendre des changements locaux dans la conformation de l’analogue 13,
mais sans grandes modifications de la structure générale. Cependant, l’analogue 14 contenant le diastéréoisomère (6R)-l2aa (6R)-(f O) a une structure globale moins semblable à celle de la GS, mais comporte une structure en feuillet f3. Les activités biologiques de 12 et 13 sont a peu près identiques à celle de la GS, mais avec une activité hémolytique quatre fois plus faible. L’analogue 14 est le moins puissant avec des activités biologiques 4 à 32
H”
12n=I
14
fois plus faibles que la GS, mais avec une activité hémolytique toutefois deux à quatre fois plus faible. La GS nécessite un coude 13 pour conserver son activité biologique.32 L’analyse de ces résultats montre que le diastéréolsomère (6S)-I2aa (6S)-(10) contenu dans les analogues 12 et 13 permet d’adopter une conformation plus proche du peptide natif. Les caractéristiques structurales nécessaires à l’activité biologique de la GS sont conservées dans les analogues 12 et 13. Par contre, le composé 14 semble adopter une structure comportant des ponts hydrogènes ressemblant à un feuillet 13, mais pas suffisamment proche de celle de la GS qui lui permettrait de maintenir son activité biologique.
Les valeurs obtenues pour les angles ‘V2 et par des études par rayons X et par
modélisation moléculaire33 du (6S)-l2aa (10) indiquent qu’il pourrait stabiliser des repliements y ou
13
de type Il’ (Tableau.1).33Méthode
W2(°) Réf.
Résidu i+1 et i+2 d’un coude
13
de type Il’ idéal -120 -80 21Résidu i+1 d’un tour de coude inverse -80
H Rayons X -176 -78 34 678 H Modélisation 33
)1N9
Min. global -112.7 -60.1 H Min. local -125.5 -65.6 16 H Modélisation 33 Min. global -135.6 -76.1 H Min. local -121.1 -71.0 17 0Tableau 1 Valeurs des angles dièdresqi2et des acides aminés indolizidinones obtenues par rayons X et modélisation moléculaire
Chapitre 1 13
1.5 Les auréobasidines
Découvertes en 1991, les auréobasidines (Abs) forment une famille de puissants antifongiques qui sont produites par la variété de levures noires Aureobasidins pullulans
R106.35’36 Actuellement, une vingtaine (A à R) a été isolée et caractérisée suivant leurs différences structurales.3738
1.5.1 Structure et activité biologique
Les auréobasidines (A à R) sont des depsinonapeptides cycliques de 27 membres qui, suivant la structure, sont formés de 8 a-amino acides dont 3 à 4 sont N-méthylés, un a hydroxyacide (Hmb: acide méthylbutanoïque ou Hmp: acide 2(R)-hydroxy-3(R)-méthylpentanoïque) et, suivant les cas, des acides 3-hydroxy a-aminoacides (N méthyl-f3-hydroxyphénylalanine ou N-méthyl-3-hydroxyvaline).36384°
Parmi les membres de cette classe, les auréobasidines A, B, C et E possèdent un squelette moléculaire semblable (Figure 12 et tableau 2) et présentent une forte activité
envers les espèces telles que Candida albicans, Cryptococcus neoformans et certaines espèces d’Aspergillus. Les AbA et AbE ont montré la plus grande activité antifongique et la toxicité la plus basse.36’41 Les auréobasidines agissent comme inhibiteur de l’inositol phosphocéramide synthase, une enzyme essentielle à la synthèse de la paroi cellulaire fongique. Cette enzyme étant exclusive aux organismes fongiques, elle est une cible thérapeutique particulièrement intéressante.42 L’auréobasidine A est un substrat pour le transporteur MDR (Multy Drug Resistance) qui permet l’efflux de substances cytotoxiques en dehors de la cellule. La substitution d’acides aminés dans l’AbA a permis de diminuer son affinité pour la MDR, ce qui laisse entrevoir la possibilité d’autres modifications qui permettraient la création d’analogues qui ne pourraient pas être évacués de la cellule par ce type de pompe.41
aa7
Hmp: R=C2H5
trans-(1 8)
CIM (ua/ml) Figure 12
AbS R aa4 aa6 aa7 aa9
Candida albicans Cryptococus neoformans A B C D E F G H J K L M N
o
P Q R C2H5 CH3 C2H5 C2H5 C2H5 C2H5 C2H5 C2H5 C2H5 C2H5 CH3 C2H5 C2H5 C2H5 C2H5 C2H5 C2H5 C2H5 MePhe MePhe MePhe MePhe -HOMePhe MePhe MePhe MePhe MePhe MePhe MePhe MePhe Phe MePhe MePhe MePhe MePhe 13-HOMePhe aile aile Val aile aile aile aile aile Leu aile aile Val aile aile aile aile aile aile MeVai MeVal MeVal MeVal MeVal Val MeVai MeVai MeVai MeVai MeVai MeVai MeVal MeVal Val Val MeVai MeVai F3-HOMeVa1 j3-HOMeVaI Ç3-HOMeVaI 3-HOMeVaI F3-HOMeVaI F3-HOMeVaI MeVal Val 3-HOMeVai N, 3-MeAsp MeVal MeVal MeVal DH34MeVaai 3-HOMeVaI MeVal MePhe MeVal <0.05 0.10 0.10 0.20 <0.05 0.78 3.12 1.56 <0.05 0.78 1.56 6.25 6.25 1.56 12.5 3.12 12.5 25 0.78 1.56 1.56 25 3.12 25 >25 >25 25 25 >25 >25 >25 >25 >25 >25 >25 >25Chapitre 1 15
