THÈSE
Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées Institut de physiologie et biologie cellulaires - IPBC (Poitiers)
(Diplôme National - Arrêté du 7 août 2006) École doctorale : Biologie-santé - Bio-santé (Limoges)
Secteur de recherche : Aspects moléculaires et cellulaires de la biologie
Présentée par : Souheyla Bensalma
Voie Hedgehog et système VIP-récepteurs dans des cellules de glioblastome
Directeur(s) de Thèse :
Jean-Marc Muller, Corinne Chadeneau Soutenue le 12 décembre 2013 devant le jury
Jury :
Président Philippe Menei Professeur des Universités, Université d'Angers
Rapporteur Jean Mazella Directeur de recherche, Université de Nice-Sophia Antipolis Rapporteur Vincent Lelièvre Professeur des Universités, Université de Strasbourg Membre Jean-Marc Muller Professeur des Universités, Université de Poitiers Membre Corinne Chadeneau Maître de conférences, Université de Poitiers
Pour citer cette thèse :
Souheyla Bensalma. Voie Hedgehog et système VIP-récepteurs dans des cellules de glioblastome [En ligne]. Thèse Aspects moléculaires et cellulaires de la biologie. Poitiers : Université de Poitiers, 2013. Disponible sur Internet <http://theses.univ-poitiers.fr>
1
THESE
Pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
(Faculté des Sciences Fondamentales et Appliquées) (Diplôme National - Arrêté du 7 août 2006)
Ecole Doctorale : BioSanté
Secteur de Recherche : Aspects Moléculaires et Cellulaires de la Biologie
Présentée par :
Souheyla BENSALMA
************************
Voie Hedgehog et système VIP-récepteurs
dans des cellules de glioblastome
************************
Directeur de thèse : Professeur Jean-Marc MULLER Co-directeur de thèse : Docteur Corinne CHADENEAU
************************ Soutenue le 12 Décembre 2013 Devant la Commission d’Examen
************************
JURY
Vincent LELIEVRE, PU, Centre de Neurochimie, Université de Strasbourg ... Rapporteur Jean MAZELLA, DR CNRS, Université de Nice-Sophia Antipolis ... Rapporteur Corinne CHADENEAU, MCU, 2RCT, FRE 3511 CNRS, Université de Poitiers ... Examinatrice Philippe MENEI, PU-PH, Département de Neurochirurgie, CHU d'Angers. ... Examinateur Jean-Marc MULLER, PU, 2RCT, FRE 3511 CNRS, Université de Poitiers ... Examinateur
Remerciements
2
Je tiens à remercier mon directeur de thèse le Professeur Jean-Marc Muller pour m’avoir
accueillie au sein de son équipe, pour m’avoir accepté en stage, pour son aide pour la rédaction de ce mémoire, pour m’avoir transmis ses connaissances et pour son écoute, son soutien et sa convivialité (« ça gaze »).Un grand merci pour vos conseils et surtout pour les discussions scientifiques au cours de nos réunions d’équipe. Je suis très contente d’avoir pu travailler avec vous.
C’est avec beaucoup de reconnaissance que je souhaite remercier ma co-directrice de thèse
le Docteur Corinne Chadéneau pour m’avoir encadrée pendant mon stage de Master et le Doctorat et pour m’avoir fait partager ses connaissances et sa rigueur. Merci pour votre disponibilité et votre attention. Un grand merci aussi pour vos encouragements et votre soutien pendant les moments de doute ou de désespoir notamment relatifs à mes papiers. Travailler avec vous et apprendre à vos cotés auront été un plaisir mais surtout un honneur.
Je voudrais également remercier Mr Thierry Bergès pour m’avoir accepté en Master, pour la
qualité d’enseignement et sa gentillesse.
Je remercie les personnes, qui m’ont fait l’honneur d’accepter de juger ce travail, de croire
en l’expression de ma reconnaissance respectueuse.
Durant ces 5 années passées au laboratoire, Mme Annie Claire Balandre (ma maman au
labo) a été d’une grande aide et je la remercie vraiment pour tout que ce soit au niveau travail qu’au niveau humain. Je n’oublierai pas la quantité d’ondes positives que tu envoyais tous les matins en arrivant au labo. J’espère que je resterai toujours ta 2ème fille même si cela fait des jalouses !!!!
Je veux remercier Mme Catherine Adolphe pour son aide technique et sa gentillesse.
Je remercie les personnes de l’équipe de chimie des anticancéreux, le Dr Sébastien Papot et
Dr Brigitte Renoux, pour leur collaboration à ce projet.
Je remercie également les personnes de l’équipe LNEC, plus particulièrement Dr Afsaneh
Gaillard pour son aide pour les expériences in vivo et Dr Marianne Benoit-Marand pour nous avoir montré et laissé utiliser le vibratome. Je remercie aussi Clément, Maureen et Céline.
