Prédire l’impact des ensemencements à l’aide
d’outils génomiques : Le cas de l’omble de
fontaine (Salvelinus fontinalis, Mitchill) au
Québec
Mémoire
Justine Létourneau
Maîtrise en biologie
Maîtrise ès sciences (M. Sc.)
Québec, Canada
© Justine Létourneau, 2017
Prédire l’impact des ensemencements à l’aide d’outils
génomiques : Le cas de l’omble de fontaine (Salvelinus
fontinalis, Mitchill) au Québec
Mémoire
Justine Létourneau
Sous la direction de :
Louis Bernatchez, directeur de recherche
Dany Garant, codirecteur de recherche
iii
Résumé
L’Omble de fontaine est l’espèce la plus pêchée de façon récréative au Québec. Afin de supporter cette activité économique, des programmes intensifs d’ensemencement ont été mis en place et visent à maintenir la productivité des stocks. Cependant, de plus en plus d’études démontrent les effets négatifs des ensemencements sur la génétique des populations sauvages. Il est donc primordial de mieux comprendre la dynamique d’hybridation introgressive entre les populations sauvages et domestiques et ce à l’aide de plusieurs milliers de marqueurs génétiques. L’état génétique des populations suite à l’arrêt des ensemencements et les facteurs modulant les niveaux d’hybridation introgressive doivent également être mieux définis. Pour ces raisons, cette étude vise à tester et proposer un modèle capable d’expliquer et de prédire la variation observée dans les niveaux d’hybridation introgressive dans les populations sauvages d’omble de fontaine en utilisant des variables d’intensité d’ensemencement et des paramètres environnementaux. Ainsi, nous avons récolté des échantillons d’ADN (n = 862, moyenne = 30 poissons par lac) provenant de 29 lacs ayant subi différentes intensités d’ensemencement en plus de recueillir des données environnementales. En moyenne, 4 750 SNPs ont été obtenus pour chacun des lacs à l’aide du Génotypage-Par-Séquençage et ont été utilisés afin de déterminer l’appartenance moyenne à la population domestique. Un processus exhaustif de sélection de modèles a ensuite été réalisé pour obtenir un meilleur modèle permettant d’expliquer 56% de la variance observée entre les valeurs d’appartenance moyenne à la population domestique. Les prédictions basées sur notre meilleur modèle ont révélé que chaque population échantillonnée pouvait, potentiellement, retourner à un état génétique d’origine après l’arrêt des ensemencements. En somme, notre étude suggère que les impacts des ensemencements pourraient être réversibles avec le temps et propose un modèle aux gestionnaires de la faune permettant de prédire les niveaux d’hybridation entre populations sauvages et domestiques.
iv
Abstract
In fisheries management, intensive stocking programs are commonly used to enhance population abundance and maintain stock productivity. However, such practices are increasingly raising concerns since multiple studies exposed adverse stocking impacts on wild population genetic. Improvement of stocking management relies on a better understanding of the dynamic of introgressive hybridization between wild and domestic populations and on assessment of the genetic state of wild populations after stocking cessation. In Québec, Canada, five million captive reared Brook Charr (Salvelinus fontinalis) are stocked every year to support recreational fishing activities. Here, we investigated the impact of stocking history and environmental conditions on the genetic integrity of Brook charr wild populations. We collected DNA samples (n = 862, average of 30 individuals per lake) from 29 lakes that underwent different stocking intensities through time and also collected environmental parameters for each sampled lake. An average of 4,750 high quality filtered SNPs was obtained for each lake using Genotyping-By-Sequencing which were then used to quantify the mean domestic membership of each sampled population. An exhaustive process of model selection was conducted to obtain a best-fitted model that explained 56% of the variance observed in mean domestic genetic membership. The number of years since the mean year of stocking was the best explanatory variable to predict variation in mean domestic genetic membership. Our model predictions also revealed that each sampled wild population could potentially return to a wild genetic state (absence of domestic genetic background) after stocking cessation. Overall, our study provides new insights on factors determining introgressive hybridization level and suggests that stocking impacts could be reversible with time.
v
Table des matières
RÉSUMÉ ... III ABSTRACT ... IV TABLE DES MATIÈRES... V LISTE DES TABLEAUX ... VII LISTE DES FIGURES ... VIII REMERCIEMENTS ... IX AVANT-PROPOS ... XI
INTRODUCTION ... 1
PROBLÉMATIQUE GÉNÉRALE ... 1
CONSERVATION GÉNÉTIQUE ... 2
IMPACTS GÉNÉTIQUES DES ENSEMENCEMENTS SUR LES POPULATIONS SAUVAGES ... 4
Hybridation et introgression ... 5
Cas des salmonidés ... 7
Facteurs influençant l’hybridation introgressive ... 8
ÉTUDE DE LA GÉNOMIQUE DES POPULATIONS ... 9
HISTOIRE DES ENSEMENCEMENTS AU QUÉBEC ... 11
OMBLE DE FONTAINE ... 12
CONTEXTE DU PROJET ... 14
OBJECTIFS ... 14
CHAPTER I: PREDICTING THE GENETIC IMPACT OF STOCKING IN BROOK CHARR (SALVELINUS FONTINALIS) BY COMBINING RAD SEQUENCING AND MODELING OF EXPLANATORY VARIABLES ... 16
RÉSUMÉ ... 17
ABSTRACT ... 18
INTRODUCTION ... 19
MATERIAL AND METHODS ... 22
Sampling strategy ... 22
Environmental and stocking intensity data ... 25