L’étude RMN de l’auréobasidine A (AbA) (18) a montré l’existence dans le CDCI3 de deux conformations possibles.37 La labilité conformationelle du N-terminal de l’amide de la proline (lien MePhe4-Pro5) est dépendante des conditions environnementales.35’3643 Les changements de géométrie de ces rotamères ont des effets sur le reste de la conformation du peptide (Figure 13). Le rotamère trans-18 présente une structure rigide et allongée, tandis que le rotamère cis-18 a une structure beaucoup plus flexible et arrondie.35 L’auréobasidine A présente dans l’eau une conformation prédominante rotamère trans-18 qui induit les résidus MePhe4-Pro5 dans un tour de type f3 Dans le CDCI3 ou le DMSO, la conformation prédominante est le rotamère cis-18 qui présente un coude de type f3 Vl.35
Figure 13: Rotamère trans-18 et cis-18 de l’auréobasidine A
La structure cristalline de l’AbA (19) obtenue par évaporation d’une solution homogène dans l’éther à température ambiante (Figure 14) a montré que le peptide adopte une structure en forme de “tête de flèche”.45 Cette conformation consiste en trois structures secondaires : un feuillet
f3,
un coudef3
de type Il’ et un tour y. La forme tridimensionnelle de ce peptide est stabilisée par trois ponts hydrogène intramoléculaires et trans-annulaires NHO=C.
L’étude RMN structurale de I’AbA a montré que tous les liens amide, incluant la Pro5, sont dans la forme
trans
et toujours planaires.35’45 Le feuilletf3
antiparallèle entre les séquences Hmp1-MeVal2-Phe3 et aIIo-lle6-MeVal7-Leu8 est stabilisé par trois liaisons hydrogène. Le tour de type f3 Il’, formé par la séquence Phe3-MePhe4-Pro5-allo-11e6, est stabilisé par un pont hydrogène entre [allo-lle6]NH O=C{Phe3J. Le tour y, formé par la séquence Leu8-HOMeVaI9-Hmp1, est stabilisé par un pont hydrogène entre le groupement OH de HOMeVaI9 et le carbonyle de la Leu8. Cette structure, où le résidu f3-HOMeVaI9 est situé à l’extrémité de la forme en “tête de flèche”, est suggérée être nécessaire à l’acticité biologique.45 L’absence du groupe OH entraîne une diminution significative de l’activité biologique.36 D’où l’hypothèse de l’importance de ce groupe dans l’activité ou pour la liaison au récepteur.45Des études de modélisation moléculaire de l’AbA comparées à sa structure cristalline45 ont suggéré deux rôles possibles pour la présence des résidus N-méthylés: ils aident à bien définir la conformation moléculaire et à inhiber des interactions intramoléculaires NHX (X accepteur d’atome) via des liaisons hydrogènes.45
Une étude a été portée sur l’exploration des effets stéréoélectroniques de la f3-HOMeVaI9 par rapport à l’activité biologique.46 Pour ce faire, ce résidu est modifié en sarcosine par une méthode impliquant une réaction rétro-aldol dans laquelle il y a une perte d’acétone pour donner le dérivé de l’auréobasidine 20 qui ne présente aucune activité biologique. L’incorporation de diverses chaînes alkyles dans le peptide 20 pour donner l’analogue 21 se fait par une réaction aldol impliquant diverses cétones telles que 2-butanone, 3-pentanone et cyclopentanone avec une épimérization du résidu 94647 Mis à part le peptide comportant le cyclopentane où il y a une activité comparable, les autres dérivés sont dix fois moins actifs par rapport au peptide natif. Les résultats obtenus indiquent que le conformère le plus favorisé est celui dont le groupe hydroxy tertiaire n’est
19
Chapitre 1 17
pas impliqué dans un pont hydrogène, d’où l’hypothèse de son importance sur l’activité biologique.46 R=Me R= CF3 R=Me R’Et R=Et R’=Et R=R’=cyclopentanone R=Me R =H R=Pr R’=H ‘Rétro-AIdo’ Acétone 20 21
1.5.2 Analogues de I’Aureobasidine restreïnts conformationnellement proposés
Pour étudier la relation entre l’activité antifongique et la flexibilité conformationnelle, des peptidomimétiques de l’aureobasidine peuvent être synthétisés. Pour ce faire, il est possible de favoriser la rigidité du N-terminal de l’amide de la proline (lien MePhe4-Pro5) qui a un effet sur le reste de la conformation du peptide.35 L’incorporation de l’indolizidinone (6S)-l2aa (6S)-(1O) pourrait stabiliser le rotamère trans-(23), tandis que la 5-tert-butylproline48 pourrait augmenter la population en rotamère cis-(22) (Figure 16).