Je remercie Madryssa de Boisvilliers pour la colocation dans notre royaume de doctorantes.
Tu n’es pas seulement une collègue mais surtout une sœur ! J’espère que ce royaume restera toujours malgré le départ du roi Jean-Marc dans 5 ans. Je te remercie pour ta joie de vivre, ton aide, ta présence, ton soutien, pour tout et surtout pour la bibliographie et vive Endnote !!!!
Je remercie Stéphanie Cochaud pour son encadrement pour mon Master 1 et pour m’avoir
initié à toutes les expériences de biologie cellulaire et moléculaire. Merci pour ta bonne humeur et la bonne ambiance.
Remerciements
3
Je remercie Loubna Abaamrane (Loubloub ghzalla) pour sa gentillesse, sa bonne humeur,
son soutien, son écoute.
Je tiens à remercier très chaleureusement l’ensemble de l’équipe 2RCT et l’équipe STIM pour
l’aide et le soutien qu’ils ont pu m’apporter au cours de ces années.
Merci à tous les doctorants Mahmoud, Amandine, Aurélie et the post doctorante de l’équipe
Alice pour leur bonne humeur.
Au cours de ce travail, il m’a été offert d’encadrer des étudiants dans la réalisation de leur
travail de fin d’année. Chacun d’entre eux m’a apporté une aide précieuse. Leur implication dans leur travail a toujours été totale et à la hauteur de mes attentes. Cette expérience de supervision m’a été d’un grand bénéfice. Je vous remercie donc Soumaya, Naïma, Marion, Pauline, Jinnifer (lol), Sybiel, Anthony et Adèle d’avoir partagé un bout de chemin avec moi. Merci également à Paul Takam et Agnès Garnier pour leur gentillesse et leur bonne humeur.
Mon parcours n’aurait jamais pu rencontrer le succès sans le soutien à toute épreuve que
mes parents m’ont apporté. Il n’existe aucun mot pour traduire ma reconnaissance, mon amour et mon affection. Ces années de thèse ont été très dures à gérer mais vous étiez toujours là pour m’encourager et me motiver sans jamais se plaindre. Un énorme merci à toi maman d’avoir toujours pensé à moi avant même de penser à toi. Un énorme merci à toi mon cher papa, sans toi je n’aurais jamais pu arriver jusque-là. Merci de m’avoir poussé et soutenu jusqu'au bout. Un grand merci à mes frères pour leur aide, ma belle sœur et ma petite nièce Inès (Nana la classe) pour leur bonne humeur.
Sommaire
4
Abréviations ... 8
INTRODUCTION ... 11
Chapitre I. Les glioblastomes et leurs traitements ... 12
Partie I : Les glioblastomes ... 12
1. Généralités ... 12
2. Le glioblastome ... 12
3. Origine des gliomes ... 13
3.1. Dédifférenciation des astrocytes ... 13
3.2. Les cellules souches neurales ... 14
3.3. Les progéniteurs oligodendrocytaires ... 15
4. Classification des GBM ... 15
4.1. GBM primaire et secondaire ... 16
4.2. Classification moléculaire TCGA ... 16
5. La biologie des GBM ... 18
5.1. Perte de contrôle du cycle cellulaire ... 18
5.1.1. Les voies p53 ... 18
5.1.1.1. La voie p14ARF/MDM2/p53 ... 18
5.1.1.2. La voie ATM/Chk2/p53 ... 19
5.1.2. La voie RB (Rétinoblastome) ... 20
5.2. Les facteurs de croissance et leur signalisation ... 20
5.2.1. Les récepteurs tyrosine-kinases ... 20
5.2.2. La signalisation des RTK ... 21
5.2.2.1. PI3K/AKT/PTEN/mTOR ... 21
5.2.2.2. RAS/MAPK ... 22
5.3. La migration et l’invasion ... 23
5.3.1. Détachement du site d’origine ... 23
5.3.1.1. Les cadhérines ... 24 5.3.1.2. Les NCAM ... 25 5.3.1.3. La connexine 43 (Cx43) ... 25 5.3.1.4. La protéine CD44 ... 25 5.3.2. L'adhérence à la MEC ... 26 5.3.3. Remodelage de la MEC ... 27 5.3.3.1. Les MMP ... 27
