HAL Id: dumas-01911279
https://dumas.ccsd.cnrs.fr/dumas-01911279
Submitted on 2 Nov 2018HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Organisation de la traçabilité de la conformité aux
référentiels des dispositifs médicaux implantables hors
groupe homogène de séjour au CHU de Toulouse
Amandine Roussière
To cite this version:
Amandine Roussière. Organisation de la traçabilité de la conformité aux référentiels des dispositifs médicaux implantables hors groupe homogène de séjour au CHU de Toulouse. Sciences pharmaceu-tiques. 2018. �dumas-01911279�
Université de Bordeaux
U.F.R. DES SCIENCES PHARMACEUTIQUES
Année 2018 N°99
Thèse pour l’obtention du
DIPLOME d’ETAT de DOCTEUR EN PHARMACIE
Présentée et soutenue publiquement Par ROUSSIERE Amandine Née le 08 août 1990 à Castres
Le 01 octobre 2018 à Toulouse
ORGANISATION DE LA TRAÇABILITE DE LA CONFORMITE AUX
REFERENTIELS DES DISPOSITIFS MEDICAUX IMPLANTABLES HORS
GROUPE HOMOGENE DE SEJOUR AU CHU DE TOULOUSE
Directeur de thèse Mme la Dr. BELLON Brigitte
Jury
Mme la Pr. BREILH Dominique Président Mme la Dr. BELLON Brigitte
Mme la Dr. BONNEFOUS Monique
Mme la Dr. JUILLARD-CONDAT Blandine M le Dr. MOREAU Jacques
1
Université de Bordeaux
U.F.R. DES SCIENCES PHARMACEUTIQUES
Année 2018 N°99
Thèse pour l’obtention du
DIPLOME d’ETAT de DOCTEUR EN PHARMACIE
Présentée et soutenue publiquement Par ROUSSIERE Amandine Née le 08 août 1990 à Castres
Le 01 octobre 2018 à Toulouse
ORGANISATION DE LA TRAÇABILITE DE LA CONFORMITE AUX
REFERENTIELS DES DISPOSITIFS MEDICAUX IMPLANTABLES HORS
GROUPE HOMOGENE DE SEJOUR AU CHU DE TOULOUSE
Directeur de thèse Mme la Dr. BELLON Brigitte
Jury
Mme la Pr. BREILH Dominique Président Mme la Dr. BELLON Brigitte
Mme la Dr. BONNEFOUS Monique
Mme la Dr. JUILLARD-CONDAT Blandine M le Dr. MOREAU Jacques
2
REMERCIEMENTS
À Madame le Professeur Dominique BREILH,
Je suis très sensible à l’honneur que vous me faites de présider ma soutenance de thèse. Veuillez trouver ici le témoignage de toute ma considération.
À Madame le Docteur Brigitte BELLON,
Je vous suis extrêmement reconnaissante d’avoir accepté de m’accompagner tout au long de ce travail. Merci pour votre grande disponibilité et vos encouragements. Je suis honorée d’avoir pu travailler avec vous pendant ces dix-huit mois. Veuillez trouver ici toute ma reconnaissance.
À Madame le Docteur Monique Bonnefous,
Je tiens particulièrement à vous remercier de m’avoir épaulée dans ce travail. Merci également de m’avoir mis le pied à l’étrier dans le grand secteur des dispositifs médicaux, pour tout ce que vous m’avez appris et vos conseils avisés. Veuillez trouver ici l’expression de ma profonde gratitude.
À Madame le Docteur Blandine Juillard-Condat,
Je suis très honorée de vous compter parmi les membres du jury de cette thèse. Je vous en remercie d’autant plus que vous êtes fortement sollicitée. Veuillez trouver ici mes sincères remerciements.
À Monsieur le Docteur Jacques Moreau,
Je suis honorée que vous ayez accepté de siéger dans ce jury de thèse. Veuillez recevoir ici le témoignage de mon profond respect.
3 À Monsieur le Docteur Mathieu Tafani,
Je n’oublierai jamais votre tolérance ni la confiance que vous m’avez accordée. Veuillez trouver ici l’expression de tout mon respect et de ma profonde gratitude.
À ma famille,
À maman, pour avoir toujours cru en moi, pour m’avoir soutenue pendant ces longues années, pour tous tes conseils et pour tout ton amour.
À Tifanny, pour ton soutien indéfectible, pour tes conseils, pour ton amour et pour m’avoir toujours aidée à prendre du recul.
À papy, mamie et Philippe, pour vos encouragements, pour votre amour et pour me proposer une bulle hors du temps quand je viens vous voir.
À Nicolas, pour être entré dans ma vie, pour le réconfort que tu m’apportes et pour ton amour si précieux.
À Alain, pour vos conseils et pour votre soutien. À Laurent, pour ta gentillesse et pour ton aide. À papa, pour n’être jamais loin malgré la distance.
Veuillez tous trouver ici l’expression de tout l’amour que je vous porte et toute ma reconnaissance pour être toujours là pour moi.
Aux personnes qui m’ont accompagnée durant ces années d’études, À Fanny, ma binôme préférée, pour tous nos fous-rires.
À Marie-Noëlle, pour m’avoir pris sous ton aile alors que je n’étais qu’un « bébé-interne ». À Marion, pour cette dernière année d’internat, toutes les connaissances que tu m’as transmises et tous tes conseils.
Aux équipes des pharmacies de Vouneuil et Lacanau Océan, pour votre confiance. À l’équipe de la pharmacie de Henri Laborit à Poitiers, pour votre bonne humeur.
Aux équipes de l’Oncopôle-IUCT, de la DNAC et de la pharmacovigilance du CHU de Toulouse, pour tout ce que vous m’avez appris.
4
Aux équipes de l’OMEDIT Occitanie, de la COMEDIMS, des achats DM et de la matériovigilance du CHU de Toulouse, pour tout ce que vous m’avez appris et pour avoir rendu ma fin d’internat si enrichissante et si agréable.
Aux pharmaciens-familles du CHU de Toulouse, pour votre disponibilité dans le cadre du travail présenté ici.