DNA extraction and sequencing ... 27
Estimation of the admixture proportions ... 28
Models construction ... 29
vi
Resilience capacity after stocking cessation ... 30
RESULTS ... 31
DNA sequencing and genotyping ... 31
Estimation of the domestic genetic membership ... 32
Selection of the best-fitted model ... 34
Composition of the most plausible models... 34
Domestic genetic membership prediction after stocking cessation ... 39
DISCUSSION ... 40
Level of mean domestic genetic membership ... 41
Key variables explaining introgression levels ... 43
Environmental variables vs. introgression levels ... 44
Resilience potential after stocking cessation ... 45
Model improvement ... 46
APPLICATIONS FOR MANAGEMENT AND CONSERVATION ... 47
ACKNOWLEDGEMENTS ... 48
CONCLUSION ... 49
UTILISATION DE LA MÉTHODE DE GÉNOTYPAGE-PAR-SÉQUENÇAGE POUR DÉTERMINER LES NIVEAUX D’HYBRIDATION INTROGRESSIVE ... 50
DÉTERMINANTS DES NIVEAUX D’HYBRIDATION INTROGRESSIVE ... 52
ÉTAT DES POPULATIONS APRÈS L’ARRÊT DES ENSEMENCEMENTS ... 53
APPLICATIONS ET PERSPECTIVES FUTURES POUR LA GESTION ET LA CONSERVATION .. 55
BIBLIOGRAPHIE ... 56
vii
Liste des tableaux
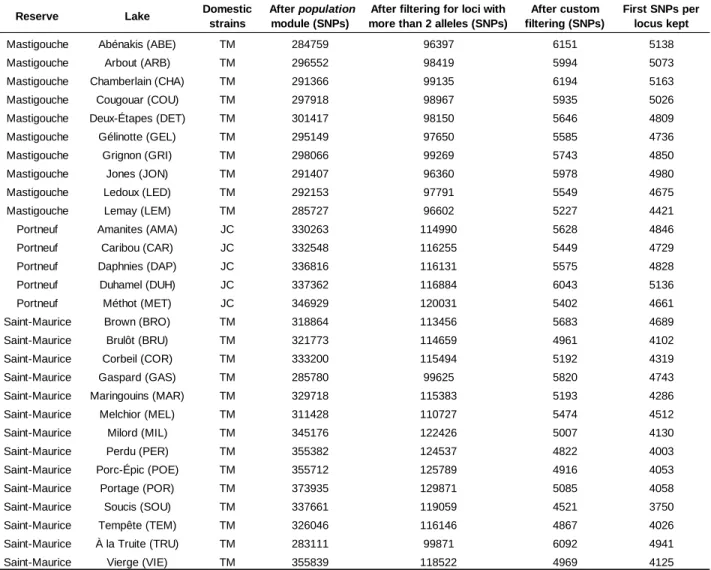
Table 1. Information on the 29 selected lakes and their associated domestic strains
included in this study on brook charr in Québec, Canada. Domestic strains TM (Truites de la Mauricie) and JC (Pisciculture Jacques-Cartier) represents the main source of the brook charr stocked in the selected lakes. The q-domestic values represent the mean membership to domestic strains for each population obtained with the software ADMIXTURE. ... 24
Table 2. Description of the environmental and stocking intensity variables used to
build models in this study on brook charr in Québec, Canada... 25
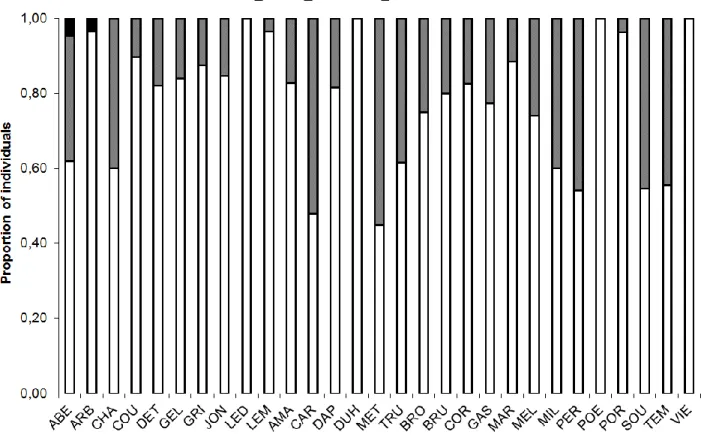
Table 3. Number of SNPs remaining after filtration steps for the 29 populations of
brook charr paired with their associated domestic strain used in this study taking place in Québec, Canada. ... 31
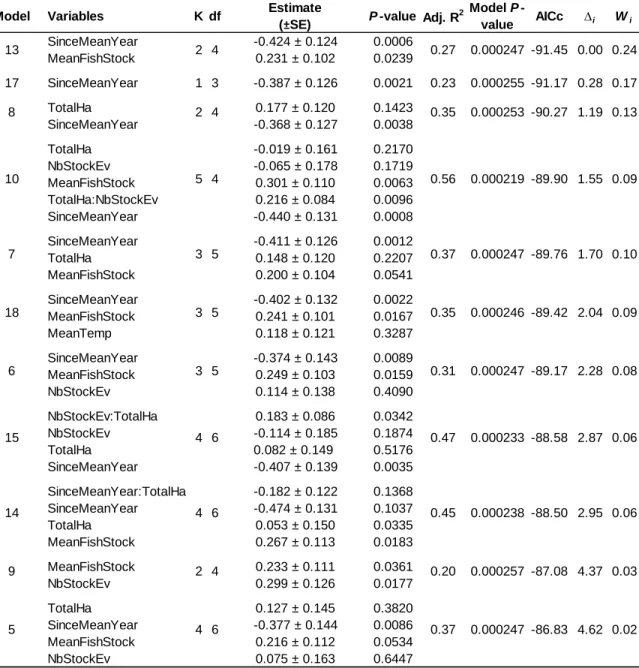
Table 4. Beta regression models built with stocking intensity variables and
environmental factors to explain the observed values of domestic membership in this study on brook charr, in Québec, Canada. The number of parameters in the models (K) include the intercepts and df represents the number of degree of freedom. ∆i corresponds to the AICc delta and models within 2 ∆i units of the best-fitted model (∆i = 0.00) are the most plausible. Wi is the AICc weight of each model. The values of the estimate are based on the centered and standardized values of the parameters. Models are classified from the smallest to the biggest values of AICc. The complete names of the variables presented here can be found in Table 2. ... 35
viii
Liste des figures
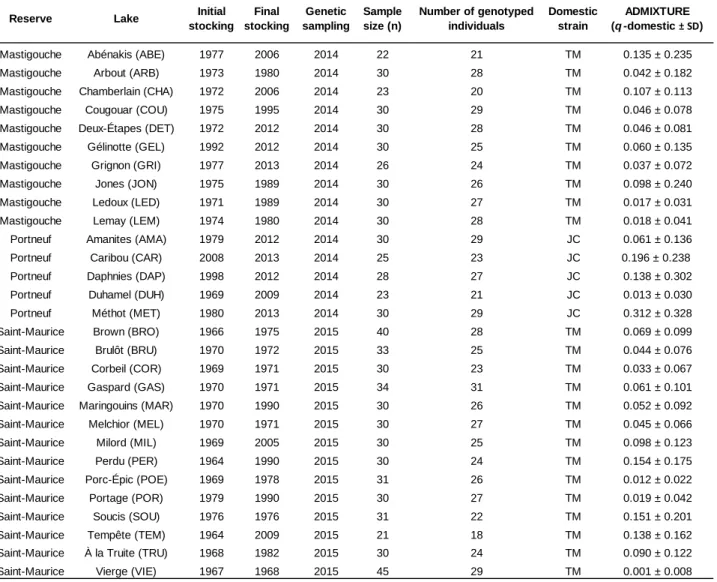
Figure 1. Geographical locations of sampled lakes in three wildlife reserves in the
province of Québec, Canada for this study on brook charr. ... 23
Figure 2. Proportion of individuals assigned to one of the three possible types (Wild: q-domestic ≤ 0.1, Admixed: 0.1 < q-domestic < 0.9 and Domestic: q-domestic ≥ 0.9)
for this study on brook charr, in Québec, Canada Complete names of the populations with the associated abbreviations can be found in Table 1. ... 32
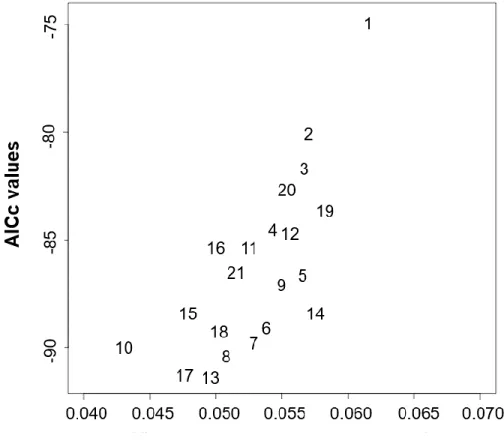
Figure 3. Values of AICc and mean difference between observed values of domestic
membership and values predicted by each model using a Jackknife approach for this study on brook charr in Québec, Canada. Every number corresponds to a model described in table 4. ... 34
Figure 4. Mean domestic membership observed in each lake as a function of the
values of mean domestic membership predicted by the three potentially best-fitted model for this study on brook charr in Québec, Canada. (A) Model 13 = SinceMeanYear + MeanFishStock, (B) Model 17 = SinceMeanYear, (C) Model 10 = SinceMeanYear + NbStockEv x TotalHa + MeanFishStock. Black dots each represent a sampled lake. Complete names and descriptions of the variables included in the models presented here can be found in Table 2. ... 37
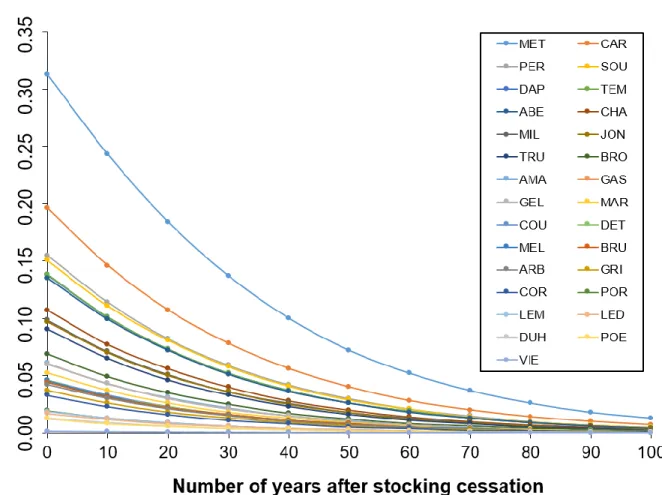
Figure 5. Mean domestic membership as a function of the number of years past
after stocking cessation. Values of mean domestic membership for the next 100 years were obtained using model 10 (SinceMeanYear + NbStockEv x TotalHa + MeanFishStock). At time 0, the values showed were obtained with ADMIXTURE for each sampled lake. Complete names of the populations showed here can be found in Table 1... 39
ix
Remerciements
Ayant commencé ma formation en biologie dès le niveau collégial en réalisant un programme technique en bioécologie, je vise depuis longtemps une carrière dans le domaine des sciences naturelles. C’est ce qui m’a poussé à poursuivre ma formation académique en m’inscrivant au baccalauréat en biologie à l’université Laval. Lors de ce parcours au premier cycle, le cours de génétique donné par le Dr. Alan Anderson m’a permis de découvrir une autre facette de la biologie qui m’a définitivement fascinée. J’ai donc eu envie de combiner ce nouvel intérêt pour le monde de l’ADN avec une autre passion que j’ai depuis longtemps, celle de la pêche. Il tombait alors sous le sens que je me devais de faire une maîtrise dans le laboratoire du Dr Louis Bernatchez, reconnu mondialement pour ces travaux sur la génomique de plusieurs espèces aquatiques.