Sommaire
5
5.3.3.2. Les ADAM ... 28
5.3.4. Motilité et contractilité cellulaire ... 28
5.3.4.1. Les Rho-GTPases ... 29
5.3.4.2. Les Rho-GTPases et les GBM ... 29
5.3.5. Régulation par les voies Akt et Hedgehog de la migration et de l’invasion des GBM 30 5.4. L’angiogenèse ... 31
5.4.1. Déstabilisation des vaisseaux sanguins existants ... 32
5.4.2. La dégradation de la membrane basale des vaisseaux et ECM environnant ... 32
5.4.3. La migration des cellules endothéliales et la formation de nouveaux vaisseaux sanguins ... 33
Partie II. Traitement des GBM ... 34
1. Traitement standard des GBM... 34
1.1. Chirurgie ... 34
1.2. La radiothérapie ... 34
1.3. La chimiothérapie ... 35
1.3.1. Carmustine (BCNU : Bis-Chloroéthyl-Nitroso-Urée) ... 35
1.3.2. Le témozolomide ... 35
1.4. Thérapie moléculaire ciblée ... 36
1.4.1. Le bevacizumab ... 37
1.4.2. Le cilengitide ... 37
1.4.3. Les inhibiteurs des RTK ... 37
1.4.4. Les inhibiteurs des kinases en aval des RTK ... 38
2. Résistance au traitement ... 38
2.1. Les deux modèles : stochastique et hiérarchique ... 38
2.2. Découverte des CSC dans les GBM (GSC : Glioma Stem Cells) ... 40
Partie III. La voie Hedgehog ... 41
1. Les protéines Hh ... 41
2. La signalisation Hh ... 41
1. La voie Hh dans les cancers ... 43
Partie IV : Les prodrogues glucuronylées ... 46
1. Généralités ... 46
2. Mise en œuvre d’enzymes endogènes sélectives dans les cellules tumorales (PMT : Prodrug Mono-Therapy) ... 47
Sommaire
6
3. Mise en œuvre d’enzymes exogènes pour des thérapies « ADEPT (antibody directed
enzyme prodrug therapy) ... 48
Chapitre II. Le système VIP-Récepteurs ... 50
1. Généralités ... 50
2. Les neuropeptides VIP et PACAP ... 50
2.1. Les polypeptides de la famille du VIP ... 50
2.2. Biosynthèse du VIP ... 51
2.3. Biosynthèse du PACAP ... 52
3. Les récepteurs du VIP et du PACAP ... 53
3.1. Le récepteur VPAC1 ... 54
1.1. Le récepteur VPAC2 ... 54
1.2. Le récepteur PAC1 ... 55
2. Place des récepteurs du VIP et du PACAP au sein de la superfamille des RCPG ... 55
3. Les variants d'épissage des transcrits primaires des récepteurs PAC1 ... 56
4. La pharmacologie des récepteurs du VIP et du PACAP ... 58
4.1. Les agonistes ... 58
4.2. Les antagonistes ... 58
5. Les principales voies de signalisations associées aux récepteurs du VIP et du PACAP ... 59
5.1. AMPc/PKA ... 59
5.2. Ca2+/PKC ... 59
5.3. Voies PI3K/Akt et Hedgehog ... 60
5.4. Les protéines accessoires ... 60
6. Les fonctions physiologiques ... 61
6.1. Les grands types de fonction des neuropeptides VIP et PACAP ... 61
6.2. Implication du système VIP-récepteurs dans le neurodéveloppement ... 62
6.2.1. Le VIP ... 62
6.2.2. Le PACAP ... 63
6.3. Implication du système VIP-récepteurs dans la neuroprotection, la différenciation et les effets neurotrophiques ... 64
6.3.1. Le système VIP-récepteurs et les cellules souches neurales (CSN) ... 64
6.3.2. Le système VIP-récepteurs et les astrocytes ... 64
6.3.3. Le système VIP-récepteurs et les neurones ... 65
7. Les fonctions tumorigènes du système VIP-récepteurs dans le système nerveux ... 67
7.1. Dans les neuroblastomes ... 67
Sommaire
7
7.3. Dans d’autres tumeurs cérébrales ... 68
7.4. Le système VIP-récepteurs dans les GBM ... 68
OBJECTIFS ... 70
Chapitre III. Objectifs des travaux de cette thèse ... 71
1. Objectif 1 : ... 71
2. Objectif 2 : ... 73
PRESENTATION DES TRAVAUX... 75
Chapitre IV. Présentation des travaux ... 76
Article n°1 ... 76