5
TABLE DES MATIERES
Table des figures ... 10
Table des tableaux... 12
Liste des abréviations ... 13
Introduction ... 16
PARTIE 1 : LES DMI ET LA CONFORMITE AUX REFERENTIELS ... 18
I. Le domaine des dispositifs médicaux ... 19
A. Les dispositifs médicaux implantables ... 19
1. Définition d’un dispositif médical ... 19
Réglementation européenne ... 19
Réglementation française ... 20
2. Définition d’un dispositif médical implantable (DMI) ... 20
Réglementation européenne ... 20
Réglementation française ... 21
Définition d’un dispositif médical implantable actif (DMIA) ... 21
B. Les classes de risque des dispositifs médicaux ... 24
1. Les différentes classes de dispositifs médicaux ... 24
2. Les classes de risques des DMI ... 26
C. La liste des Produits et Prestations Remboursables (LPPR) ... 28
D. Les dispositifs médicaux Hors GHS ... 32
II. Les différents référentiels ... 35
A. Documentation technique fournie par le fabricant ... 35
1. Notice d’utilisation ... 35
6
B. Informations fournies par la CNEDIMTS ... 39
1. Rappels sur la composition et les missions de la CNEDIMTS ... 39
Composition de la CNEDIMTS ... 39
Missions de la CNEDIMTS ... 40
2. Avis de la CNEDIMTS ... 41
3. Fiche d’information thérapeutique ... 44
C. Informations fournies par les ministères ... 46
D. Informations fournies par les sociétés savantes et revues scientifiques ... 49
III. Conformité aux référentiels ... 54
A. Sécurité ... 54
1. Evaluation clinique ... 54
2. Le suivi clinique après commercialisation ... 56
3. Surveillance effectuée par l’ANSM ... 58
B. Pertinence des soins ... 60
1. Définition de la pertinence ... 60
2. Les définitions de la non pertinence ... 61
3. Evaluation par la CNEDIMTS... 62
C. Maitrise des depenses de santé ... 64
1. Le Contrat d’Amélioration de la Qualité et de l’Efficience des Soins ... 64
2. Evaluation des engagements souscrits dans le cadre du CAQES ... 65
PARTIE 2 : APPLICATION AU CHU DE TOULOUSE ... 71
I. Etat des lieux 2017 ... 72
A. Traçabilité sanitaire ... 72
7
2. Traçabilité informatisée ... 73
3. Résultats de la cartographie des risques 2017 sur le circuit des dispositifs médicaux 74 B. Traçabilité des indications ... 75
C. Rapport d’Etape Annuel (REA) CAQES 2017 ... 76
II. Contexte et pilotage du projet ... 79
A. Exigences du CAQES pour 2019, 2020 et 2021 ... 79
B. Pilotage du projet ... 81
1. Direction Générale ... 81
2. Pharmaciens ... 82
3. Médecins ... 82
III. Traçabilité des indications dans Copilote® ... 84
A. Visite de l’hôpital Foch ... 84
1. Organisation de la traçabilité des DMI à l’hôpital Foch ... 84
2. Transposition et ajustements nécessaires pour le CHU de Toulouse ... 85
B. Principe de fonctionnement du module DMI de Copilote® ... 87
1. Appairage des codes-barres ... 87
2. Relation entre Copilote® et les logiciels métiers ... 89
3. Traçabilité des indications dans Copilote®... 93
IV. Thésaurus et argumentaires ... 98
A. Référentiels de bon usage (RBU) ... 98
B. Constitution du thésaurus des indications ... 101
1. Méthode ... 102
Partie descriptive du dispositif ... 102
8
2. Résultats du thésaurus ... 109
3. Discussion ... 111
Niveau de l’indication de la LPPR ... 111
Découpage de l’indication LPPR ... 112
Dispositifs médicaux implantables associés ... 113
Limitation du nombre de DMI ... 113
Réunions de Concertation Pluridisciplinaires ... 114
Partage du thésaurus ... 114
C. Mise à jour du thésaurus et des RBU et contrôle du thésaurus des indications ... 116
1. Méthode ... 116
Les origines de la mise à jour ... 116
Les acteurs de ce processus de mise à jour ... 116
Contrôle de cohérence... 117
Formalisation d’une procédure de mise à jour ... 119
2. Résultats : le circuit de mise à jour des RBU et du Thésaurus ... 119
a) Mise à jour à partir du Journal Officiel ... 119
b) Mise à jour lors du référencement de nouveaux DMI hors GHS ou de leur déréférencement ... 120
3. Discussion ... 122
Délais de mise à jour du thésaurus et des RBU ... 122
Sources d’informations ... 124
Sécurisation de l’outil thésaurus ... 124
V. Mise en œuvre et calendrier... 126
CONCLUSION ... 130
9
Annexe 1 : Extrait en français de la notice d’utilisation du Sir-Spheres® (décembre 2013 – version PI-EC-11 - Sirtex Medical Limited) ... 136 Annexe 2 : Extrait de l’avis de la CNEDIMTS du 24/03/2015 pour le Sir-spheres®... 139 Annexe 3 : Specimen d’ordonnance de medicaments, de produits ou de prestations d'exception ... 148 Annexe 4 : Fiche d’Information Thérapeutique du Sir-spheres® publiée par l’arrêté du 14/02/2017 ... 149 Annexe 5 : Extrait de la LPPR : Nomenclature du Sir-spheres® du 16/08/2018 ... 151 Annexe 6 : Vademecum pour la prescription/implantation des dispositifs médicaux implantables (DMI) ... 152 Annexe 7 : Référentiel de Bon Usage DMI Hors-GHS pour les Implants exovasculaires pour fermeture de l’appendice auriculaire gauche (AAG) ... 154 Annexe 8 : Extrait du thésaurus du CHU de Toulouse du 30/08/2018 pour le code 3452694 (Sir-spheres®) ... 156 Annexe 9 : Procédure pour la veille du Journal Officiel et la mise à jour du thésaurus des indications des DMI hors-GHS ... 157
10
TABLE DES FIGURES
Figure 1 : Exemples de dispositifs médicaux de chaque classe de risque ... 25 Figure 2 : Exemple d’inscription sous description générique d’après la LPPR du 16/08/2018 30 Figure 3 : Exemple d'inscription sous nom de marque d’après la LPPR du 16/08/2018 ... 31 Figure 4 : Extrait de la notice d’utilisation du SIR-Spheres® - décembre 2013 – version PI-EC-11 - Sirtex Medical Limited ... 37 Figure 5 : Extrait de l’avis de la CNEDIMTS du 24/03/2015 pour le SIR-Spheres® (Evaluation du SA et de l’ASA) ... 42 Figure 6 : Extrait de l’avis de la CNEDIMTS du 24/03/2015 pour le SIR-Spheres® (Conditions de renouvellement d’inscription) ... 42 Figure 7 : Extrait de l’avis de la CNEDIMTS du 24/03/2015 pour le SIR-Spheres® (Modalités de prescription et d’utilisation) ... 43 Figure 8 : Extrait de la FIT du SIR-Spheres® publiée par l’arrêté du 14/02/2017 ... 44 Figure 9 : Extrait de la nomenclature de la LPPR du SIR-Spheres® du 16/08/2018 ... 48 Figure 10 : Résultats de la cartographie des risques 2017 au CHU de Toulouse pour le circuit des DMI ... 74 Figure 11 : Schéma du parcours des informations d'identification du DMI ... 88 Figure 12 : Schéma des interconnexions entre les logiciels métiers Opéra®, Copilote®, Magh2® et Orbis® ... 91 Figure 13 : Schéma des interconnexions entre les logiciels métiers Xplore®, Copilote®, Magh2® ... 91 Figure 14 : Schéma des interconnexions entre Hémolia® et Magh2® ... 92 Figure 15 : Aperçu de l'écran de saisie des indications du module DMI de Copilote®. Exemple du SIR-spheres®. ... 93 Figure 16 : Aperçu du menu déroulant du module DMI de Copilote® disponible lors de la saisie des indications. Exemple du SIR-spheres®. ... 94 Figure 17 : Aperçu de la fenêtre "Commentaire" du module DMI de Copilote® disponible lors de la saisie des indications ... 94
11
Figure 18 : Aperçu du menu déroulant des indications dans Copilote® pour un DMI avec plusieurs indications. Exemple d’endoprothèses vasculaires périphériques. ... 95 Figure 19 : Nomenclature de la LPPR du 30/08/2018 pour le code 3130016 (endoprothèses couvertes extensibles sur ballonnet) ... 107
12
TABLE DES TABLEAUX
Tableau 1 : Tableau comparatif des définitions de DM, DMI et DMI actifs dans les réglementations européenne et française ... 23 Tableau 2 : Description des titres de la LPPR (13) ... 29 Tableau 3 : Tableau représentant les différentes indications du SIR-spheres® en fonction du référentiel ... 53 Tableau 4 : Tableau comparatif des trois logiciels de traçabilité des DMI au CHU de Toulouse ... 75 Tableau 5 : Tableau détaillant les résultats du CAQES 2017 pour chaque famille de DMI concernés ... 77 Tableau 6 : Tableau comparatif des activités de chirurgie entre le CHU de Toulouse et l'hôpital Foch ... 85 Tableau 7 : Tableau récapitulatif des RBU disponibles au CHU de Toulouse au 08/09/2018 100 Tableau 8 : Extrait du thésaurus du CHU de Toulouse du 30/08/2018 pour le code 3452694 (SIR-spheres®) ... 103 Tableau 9 : Extrait du thésaurus du CHU de Toulouse du 30/08/2018 pour la partie descriptive du dispositif du code 3130016 (endoprothèses couvertes extensibles sur ballonnet) ... 103 Tableau 10 : Extrait du thésaurus du CHU de Toulouse du 30/08/2018 pour le code 3130016 (endoprothèses couvertes extensibles sur ballonnet) ... 108 Tableau 11 : Tableau représentant les résultats chiffrés du thésaurus du 14/08/2018 ... 110 Tableau 12 : Tableau représentant l'estimation de l'activité sur les modifications de codes LPPR au CHU de Toulouse ... 119 Tableau 13 : Calendrier de mise en place du projet DMI dans les services du CHU de Toulouse au 11/09/2018 ... 129