Figure 16 : Auréobasidines: Rotamères trans-(21) et cis -(21) et analogues trans (23) et cis (24)
Il est proposé dans cette étude de développer une méthode de synthèse d’un analogue de l’auréobasidine (23) contenant le (6S)-l2aa (6S)-(1O) dont le rôle est de remplacer un tour 13 de type Il’ formé par les résidus 4 et 5, soit une Phe4 N-méthylée et une Pro5. Cependant dans des buts de simplification synthétique certains résidus sont remplacés par des composés plus accessibles. Premièrement, l’allo-11e6 en position 6 est substituée par la valine comme dans l’AbC. Deuxièmement, le D-Hmp1 en position 1 est remplacé par le D-Hmb1 comme dans l’AbB. Ce résidu peut facilement être obtenu par la substitution de l’amine de la valine par la fonction alcool avec une rétention de la configuration. L’AbB et ‘AbC sont parmi les Abs les plus actives et le remplacement de ces
Indolizidinone (6)-l2aa tmns-{22) cis-(22) 5-tert-butylproline
(23) aa1=Hmp (24)
aa6=Val
Chapitre 1 19
1.6 Synthèses de I’Auréobasidine
La synthèse totale de l’auréobasidine A (AbA) (18) a déjà été publiée par d’autres
groupes de recherche.4952 Diverses stratégies ont été utilisées pour le couplage des acides aminés N-méthylés, pour la formation du lien depsi ainsi que pour la synthèse des acides aminés non naturels. Trois des méthodologies4952 rapportées s’effectuent en solution et utilisent la chimie Boc (Figures 17 à 22). Une autre méthode53 prépare une série d’analogue de l’AbA (57) contenant l’HOVaI, elle combine une synthèse sur support solide et en solution. Elle utilise la chimie Fmoc et l’étape de macrocyclisation s’effectue en solution (Figure 23).