Article n°2 ... 103
Les données annexes de l’article n°2 ... 131
DISCUSSION, CONCLUSION ET PERSPECTIVES ... 137
Chapitre V. Discussion, conclusion et perspectives ... 138
1. Evaluation de la cytotoxicité d’une prodrogue glucuronylée de la cyclopamine et de son efficacité à cibler la voie Hh ... 139
2. Régulation de la migration et de l’invasion des cellules de GBM par le système VIP-récepteurs ; implication d’un « cross-talk » entre les voies Hh/Gli1 et PI3K/Akt ... 142
2.1.1. Mécanismes moléculaires régulés par le système VIP-récepteurs et impliqués dans la migration des cellules de GBM ... 144
2.1.2. Régulation de l’activation de la protéine GLI1 par VIP, PACAP-38 et VIP10-28 dans les cellules de GBM. ... 144
2.1.3. Régulation de l’expression des ARNm de GLI1 et MMP2 par VIP, PACAP-38 et VIP10-28 dans les cellules C6 de GBM (Cf Annexe de l’article 2) ... 145
2.1.4. Régulation par la voie PI3K/Akt de la migration et de l’invasion des cellules de GBM 146 Chapitre VI. Annexes ... 150
Article n°3 ... 150
Article n°4 ... 151
BIBLIOGRAPHIE ... 152
Abréviations
8
Abréviations
Les termes suivis d’un astérisque sont en anglais AC Adenylyl Cyclase*
ADEPT (antibody directed enzyme prodrug therapy)
ARF ADP Ribosylation Factor*
CNF Ciliary Neurotrophic Factor*
CSC Cancer stem cells*
E jour de vie embryonnaire
EC boucle extracellulaire
EGF Epidermal Growth Factor*
EGFR Epidermal Growth Factor Receptor*
ERK1/2 Extracellular-Regulated Kinase ½*
FAK Focal Adhesion Kinase*
GAP GTPase-Activating Protein*
GBM Glioblastome Multiforme
GDI Guanine nucleotide Dissociation Inhibitor*
GEF Guanine nucleotide exchange Factor*
GFAP Glial Fibrillary Acidic Protein*
GHF-1 Growth Hormone Factor-1*
GRF Growth hormone Releasing Factor*
GSC Glioma Stem Cells*
GTP Guanosine TriPhosphate*
Hh Hedgehog*
IA Inhibiteur d’Akt
IC boucle intracellulaire
IL InterLeukine
Abréviations
9 IRM Imagerie par Résonance Magnétique
IDH1 et 2 Isocitrate déshydrogénase 1 et 2
LIF Leukemia Inhibitory Factor*
MAPK Mitogen-Activated Protein Kinase*
MMP Matrix MetalloProteinase*
NF-κB Facteur Nucléaire κB
NRA Astrocytes normaux de rats
NSCLC cancer du poumon non à petites cellules
OMS Organisation Mondiale de la Santé
OPC Cellules précurseurs d’oligodendrocytes
P jour post-natal
PAC1 récepteur spécifique du PACAP
PACAP Pituitary Adenylate Cyclase-Activating Polypeptide*
PDGFR Plateled Derived Growth Factor Receptor*
PHI Peptide Having Carboxyterminal Isoleucine*
PHM Peptide Having Carboxyterminal Methionine*
PHV Peptide Histidine Valine
PI3K Phospho-Inositide 3 Kinase*
PIP2 PhosphatidylInositol-4,5-bisPhosphate
PIP3 PhosphatidylInositol-3,4,5-triPhosphate
PKA Protéine kinase A
PKC Protéine kinase C
PLC Phospholipase C
PMA Phorbol-12-Myristate-13-Acetate (ou Acétate de Phorbol Myristate)
PMT Prodrug mono-therapy
PrP PACAP related Peptide*
PTEN Phosphatase and TENsin homologue deleted on chromosome 10*
Abréviations
10 RE-1 Elément répresseur de type 1
REST RE-1 Silencing transcription factor*
RTK Récepteur Tyrosine Kinase
SNC Système nerveux central
SVZ Zone sous ventriculaire
TGF α Transforming Growth Factor α*
TM domaine transmembranaire
TMZ Témozolomide
TPA 12-O-Tétra-décanoylPhorbol 13-Acétate*
TRE TPA/PMA-Responsive Element*
VEGF Vascular Endothelial Growth Factor*
VIP Vasoactive Intestinal Peptide*
11
Tableau 1 : Classification des astrocytomes selon l’Organisation mondiale de la santé
(D’après Kleihues et al., 2002).