Ainsi, j’aimerais tout d’abord remercier Louis qui m’a permis de m’épanouir et de me surpasser dans son laboratoire tout étant toujours à l’écoute des tracas et des soucis qui accompagnent le parcours d’une étudiante aux études graduées. Merci aussi à mon co-directeur, Dany Garant, qui a toujours été prêt à répondre à mes questions. Je souhaite également remercier tous les membres du laboratoire Bernatchez pour leur aide, plus particulièrement Anne-Laure Ferchaud (confidente et amie), Jérémy Le Luyer (maître zen des supers boucles) et Martin Laporte (expert de la sélection de modèles et de la rigolade) qui m’ont donné beaucoup de leur temps pour l’analyse et l’interprétation de mes données. J’aimerais également remercier Éric Normandeau pour le support en ce qui a trait à l’analyse des données de séquençage et Cécilia Hernandez pour l’aide au laboratoire. Également, un grand merci à tous ceux et celles qui m’auront donné un coup de main pour la réalisation de l’échantillonnage de ce projet sur le terrain, plus précisément Damien Boivin-Delisle, Guillaume Côté, Alysse Perreault-Payette et Philippine Gossieaux. La récolte de plus de 850 échantillons n’aurait pas été possible sans eux.
x
Une mention spéciale aux équipes de la Société d’Établissement de Plein Air du Québec (SÉPAQ) des réserves Mastigouche, Portneuf et Saint-Maurice et à Amélie Gilbert pour l’aide incroyable qu’ils ont fourni lors des campagnes d’échantillonnage. Merci également aux personnels du Ministère de la Faune, de la Forêt et des Parcs pour m’avoir fourni les bases de données nécessaires à la réalisation de cette étude (en particulier Isabel Thibault et Stéphanie Gagné).
Enfin, un grand merci à mes parents qui m’ont toujours supportée et qui croient en moi depuis le début de ce parcours. Ils m’ont donné tous les outils nécessaires pour affronter avec détermination les défis qui se sont mis sur mon chemin.
xi
Avant-propos
Ce mémoire est principalement composé d’un article intitulé « Predicting the genetic impact of stocking in brook Charr (Salvelinus fontinalis) by combining RAD sequencing and modeling of explanatory variables ». Cet article a été soumis à la revue « Evolutionary Applications » et est en attente de révision par les paires.
Les co-auteurs sont madame Anne-Laure Ferchaud et messieurs Jérémy Le Luyer, Martin Laporte, mon co-directeur Dany Garant et mon directeur Louis Bernatchez.
Anne-Laure Ferchaud et Jérémy Le Luyer ont pris part dans l’analyse et l’interprétation des résultats de séquençage, tandis que Martin Laporte a apporté son expertise au niveau de l’analyse de la sélection de modèle. Pour ma part, j’ai réalisé le travail sur le terrain et au laboratoire, rédigé l’article (auteure principale) et effectué l’analyse et l’interprétation de l’ensemble des données et des résultats avec l’aide de mes co-auteurs. Tous les co-auteurs ont également collaboré à la révision du manuscrit.
Ce projet a été financé par le Conseil de recherches en sciences naturelles et en génie (CRSNG), le Fonds de recherche du Québec – Nature et technologies (FRQNT) ainsi que par le regroupement Ressources Aquatiques Québec (RAQ).
1
Introduction
Problématique générale
Au cours du dernier siècle, les cas où les méthodes d’exploitation des ressources ont eu des effets néfastes sur des espèces animales et végétales se sont multipliés. Selon l’Union Internationale pour la Conservation de la Nature (UICN), la surexploitation des populations par la chasse, la trappe et le commerce d’animaux exotiques est responsable du statut de 14 % des espèces de Mammifères menacées et de 11% des espèces d’Oiseaux menacées (Rosser and Mainka 2001). De plus, il a été observé que l’exploitation commerciale et la chasse de subsistance ont causé l’extinction locale ou l’épuisement de plusieurs espèces animales (Rosser and Mainka 2001). De nombreuses espèces végétales récoltées pour l’alimentation, la médecine et la production de matériaux de construction souffrent également de ces méthodes de prélèvement intensif (Chivian and Bernstein 2008). Cependant, les cas les mieux documentés relatant les effets négatifs d’une gestion inadéquate des populations exploitées sont probablement reliés à l’industrie de la pêche. Présentement, l’exploitation commerciale et récréative des populations sauvages de plusieurs espèces aquatiques a atteint et même dépassé le seuil du rendement maximal durable. Plusieurs stocks montrent un déclin marqué de productivité principalement en raison de la surpêche et des conditions environnementales changeantes (Allan et al. 2005; Waples & Hendry 2008; Dunham 2011; Hoegh-guldberg & Bruno 2016).
En conséquence, dans le but de contrecarrer les problèmes de diminution de productivité induits par la pêche, des programmes de repeuplement (ensemencements) basés sur le relâchement d’individus élevés en captivé (domestiqués) ont été mis sur pied dans plusieurs régions du monde (Laikre & Ryman, 1996; Laikre et al. 2010). Ce type de solution vise à contrer les effets négatifs de la surexploitation en augmentant la taille absolue des stocks de poissons
2
pour en faire accroître la productivité (Ritter 1997; Laikre et al. 2010). Cependant, plusieurs études menées au cours des dernières années ont mis l’emphase sur les impacts potentiellement négatifs qu’ont les ensemencements à grande échelle sur la génétique des populations sauvages (Ryman & Laikre 1991; Laikre & Ryman 1996; Rhymer & Simberloff 1996; Fraser 2008; Araki et al. 2008; Laikre et al. 2010).
Conservation génétique
Au cours du siècle dernier, les gestionnaires de ressources naturelles ont déplacé et ensemencé de nombreuses populations de poissons sans égard aux conséquences possibles de telles actions sur la génétique des populations. C’est au début des années 1970 que le concept de conservation génétique a fait son apparition (Soulé 1986; Dunham 2011). Ce champ de recherche permet d’allier biologie moléculaire et évolutive afin de réduire le risque d’extinction des populations et de reconnaître les espèces en tant qu’entités dynamiques possédant le bagage nécessaire pour répondre et s’adapter aux changements environnementaux (Frankham et al., 2010). Ce bagage porté par les espèces est l’ensemble des variations présentes dans l’ADN de chaque individu et est directement relié à la diversité génétique (Toro & Caballero 2005; Frankham et al. 2010). Celle-ci constitue la base du potentiel évolutif.
Quatre grandes forces évolutives sont reconnues pour façonner la diversité au sein des espèces, soit les mutations, la dérive génétique, la sélection et la migration (Hamilton 2009; Frankham et al. 2010; Thomas et al. 2010). Les mutations correspondent à un changement dans le matériel génétique d’un individu. Il s’agit la plupart du temps d’un changement ponctuel au niveau d’un nucléotide. Lorsque ce phénomène se produit, on affirme alors qu’un nouvel allèle est apparu, créant ainsi un polymorphisme au locus correspondant. Les allèles désignent donc les différentes versions qu’un individu peut posséder pour une région génomique donnée. Toutefois, l’avenir de cet allèle est incertain puisqu’il peut disparaître
3
complètement si l’individu muté n’arrive pas à se reproduire et à le transmettre à la génération suivante. Cette perte peut également se produire dans les générations subséquentes. En contrepartie, l’allèle peut aussi éviter la disparition et se répandre dans la population, en particulier s’il présente un avantage évolutif (Hamilton 2009; Frankham et al. 2010; Thomas et al. 2010). La dérive génétique aura une influence sur la persistance ou la perte d’une mutation dans une population. Effectivement, celle-ci équivaut à un changement aléatoire de la fréquence allélique au sein des populations d’une génération à l’autre, en raison d’un nombre fini d’individus, de gamètes et ultimement d’allèles qui peuvent contribuer à la génération suivante. L’ampleur de la dérive génétique augmente à mesure que le nombre d’individus retrouvés dans les prochaines générations diminue. Donc, en absence de sélection ou de migration, à plus ou moins long terme dans un système, on finira par observer la fixation et la perte d’un allèle (Hamilton 2009). En plus, lorsqu’une mutation attribue un effet avantageux ou délétère à ses porteurs, le processus de sélection entre en jeu. La sélection peut, entre autres, avoir un effet directionnel sur la fréquence allélique, et peut mener à la fixation les mutations avantageuses et à l’élimination les mutations délétères. Enfin, la migration qui peut être décrite par le déplacement physique d’individus dans l’espace permet un flux génique entre des populations qui seront plus ou moins différenciées en fonction de l’intensité de cette force évolutive (Hamilton 2009; Frankham et al. 2010; Thomas et al. 2010). Ces processus évolutifs peuvent entraîner des différences morphologiques, physiologiques et comportementales entre les individus et les populations d’une même espèce. Ces dissimilitudes découlent de la variété d’allèles et de génotypes présents dans une population (Toro & Caballero 2005; Frankham et al. 2010). La maintien de l’intégrité de la diversité génétique est donc un objectif majeur pour la conservation des espèces puisque celle-ci est nécessaire pour permettre aux populations d’évoluer et de s’adapter, mais surtout de conserver l’association adaptative génotype-environnement local (Frankham et al. 2010).