1.6.1 Synthèses en solution
Dans un premier temps, les divers fragments sont synthétisés en solution puis, dans un deuxième temps, ils sont assemblés entre eux. La macrocyclisation s’effectue en milieu dilué à l’aide d’un agent de couplage.
Synthèse N°1 49,50
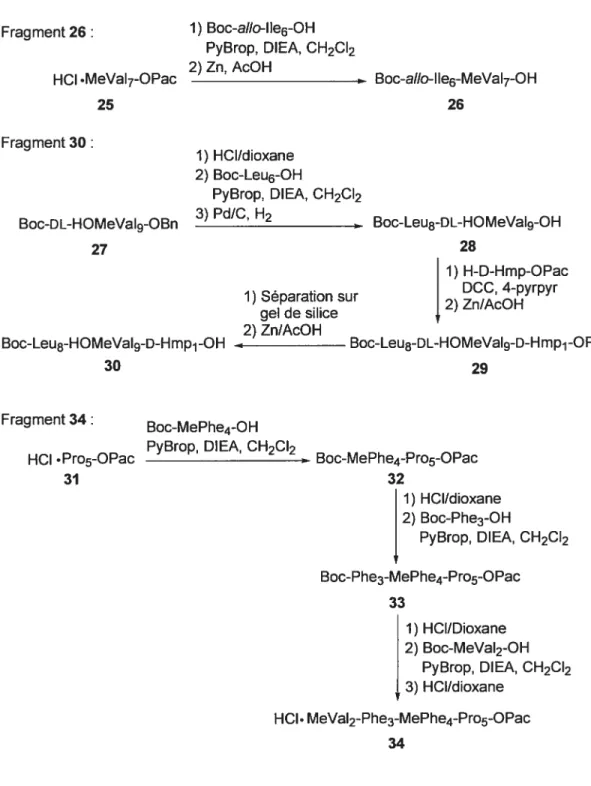
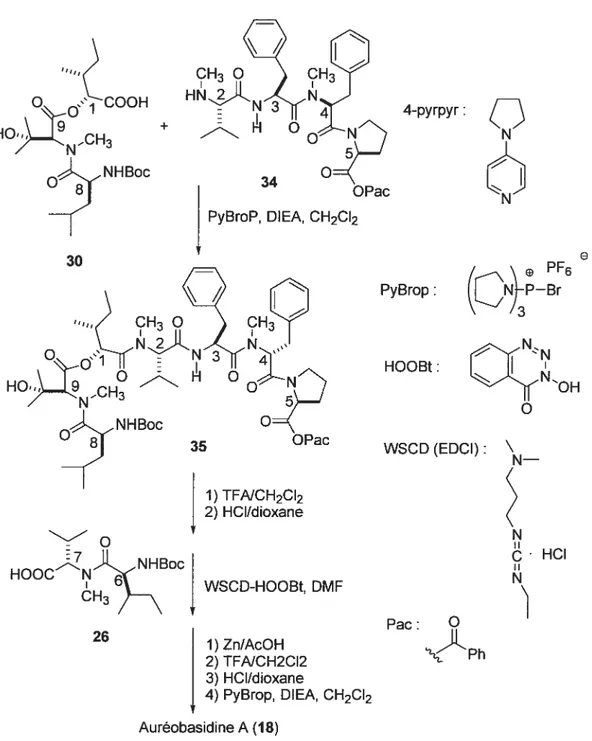
La première synthèse totale de l’AbA (18) a été rapportée par l’équipe de Takesoka (Figure 17 et 18). La synthèse du peptide linéaire utilise la condensation successive selon l’acide aminé concerné avec du PyBrop ou du EDCI (WSCD: “Water Soluble Carbodiimide”)-HOOBt comme agent de couplage. La 13-HOMeVaI est préparée sous sa forme racémique selon la méthode d’lzumiya54 et incorporée dans le tripeptide Boc-L-Leu DL-HOMeVaI9-D-Hmp1-OPac (30). Le fragment 30 contenant la L-Ç3-HOMeVaI9 a été séparé de l’isomère indésirable par chromatographie sur gel de silice (Figure 17). Le lien ester formé entre l’HOMeVal9 et le D-Hmp1 est effectué avec du DCC en présence du 4-pyrrolidinopyridine (4-pyrpyr). La macrocyclisation s’effectue entre l’alto-lie6 et la Pro5 avec du PyBrop/DIEA pour obtenir l’AbA (18) avec un rendement de 45% et un produit épimérizé avec un rendement de 7.6% (Figure 18).
Fragment 26: 1) Boc-aio-11e6-OH PyBrop, DIEA, CH2CI2 2) Zn, AcOH
HCI .MeVaI7-OPac Boc-allo-lle6-MeVaI7-OH
25 26
Fragment 30:
1) HCI/dioxane 2) Boc-Leu6-OH
PyBrop, DIEA, CH2CI2
Boc-DL-HOMeVaI9-OBn 3) Pd/C, H2 Boc-Leu8-DL-HOMeVaI9-OH
27 28
1) H-D-Hmp-OPac DCC 4-pyrpyr 1) Separation sur 2) Zn/AcOH
gel de silice 2) Zn/AcOH
Boc-Leu8-HOMeVaI9-D-Hmp1 -OH Boc-Leu8-DL-HOMeVaI9-D-Hmp1 -OPac