13
LISTE DES ABREVIATIONS
AM Assurance Maladie
ANAP Agence Nationale d’Appui à la Performance
ANSM Agence Nationale de Sécurité du Médicament et des produits de santé
ARS Agence Régionale de Santé
ASA Amélioration du Service Attendu
ASN Autorité de Sureté Nucléaire
ASR Amélioration du Service Rendu
ATIH Agence Technique de l’Information sur l’Hospitalisation
AVC Accident Vasculaire Cérébral
CAQES Contrat d’Amélioration de la Qualité et de l’Efficience des Soins CBUMPP Contrat de Bon Usage des Médicaments, Produits et Protestations CHU Centre Hospitalier Universitaire
CME Commission Médicale d’Etablissement
CNAM Caisse Nationale d'Assurance Maladie
CNEDIMTS Commission Nationale d’Evaluation des Dispositifs Médicaux et des Technologies de Santé
CNP Conseils Nationaux Professionnels
CNRTL Centre National de Ressources Textuelles et Lexicales
COMEDIMS Commission du Médicaments et des Dispositifs Médicaux Stériles
COPIL Comité de Pilotage
CRO Compte Rendu Opératoire
CSP Code de la Santé Publique
CSS Code de la Sécurité Sociale
DAI Défibrillateur Automatique Implantable
DG Direction Générale
DM Dispositif Médical
14
DSIO Direction du Système d’Information et de l’Organisation
ENEIS Enquête Nationale sur les Evènements Indésirables graves associés aux Soins
EIG Evènements Indésirables Graves
FMH Fédération des Médecins Suisses
GHM Groupe Homogène de Malades
GHS Groupe Homogène de Séjour
HAS Haute Autorité de Santé
I3LM Inflammation, Infection, Immunologie, Loco-Moteur IBODE Infirmier de Bloc Opératoire Diplômé d’Etat
IEP Identification Externe du Patient
IF Impact Factor
InVS Institut de Veille Sanitaire
IRM Imagerie par Résonnance Magnétique
ISFM Institut Suisse pour la Formation Médicale post graduée et continue ISI Institute for Scientific Information
IV Intra-Veineuse
FIT Fiche d’Information Thérapeutique
LPPR Liste des Produits et Prestations Remboursables
MCO Médecine-Chirurgie-Obstétrique
OMEDIT Observatoire du Médicament, des Dispositifs Médicaux et de l'Innovation Thérapeutique
OMS Organisation Mondiale de la Santé
PDF Format de Document Portable
PPH Préparateur en Pharmacie Hospitalière
PSUR Periodic Safety Update Report
PUI Pharmacie à Usage Intérieur
RCP Résumé des Caractéristiques du Produit RCP Réunion de Concertation Pluridisciplinaire
15
RSS Résumé de Sortie Standardisé
SA Service Attendu
SCAC Suivi Clinique Après Commercialisation
SR Service Rendu
T2A Tarification A l’Activité
UA Unité Administrative
UNCAM Union Nationale des Caisses d’Assurance Maladie
16
INTRODUCTION
Une des missions fondamentales des établissements de santé est d’assurer la qualité et la sécurité des soins. Les résultats d’études sur les événements indésirables associés aux soins montrent que des axes d’amélioration sont encore possibles et notamment dans le domaine des dispositifs médicaux.
En 2009, l’Enquête Nationale sur les Evènements Indésirables graves associés aux Soins (ENEIS) a mis en évidence qu’en moyenne 1,1 Evènement Indésirable Grave (EIG) évitable pour 1000 journées d’hospitalisation sur 2,6 sont liés aux produits de santé. De plus, sur 1,6% des séjours causés par des EIG évitables associés à des produits de santé, 0,2% concernent des dispositifs médicaux implantables (1).
Le bon usage des dispositifs médicaux implantables est donc un des axes d’amélioration de la qualité et de la sécurité des soins.
Plus particulièrement, le bon usage des dispositifs médicaux implantables (DMI) hors Groupe Homogène de Séjour (GHS) fait l’objet d’un suivi spécifique dans le cadre du Contrat d’Amélioration de la Qualité et de l’Efficience des Soins (CAQES) qui lie les établissements de santé aux Agences Régionales de Santé (ARS) et à l’Assurance Maladie. En effet, optimiser la pertinence des prescriptions de tels dispositifs innovants et/ou onéreux permettrait non seulement une amélioration de la qualité et de la sécurité des soins, mais aussi une meilleure maitrise des dépenses de santé.
Dans cet objectif d’amélioration des pratiques, les professionnels de santé ont à leur disposition des outils, appelés référentiels, afin d’assurer la pertinence des prescriptions des dispositifs médicaux implantables hors-GHS. La traçabilité de la conformité à ces référentiels est une exigence du CAQES.
17
Afin d’assurer le respect des référentiels, sa traçabilité et l’extraction des données de traçabilité demandées par le CAQES, les établissements de santé doivent disposer d’outils et d’une organisation adaptés.
En 2017, le Centre Hospitalier Universitaire (CHU) de Toulouse ne disposait pas de tels outils. La traçabilité de la conformité aux référentiels n’y était assurée que pour certains DMI hors-GHS. Or, nous devrons être capables d’ici 2021 de transmettre des données de traçabilité exhaustives dans le cadre du CAQES.
Dans un premier temps, après avoir posé le champ de notre travail, nous ferons un état des lieux des référentiels existants et expliquerons les raisons pour lesquelles le respect de ces référentiels est indispensable. Nous nous appuierons sur l’exemple d’un dispositif médical implantable hors-GHS : le SIR-spheres®. Les exigences réglementaires européennes décrites dans cette thèse sont issues du nouveau règlement européen (paru en 2017) qui sera applicable en 2020.
Dans un deuxième temps, nous présenterons la solution choisie par le CHU de Toulouse pour s’assurer du respect des référentiels des dispositifs médicaux implantables hors-GHS ainsi que de la traçabilité de cette conformité aux référentiels dans le cadre du CAQES.
18
PARTIE 1 : LES DMI ET LA CONFORMITE AUX
REFERENTIELS
19
I. LE DOMAINE DES DISPOSITIFS MEDICAUX
A. LES DISPOSITIFS MEDICAUX IMPLANTABLES
1. Définition d’un dispositif médical
Réglementation européenne
Dans le nouveau règlement européen, un dispositif médical (DM) est défini comme (2) : « tout instrument, appareil, équipement, logiciel, implant, réactif, matière ou autre article,
destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs des fins médicales précises suivantes:
— diagnostic, prévention, contrôle, prédiction, pronostic, traitement ou atténuation d'une maladie,
— diagnostic, contrôle, traitement, atténuation d'une blessure ou d'un handicap ou compensation de ceux-ci,
— investigation, remplacement ou modification d'une structure ou fonction anatomique ou d'un processus ou état physiologique ou pathologique,
— communication d'informations au moyen d'un examen in vitro d'échantillons provenant du corps humain, y compris les dons d'organes, de sang et de tissus,
et dont l'action principale voulue dans ou sur le corps humain n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens.
« Les produits ci-après sont également réputés être des dispositifs médicaux :
— les dispositifs destinés à la maîtrise de la conception ou à l'assistance à celle-ci,
— les produits spécifiquement destinés au nettoyage, à la désinfection ou à la stérilisation des dispositifs [médicaux et des accessoires] ».
20 Réglementation française
Le code de la santé publique, propose une définition semblable d’un dispositif médical. Cependant, à la différence du règlement européen, elle exclut explicitement les produits d’origine humaine du champ des dispositifs médicaux, mais y inclut les accessoires (3). En effet, le règlement européen précise que les accessoires ne sont pas considérés comme des dispositifs médicaux puisqu’il les définit ainsi (2) :
« tout article qui, sans être lui-même un dispositif médical, est destiné par son fabricant à être utilisé avec un ou plusieurs dispositifs médicaux donnés pour permettre une utilisation de ce ou ces derniers conforme à sa ou leur destination ou pour contribuer spécifiquement et directement à la fonction médicale du ou des dispositifs médicaux selon sa ou leur destination ».