4
Impacts génétiques des ensemencements sur les populations
sauvages
À la lumière de ces informations, il est plus aisé d’entrevoir l’importance des processus évolutifs et de la diversité génétique qui en découle pour la persistance des espèces dans le temps. Ainsi, même si les tentatives de repeuplement paraissent avoir à prime abord des impacts positifs sur la démographie des populations et favoriser des bénéfices sociaux et économiques, elles semblent aussi avoir des effets délétères sous-jacents. Ces tentatives de repeuplement pourraient ainsi influer de manière négative sur l’intégrité de la diversité génétique des populations sauvages (Ryman & Laikre 1991; Wang & Ryman 2001; Laikre et al. 2010; Marie et al. 2010). En effet, les repeuplements à grande échelle peuvent influencer la dynamique des populations naturelles à plusieurs niveaux.
Tout d’abord, il est possible d’entrevoir des conséquences n’impliquant pas nécessairement de flux génique, mais qui ont plutôt des effets indirects sur la perte de diversité génétique. On dénote notamment la transmission de maladies absentes auparavant dans l’environnement et l’augmentation de la compétition intraspécifique (Laikre & Ryman 1996; Laikre et al. 2010). À ce moment, ces effets collatéraux auront comme résultat de réduire la taille efficace de la population ce qui se solde au final en une perte de diversité génétique.
De plus, des effets influençant directement le flux génique entre les populations ont été observés au cours des dernières années chez des espèces faisant l’objet de programme de repeuplement (Laikre & Ryman 1996). Il est important de mentionner qu’une bonne partie de ces tentatives de rehaussement des populations consistent typiquement en la capture d’un petit nombre de géniteurs sauvages qui serviront par la suite à créer des lignées de poissons sur plusieurs générations qui seront élevées en captivité pour la reproduction. C’est ce qu’on appelle de l’élevage de soutien. En effet, il n’est pas raisonnable de penser qu’il est possible d’utiliser des géniteurs de
5
chacun des lacs ensemencés pour produire une progéniture indigène étant donné le trop grand nombre de populations existantes et ce en particulier au Québec.
Hybridation et introgression
Dans le cadre de cette étude, nous nous intéresserons plus particulièrement aux processus d’hybridation et d’introgression qui ont un rôle très important à jouer dans la dynamique des repeuplements à grande échelle (Allendorf et al. 2001; Araguas et al. 2004; Hansen et al. 2006; Eldridge & Naish 2007; Marie et al. 2010). Tout d’abord, l’hybridation peut être définie comme étant la reproduction d’individus provenant de populations ou de groupes de populations distinctes. Les individus de ces populations doivent être différenciés sur la base d’un ou de plusieurs traits héritables (Harrison 1993; Harrison & Larson 2014). En effet, il est vrai que l’hybridation réfère le plus souvent à la reproduction entre individus d’espèces différentes, mais ce terme peut aussi être utilisé pour décrire le croisement entre des populations de la même espèce qui divergent du point de vue génétique en raison de divers facteurs tels que l’isolement géographique ou un régime de sélection différent par exemple (Rhymer and Simberloff 1996). Ce phénomène peut également être accompagné par de l’introgression. On observera alors l’incorporation des allèles d’une entité (espèce ou population) à l’intérieur du bassin génique d’une seconde entité différente de la première (Allendorf et al. 2010). Ce flux de gènes se produira dans des populations où les hybrides réussissent à se reproduire avec des individus possédant le génotype des populations parentales. C’est ce qu’on appelle l’hybridation introgressive (Rhymer & Simberloff 1996; Frankham et al. 2010; Harrison & Larson 2014).
Même si l’hybridation est un processus naturel qui peut contribuer à la diversification et à l’adaptabilité des espèces animales (Mallet 2005) tout en jouant un rôle significatif dans le parcours évolutif de celles-ci (Dowling & Secor 1997; Allendorf et al. 2001), ce processus peut aussi mener à la perte de populations sauvages et même, éventuellement, à l’accélération de l’extinction d’espèces (Rhymer &
6
Simberloff 1996; Seehausen et al. 2008; Kelly et al. 2010; Gozlan et al. 2010; Andrews et al. 2016). Il semble que le mélange des patrimoines génétiques et la perte de populations génotypiquement différentiées puissent être délétères, notamment, pour les espèces plus rares en raison des tailles efficaces souvent plus faibles chez celles-ci créant une perte accentuée des adaptations locales (Rhymer and Simberloff 1996). Cependant, il est important de mentionner que dans certains cas, une population menacée d’extinction peut bénéficier d’une hybridation avec une autre population distincte puisque ce processus permettrait de restaurer la valeur sélective de la population en apportant de la diversité génétique. C’est ce qu’on appelle alors le sauvetage génétique (ou « genetic rescue ») (Whiteley et al. 2014).
Dans les cas où l’on procède au rehaussement des populations par les ensemencements, il semble qu’une certaine sélection survenant dans les élevages soit délétère puisque les traits qui sont avantageux dans un environnement domestique peuvent ne pas être avantageux dans le milieu naturel (Laikre & Ryman 1996; Ford 2002; Laikre et al. 2010; Dunham 2011; Christie et al. 2012). Ainsi, le relâchement d’individus qui ont une distribution de traits différente en raison de la sélection imposée en captivité peut résulter en la réduction de la valeur sélective de la population par l’hybridation introgressive. (Laikre & Ryman 1996; Ford 2002; Laikre et al. 2010). C’est ce qu’on appellera alors la dépression de croisement. La valeur sélective des générations F1 et/ou F2 plus particulièrementsera diminuée en
raison d’incompatibilités génétiques résultant d’un réarrangement des complexes de gènes coadaptés qui ne peuvent presque jamais être recréés (Ryman et al. 1995; Rhymer & Simberloff 1996; Laikre & Ryman 1996; Ford 2002; Tallmon et al. 2004; Edmands 2007; Allendorf et al. 2010; Dunham 2011) et/ou de la détérioration des adaptations locales (Mcginnity et al. 2003; Araki et al. 2007, 2008; Finnegan & Stevens 2008; Hansen et al. 2009).
7
Cas des Salmonidés
Plusieurs études démontrent d’ailleurs que les effets délétères reliés à l’élevage de soutien soulevés précédemment ont été observés chez différentes populations de salmonidés exploitées tant pour le commerce que pour les activités récréatives partout dans le monde (Leary et al. 1995; Dunham 2011). Une étude portant sur la Truite arc-en-ciel (Onchorhynchus mykiss) réalisée par Reisenbichler & McIntyre (1977) a montré que les truites sauvages provenant d’une rivière en Oregon (États-Unis) étaient génétiquement différentes des truites de pisciculture ensemencées et que leur croisement causait potentiellement une diminution notable du nombre de jeunes produits. Hansen et al. (2000), quant à eux, ont observé dans une étude impliquant trois populations de Truite brune (Salmo trutta L.) largement ensemencées, plusieurs indications suggérant la réduction significative de la taille efficace de deux de ces trois populations. De plus, une réduction de la différenciation génétique entre les populations (Eldridge and Naish 2007; Eldridge et al. 2009; Hansen et al. 2009; Marie et al. 2010; Lamaze et al. 2012; Perrier et al. 2013) et une perte de diversité génétique dans différentes populations sauvages ont été observées (Araguas et al., 2004; Eldridge & Naish, 2007; Hansen et al., 2006). Selon Araki et al. (2008), Finnegan & Stevens (2008) et Hansen et al. (2009) ces effets potentiels des ensemencements peuvent ultimement mener à la perte d’adaptation locale et à la réduction de la valeur sélective chez des populations sauvages de Salmonidés. Plus récemment, Ozerov et al. (2016) ont utilisé une approche ciblant le génome entier afin de mieux comprendre les impacts génétiques du flux génique entre les saumons atlantiques (Salmo salar) sauvages du Golfe de la Finlande et de leurs conspécifiques échappés des fermes d’aquaculture. Les auteurs ont conclu que l’introgression change la structure génétique des populations sauvages en augmentant leur diversité génétique, mais en diminuant la divergence génétique entre les populations. Ils observent aussi une hétérogénéité importante dans les patrons d’introgression sur l’ensemble du génome. Ils concluent que l’hybridation entre des populations auparavant séparées apporte une dynamique complexe et que les patrons d’introgression ne sont pas aléatoires et peuvent avoir des
8
conséquences fonctionnelles sur les populations indigènes. Ceci est également soutenu par Pritchard et al. (2016) qui dénote l’importance d’établir une liste de marqueurs diagnostiques permettant de distinguer des populations de saumon atlantiques sauvages des individus élevés en pisciculture afin de mieux investiguer les interactions génétiques entre ceux-ci.