30 29
Fragment 34: Boc-MePhe4-OH PyBrop DIEA CH2CI2
HCI .Pro5-OPac Boc-MePhe4-Pro5-OPac
31 32
1) HCI/dioxane 2) Boc-Phe3-OH
PyBrop, DIEA, CH2CI2 Boc-Phe3-MePhe4-Pro5-OPac
33
1) HCI/Dioxane 2) Boc-MeVal2-OH
PyBrop, DIEA, CH2CI2 3) HCI/dioxane
HCI. MeVal2-Phe3-MePhe4-Pro5-OPac 34
Chapitre I 21 26 WSCD-HOOBt, DMF 1) Zn/AcOH 2) TFNCH2CI2 3) HCI/dioxane
4) PyBrop, DIEA, CH2CI2 Auréobasidine A (1$) 4-pyrpyr:
Ç)
PF6 PyBrop: HOOBt: WSCD (EDCI): e 34 30PyBroP, DIEA, CH2CI2
1) TFNCH2CI2
I
2) HCI/dioxane HOOCNFFBoc CH3 HCI Pac: O PhSynthèse en solution N°2 51
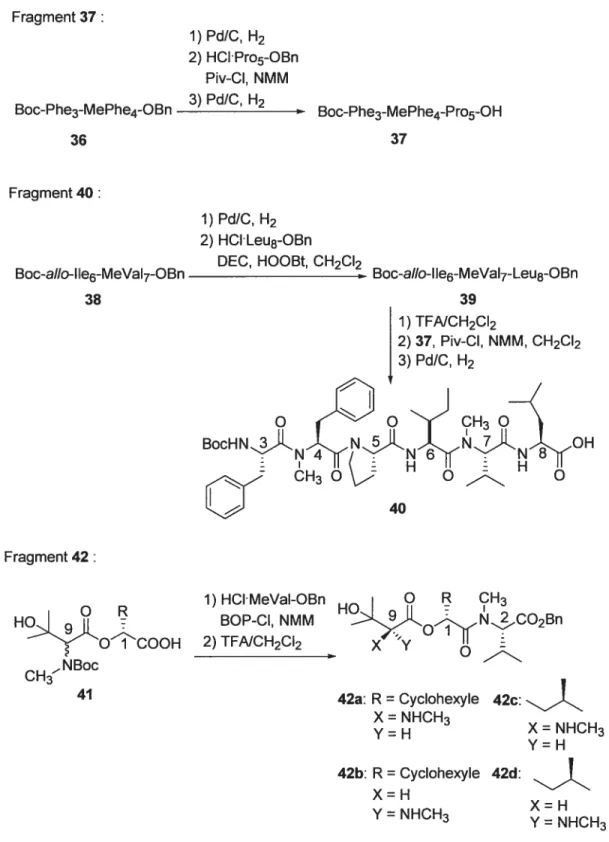
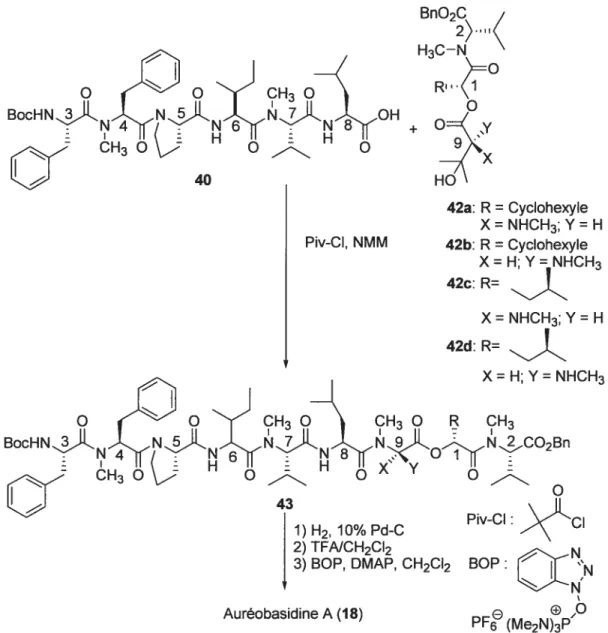
Le groupe de Jao a utilisé une approche convergente pour obtenir l’AbA (18) (Figures 19 et 20). La stratégie implique la synthèse de trois tripeptides utilisant le couplage DCC/HOBt ou BOP-Cl pour les acides aminés naturels et la méthode des anhydrides mixtes (Piv-Cl) pour les acides aminés N-méthylés. Le couplage des différents fragments a été effectué également par la méthode des anhydrides mixtes (Piv-Cl). La 3-HOMeVaI a été obtenue sous la forme d’un mélange racémique par une réaction aldolique sur la Boc Sarcosine benzylester avec de l’acétone.55 Elle a été incorporée dans le tripeptide N déprotégé DL-HOMeVal9-D-Hmb1-Va12-OBn. Le fragment 42 contenant la L-13-HOMeVaI9 est séparé de l’isomère indésirable par chromatographie sur gel de silice (Figure 19). Le lien ester formé entre l’HOMeVal9 et le D-Hmp1 est effectué avec du DCC/DMAP. La macrocyclisation s’effectue entre la Phe3 et la MeVal2 avec du BOP et DMAP pour obtenir I’AbA (18) avec un rendement de 21% (Figure 20).