Grade Tumeurs Critères Taux de survie relatif à 10 ans
I Astrocytome pilocytique Prolifération cellulaire anarchique 89,30 % II Astrocytome diffus
Atypies nucléaires discrètes 38,70 %
III Astrocytome
anaplasique
Atypies importantes avec
mitoses 22,20 %
IV Glioblastome
Atypies importantes avec mitoses, nécrose et/ou
prolifération endothéliale 2,30 %
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
12
Chapitre I. Les glioblastomes et leurs
traitements
Partie I : Les glioblastomes
1. Généralités
Les gliomes sont des tumeurs cérébrales primitives. Elles représentent plus de 70% des tumeurs primaires du système nerveux central chez l’adulte. La classification de l’Organisation Mondiale de la Santé (OMS) de 2007 distingue quatre types de gliomes en fonction de critères cytologiques : les astrocytomes, les oligodendrogliomes, les oligoastrocytomes et les épendymomes (Louis et al., 2007 ; Walker et al., 2011). Les astrocytomes sont les tumeurs cérébrales les plus fréquentes, constituant 90% des gliomes, soit plus de la moitié de toutes les tumeurs cérébrales primitives (Behin et al., 2003 ; Ricard et al., 2012). L’OMS a attribué pour les astrocytomes quatre grades tumoraux (Tableau 1). Cette sous-classification est basée sur cinq critères histologiques: la différenciation et la densité cellulaire, la présence d'atypies cytonucléaires, l'activité mitotique, l'aspect nécrotique et la prolifération microvasculaire. Parmi les astrocytomes, la forme la plus agressive (grade IV) est le glioblastome (GBM) constituant environ 50% des gliomes.
2. Le glioblastome
Avec une incidence annuelle de 3 à 5 cas pour 100 000 individus selon les sources, le GBM est une forme rare de cancer. Cependant sa localisation, son comportement envahissant, et son pronostic extrêmement faible font de lui une des formes les plus redoutées de cancer. Même si ils peuvent survenir à tout âge, les GBM ont un pic d'incidence entre 50 et 70 ans. Les hémisphères cérébraux sont le site le plus commun pour les GBM, avec 95% se développant dans la région sus-tentorielle et 5% dans le tronc cérébral, le cervelet et la moelle épinière (Walker et al. 2011).
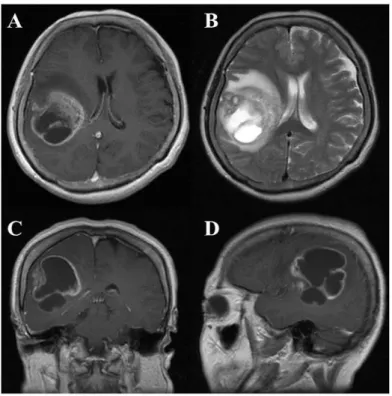
Le diagnostic des GBM est généralement effectué par un scanner à rayon X (la tomodensitométrie TDM) et l’imagerie par résonance magnétique (IRM). Ces techniques ont révélé que le GBM présente un aspect typique sous forme d’un anneau constitué d’une zone nécrotique centrale entourée par des cellules viables de la tumeur (Figure 1). L’IRM associée
Figure 1 : Résultats radiologiques représentatifs des GBMs. IRM en pondération T1 après injection de l’agent de contraste «Gadolinium» (A, coupe axiale; C, coupe coronale ; D, coupe sagittale) montre « ring enhancement lesion » dans le lobe droit temporo-pariétal. IRM en pondération T2 (B) a montré un important œdème péri-tumoral (D’après Nakada et al., 2011)
Figure 2: Analyse histopathologique d’un GBM. Ces images montrent l'aspect histologique d'un glioblastome, caractérisé par un polymorphisme nucléaire, une densité cellulaire, et une nécrose pseudopalisadique (astérisque en A), ainsi que la prolifération endothéliale vasculaire (astérisque en B) et la division mitotique (flèches en B) (D’après Wen et al., 2008).
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
13
au gadolinium, un produit de contraste, permet d’obtenir une excellente qualité d’images de cerveau en 3D, ce qui permet une analyse beaucoup plus précise de la tumeur. Un œdème péri-tumoral, et fréquemment une distorsion des ventricules et de toutes les parties du cerveau entourant la tumeur (effet de masse), sont bien visualisés en IRM (Tanaka et al., 2013)
Etant donné l’hétérogénéité des GBM, le diagnostic par radiologie n’est pas suffisant. Afin de poser le diagnostic, une étude histologique du tissu tumoral doit être réalisée après une biopsie ou une craniotomie. Une coloration Hématoxyline/Eosine de tissu tumoral d’un GBM (Figure 2) permet d’observer la présence de cellules anaplasiques avec une forte activité mitotique, des zones de prolifération vasculaire et des zones nécrotiques. L’emplacement de ces éléments au niveau de la tumeur est variable, mais généralement les zones nécrotiques occupent le centre de la masse tumorale. Le reste des cellules viables de la tumeur s’accumulent autour de la nécrose pour former une structure dite « zone pseudo-palissadique » (Wen et al., 2008).
3. Origine des gliomes
Le type cellulaire à l’origine des tumeurs gliales reste encore controversé. En se basant sur des informations provenant de patients et de modèles expérimentaux de gliomes, les cellules les plus susceptibles d’être à l’origine des gliomes sont : les astrocytes, les cellules souches neurales (CSN) ou encore les cellules précurseurs d’oligodendrocytes (OPC) (Figure 3) (Jiang et al., 2012).