Facteurs influençant l’hybridation introgressive
Par ailleurs, d’autres études ont également démontré que le niveau d’hybridation introgressive entre des populations sauvages et domestiques de salmonidés pouvaient être influencé par plusieurs facteurs. Entre autres, il a été documenté que l’intensité des ensemencements, représentée par le nombre de poissons ensemencés par hectare et/ ou le nombre d’évènements d’ensemencements, avait tendance à être corrélés positivement avec le niveau d’hybridation introgressive (Almodódovar et al. 2006; Finnegan & Stevens 2008; Hansen & Mensberg 2009; Marie et al. 2010; Lamaze et al. 2012). Par contre, la taille de la population sauvage (Hansen et al. 2009; Perrier et al. 2012) et le taux de survie de la population domestique (Araki et al. 2008) peuvent aussi influencer le niveau d’hybridation introgressive indépendamment de l’intensité des ensemencements. De plus, il a été suggéré que le temps passé depuis l’arrêt des ensemencements est aussi un facteur régulateur important (Harbicht et al. 2014; Valiquette et al. 2014). Il a été observé qu’après l’arrêt des ensemencements, l’appartenance génétique des populations ensemencées aux populations domestiques sources diminue avec le temps jusqu’à atteindre une valeur quasi nulle (Valiquette et al. 2014) et que ceci pouvait se produire au cours de 5 à 11 générations (Harbicht et al. 2014b). Ainsi, il est possible que la sélection en milieu naturel élimine des populations sauvages les variantes géniques provenant des populations domestiques.
Il a aussi été démontré que les paramètres abiotiques d’un milieu pouvaient jouer un rôle dans les niveaux d’hybridation introgressive entre les populations sauvages et domestiques. Une étude précédente menée par Marie et al. (2012) sur l’Omble
9
de fontaine (Salvelinus fontinalis) laisse croire que les niveaux d’hybridation pourraient être plus prononcés dans les petits lacs peu profonds et auraient tendance à augmenter avec la température de l’eau et le pH, mais à diminuer en fonction de la quantité d’oxygène dissout (Marie et al. 2012; mais voir aussi Harbicht et al. 2014a). Ces résultats suggèrent que des conditions moins favorables pour une espèce en milieu naturel pourraient faire augmenter les niveaux d’hybridation (Seehausen 2004).
Toutes ces informations démontrent donc qu’une connaissance du milieu et de l’intensité des ensemencements pourraient éventuellement aider à prédire les effets des repeuplements sur l’intégrité des populations sauvages d’Omble de fontaine.
Étude de la génomique des populations
Grâce aux avancées technologiques des dernières années, il est maintenant plus facile de comprendre les effets des ensemencements sur les populations sauvages. En effet, la génomique des populations permet l’étude d’un grand nombre de marqueurs génétiques ou de régions d’un génome afin d’améliorer la compréhension des processus évolutifs qui influencent la variation au sein des populations (Luikart et al. 2003). Les deux principes de base de la génomique des populations sont que les loci (emplacements sur le génome) neutres dans le génome seront similairement affectés par la démographie et l’histoire évolutive des populations et que les loci sous sélection se comporteront souvent différemment et révéleront des patrons de variations (Luikart et al. 2003). Ainsi, la génomique des populations facilite l’identification de variations adaptatives au niveau moléculaire et améliore les estimations de paramètres populationnels primordiaux tels que la taille efficace des populations et le taux de migration (Luikart et al. 2003; Allendorf et al. 2010). Les approches génomiques peuvent donc permettre une meilleure compréhension des bases génétiques de l’adaptation et de la dépression des
10
populations causée par la consanguinité. Ces informations peuvent ensuite être appliquées à des problèmes de conservation majeurs et être utilisées pour prédire les réponses évolutives des espèces face aux changements climatiques (Allendorf et al. 2010).
Également, grâce à l’avènement des technologies de séquençage de nouvelle génération, il existe maintenant plusieurs approches capables de découvrir, séquencer et génotyper des milliers de marqueurs moléculaires au sein de presque n’importe quel génome, même dans les populations où les informations génétiques sont inexistantes (Davey et al. 2011). Les marqueurs moléculaires utilisés par ces méthodes sont les polymorphismes nucléotidiques simples, communément appelés les SNPs. Ceux-ci correspondent à un changement d’un seul nucléotide à une position précise sur l’ADN (« Single-nucleotide polymorphism ») et se retrouvent en grande abondance dans le génome. Ils peuvent être présents autant dans les régions codantes que non-codantes de l’ADN. Ils sont donc d’une grande utilité puisqu’ils permettent une vision très élargie du génome (Hamilton 2009). Ces caractéristiques en font des marqueurs de prédilection pour mesurer le niveau d’hybridation et d’introgression dans une population (Allendorf et al. 2010).
En effet, en ce qui a trait à la détection de l’hybridation introgressive entre populations sauvages et domestiques, celle-ci peut être accomplie en utilisant un petit nombre de loci, mais une plus grande quantité de marqueurs est nécessaire pour évaluer la proportion d’hybridation à l’intérieur même d’un individu (Allendorf et al. 2010). Il a été suggéré que d’utiliser un nombre réduit de marqueurs génétiques peut être trompeur lorsque les populations étudiées qui s’hybrident sont fortement apparentées (comme dans le cas des ensemencements) puisque les hybrides de ces populations peuvent être difficiles à identifier (Vähä & Primmer 2006; Hansen & Mensberg 2009; Ozerov et al. 2016). De plus, des taux d’introgression différents en fonction de l’emplacement sur le génome des marqueurs génétiques étudiés ont été observés chez des espèces de salmonidés (Lamaze et al. 2012; Ozerov et al. 2016). Ces observations mettent donc en lumière les avantages potentiels d’utiliser un
11
grand nombre de marqueurs génétiques de type SNPs afin de mieux comprendre la dynamique de l’hybridation introgressive entre populations sauvages et domestiques.
Histoire des ensemencements au Québec
Au Québec, les ensemencements en eau douce ont débuté en 1857 avec l’élevage de jeunes saumons atlantiques (Salmo salar) et d’omble de fontaine (MAPAQ, 2012). Ces pratiques ont été mises en place dans le but de reconstituer les populations de certains lacs et rivières afin de favoriser l’industrie de la pêche sportive et compenser pour la diminution de la qualité des habitats et la réduction de la capacité de support des milieux naturels (MRNF, 2008). Les entreprises piscicoles privées ont ensuite fait leur apparition au cours des années 1950 et produisent actuellement une grande quantité des poissons ensemencés afin de répondre à la demande du marché de la pêche récréative (MRNF 2008 et MAPAQ 2012). Le Ministère de l’Agriculture, des Pêcheries et de l’Alimentation (MAPAQ) estime d’ailleurs que des 140 piscicultures détentrices d’un permis d’aquaculture, 80% de ces entreprises se consacrent entièrement à la production de poissons pour l’ensemencement. Ce qui représente en moyenne 800 tonnes de poissons produits par année. Environ 70% de cette production est composée d’Omble de fontaine et on estime que la Truite arc-en-ciel quant à elle, occupe près de 30% du marché. Certaines autres espèces de Salmonidés telles que l’Omble chevalier (Salvelinus
alpinus), la Truite brune et les hybrides représentent moins de 1% des parts des
ensemencements (Morin 2007 et MRNF 2008). En plus des entreprises piscicoles privées, il existe présentement au Québec trois stations piscicoles gouvernementales gérées par la Direction générale des pépinières et des stations piscicoles. La station piscicole de Tadoussac (région administrative de la Côte-Nord) produit presque exclusivement du Saumon atlantique servant à la conservation de l’espèce. La station de Baldwin-Mills (région administrative de l’Estrie) se spécialise également dans la production de poissons pour des fins de conservation. On y élève
12
principalement du Bar rayé (Morone saxatilis), du Chevalier cuivré (Moxostoma
hubbsi) et du Doré jaune (Sander vitreus). Enfin, la station du Lac-des-Écorces
(région administrative des Laurentides) oriente ses activités vers la production d’espèces telles que l’Omble de fontaine, la Truite arc-en-ciel et la Truite brune à des fins de mise en valeur de la pêche sportive (Morin 2007 et MRNF 2008).