Chapitre 1 23 Fragment 37: 1) Pd/C, H2 2) HCIPro5-OBn Piv-CI, NMM 3) Pd/C, H2 Boc-Phe3-MePhe4-OBn 36 Boc-Phe3-MePhe4-Pro5-OH 37 Fragment 40: Boc-aIIo-I 1e6-MeVaI7-OBn 38 1) Pd/C, H2 2) HCILeu8-OBn
DEC, HOOBt, CH2CI2
Boc-aio-11e6-MeVal7-Leu8-OBn 39 Fragment 42: HO.j OîCOOH CH30C 41 1) HCIMeVaI-OBn BOP-CI, NMM 2) TFNCH2CI2 F.JCO2Bn 42a: R= Cyclohexyle 42c: X= NHCH3 Y=H X = NHCH3 Y=H 42b: R= Cyclohexyle 42d: X=H Y = NHCH3 X= H Y= NHCH3 1) TFNCH2CI2 2) 37, Piv-CI, NMM, CH2CI2 3) PdJC, H2 40
BocHNLN. 4 CH3 Piv-CI, NMM Auréobasidine A (18) BnO2C 2)1K H3C—N RI O + O<y 9) -7(X HO
\
42a: R = Cyclohexyle X= NHCH3; Y = H 42b: R= Cyclohexyle X = H Y = NHCH3 42c: R= X= NHCH3;Y= HFigure 20 : Synthèse totale de l’Auréobasidine A (18) en solution par le groupe Jao51
Synthèse en solution N°3 : 52
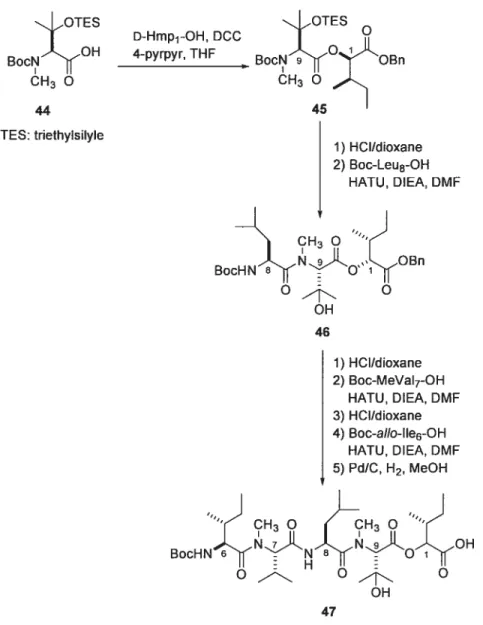
Le groupe de Schmidt a synthétisé le peptide linéaire en utilisant la condensation successive d’acides aminés en commençant pat le lien ester formé entre l’HOMeVaI3 et le D-Hmp1 (Figure 21 et 22). L’agent de couplage utilisé tant pour les acides aminés naturels que pour les N-méthylés est l’HATU. La 3-HOVaI est préparée sous sa forme racémique selon la méthode de Schrauth.56 La L-13-HOVal est obtenue par une cristallisation fractionnelle des sels diastéréomériques de la (S)-Œ-phényléthylamine. Celle recristallisation a donné la L--HOVal avec un excès énantiomérique >97%57 La Boc-L-3-HOVal-OH, où l’acide est protégé par un benzyle et l’alcool tertiaire par un triethylsilyle (TES), subit une
40 42d: R= X = H; Y= NHCH3 O R 43 1) H2, 10% Pd-C 2) TFNCH2CI2 N
3) BOP, DMAP, CH2CI2 BOP:
e
Chapitre 1 25
méthylation de l’azote avec du Mel en présence de Ag20 pour donner la Boc-L-f3-HOMeVaI-OBn (Figure 21). Le lien ester formé entre la TESOMeVaI9 (44) et le D-Hmp1 est effectué avec du DCC et 4-pyrrolidinopyridine. La macrocyclisation s’effectue entre l’aIIo-l le6 et la Pro5 avec la formation d’un ester pentafluorophényle sur la Pro5 en présence DCC et de DMAP pour obtenir l’AbA (18) avec un rendement de 27% et un produit épimérisé avec un rendement de 9% (Figure 22). JOTES BocN CH3 O 44 TES: triethylsilyle D-Hmp1-OH, DCC 4-pyrpyr, THF OTES BocHNN (H3 O J0oH O OH 47 45 1) HCI/dioxane 2) Boc-Leu8-OH HATU, DIEA, DMF 46 1) HCI/dioxane 2) Boc-MeVal7-OH HATU, DIEA, DMF 3) HCI/dioxane 4) Boc-aIIo-11e6-OH HATU, DIEA, DMF 5) Pd/C, H2, MeOH
BocN CH3 CH3 OBn 48 1) HCI/dioxane 2) 47, HATU, DIEA, DMF CH3 O 7H3 O “ CH3 Ph CH3 O O O / OH Ph o OBn 49 1) PdIC, H2, MeOH
2) Pfp, EDCI, DMAP, CH2CI2 3) HCI/dioxane 4) iN aq. NaHCO3/CHCI3 Auréobasidine A (18) Pfp DCC: HATU: FF NC=N N PF O NMe2 F NMe2 Figure 22 : Synthèse totale de l’Auréobasidine A (18) en solution par le groupe Schmidt52
Chapitre 1 27
1.6.2 Synthèse mixte sur support solide et solution
La stratégie du Groupe Lajoie utilise la résine 2-chlorotrityl (Trt-Cl) et la chimie Fmoc pour la synthèse d’une série d’analogues de l’AbA (57) (Figures 23 et 24). L’acide aminé non naturel L-aIIo-11e6 a été isolé d’un mélange racémique de quatre diastéréoisomères via une recristallisation fractionnée et une résolution enzymatique à l’aide d’une acylase rénale porcine.58 Dans cette synthèse, la méthode développée n’a pas permis d’obtenir l’HOMeVal sous une forme énantiomériquement pure, cet acide aminé non naturel a donc été remplacé par l’HOVaI (Chapitre 2.5, Figure 32). Le lien ester entre la Cbz-HOVal9 (50) et le D-Hmp1-allyl (51) est formé avec du DIC/DMAP et les acides aminés sont couplés avec du HOAUDIC (Figure 23). L’élongation jusqu’à l’obtention du peptide linéaire 56 est effectuée sur support solide en utilisant HOAtIDIC pour les acides aminés N-méthylés et BOP/HOBt pour les acides aminés naturels.53 Le fragment 54 est couplé sur la Phe3-Phe4-Pro5-Résine (55) à l’aide de HOAUDIC. L’élongation du peptide est poursuivie selon les conditions préalables. Le clivage du peptide linéaire est fait avec HIFIP (1,1,1,3,3,3-Hexafuoroisopropanol) et la cyclisation s’effectue avec du PyBrop/DIEA entre l’aIIo-11e6 et la Pro5 pour donner une série d’analogue de l’AbA (57) avec un rendement maximal de 58% (Figure 24).