3.1. Dédifférenciation des astrocytes
Une première hypothèse concernant le développement des cancers en général, a été que les cellules cancéreuses provennaient de l'accumulation d'altérations qui se produisaient dans les cellules matures différenciées, entrainant une dédifférenciation de ces cellules au cours du processus de cancérogenèse. Cette hypothèse est également valable en ce qui concerne les gliomes, puisque des similitudes histologiques sont observées entre les astrocytes et les cellules de gliome. De plus au niveau moléculaire, le marqueur astrocytaire GFAP est fréquemment exprimé par les gliomes (Jones et al., 1981). Des études plus récentes supportent aussi cette théorie de la dédifférenciation des astrocytes. Ainsi, l’étude de Bachoo et ses collaborateurs a montré que des cellules différenciées, des astrocytes primaires obtenus à partir de souris nouveau-nées doublement invalidées pour p16Ink4a et p14Arf (Ink4a/Arf-/), et
Figure 3 : Les cellules les plus susceptibles d’être à l’origine des gliomes. A: une représentation schématique simplifiée de développement des cellules gliales, des cellules souches multipotentes du CNS à des astrocytes et des oligodendrocytes, où chaque étape est définie par l’expression de marqueurs. Les éclairs rouges indiquent l’apparition d’un événement oncogénique qui va favoriser le développement d’un gliome. B: Le rôle de l’origine cellulaire dans l’initiation, la progression, la récurrence et la réponse des gliomes à la thérapie reste largement inconnu. La recherche de biomarqueurs qui permettraient de distinguer des sous-groupes de gliome phénotypiquement distincts, basés sur la cellule d'origine, pourrait être une voie pour élucider ce rôle. NSC = cellules souches neurales; GRPC = cellules progénitrices gliales restreinte; APC = cellule précurseur astrocytes; OPC = cellules précurseurs d'oligodendrocytes (D’après Jiang et al., 2012).
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
14
cultivés dans un milieu sans sérum supplémenté avec de l’EGF, pouvaient être transformés en neurosphères avec un phénotype de cellules souches pluripotentes (pas d’expression de GFAP, de la nestine et d’A2B5). Ces cellules se sont avérées sensibles à la transformation avec un récepteur muté de l’EGF, et leur transplantation orthotopique dans des souris provoque le développement d’un gliome de haut-grade (Bachoo et al., 2002). Une longue incubation d’astrocytes matures dans du milieu contenant du TGFα permet leur transformation en cellules gliales radiales en premier, et plus tard en cellules semblables aux CSN (Sharif et al., 2007). Lorsque ces astrocytes dédifférenciés ont été exposés à des rayonnements gamma, ils sont devenus des cellules immortalisées et capables d’induire des gliomes de haut grade après greffe dans le cerveau (Dufour et al., 2009).
3.2. Les cellules souches neurales
Une autre hypothèse est que les CSN de la SVZ seraient les cellules d’origine de gliomes. Les CSN sont principalement situées dans la SVZ des ventricules latéraux et dans le gyrus denté de l’hippocampe. Il a été montré que ces CSN expriment plusieurs marqueurs de surface cellulaire tels que la nestine, Sox2, CD133, et la GFAP et la delta GFAP, mais aucun de ces marqueurs n’est unique aux CSN. Les premières observations chez des patients ont montré que les tumeurs cérébrales humaines étaient souvent situées près de la SVZ, ce qui suggérait qu’elles étaient originaires de cette zone. Cette notion a été récemment confirmée dans une large série de patients qui a montré que dans 93% des cas la tumeur est en contact avec la paroi du ventricule latérale (Barami et al., 2009). Une indication supplémentaire que les CSN pourraient être la cellule d’origine des gliomes est que les voies de signalisation impliquées dans la régulation de l’autorenouvellement, la prolifération et la différentiation des CSN sont couramment altérées dans les gliomes telles que la voie de signalisation liée au récepteur EGFR (TCGA, 2008). D’autres éléments en faveur de cette hypothèse viennent des modèles de souris. Des souris pourtant les gènes p53 et Nf-1 inactivés ont développé des astrocytomes avec une pénétrance de 100% (Liu et al., 2011). L’analyse histopathologique de ces tumeurs a démontré que les lésions pré-symptomatiques étaient situées dans la SVZ, ce qui suggère que les cellules souches de la SVZ peuvent servir de cellules d’origine des GBM. Dans ce modèle de souris, l’addition de mutations supplémentaires au niveau du gène PTEN a permis une progression tumorale vers des GBM. En revanche, la même combinaison de mutations dans les régions non-neurogéniques de souris adulte n’induit pas la formation de tumeurs (Zhu et al., 2005 ; Kwon et al., 2008 ; Alcantara et al., 2009).