Omble de fontaine
L’Omble de fontaine est une espèce de poissons de la famille des Salmonidés, plus précisément de la sous-famille des Salmoninés (Scott and Crossman 1973). Celle-ci est largement répartie dans l’est du Canada et fréquente les eaux fraîches, claires et bien oxygénées des ruisseaux, des rivières et des lacs. L’Omble de fontaine étant une espèce combative ayant une chaire de qualité, celle-ci est l’une des espèces les plus recherchées pour la pêche sportive. En effet, dans l’Est du Canada, la pêche récréative de l’Omble de fontaine crée des retombées économiques de 600 millions $ par année (Pêches et Océans Canada 2013). Celles-ci surpassent les profits générés par l’ensemble de toutes les autres espèces pêchées sportivement. Ainsi, au Québec, pour soutenir cette activité économique d’une grande importance, des ensemencements massifs sont entrepris depuis les années 1970. Le ministère de L’Agriculture, des Pêcheries et de l’Alimentation du Québec (MAPAQ) estime les ventes des stations piscicoles privées aux fins d’ensemencement à près de 654 tonnes d’Ombles de fontaine par an (Ministère du Développement Durable, de l'Environnement, de la Faune et des Parcs 2013). Les individus ensemencés peuvent provenir de plusieurs piscicultures différentes, par contre il est à noter que les lignées d’omble de fontaine domestiques présentes au Québec proviendraient d’une même origine et que celles-ci seraient génétiquement similaires entre elles (Martin et al. 1997). Donc, l’ensemencement intensif dont elle fait l’objet et son importance sur le plan économique dans l’Est du Canada font de l’Omble de fontaine une espèce cible pour l’étude des impacts des ensemencements.
13
En effet, l’étude de Marie et al. (2010; 2012) avait pour but général de déterminer l’impact de l’intensité des ensemencements et des facteurs environnementaux sur les populations d’Omble de fontaine au Québec. En plus d’observer les effets des ensemencements sur la structure génétique des populations sauvages, cette étude montrait également que les paramètres abiotiques du milieu pouvaient avoir un rôle à jouer sur les niveaux d’hybridation introgressive entre populations sauvages et domestiques. Les niveaux d’hybridation étaient plus élevés dans les petits lacs peu profonds et augmentaient avec la température et le pH, mais diminuaient avec l’augmentation de la quantité d’oxygène dissout. Ces résultats suggèrent que des conditions moins favorables pour l’Omble de fontaine pourraient favoriser l’hybridation. Cependant, cette étude, en raison de l’échantillonnage effectué, avait une couverture limitée du nombre d’années passées depuis l’arrêt des ensemencements et ne prenait pas en compte le possible effet du temps sur les niveaux d’hybridation. Cette étude ne visait également pas à bâtir un modèle pouvant être utilisé comme outil concret pour prédire l’impact génétique des ensemencements. Une étude de Harbicht et al. (2014a) réalisée sur des populations d’Omble de fontaine en Ontario a visé, de façon différente, à identifier des variables clés de la détermination des niveaux d’hybridation introgressive par l’utilisation de modèles, mais eux non plus n’ont pas fourni un outil utilisable par les gestionnaires qui servirait à prédire l’évolution des niveaux d’hybridation avec le temps. De plus, ces études étaient basées sur un petit nombre de marqueurs génétiques neutres rendant l’évaluation des niveaux d’hybridation potentiellement moins sure. Ainsi, il y a donc un besoin de fournir aux gestionnaires de la faune un modèle pouvant être utilisé facilement et permettant la prédiction des impacts des ensemencements pour les populations d’Omble de fontaine au Québec afin d’améliorer les stratégies d’ensemencement et de viser une exploitation plus durable de l’espèce.
14
Contexte du projet
Ainsi, le présent projet de maîtrise fait partie d’une étude de grande envergure qui a pour objectif général de recueillir de nouvelles connaissances qui serviront à long terme à maintenir les activités économiques reliées à l’Omble de fontaine. Le but est principalement de conserver l’intégrité génétique et écologique des populations sauvages tout en promouvant la mise en valeur du territoire en plus de la durabilité économique des industries récréo-touristiques et aquicoles qui dépendent de cette espèce. Cette étude inclut la participation active de deux organismes d’appui gouvernementaux qui sont directement concernés par les retombées de cette recherche soit le ministère des Forêts, de la Faune et des Parcs (MFFP) et la Société des établissements de plein air du Québec (SÉPAQ). En effet, au cours des prochaines années, le MFFP élaborera le premier plan de gestion de l’Omble de fontaine. Celui-ci devra être basé sur les meilleures informations scientifiques possibles et sur une conception juste des enjeux socio-économiques impliqués. Ainsi, de meilleures connaissances sur les impacts des ensemencements en termes de coûts et de bénéfices et sur les facteurs les modulant sont primordiales pour la mise en place de ce plan de gestion.
Objectifs
Dans le cadre de ce projet de maîtrise, il sera question de mettre en relation des paramètres environnementaux et des variables d’intensité d’ensemencement avec les niveaux d’hybridation introgressive de populations ensemencées d’Omble de fontaine du Québec afin de trouver le meilleur modèle pouvant prédire l’impact des ensemencements sur les populations. L’objectif final étant de fournir un outil utilisable par les gestionnaires de la faune, les buts de cette étude étaient 1) de déterminer les niveaux d’hybridation entre populations naturelles et domestiques grâce à des outils basés sur l’étude de plusieurs milliers de marqueurs répartis sur le génome, 2) de proposer et de tester un modèle plausible basé sur des données
15
d’historique d’ensemencement et des facteurs environnementaux afin d’expliquer les niveaux d’hybridation observés chez les populations échantillonnées et 3) d’utiliser ce même modèle pour évaluer les niveaux d’hybridation des populations d’Omble de fontaine après la cessation des ensemencements afin de prédire le nombre d’années nécessaires pour un retour à un état naturel. Cet outil permettra une gestion plus adéquate des populations d’omble de fontaine ensemencées dans un optique de développement durable de la ressource.
16
Chapter I: Predicting the genetic impact of stocking
in Brook Charr (Salvelinus fontinalis) by combining
RAD sequencing and modeling of explanatory
variables
Chapitre I: Prédire l’impact génétique des ensemencements chez
l’Omble de fontaine (Salvelinus fontinalis) en utilisant une approche
combinée de Séquençage-par-Génotypage et de modélisation de
variables explicatives.
17
Résumé
Dans l’industrie des pêcheries, les programmes d’ensemencements intensifs sont largement utilisés afin de rehausser l’abondance et la productivité des populations. Cependant, ce type de pratiques soulève désormais de nombreux questionnements puisque de multiples études ont révélé les effets néfastes des ensemencements sur la génétique des populations sauvages. L’amélioration de la gestion de ces ensemencements dépend d’une meilleure compréhension de la dynamique d’hybridation introgressive entre les populations sauvages et domestiques et de l’acquisition de connaissances sur l’état génétique des populations suite à l’arrêt des ensemencements. Au Québec, cinq millions d’Ombles de fontaine (Salvelinus
fontinalis) sont ensemencés chaque année pour supporter la pêche récréative. Ce
contexte particulier nous a permis d’étudier l’impact de l’historique des ensemencements et des facteurs environnementaux sur l’intégrité génétique des populations sauvages d’Omble de fontaine. Pour ce faire, nous avons récolté des échantillons d’ADN (n = 862, moyenne = 30 poissons par lac) provenant de 29 lacs ayant subi différentes intensités d’ensemencement en plus de recueillir des données environnementales. En moyenne, 4 750 SNPs ont été obtenus pour chacun des lacs à l’aide du Génotypage-Par-Séquençage et ont été utilisés afin de déterminer l’appartenance moyenne à la population domestique. Un processus exhaustif de sélection de modèles a ensuite été réalisé pour obtenir un meilleur modèle permettant d’expliquer 56% de la variance observée entre les valeurs d’appartenance moyenne à la population domestique. Le nombre d’années écoulées depuis l’année moyenne des ensemencements était la variable ayant le plus de poids dans la prédiction de la variation observée. Les prédictions de notre meilleur modèle ont aussi révélé que chaque population échantillonnée pouvait, potentiellement, retourner à un état génétique d’origine après l’arrêt des ensemencements. En somme, notre étude apporte de nouvelles informations sur les facteurs déterminants les niveaux d’hybridation introgressive et suggère que les impacts des ensemencements pourraient être réversibles avec le temps.
18
Abstract
In fisheries management, intensive stocking programs are commonly used to enhance population abundance and maintain stock productivity. However, such practices are increasingly raising concerns since multiple studies exposed adverse stocking impacts on wild population genetic. Improvement of stocking management relies on a better understanding of the dynamic of introgressive hybridization between wild and domestic population and on assessment of the genetic state of wild populations after stocking cessation. In Québec, Canada, five million captive reared brook Charr (Salvelinus fontinalis) are stocked every year to support recreational fishing activities. Here we investigated the impact of stocking history and environmental conditions on the genetic integrity of brook charr wild populations. To achieve this, we collected DNA samples (n = 862, average of 30 individuals per lake) from 29 lakes that underwent different stocking intensity through time and also collected environmental parameters for each sampled lake. An average of 4,750 high quality filtered SNPs was obtained for each lake using Genotyping-By-Sequencing which were then used to quantify the mean domestic membership of each sampled population. An exhaustive process of model selection was conducted to obtain a best-fitted model that explained 56% of the variance observed in mean domestic genetic membership. The number of years since the mean year of stocking was the best explanatory variable to predict variation in mean domestic genetic membership. Our model predictions also revealed that each sampled wild population could potentially return to a wild genetic state (absence of domestic genetic background) after stocking cessation. Overall, our study provides new insights on factors determining introgressive hybridization level and suggests that stocking impacts could be reversible with time.