Les dispositifs médicaux sont donc une catégorie hétérogène de produits qui comprend à la fois les stéthoscopes, les appareils à IRM et les implants cardiaques par exemple. Au sein de
cette diversité de produits, il existe un groupe de dispositifs médicaux caractérisés d’ « implantables », objet de cette thèse.
2. Définition d’un dispositif médical implantable (DMI)
Réglementation européenne
Un dispositif médical implantable (DMI) est défini ainsi dans la nouvelle réglementation européenne (2) :
« tout dispositif, y compris ceux qui sont absorbés en partie ou en totalité, destiné à être
introduit intégralement dans le corps humain, ou à remplacer une surface épithéliale ou la surface de l'œil, par une intervention clinique et à demeurer en place après l'intervention.
21
« Est également réputé être un dispositif implantable tout dispositif destiné à être introduit
partiellement dans le corps humain par une intervention clinique et à demeurer en place après l'intervention pendant une période d'au moins trente jours. »
Dans cette définition, trois notions sont donc à prendre en compte :
- le lieu d’implantation (en totalité ou en partie dans le corps humain, en remplacement d’une surface épithéliale ou de l’œil),
- la durée de l’implantation (au moins 30 jours s’il s’agit d’une implantation partielle), - les modalités de pose du dispositif (par une intervention clinique).
Le nouveau règlement européen élargit donc la définition des dispositifs médicaux implantables. En effet, la définition européenne actuellement applicable implique une pose lors d’une intervention chirurgicale (4). La nouvelle réglementation intègre dans le champ des DMI, les dispositifs médicaux implantés également au cours d’une intervention clinique.
Réglementation française
La définition d’un dispositif médical implantable disponible dans le code de la santé publique est plus succincte que celle du nouveau règlement européen. En effet, elle ne fait mention ni d’une durée d’implantation ni de modalité de pose. De plus, le lieu d’implantation qu’elle décrit n’inclut ni une surface épithéliale ni une surface de l’œil. La définition du code de la santé publique est la suivante (3) :
« Les dispositifs médicaux qui sont conçus pour être implantés en totalité ou en partie dans le corps humain ou placés dans un orifice naturel, […] sont dénommés dispositifs médicaux implantables […]. »
Définition d’un dispositif médical implantable actif (DMIA)
Les dispositifs implantables peuvent être dits « actifs ». C’est-à-dire qu’ils peuvent dépendre pour leur fonctionnement « d'une source d'énergie autre que celle générée par le
22
corps humain à cette fin ou par la pesanteur et agissant par modification de la densité de cette énergie ou par conversion de celle-ci » (2).
A titre d’exemple, un Défibrillateur Automatique Implantable (DAI) ou un implant cochléaire sont des dispositifs médicaux implantables actifs car ils ont besoin d’une batterie pour fonctionner. En revanche, les implants orthopédiques par exemple sont des dispositifs médicaux implantables non actifs car ils n’ont pas besoin d’énergie artificielle pour fonctionner.
Le tableau ci-dessous reprend les principales différences entre les définitions des réglementations européenne et française.
Règlement UE Code Santé Publique
DM « Tout instrument, appareil, équipement, logiciel, implant,
réactif, matière ou autre article,
destiné par le fabricant à être utilisé, seul ou en association, chez l'homme pour l'une ou plusieurs […] fins médicales précises […] ». (2)
« Tout instrument, appareil, équipement, matière, produit, à
l'exception des produits d'origine humaine, ou autre article utilisé seul ou
en association, y compris les accessoires et logiciels nécessaires au bon
fonctionnement de celui-ci, destiné par
le fabricant à être utilisé chez l'homme à des fins médicales et dont l'action
principale voulue n'est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tels moyens. Constitue
également un dispositif médical le logiciel destiné par le fabricant à être utilisé spécifiquement à des fins diagnostiques ou thérapeutiques. » (3)
DMI « Tout dispositif, y compris ceux qui
sont absorbés en partie ou en totalité, destiné à être introduit
intégralement dans le corps humain,
ou à remplacer une surface épithéliale ou la surface de l'œil, par
« Les dispositifs médicaux qui sont conçus pour être implantés en totalité ou en partie dans le corps humain ou
placés dans un orifice naturel, […] sont
dénommés dispositifs médicaux implantables […]. » (3)
23
une intervention clinique et à
demeurer en place après l'intervention.
Est également réputé être un dispositif implantable tout dispositif destiné à être introduit partiellement dans le corps humain par une
intervention clinique et à demeurer en place après l'intervention pendant une période d'au moins trente jours. » (2)
DMI actifs
DMI dont le fonctionnement dépend
« d'une source d'énergie autre que celle générée par le corps humain à cette fin ou par la pesanteur et
agissant par modification de la densité de cette énergie ou par conversion de celle-ci. » (2)
DMI « qui dépendent pour leur bon
fonctionnement d'une source d'énergie
électrique ou de toute source d'énergie
autre que celle qui est générée
directement par le corps humain ou la pesanteur ». (3)
24
B. LES CLASSES DE RISQUE DES DISPOSITIFS MEDICAUX
1. Les différentes classes de dispositifs médicaux
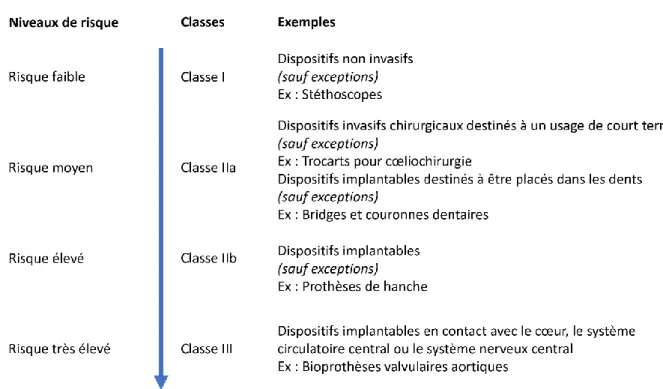
Les dispositifs médicaux sont répartis selon quatre classes de risque : I, IIa, IIb et III. La notion de risque prend en compte la probabilité de survenue d’un évènement indésirable ainsi que la gravité de cet évènement indésirable. (2) De façon générale, les dispositifs de classe I sont des dispositifs de faible risque tandis que les dispositifs de classe III sont des dispositifs qui présentent un risque très élevé.
La nouvelle règlementation européenne établit vingt-deux règles de classification des dispositifs médicaux. (2)
Ces règles prennent en compte : - la durée d’utilisation du dispositif.
o Un dispositif médical qui a une durée d’utilisation temporaire est utilisé pendant moins de soixante minutes.
o Un dispositif qui a une durée d’utilisation de court terme est utilisé en continu pendant soixante minutes à trente jours.
o Un dispositif médical qui a une durée d’utilisation de long terme est utilisé en continu pendant plus de trente jours.
- son caractère invasif ou non,
- les tissus avec lesquels il entre en contact. Notamment, les dispositifs qui sont en contact avec le système circulatoire central ou avec le système nerveux central sont généralement des dispositifs à risque très élevé (classe III).
25
Figure 1 : Exemples de dispositifs médicaux de chaque classe de risque
Ces classes de risque sont utilisées afin de définir les modalités d’évaluation de la conformité à la réglementation européenne pour un dispositif médical donné. En effet, plus le niveau de risque d’un dispositif est élevé, plus la conformité aux exigences européennes sera évaluée de façon « stricte ». Par exemple, la conformité d’un dispositif de classe I sera évaluée par le fabricant tandis que pour un dispositif de classe de risque plus élevée, la conformité sera évaluée par un Organisme Notifié indépendant. Nous reviendrons sur l’évaluation de la conformité à la réglementation européenne d’un dispositif médical plus loin dans cette thèse.
26 2. Les classes de risques des DMI
Les dispositifs médicaux implantables appartiennent aux classes IIa, IIb ou III.
En effet, de par leur nature, ce sont des dispositifs à risque moyen, élevé ou très élevé. Parmi les complications associées aux dispositifs médicaux implantables, peuvent être cités :
- le risque d’infection (5),
- le risque de complications mécaniques (sur la période 2010-2012, en France, 65% des déclarations d’évènements indésirables associés à des prothèses mammaires en silicone concernaient une rupture de prothèse) (6).