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
15 3.3. Les progéniteurs oligodendrocytaires
Récemment une nouvelle hypothèse a été émise. Liu et ses collaborateurs (2011) ont montré avec le modèle murin de gliome spontané (p53-/- ; Nf1-/-) que si les mutations oncogéniques étaient induites dans les cellules souches neuronales, seuls les cellules progénitrices des oligodendrocytes (OPC) mais pas les autres dérivés des CSN, présentaient une prolifération anarchique conduisant au développement d’une tumeur (Liu et al., 2011). Ceci est en accord avec la théorie qui soutient que « la cellule touchée par la mutation (COM ou « cell of mutation ») et la cellule à l’origine de la tumeur (COT ou « cell of origin») peuvent être différentes» (Visvader, 2011). Ainsi les gliomes peuvent provenir des OPC. Ces cellules appelées progéniteurs ou cellules précurseurs sont des cellules en partie différenciées, présentes dans le cerveau fœtal et adulte et qui se divisent afin de produire des cellules matures, les oligodendrocytes. L’altération génique de certains marqueurs moléculaires des OPC tels que NG2 (Dawson et al., 2003), A2B5 (Baracskay et al., 2007), CNPase (Scherer et al.,1994) et PDGFR (Wilson et al., 2006), a permis d’obtenir plusieurs modèles murins de glioblastome spontané. Un argument supplémentaire en faveur de cette hypothèse des OPC est que la voie de signalisation PDGFR, impliquée dans le développement normal des oligodendrocytes en contrôlant la prolifération et la migration des OPC, est aussi couramment activée dans les gliomes (Parsons et al., 2008 ; TCGA., 2008).
4. Classification des GBM
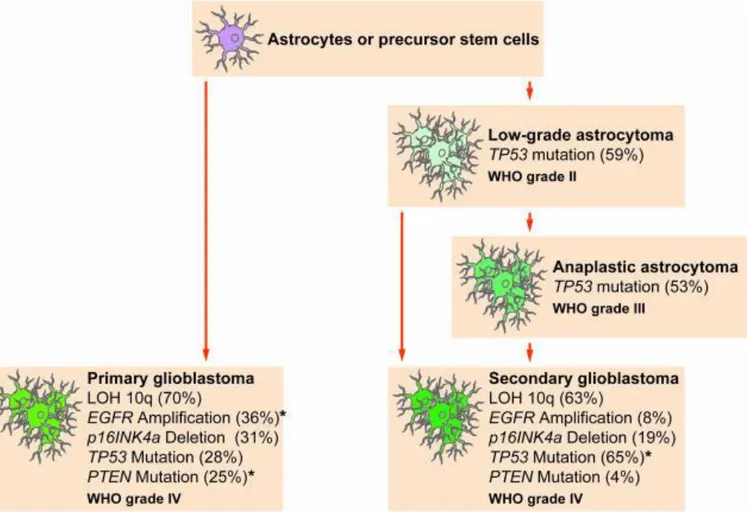
L’hétérogénéité cellulaire et moléculaire des GBM rend l’établissement de sous catégories difficile. Cependant ces dernières années des progrès importants ont été réalisés dans le recensement des altérations moléculaires présentes dans les GBM. Des études visant à lier les caractéristiques histologiques et les altérations moléculaires ont permis de subdiviser les GBM en deux types (Hoang-Xuan et al., 2005) : les GBM primaires ou dits de novo (GBM I) et les GBM secondaires (GBM II) (Figure 4) (Ohgaki et Kleihues, 2009 ; 2013). Cependant, malgré la distinction GBM I/GBM II, les GBM I nécessitent une caractérisation plus précise. En effet, l’analyse du transcriptome de GBM I a permis de déterminer 4 sous-classes définies selon les altérations majeures observées lors de l’étude TCGA « The Cancer Genome Atlas » (Verhaak et al., 2010). TCGA est un projet qui a débuté en 2005 pour cataloguer les mutations génétiques responsables de cancers en utilisant le séquençage du génome.