Cbz-HOVaI9-OH + D-Hmp1-allyl
50 51
1) DIC, DMAP, CH2CI2 2) Pd(PPh3)4, PPh3, CH2CI2 Cbz-HOVaI9-D-Hmp1 -OH
52
MeVal2-O-t-Bu,
DIC, HOAt, DIEA, CH2CI2
Cbz-HOValg-D-H mp1 -MeVal2-O-t-Bu 53
1) Pd/C, H2
2) Fmoc-Leu8-OH, HOAt, DIC, CH2CI2 3) H
Fmoc-Leu8-HOVaI9-D-Hmp1 -MeVal2-OH 54
Figure 23 : Synthèse du fragmentsFmoc-Leu8-HOVaI9-D-Hmpl-MeVal-0H2 (42) de l’analogue de l’Auréobasidine A (57) en solution par le groupe Lajoie52
Fmoc-Phe3-MePhe4-Pro—Q Trt-Cl Résine 55
1) Couplage: 54, HOAt, DIC, DIEA, NMP
2) Couplage Fmoc-MeVal7-OH: HOAt, DIC, DIEA, NMP 3) Couplage Fmoc-a/Io-11e6-OH: BOP, HOBt, NMM, DMF 4) Clivage: HIFIP, CH2CI2
(Déprotection: Piperidine, CH2CI2) aio-11e6-MeVal7-Leu8-HOVaI9-D-Hmp1 -MeVal2-Phe3-MePhe4-Pro5 56 PyBrop DIEA, CH2CI2 (Haute dilution) Cyclo(aIIo-lle6-MeVal7-Leu8-HOVaI9-D-Hmp1 -MeVa2l-Phe3-MePhe4-Pro5) Analogue de l’AbA (57)
PyBrop: BOP: HOAt: HOBt:
(LN>_°
cN JC”N3 PF6 bH OH
P(NMe2)3 HIFIP DIC:
NCN
Figure 24 : Synthèse d’un analogue de l’auréobasidine A (57) utilisant une stratégie solution/support solide Crrt-Cl) par le groupe Lajoie52
Chapitre 1 29
1.7 Conclusion
La littérature ne rapporte que trois méthodes de synthèse totale de l’AbA (18) en solution avec la chimie Boc.4952 Pour chaque méthodologie, la L-HOMeVaI est préparée sous forme d’un produit racémique qui est soit séparé de l’isomère indésirable une fois incorporé dans un fragment peptidique, soit isolé par une cristallisation fractionnelle. Chaque stratégie utilise, une fois leurs peptides linéaires déprotégés, un protocole de macrocyclisation différent. Deux des méthodes cyclisent entre l’aIIo-lle6 et la Pro5. La première utilise du PyBrop/DIEA pour donner un rendement de45%•495)La deuxième utilise la formation d’un ester pentafluorophényle sur la Pro5 en présence de DCC et de DMAP pour donner un rendement de 27%.51 La dernière méthode cyclise entre la Phe3 et la
MeVal2 avec du BOP/DMAP pour donner un rendement de 21%.52
Une autre méthode proposée qui est reportée dans un mémoire de maîtrise propose la préparation d’une série d’analogues de l’AbA (57)53Cette méthodologie utilise la chimie Fmoc et combine une synthèse sur support solide et en solution. Dans le cadre de ce travail, l’HOMeVal n’a pu être obtenue sous une forme énantiomériquement pute, elle a été remplacée par l’HOVal (Chapitre 2.5, Figure La macrocyclisation s’effectue sur le peptide linéaire déprotégé entre l’aIIo-lle6 et la Pro5 avec du PyBrop/DIEA avec un rendement maximale de 58%.