Lors de la mise en service en France de dispositifs médicaux de classe IIa, IIb, III, des informations permettant d’identifier ces dispositifs doivent être fournies à l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) (7)(8) :
- leur dénomination commerciale, - leur étiquetage,
- leur notice d’utilisation (sur laquelle figure le marquage CE) (2), - le nom et l’adresse de la personne qui effectue cette communication.
A l’occasion de cette communication, il doit également être précisé si la fabrication du dispositif fait intervenir un produit d’origine animale et l’espèce en question (8).
L’ensemble de ces informations alimente une liste tenue par l’ANSM qui à ce jour comprend plus de 54 000 références de dispositifs médicaux. Il ne s’agit pas d’une liste exhaustive des dispositifs médicaux de classe IIa, IIb, III commercialisés en France puisque l’inscription sur cette liste est effective depuis 2010 pour la classe IIa (9) et 2002 pour les autres catégories de dispositifs (10).
27
Par ailleurs, la traçabilité sanitaire des DMI est une obligation réglementaire (11) du fait des risques particuliers qui leurs sont associés. L’objectif de cette traçabilité est de pouvoir identifier à tout moment (12) :
« 1° Les patients pour lesquels les dispositifs médicaux d'un lot ont été utilisés ;
2° Les lots dont proviennent les dispositifs médicaux utilisés chez un patient. »
L’identification rapide des patients ou des lots de DMI concernés par un risque identifié permet de mettre en place rapidement des actions correctives et/ou préventives du risque.
Les dispositifs médicaux implantables sont des produits de santé pouvant être pris en charge par l’Assurance Maladie. Nous allons maintenant décrire ces modalités de prise en charge.
28
C. LA LISTE DES PRODUITS ET PRESTATIONS REMBOURSABLES (LPPR)
La LPPR est la Liste des Produits et Prestations qui peuvent être pris en charge par l’assurance maladie.
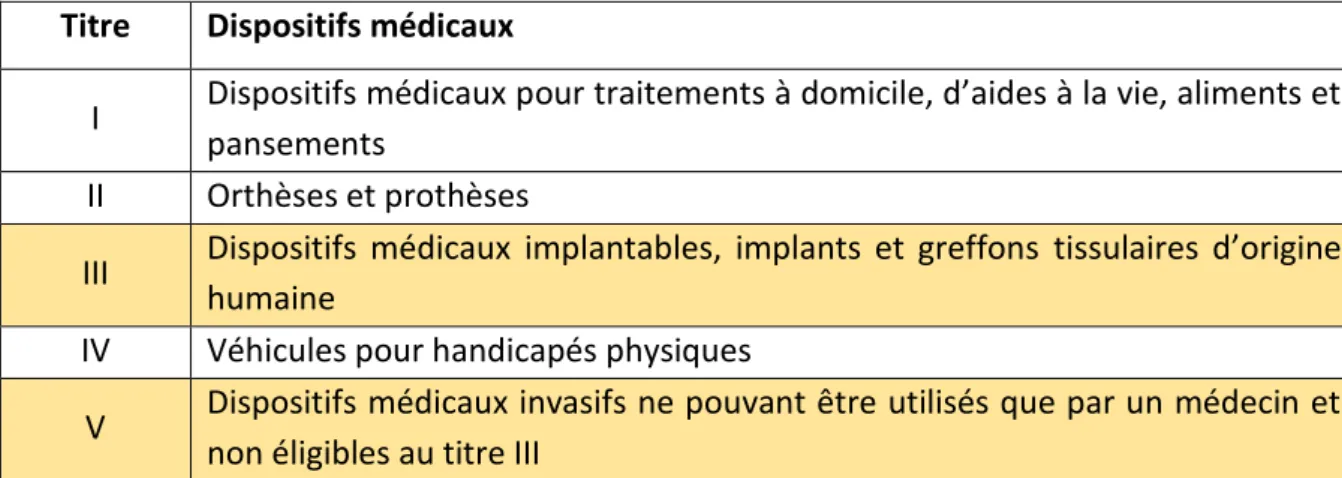
Elle est divisée en cinq titres distincts (13) :
- Le titre I correspond aux dispositifs médicaux utilisés pour les traitements à domicile, les dispositifs médicaux d’aides à la vie, les aliments et les pansements.
- Le titre II regroupe les orthèses et les prothèses.
- Le titre III correspond aux dispositifs médicaux implantables, aux implants et aux greffons tissulaires d’origine humaine. C’est celui sur lequel nous allons nous focaliser dans ce travail.
- Le titre IV regroupe les véhicules pour handicapés physiques.
- Enfin, le titre V, qui est le plus récent (il a été créé en 2017) (14), correspond aux dispositifs médicaux invasifs qui ne peuvent être utilisés que par un médecin et qui ne sont pas éligibles au titre III.
C’est le cas par exemple de stents Retriever (14) (15), destinés à la prise en charge des Accidents Vasculaires Cérébraux (AVC) ischémiques. Ces stents permettent de « capturer » le caillot et ainsi de le retirer. Ils ne sont pas des DMI puisqu’ils ne restent pas en place après l’intervention. Ils doivent être utilisés par une équipe comprenant au moins deux médecins (dans ce cas précis : un neuroradiologue interventionnel qualifié et un médecin anesthésiste réanimateur). Ils sont donc éligibles au titre V.
29 Titre Dispositifs médicaux
I Dispositifs médicaux pour traitements à domicile, d’aides à la vie, aliments et pansements
II Orthèses et prothèses
III Dispositifs médicaux implantables, implants et greffons tissulaires d’origine humaine
IV Véhicules pour handicapés physiques
V Dispositifs médicaux invasifs ne pouvant être utilisés que par un médecin et non éligibles au titre III
Tableau 2 : Description des titres de la LPPR (13)
L’inscription sur la LPPR peut être effectuée soit par description générique, soit par nom de marque. (16)
L’inscription sous description générique n’identifie pas le produit par son nom de marque mais à travers un groupe de produits qui ont tous les mêmes indications et les mêmes spécifications techniques minimales. Dans ce cas, l’inscription est sous la responsabilité du fabricant et le produit n’est pas évalué a priori par la commission de la Haute Autorité de Santé (HAS) appelée Commission Nationale d’Evaluation des Dispositifs Médicaux et des Technologies de Santé (CNEDIMTS). L’inscription d’un dispositif médical sous cette forme est valable pendant maximum dix ans renouvelables, à l’issue desquels la CNEDIMTS réévalue les descriptions génériques.
30
Pour certains dispositifs médicaux, l’inscription est réalisée sous nom de marque. Les dispositifs ainsi concernés peuvent par exemple :
- être des produits innovants (17),
- nécessiter un suivi particulier du fait de leur impact sur les dépenses de l’assurance maladie, d’impératifs de santé publique ou bien du contrôle des spécifications techniques minimales (17),
- être des produits uniques ne permettant pas de description générique (13),
Dans le cadre de cette inscription, le fabricant émet une demande de remboursement à la CNEDIMTS qui évalue son dossier et publie un avis. L’avis de la CNEDIMTS peut être favorable ou non à l’inscription du dispositif sur la LPPR. En cas d’avis favorable, l’inscription sous nom de marque peut être temporaire. En effet, dès que le dispositif est soumis à la concurrence, une inscription sous description générique est possible (13).
31
Figure 3 : Exemple d'inscription sous nom de marque d’après la LPPR du 16/08/2018
Depuis 2015, il existe un troisième mode d’inscription d’un dispositif médical sur la LPPR : la description générique renforcée. Comme pour la description générique, les produits inscrits selon ce mode doivent répondre aux spécifications techniques d’un groupe générique. Mais à la différence de la description générique, leur inscription sur la LPPR est soumise au dépôt d’une déclaration de conformité à ces spécifications techniques auprès de l’ANSM. Cette déclaration est établie par un Organisme Notifié. Leur inscription sur la LPPR permet l’identification individuelle des dispositifs au sein du groupe générique. (16) Les dispositifs qui doivent suivre ce mode d’inscription sont sélectionnés en fonction de (16) :
- leur intérêt pour la santé publique,
- leur impact sur les dépenses de santé de l’assurance maladie.
L’inscription sous description générique renforcée n’a pas encore été utilisée à ce jour (08/09/2018).