19
Introduction
Commercial and recreational exploitation of many wild fish populations have reached and even exceeded the threshold for maximum sustainable yield (Dunham 2011). Many populations are showing important declines because of overfishing and changing environmental conditions (Allan et al. 2005; Dunham 2011; Hoegh-guldberg & Bruno 2016). As a result, supplementation (hereafter stocking) programs based on releases of captively reared (domesticated) fish are now used world-wide to counteract the negative effects of overexploitation by increasing the absolute size of fish stocks (NASCO 1992; Ritter 1997). Yet, numerous studies have documented the potentially negative effects of stocking on the genetic integrity of wild populations as well as on their evolutionary potential (Ryman & Laikre 1991; Rhymer & Simberloff 1996; Laikre & Ryman 1996; Fraser 2008; Araki et al. 2008; Laikre et al. 2010). Possible impacts of stocking include a significant decrease of wild populations effective size, due to the low number of reproducers used to perform supportive breeding (Ryman & Laikre 1991; Laikre & Ryman 1996; Hansen et al. 2000; Wang & Ryman 2001; Laikre et al. 2010), a loss of genetic diversity in stocked populations (Eldridge et al. 2009) and a loss of genetic differentiation between populations (Eldridge & Naish 2007; Eldridge et al. 2009; Hansen et al. 2009; Marie et al. 2010; Lamaze et al. 2012; Perrier et al. 2013).
The incorporation of alleles from a population into the gene pool of another genetically distinct populations, (e.g. introgressive hybridization), is another threat to the genetic integrity of stocked populations (Araguas et al. 2004; Hansen et al. 2006; Eldridge and Naish 2007; Marie et al. 2010). Indeed, although hybridization is a natural process sometimes contributing to diversification and adaptability (Dowling and Secor 1997; Allendorf et al. 2001), it can also lead to loss of genotypically different populations and accelerate extinction of species (Rhymer and Simberloff 1996; Seehausen et al. 2008; Kelly et al. 2010; Gozlan et al. 2010; Andrews et al. 2016). In the case of stocking, since domestic fish and their wild counterparts undergo drastically different selection regimes, captive individuals often tend to do
20
poorly in natural environment (Laikre & Ryman 1996; Ford 2002; Fraser 2008; Christie et al. 2012). Furthermore, in addition to domestication selection, genetic load due to inbreeding and relaxed sexual selection in captive stocks could also explain the lower fitness of domestic fish when released in the wild (Ford 2002; Mcginnity et al. 2003; Araki et al. 2007; Araki et al. 2008; Frankham et al. 2010; Christie et al. 2012). Therefore, reproduction between domestic and wild fish can result in the loss of local adaptation to the environmental conditions of wild populations (Mcginnity et al. 2003; Araki et al. 2007, 2008; Finnegan & Stevens 2008; Hansen et al. 2009) or the disruption of co-adapted genes complex through introgression (Laikre & Ryman 1996; Allendorf et al. 2001; Ford 2002; Tallmon et al. 2004; Edmands 2007; Laikre et al. 2010; Allendorf et al. 2010).
In salmonid fishes, several factors were also shown to affect introgressive hybridization between wild and domestic populations. Namely, it has been documented that admixture level tends to be correlated with stocking intensity variables such as the number of fish stocked par hectare and/or the number of stocking events (Almodódovar et al. 2006; Finnegan & Stevens 2008; Hansen & Mensberg 2009; Marie et al. 2010; Lamaze et al. 2012). However, the size of wild populations (Hansen et al. 2009; Perrier et al. 2012) and the survival and reproductive success of domestic fish could also influence admixture rates independently from stocking intensity. Also, it has been suggested that time spent following stocking events may be an important factor influencing admixture proportion (Hansen & Mensberg 2009; Hansen et al. 2009; Perrier et al. 2013; Valiquette et al. 2014; Harbicht et al. 2014b). For instance, after stocking cessation, the genetic background originating from the populations used for stocking tends to decrease with time and eventually almost disappears in lake Trout populations (Salvelinus namaycush) (Valiquette et al. 2014).
The detection of introgressive hybridization between two different populations can be accomplished using fewer loci, but many more markers are required to assess the proportion of admixture within individuals, as suggested by Allendorf et al.
21
(2010). Indeed, using a reduced number of markers can be misleading when hybridizing populations are closely related, since hybrids in those populations can be difficult to identify correctly (Vähä & Primmer 2006; Hansen & Mensberg 2009; Ozerov et al. 2016) . Furthermore, differential rates of introgression among parts of the genome have also been observed for some salmonid species (Lamaze et al. 2012; Ozerov et al. 2016). These observations emphasize the potential benefit of using a larger number of Single Nucleotide Polymorphisms (SNPs) towards better understanding the dynamics of introgressive hybridization.
The brook charr is a salmonid species native from Eastern North America. It is widely distributed in East Canada and populations are found in clear and well oxygenated water of rivers and lakes (Scott and Crossman 1973). It is a combative species strongly targeted by anglers. In Québec, Canada, brook charr recreational fishing supports an industry generating 600 millions$/year (Fisheries and Oceans Canada 2013). To support this economically important activity, intensive stocking programs have been conducted since 1970. Hence, every year, 654 tons of brook charr are stocked, which represents 70% of the annual production of Québec fish farming (Ministère du Développement Durable, de l'Environnement, de la Faune et des
Parcs 2013). Stocking history of several lakes has been recorded by governmental
institutions, thus providing an excellent context to study the influence of stocking intensity along with environmental variables on the extent of introgressive hybridization between wild and domestic populations. Indeed, a previous study on brook charr populations in Québec conducted by Marie et al. (2010;2012) aimed to assess the impact of stocking practices and environmental factors on hybridization level. In addition to observing the effect of intense stocking on the genetic structure of wild population (Marie et al. 2010), they also showed that abiotic parameters seemed to play a role in the admixture rates between wild and domestic populations (Marie et al. 2012). Hybridization was pronounced in smaller and shallower lakes and increased with water temperature and pH, but decreased with dissolve oxygen (Marie et al. 2012; but see Harbicht et al. 2014a). These results suggest that less favorable environmental conditions for the species could increase the hybridization
22
level. Nevertheless, this previous study had a limited time coverage of the number of years since the last stocking events and did not take into consideration the resilience capacity of wild populations. It also did not specifically aim to build a model able to predict the genetic impact of stocking using stocking intensity and environmental variables. Harbicht et al. (2014a) did aim to identify key variables determining hybridization in brook charr populations in Ontario by using model selection, but they did not provide a usable tool able to predict the level of hybridization trough time. Furthermore, both studies relied on relatively small sets of genetic markers. Thus, there is a need to provide wildlife managers with a concrete model predicting the impact of stocking in order to improve stocking strategies and to achieved a more sustainable exploitation of brook charr populations in Québec.
In this context, the purpose of this study was to provide a tool allowing the prediction of admixture proportion between wild and domestic populations of brook charr combining a set of variables describing the stocking history and environmental factors for each individual population being studied. We specifically aimed to (i) assess admixture proportion in stocked populations of brook charr using a genome-wide approach, (ii) test and define a best-fitted model able to explain observed variation in admixture proportions between wild and domestic populations sampled using stocking history and environmental variables and to (iii) investigate the resilience capacity of populations by determining the number of years needed for the populations to go back to a state of origin after the stocking cessation.
Material and methods
Sampling strategy
Sampling was conducted in three different wildlife reserves (e.g. Portneuf, Mastigouche and St-Maurice) in Québec, Canada which were created in 1971 and where fishing is strictly regulated and managed. Stocking was used in many lakes
23
at various intensities over time to support angling and to reduce fishing pressure on natural populations and the history of stockings has been well recorded since the creation of the reserves. We selected 29 lakes according to their different stocking histories representing a continuum of diverse stocking intensities based on: i) the stocking frequency, ii) the year of the last stocking event and iii) the quantity of stocked fish. Five lakes were sampled in Portneuf, ten in Mastigouche and fourteen in Saint-Maurice reserve, respectively (Figure 1). A total of 862 brook charr (from 21 to 45 individuals per lake, mean = 30) was sampled in summer (June to August) 2014 and 2015 using experimental gillnets with different mesh sizes (Table 1). The stocked brook charr used to supply the selected lakes came from different hatcheries: fish originated from Jacques-Cartier hatchery for the Portneuf reserve and Truites de la Mauricie aquaculture center for the Mastigouche and Saint-Maurice reserves. However, note that domestic strains are derived from the same origin and are therefore genetically similar (Martin et al. 1997). Fin tissues from 91 individuals were obtained from these two domestic strains (56 individuals and 35 individuals respectively for the Portneuf and Mastigouche/Saint-Maurice hatchery strains sources). All samples were preserved in ethanol 95% until DNA extraction.