La synthèse de l’auréobasine présente certaines difficultés qui rendent les étapes de couplage plus difficiles s un lien ester (depsi) et la présence d’acides aminés N-méthylés. La préparation d’acides aminés non naturels comme l’aIIo-lle ou l’HOMeVaI (Figure 13) présente également un défi pour une synthèse effective. Avant le développement de notre propre méthodologie de synthèse, il n’existait aucune méthode de synthèse énantiosélective de la HOMeVal garantissant l’obtention d’un produit avec une pureté énantiomérique élevée (Chapitre 2.5). Actuellement, il n’est rapporté aucune méthodologie de synthèse de l’aurèobasidine effectuée entièrement sur support solide, Il serait intéressant de relever le défi et développer une telle stratégie efficace qui permettrait l’obtention rapide de toute une famille d’analogues. Ceci permettrait de faire une étude approfondie de la relation entre la structure et l’activité biologique (Chapitre 1.5.2, Figure 16).
1.8 Références du chapitre I
(1) ElIis, M. Molecular lmmunology 2001, 38, 947.
(2) McNeiI, M. M.; Nash, S. L.; Hajjeh, R. A.; Phelan, M. A.; Conn, L. A.; Plikaytis, B. D.; Warnock, D. W. Clinical lnfectious Diseases 2001, 33, 641.
(3) Walsh, T. J.; Groli, A. H. Transplant lnfectious Disease 1999, 1, 247. (4) Wade, J. C. Leukemia 1997, 11, S38.
(5) Odds, F. C.; Brown, A. J. P.; Gow, N. A. R. Trendsin Microbiology 2003, 11,272. (6) Bennett, J. In Harrisons principles of internai medicine; Braunwald, E. F., A.; Kasper, D.;
Hauser, S. et al., Ed.; McGraw-Hill: NewYork, 2001, pp 1168.
(7) Husaïn, S.; Alexander, B. D.; Munoz, P. e. a. Ciinical lnfectious Diseases 2003, 37, 221. (8) Deresinski, S. C.; Stevens, D. A. Reviews ofAnti-Infective Agents 2003, 36, 1445. (9) Barrett, D. Biochimica et Biophysica Acta 2002, 224.
(10) Maschmeyer, G. J. Antimicrob. Chemother. 2002, 49, 23.
(11) Wieland, T.; Bodanszky, M. The World of Peptides, a BriefHistrory of Peptide Chemistry; Sringer- Verlag: Berlin, Heidelberg., 1991.
(12) Percheron, F.; Perlès, R.; Foglietti, M. J. Biochimie structurale et métabolique; Masson: Paris, 1991.
(13) Stem, R. L. Adv. Protein Chem. 1993, 44, 1.
(14) Voet, J.; Voet, D. Biochimie; 2e édition eu.; Éditions de Boeck Université: Paris, 1998. (15) Pure Appl. Chem. 1974, 401, 291.
(16) Creighton, T. E. Proteins, New York 199. Freehan, W.H. & Co., 1993.
(17) Moore, M. L. “Peptide Design Consideration” in “Synthetic Peptides: A userss Guide”, Grant G.Freeman, W.H. and Compagny Eds, New YorK 1933, p.22.
(18) Linderstrôm-Lang, K. U.; Sheliman, J. A. Protein Structure and Enzyme Activity. The Enzymes Boyer; Lardy, Myrbaks; Eds., Academic Press, New York, 1959 Vol. 1, Second Ed.;pp443-510, 1959.
(19) Rose, G. D.; Gierarsch, L. M.; Smith, J. A. Adv. Protein Chem. 1985, 37, 1.
(20) Milner-White, E. J.; Ross, B. M.; Ismail, R.; Belhadj-Mostefa, K.; Poet, R. J. Mol. Biol. 1988, 204, 777.
(21) Venkatachalam, C. M. Biopolymers 1968, 6, 1425.
(22) Lewis, P. N.; Momany, F. A.; Scheraga, H. A. Biochim. Biophy. Acta 1973, 303, 211. (23) Patch, J. A.; Barron, A. E. Curr. Opin. Chem. Biol. 2002, 6, 872.
(24) Belley, N. R. A. Drug Discovery Today 2000, 5, 354.
(25) Giannis, A.; Kolter, T. Angew. Chem. Int. Ed. Engi. 1993, 32, 1244. (26) Ripka, A. S.; Rich, D. H. Curr. Opin. Chem. Biol. 1998, 2, 441. (27) Halab, L.; Gosselin, F.; Lubeil, W. D. Biopolymers 2000, 55, 101.