Les fabricants doivent déclarer à l’ANSM tous les dispositifs médicaux inscrits à la LPPR qu’ils commercialisent ainsi que toute modification des codes LPPR concernés. (18) L’ANSM peut effectuer un contrôle ou demander à un organisme d’effectuer un contrôle du respect des spécifications techniques qui conditionnent l’inscription sur la LPPR. Si une de ces spécifications techniques n’est pas respectée, le fabricant risque des pénalités. (19)
Nous allons maintenant nous arrêter sur les modalités de prise en charge d’un dispositif médical dans un établissement de santé.
32 D. LES DISPOSITIFS MEDICAUX HORS GHS
Depuis 2005, la Tarification À l’Activité (T2A) est la règle dans les établissements de santé publics. Pour cela, sont définis des GHM (Groupe Homogène de Malades) et des GHS (Groupe Homogène de Séjours).
Tout séjour hospitalier dans le secteur Médecine-Chirurgie-Obstétrique (MCO) d’un établissement de santé doit donner lieu à la production d’un Résumé de Sortie Standardisé (RSS) constitué (20) :
- d’informations administratives (âge et sexe du patient, durée du séjour…),
- d’informations médicales (diagnostic principal, diagnostics associés, actes médicaux …).
Le RSS est alors classé dans un Groupe Homogène de Malades (GHM). L’homogénéité est à la fois médicale et économique (20) :
- médicale car fondée sur des critères médicaux (diagnostic, actes médicaux …),
- économique car les séjours classés dans un même groupe ont des consommations de ressources voisines.
C’est-à-dire que des patients avec des caractéristiques pathologiques communes sont virtuellement regroupés dans un même ensemble. En effet, ces patients auront sensiblement besoin des mêmes soins. Par conséquent, le coût de leur séjour en établissement hospitalier sera sensiblement le même.
La valorisation en coût du GHM se traduit dans le Groupe Homogène de séjour (GHS) correspondant.
Le GHS est un tarif qui comprend tous les coûts nécessaires à la prise en charge du GHM auquel il est rattaché. Le plus souvent, un GHM est associé à un GHS.
33
Avec ce système de tarification, c’est donc l’activité de l’établissement qui définit le niveau des ressources qui lui sont attribuées.
Le droit commun prévoit que les dépenses liées à l’utilisation des dispositifs médicaux (DM) soient intégrées dans les GHS. Mais pour certains dispositifs médicaux, tels que des dispositifs médicaux implantables, qui peuvent être des dispositifs innovants, sur lesquels le recul est encore faible et qui sont souvent coûteux, cette tarification intra-GHS est impossible.
En effet, s’ils ne sont pas utilisés systématiquement dans le GHM, en tant que produits onéreux, ils déséquilibreraient les coûts de séjour qui ne seraient donc plus « homogènes ».
De plus, en tant que produits innovants ils nécessitent un suivi attentif de leur utilisation afin de garantir la qualité et la sécurité des soins.
Ainsi, dans un objectif de soutien et de diffusion de l’innovation dans les établissements de santé, ces dispositifs médicaux peuvent, après une évaluation appropriée par la CNEDIMTS, être inscrits sur la liste de facturation en sus des GHS. (21) Cela signifie que leur coût n’est pas inclus dans le GHS, mais qu’ils sont pris en charge sous certaines conditions en plus du GHS.
Ces conditions nécessitent l’engagement des établissements de santé sur (22) : - le bon usage des DMI hors-GHS, avec le respect des indications de la LPPR,
- l’argumentation du choix thérapeutique dans le dossier du patient en cas de prescription hors référentiel d’un DMI hors-GHS,
- la sécurisation du circuit des DMI qui implique une informatisation de ce circuit et la traçabilité sanitaire des DMI,
- une évaluation annuelle remise à l’ARS et à l’Assurance Maladie
Ces dispositifs pris en charge en sus des GHS sont inscrits sur la LPPR au titre III ou V.
Tous les DMI ne sont pas forcément inscrits sur la LPPR. Par exemple, les implants intraoculaires monofocaux utilisés dans le traitement chirurgical de la cataracte sont pris en
34
charge par l’assurance maladie dans le cadre d’un GHS et ne sont pas disponibles en ville : ils ne figurent donc pas sur la LPPR.
Les informations transmises à l’ARS et l’Assurance Maladie leur permettent d’évaluer le respect des engagements de l’établissement de santé à garantir le bon usage et la sécurisation de l’utilisation des DMI. Dans le cadre de cette évaluation, le respect de différents référentiels, notamment la LPPR, est pris en compte. Nous allons maintenant décrire ces différents référentiels.
35
II. LES DIFFERENTS REFERENTIELS
Les référentiels concernés par ce travail sont des sources d’informations mises à la disposition des professionnels de santé. Ils contiennent les indications et/ou les conditions d’utilisation des dispositifs médicaux et permettent ainsi d’assurer la qualité et la sécurité des soins. Ils sont créés et diffusés par différents acteurs du domaine des dispositifs médicaux en France et dans le monde (fabricants, agences nationales de santé, sociétés savantes). Comme nous allons le voir, la création de certains de ces référentiels est une obligation réglementaire, tandis que d’autres ont un rôle de diffusion des connaissances actuelles de la science.
A. DOCUMENTATION TECHNIQUE FOURNIE PAR LE FABRICANT
La documentation technique d’un dispositif médical est composée entre autres de la notice d’utilisation et du résumé des caractéristiques de sécurité et des performances cliniques.
1. Notice d’utilisation
La notice d’utilisation correspond aux (2) :
« indications fournies par le fabricant pour informer l'utilisateur de la destination et de la
bonne utilisation d'un dispositif et des précautions à prendre. »
La destination est déterminée pour un dispositif médical par son fabricant. La définition de ce terme dans le nouveau règlement européen reste évasive (2) :
« l'utilisation à laquelle un dispositif est destiné d'après les indications fournies par le fabricant sur l'étiquette, dans la notice d'utilisation ou dans les documents ou indications publicitaires ou de vente, et comme celles présentées par le fabricant dans l'évaluation clinique ».
36
La création d’une notice d’utilisation est obligatoire pour tous les dispositifs médicaux. Son rôle est d’informer l’utilisateur du dispositif médical pour lui permettre d’utiliser correctement celui-ci. Elle doit être adaptée aux connaissances techniques et au niveau de formation des utilisateurs auxquels le dispositif est destiné. (2)
La notice comprend les informations nécessaires pour l’identification du dispositif auquel elle se rapporte et des informations relatives à la sécurité et aux performances du produit. Doivent y figurer :
- la destination du dispositif,
- une description précise de ses indications et contre-indications, - les groupes cibles de patients et d’utilisateurs auxquels il est destiné.
Elle peut également contenir des liens vers le résumé des caractéristiques de sécurité et des performances que nous décrirons plus loin. (2)
Elle doit bien sûr être tenue à jour par le fabricant.
La notice d’utilisation est tenue à disposition des utilisateurs sous forme papier ou sous forme dématérialisée et dans leur langue officielle (2). Elle accompagne chaque dispositif auquel elle se réfère à l’exception des dispositifs de classe I et IIa (dispositifs médicaux de risque faible ou moyen) si ceux-ci peuvent être utilisés en toute sécurité même en l’absence de la notice. Elle peut être fournie en un seul exemplaire si elle accompagne plusieurs dispositifs médicaux, mais l’acheteur peut à tout moment demander à recevoir des exemplaires supplémentaires. Il est également possible de la trouver sur le site internet du fournisseur du dispositif médical en question. Enfin, le fabricant se doit de la fournir à l’utilisateur si celui-ci le demande. (2)
37
Figure 4 : Extrait de la notice d’utilisation du SIR-Spheres® - décembre 2013 – version PI-EC-11 - Sirtex Medical Limited
Notice d’utilisation du SIR-spheres® (Annexe 1)
Parmi les informations contenues dans la notice de SIR-sphere®, peuvent être cités : - la description du dispositif : « SIR-Spheres microsphères se compose de
microsphères biocompatibles de diamètre compris entre 20 et 60 microns et contenant de l’yttrium-90. […] Le parcours maximal de la particule [bêta] est de 11 mm dans les tissus avec un parcours moyen de 2,5 mm. […] Les microsphères de SIR-Spheres microsphères sont implantées dans les tumeurs hépatiques […]. La région tumorale reçoit une plus grande densité par unité de distribution de SIR-Spheres microsphères que le foie normal. »
- l’indication : « le traitement de patients souffrant d’un cancer hépatique avancé
non-opérable ».