24
Figure 1. Geographical locations of sampled lakes in three wildlife reserves
25
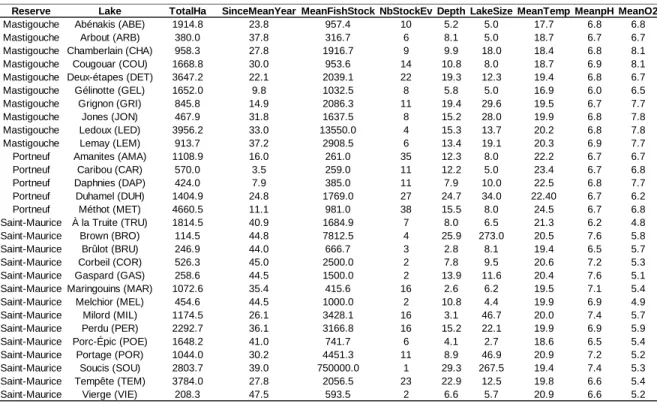
Table 1. Information on the 29 selected lakes and their associated domestic strains
included in this study on brook charr in Québec, Canada. Domestic strains TM (Truites
de la Mauricie) and JC (Pisciculture Jacques-Cartier) represents the main source of
the Brook Charr stocked in the selected lakes. The q-domestic values represent the mean membership to domestic strains for each population obtained with the software ADMIXTURE.
Environmental and stocking intensity data
Two types of variables were selected in this study: environmental and stocking intensity variables (see Table 2 for a detailed description and Table S1 in Supporting information for the parameters values for each lake). Firstly, the selection of five environmental parameters with a putative effect on admixture proportion was based
Reserve Lake Initial stocking Final stocking Genetic sampling Sample size (n) Number of genotyped individuals Domestic strain ADMIXTURE (q -domestic ± SD)
Mastigouche Abénakis (ABE) 1977 2006 2014 22 21 TM 0.135 ± 0.235
Mastigouche Arbout (ARB) 1973 1980 2014 30 28 TM 0.042 ± 0.182
Mastigouche Chamberlain (CHA) 1972 2006 2014 23 20 TM 0.107 ± 0.113
Mastigouche Cougouar (COU) 1975 1995 2014 30 29 TM 0.046 ± 0.078
Mastigouche Deux-Étapes (DET) 1972 2012 2014 30 28 TM 0.046 ± 0.081 Mastigouche Gélinotte (GEL) 1992 2012 2014 30 25 TM 0.060 ± 0.135
Mastigouche Grignon (GRI) 1977 2013 2014 26 24 TM 0.037 ± 0.072
Mastigouche Jones (JON) 1975 1989 2014 30 26 TM 0.098 ± 0.240
Mastigouche Ledoux (LED) 1971 1989 2014 30 27 TM 0.017 ± 0.031
Mastigouche Lemay (LEM) 1974 1980 2014 30 28 TM 0.018 ± 0.041
Portneuf Amanites (AMA) 1979 2012 2014 30 29 JC 0.061 ± 0.136
Portneuf Caribou (CAR) 2008 2013 2014 25 23 JC 0.196 ± 0.238
Portneuf Daphnies (DAP) 1998 2012 2014 28 27 JC 0.138 ± 0.302
Portneuf Duhamel (DUH) 1969 2009 2014 23 21 JC 0.013 ± 0.030
Portneuf Méthot (MET) 1980 2013 2014 30 29 JC 0.312 ± 0.328
Saint-Maurice Brown (BRO) 1966 1975 2015 40 28 TM 0.069 ± 0.099
Saint-Maurice Brulôt (BRU) 1970 1972 2015 33 25 TM 0.044 ± 0.076
Saint-Maurice Corbeil (COR) 1969 1971 2015 30 23 TM 0.033 ± 0.067 Saint-Maurice Gaspard (GAS) 1970 1971 2015 34 31 TM 0.061 ± 0.101 Saint-Maurice Maringouins (MAR) 1970 1990 2015 30 26 TM 0.052 ± 0.092 Saint-Maurice Melchior (MEL) 1970 1971 2015 30 27 TM 0.045 ± 0.066
Saint-Maurice Milord (MIL) 1969 2005 2015 30 25 TM 0.098 ± 0.123
Saint-Maurice Perdu (PER) 1964 1990 2015 30 24 TM 0.154 ± 0.175
Saint-Maurice Porc-Épic (POE) 1969 1978 2015 31 26 TM 0.012 ± 0.022 Saint-Maurice Portage (POR) 1979 1990 2015 30 27 TM 0.019 ± 0.042
Saint-Maurice Soucis (SOU) 1976 1976 2015 31 22 TM 0.151 ± 0.201
Saint-Maurice Tempête (TEM) 1964 2009 2015 21 18 TM 0.138 ± 0.162 Saint-Maurice À la Truite (TRU) 1968 1982 2015 30 24 TM 0.090 ± 0.122
26
on previous studies (Marie et al. 2012; Harbicht et al. 2014a) and on current knowledge of factors influencing physiological conditions of brook Charr (Power 1980; Warren et al. 2010). Thus, data for surface area (ha) and maximum depth (m) for the selected lakes were provided by the Ministère des Forêts, de la Faune et des
Parcs (MFFP). Temperature (o C), dissolved oxygen (mg.L-1) and pH were measured
at 1 m below the water surface at the deepest point of each lake. Temperature and dissolved oxygen data were collected with a multi-probe (Seabird, SBE 19plus SeaCat CTD Profiler) and the pH values were obtained using a phTestr 20 (Eutech Instruments). Physico-chemical parameters were measured twice before the breeding period (June and end of July) in summer 2014 or 2015 and were averaged for each lake, except for pH data for lakes sampled in 2015 that were collected only once during the summer. Secondly, our stocking intensity variables were all determined from data provided by the MFFP and the Société d’Établissement de
Plein Air du Québec (SEPAQ) and included: i) the number of stocking events, ii) the
quantity of stocked fish per stocking event, and iii) the mean number of fish stocked per stocking event (Table 2). We also included a time variable represented by the number of years since the mean year over all stocking events.
Table 2. Description of the environmental parameters and stocking intensity variables used to
27
DNA extraction and sequencing
Total DNA was extracted from adipose fin tissue (5 mm2) using a slightly modified
version of Aljanabi et al. (1997) salt extraction protocol. Sample concentration and quality were checked using 1% agarose gel and a NanoDrop 2000 spectrophotometer (Thermo Scientific). DNA quantification was completed using the Picogreen assay (Fluoroskan, Ascent FL, Thermo Labsystems). Genomic DNA was normalized to obtain 20 ng/µL in 10 µL (200 ng) for each individual. The libraries were created accordingly to Mascher et al. (2013) protocol. Namely, in each sample, a digest buffer (NEB4) and two restriction enzymes (PstI and MspI) were added. After a two hours incubation period at 37oC, enzymes were inactivated by a 20 min
incubation period at 65oC. Then, the ligation of two adaptors was performed using a
ligation master mix followed by the addition of T4 ligase and completed for each sample at 22oC for 2 hours. Enzymes were again inactivated by a 20 min. incubation
period at 65oC. Finally, samples were pooled in 48-plex and QIAquick PCR
purification kits was used to clean and purify the DNA. After library PCR amplification, sequencing was performed on the Ion Torrent Proton P1v2 chip. Subsequently, FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to check raw reads for overall quality and presence of adapters. All the bioinformatic steps, options and software versions employed in the subsequent GBS pipeline are detailed in Table S2 in Supporting Information. Briefly, we used cutadapt v1.8.1 (Martin 2011) to remove the adapter from raw sequences and STACKS v1.40
process_radtags to demultiplex the samples and do the quality trimming (Catchen et al. 2013). Sequence reads were aligned on the Rainbow Trout (Oncorhynchus mykiss) reference genome (Berthelot et al. 2014) with GSnap v9 (Wu and Nacu
2010). Then, pstacks was performed to extract the stacks aligned to the reference genome and to identify SNPs at each locus. Cstacks was used to build a reference catalog with all loci identified across all the individuals. Loci from each individual were then matched against the catalog to determine the allelic state in each individual (sstacks). Thereafter, the module populations was run independently for each lake with the domestic strain used for stocking. Hence, SNPs were defined and