- les groupes cibles de patients : « souffrant de tumeurs non-résécables […]La
sélection des patients […] nécessite un avis médical assurant que le traitement de la tumeur intra-hépatique sera bénéfique pour ces patients »
- les modalités de calcul de la dose - le mode d’emploi
- les contre-indications comme par exemple un antécédent de radiothérapie externe du foie ou, une ascite ou insuffisance hépatique clinique.
En plus de la notice d’utilisation, un résumé des caractéristiques de sécurité et des performances cliniques sera mis à la disposition des professionnels de santé par le fabricant.
2. Résumé des caractéristiques de sécurité et de performances cliniques
Seuls les dispositifs de classe III (dispositifs médicaux à très haut risque) et les dispositifs médicaux implantables sont concernés par ce document.
38
Le résumé des caractéristiques du dispositif a été prévu dans la réglementation française en 2016. (23) Il devait être l’équivalent pour les dispositifs médicaux du Résumé des Caractéristiques du Produit (RCP) pour les médicaments. Comme la notice d’utilisation, il devait contenir par exemple (24) :
- des informations sur la destination du dispositif et ses indications, - ses éventuelles contre-indications,
- les populations cibles de patients.
En plus de ces informations, il devait comprendre notamment (24) : - la place du DM dans la stratégie diagnostique ou thérapeutique, - le résumé de l’évaluation clinique du dispositif,
- les informations sur le suivi après commercialisation.
Le décret qui déterminait le contenu et les modalités de transmissions de ce résumé a finalement été annulé en 2018 (24), suite à la publication du nouveau règlement européen relatif aux dispositifs médicaux.
En effet, la nouvelle règlementation européenne de 2017 introduit à son tour l’existence d’un tel document. Il s’agit du résumé des caractéristiques de sécurité et performances cliniques dont l’existence ne sera obligatoire qu’à partir de 2020. Il doit être fourni à l’Organisme Notifié qui le valide. L’objectif de ce document étant d’améliorer la transparence des procédures de mise sur le marché d’un dispositif médical, il est mis à la disposition du public via Eudamed (la base de données Européenne sur les dispositifs médicaux). (2)
Le fabricant doit mentionner sur l’étiquette ou sur la notice d’utilisation où ce résumé est disponible. (2)
39
B. INFORMATIONS FOURNIES PAR LA CNEDIMTS
1. Rappels sur la composition et les missions de la CNEDIMTS
La Haute Autorité de Santé (HAS) est une instance nationale dont les missions sont multiples (25) :
- elle évalue les produits de santé en vue de leur remboursement,
- elle élabore des guides de bon usage ainsi que des recommandations de bonnes pratiques,
- elle apprécie la qualité de la prise en charge sanitaire de la population.
Pour ce faire elle s’appuie sur des commissions spécialisées (26) comme la CNEDIMTS.
Composition de la CNEDIMTS
La Commission Nationale d’Evaluation des Dispositifs Médicaux et des Technologies de Santé (CNEDIMTS) est la commission spécialisée de la HAS qui évalue les dispositifs médicaux et les technologies de santé. C’est une commission indépendante et multidisciplinaire. (27) Elle est composée de professionnels de santé, de représentants de patients mais aussi de représentants du ministère de la Santé, de l’ANSM et des trois principaux régimes de l’assurance maladie.
Elle a vingt membres titulaires (« de droit ») désignés pour leurs compétences techniques ou scientifiques dans le domaine des dispositifs médicaux dont au moins un infirmier et un auxiliaire médical. Deux autres membres titulaires sont choisis parmi les adhérents d’associations de malades et d’usagers du système de santé. Ces vingt-deux membres ont voix délibérative.
Cette commission a également sept membres suppléants (ou « invités »). Ces membres sont choisis comme les précédents. Ils assistent aux séances avec une voix consultative et peuvent être amenés à remplacer un membre titulaire.
40
Sept autres membres ont également une voix consultative lors des séances. Il s’agit des directeurs généraux de la santé, de l’offre de soin, de l’ANSM, mais aussi des directeurs de la sécurité sociale, de la Caisse Nationale d’Assurance Maladie (CNAM), de la caisse centrale de la mutualité sociale agricole et de l’Union Nationale des Caisses d’Assurance Maladie (UNCAM).
Enfin, le directeur général de l’agence de la biomédecine et le directeur central du service de santé des armées peuvent être amenés à participer aux travaux de la CNEDIMTS avec voix consultative.
Elle peut faire appel à des rapporteurs extérieurs à la Commission. Ces rapporteurs sont choisis le plus souvent sur proposition des Collèges ou sociétés savantes. Leur déclaration d’intérêt est vérifiée et validée par le comité de déontologie de la HAS.
La CNEDIMTS peut également interroger les Conseils Nationaux Professionnels (CNP) afin de connaitre leur positionnement en termes de recommandations et de pratiques professionnelles. (28)
En plus des représentants de patients et d’usagers du système de santé qui font partie de ses membres, la CNEDIMTS peut faire appel à des associations de patients et un processus de contribution spontanée des patients a été instauré. (28)
Enfin, toute personne qualifiée peut être entendue par la Commission. (27)
La CNEDIMTS est donc constituée de nombreux experts dans différentes disciplines afin de mener à bien ses missions.
Missions de la CNEDIMTS
La CNEDIMTS contribue à améliorer la qualité des pratiques professionnelles en formulant des recommandations dans le domaine des dispositifs médicaux et des technologies de santé. Elle détermine les conditions de bon usage et la place dans la stratégie
41
thérapeutique des dispositifs qu’elle évalue. Elle élabore ainsi des documents d’informations à destination des professionnels de santé. (29)
Elle évalue les dossiers de demande de remboursement de dispositifs médicaux soumis par les fabricants et rend des avis sur ces dossiers. Cette mission d’évaluation scientifique pour un dispositif médical intervient donc après que celui-ci ait obtenu le marquage CE. Son rôle n’est pas de valider ou d’invalider les indications que le fabricant a défini pour son dispositif médical, mais d’émettre un avis sur l’ouverture de certaines indications au remboursement par la sécurité sociale. Cet avis consultatif est transmis aux autorités de santé. (28)
2. Avis de la CNEDIMTS
Après avoir évalué les dossiers de demande de remboursement des dispositifs médicaux, la CNEDIMTS rend un avis qui est publié sur le site de la HAS.
Les avis de la CNEDIMTS proposent une évaluation complète de l’intérêt d’un dispositif médical en prenant en compte notamment :
- les études cliniques
- la place dans la stratégie thérapeutique - l’intérêt pour la santé publique
Les avis de la CNEDIMTS comportent l'appréciation du Service Attendu (SA) du dispositif évalué pour chaque indication et si besoin pour chaque groupe de population. Le SA mesure l’amélioration clinique attendue de l’état des patients. (30) Il peut être suffisant ou insuffisant pour justifier l'inscription au remboursement. (31)
Lorsque le service attendu est suffisant pour justifier l'inscription au remboursement, l’avis de la CNEDIMTS comporte également l'appréciation de l'Amélioration du Service Attendu (ASA) par rapport à un produit, une prestation ou un acte comparable, considérés comme références selon les données actuelles de la science. L'ASA peut être définie comme majeure,
42
Figure 5 : Extrait de l’avis de la CNEDIMTS du 24/03/2015 pour le SIR-Spheres® (Evaluation du SA et de l’ASA)
Figure 6 : Extrait de l’avis de la CNEDIMTS du 24/03/2015 pour le SIR-Spheres® (Conditions de renouvellement d’inscription)
importante, modérée, mineure ou absente et est évaluée pour chaque indication qui a un SA suffisant. (31)
Avis de la CNEDIMTS pour le SIR-spheres® (Annexe 2)
Pour le SIR-spheres®, la CNEDIMTS retient l’indication suivante : « Métastases
hépatiques du cancer colorectal en échappement thérapeutique ».
Elle évalue :
- le SA comme suffisant pour justifier l’inscription du SIR-sphere® au remboursement et propose que l’inscription soit effectuée sous nom de marque du dispositif, - l’ASA comme étant de niveau IV (c’est-à-dire mineure) (13) par rapport à un
traitement symptomatique adapté.
En cas de demande de renouvellement de l’inscription au remboursement d’un dispositif médical, l’évaluation de cette demande suit la même méthodologie qu’en cas de première inscription. La principale différence est la possibilité de prendre en compte les données récoltées depuis la première inscription et d’actualiser ainsi les informations figurant dans l’avis rendu lors de la primo-évaluation. Ce n’est alors plus le SA (Service Attendu) ni le ASA (Amélioration du Service Attendu) qui sont évalués pour chaque indication mais le SR (Service Rendu) et le ASR (Amélioration du Service Rendu). (32)
Avis de la CNEDIMTS pour le SIR-spheres® (Annexe 2)
Pour le SIR-spheres®, la CNEDIMTS propose que le renouvellement d’inscription sur la LPPR soit conditionné à la transmission d’un registre de recueil des données :
« La Commission subordonne le renouvellement d’inscription à la transmission des
résultats d’un registre portant sur l’ensemble des patients traités par SIRSPHERES. »
43
Figure 7 : Extrait de l’avis de la CNEDIMTS du 24/03/2015 pour le SIR-Spheres® (Modalités de prescription et d’utilisation)
Dans les avis de la CNEDIMTS, figurent également des recommandations sur les modalités de prescription et d'utilisation du dispositif.
Avis de la CNEDIMTS pour le SIR-spheres® (Annexe 2)
Pour le SIR-spheres®, la CNEDIMTS propose :
- Des modalités de prescription, par exemple : « la décision thérapeutique et du
suivi post-thérapeutique devra être prise en Réunion de Concertation Pluridisciplinaire »
- Des modalités d’utilisations, par exemple : « dans des centres disposant de
l’infrastructure suffisante pour être autorisé par l’Autorité du Sureté Nucléaire (ASN) à réaliser des activités de radiothérapie interne »
- Un groupe cible de patients pour lesquels un certain nombre de critères cliniques doivent être respectés à savoir : « les patients répondant à l’ensemble des critères
suivants :
o état général conservé [score ECOG (Eastern Cooperative Oncology Group)
≤ 2];
o absence d'envahissement tumoral hépatique important (<25%) ; o absence de localisation extra-hépatique ;
o réfractaires à l’ensemble des thérapeutiques [intra-veineuses] IV et orales
reconnues. »
Les avis de la CNEDIMTS peuvent parfois proposer de soumettre les produits à la procédure des produits d’exception, impliquant la création d’une fiche d’information thérapeutique. (31)
44
Figure 8 : Extrait de la FIT du SIR-Spheres® publiée par l’arrêté du 14/02/2017
3. Fiche d’information thérapeutique
Les dispositifs médicaux appartenant au groupe des produits d’exception sont des dispositifs particulièrement couteux pour lesquels sont spécifiées une ou plusieurs indications précises. Pour ces produits, l'inscription sur la LPPR peut être assortie d'une clause prévoyant qu'ils ne soient pris en charge que selon une procédure fixée par le ministère des affaires sociales et de la santé. (33)
Une Fiche d’Information Thérapeutique (FIT) établie par la CNEDIMTS accompagne alors l’arrêté d’inscription sur la LPPR du dispositif. Cette fiche mentionne notamment (33) :
- les indications prises en charge,
- les modalités de prescription et d'utilisation.
Elle peut également préciser les informations suivantes : - la place du dispositif dans la stratégie thérapeutique, - l’évaluation du SA et de l’ASA,
- des spécifications économiques et médico-sociales.
L’existence d’un tel document implique un engagement éclairé et volontaire du prescripteur. En effet, à chaque prescription de produits d’exception, le médecin doit confirmer qu’il respecte les indications de la FIT en cochant la case correspondante sur l’ordonnance des produits d’exception (Annexe 3).
Fiche d’informations thérapeutiques du SIR-spheres® (Annexe 4)
L’arrêté d’inscription sur la LPPR du dispositif implantable SIR-Spheres®, est accompagné d’une fiche d’informations thérapeutiques. Le SIR-Spheres® a obtenu le statut de produit d’exception car il s’agit d’un produit innovant, indiqué précisément dans le traitement des métastases hépatiques du cancer colorectal en échappement thérapeutique,et il s’agit par ailleurs d’un produit onéreux (12 660 €).
45
Les avis de la CNEDIMTS et les FIT sont transmis aux autorités de santé (ministère de la santé et chargé de la sécurité sociale), pour les aider dans leur travail d’élaboration de la LPPR. En effet, après son évaluation par la CNEDIMTS, l’inscription ou non d’un dispositif sur la LPPR est décidée par les ministres chargés de la santé et de la sécurité sociale. (33)
46
C. INFORMATIONS FOURNIES PAR LES MINISTERES
L’avis de la CNEDIMTS est transmis aux autorités de santé, constituées des ministères chargés de la santé et de la sécurité sociale. Les ministres correspondants s’appuient sur ces avis pour publier au Journal Officiel les arrêtés de modification de la LPPR. (33) Ces arrêtés peuvent concerner :
- une inscription de nouveaux codes LPPR, - une radiation de codes LPPR,
- une modification de la nomenclature, du tarif de prise en charge ou de la date de fin de prise en charge de codes LPPR.
Les référentiels de la LPPR sont les seuls référentiels opposables. Ils sont complexes de par la multitude d’éléments à prendre en compte. En effet, l’indication n’est pas le seul critère à respecter dans la nomenclature des dispositifs médicaux inscrits à la LPPR. De façon non exhaustive, celle-ci peut également préciser :
- des conditions de prescription et d’utilisation :
o par exemple : la nécessité d’une Réunion de Concertation Pluridisciplinaire avant de prendre la décision de prescrire un dispositif médical afin que les professionnels de santé les plus susceptibles d’intervenir dans le parcours de soins du patient puissent émettre un avis sur le choix de la thérapie compte tenu de leurs connaissances et leur expérience.
- une quantité maximale de dispositifs posés chez un même patient :
o pour des raisons financières (l’assurance maladie peut ne pas avoir les ressources suffisantes pour prendre en charge un plus grand nombre de ces dispositifs onéreux),
o pour des raisons de dose élevée de principe actif si le dispositif est imprégné de médicament,
o pour des raisons de stratégie thérapeutique (peut être vaut-il mieux changer de stratégie plutôt que d’accumuler les dispositifs à implanter).
47
- la qualification ou compétence des praticiens :
o certaines procédures sont très complexes et peuvent nécessiter non seulement une formation, mais aussi une pratique régulière afin que le chirurgien ait l’habitude de ces gestes.
- une liste restrictive des références prises en charge en cas d’inscription sous nom de marque,
- si la prise en charge concerne une primo-implantation ou un renouvellement,
- dans le cas d’un renouvellement, le temps qui doit s’être écoulé depuis la dernière implantation.
Nomenclature de la LPPR pour le SIR-sphères® (Annexe 5)
La nomenclature du SIR-Spheres® comporte :
- une description du dispositif : « SIR-SPHERES se compose de microsphères en
résine, biocompatibles, de diamètre compris entre 20 et 60 microns et contenant de l’yttrium-90. SIR-SPHERES est conditionné en flacon unitaire. »
- les indications de prise en charge : « Métastases hépatiques du cancer colorectal
en échappement thérapeutique. La progression sous chimiothérapie doit être documentée. »
- les caractéristiques des patients éligibles à cette indication : « Les patients
doivent répondre à l’ensemble des critères suivants :
o état général conservé [score ECOG (Eastern Cooperative Oncology Group)
≤ 2];
o absence d’envahissement tumoral hépatique important (< 25%) ; o absence de localisation extra-hépatique ;
o réfractaires ou intolérants à l’ensemble des thérapeutiques IV et orales
reconnues. »
- les qualités de l’équipe qui réalise l’implantation de ce dispositif : « Cette