Figure 4 : Les principales altérations génétiques des GBM primaires et secondaires. *Les altérations génétiques qui présentent une difference significative en fréquence entre les
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
16 4.1. GBM primaire et secondaire
Les GBM I représentent 90% à 95% de la totalité des GBM. Ils sont caractérisés par un court délai entre le début des symptômes et le diagnostic (inférieur à trois mois) et ils affectent les patients âgés en moyenne de 62 ans. Les principales altérations génétiques qui caractérisent le GBM I sont l’amplification du gène EGFR, des délétions au niveau du chromosome 10q et des mutations/délétions du gène PTEN. Les GBM II représentant 5 à 10 % des GBM et proviennent de la progression d'un gliome de bas grade chez de patients d’un âge moyen de 45 ans. Ils ont un meilleur pronostic que les GBM I. Les GBM II se caractérisent essentiellement par des mutations du gène de la protéine p53, la surexpression du récepteur au PDGF et des délétions au niveau des chromosomes 19q et 17p. (Kleihues and Ohgaki, 1999; Ohgaki and Kleihues, 2005). Récemment, d’autres mutations ont été mises en évidence récemment dans les GBM II, telles que celles des gènes codant pour les isocitrate déshydrogénase 1 et 2 (IDH1 et IDH2). Ces deux enzymes produisent le NADPH cytosolique qui est un cofacteur nécessaire à la régénération du glutathion réduit. Ce dernier est indispensable à la protection des cellules contre les dommage liés au stress oxydatif (Bleeker et al., 2010). Dans les GBM II des mutations notées R132H pour IDH1 et R172H pour IDH2 induisent l’inhibition de l’activité enzymatique de ces enzymes (Yan et al., 2009). Des études récentes ont montré que ces mutations IDH représentent un marqueur de bon pronostic pour les patients atteints d’un GBM (Sanson et al., 2009).
4.2. Classification moléculaire TCGA
En utilisant une analyse de classification hiérarchique, Verhaak et al. (2010) ont utilisé des données TCGA pour classer avec succès les GBM en quatre sous-types : classique, mésenchymateux, proneural et neural (Figure 5). Cette étude a permis de compléter les classifications précédentes des GBM (Murat et al., 2008, Liang et al., 2005 ; Mischel et al.,2003 ; Shai et al., 2003 ; Freije et al., 2004 ; Nutt et al., 2003 ;; Tso et al., 2006 ; Phillips et al., 2006).
Le sous-type classique : Ce sous-type a un profil caractéristique des cellules hautement
prolifératives. Les tumeurs de ce type montrent des amplifications dans le chromosome 7, accompagnée par des pertes sur le chromosome 10 (93%) et sur le chromosome 9p21.3 (95%). Ces événements chromosomiques conduisent à l'amplification du gène EGFR et la perte de PTEN et du locus CDKN2A. Cependant, ce sous-type ne présente pas d’altérations
Figure 5 : Schéma représentatif de l’origine des GBM et de leur classification moléculaire. TICs : tumor–initiating cells, BCPC : brain cancer–propagating cells. (D’après Van Meir et al., 2010)
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
17
dans les gènes TP53, NF1, PDGFRA ou IDH1. De plus, il présente une expression élevée du gène de marqueur des précurseurs neuraux NES et les voies de signalisation Notch (NOTCH3, JAG1, LFNG) et sonic hedgehog (SMO, GAS1, GLI2), qui sont des marqueurs des cellules souches, sont fortement actives dans ce sous-type (Verhaak et al., 2010).
Le sous-type mésenchymateux est principalement caractérisé par la surexpression des
marqueurs mésenchymateux, comme CHI3L1 (Chitinase 3-like protein 1) et MET, ainsi que les marqueurs astrocytaires CD44 et MERTK. CHI3L1 est un facteur de survie activé en réponse au stress physiologique subi par la cellule et inhibiteur de l’apoptose (Ku et al., 2011). Tandis que Met est un récepteur tyrosine kinase activé par HGF (hepatocyte growth factor) et impliqué dans la prolifération, l’adhérence et la migration cellulaires ainsi que dans les réponses anti-apoptotiques (Liu et al., 2011). Ce sous type présente une perte fréquente des gènes codant les protéines NF1 (37%), p53 (32%), et PTEN (32%). (Verhaak et al., 2010).
Le sous-type de neural est moins bien défini et présente des caractéristiques qui sont
similaires à celles trouvées dans le tissu cérébral normal, avec une expression des marqueurs neuronaux tels que NEFL (neurofilament light chain polypeptide), le récepteur de l'acide gamma-aminobutyrique (GABRA1), la synaptotagmine I (STY1) et le SLC12A5 (Verhaak et al., 2010).
Le sous-type proneural. Ses caractéristiques les plus pertinentes sont une amplification
du gène PDGFRA et des mutations ponctuelles dans le gène IDH1 (30% des cas). La présence des mutations du gène IDH1 suggère que les GBM II pourraient appartenir à ce sous type. Des mutations fréquentes des gènes TP53 (54%) et PIK3CA/PIK3R1 (19%) sont également observées, alors que des délétions au niveau des chromosomes 9 et 10 (perte de CDKN2A et de PTEN) sont importantes (50%) mais moins fréquentes que dans le sous-type classique. Des gènes du développement oligodendrocytaire, comme PDGFRA, NKX2 -2 et OLIG2, sont fortement exprimés. Une faible expression de CDKN1A a été observée, probablement due à la surexpression d’OLIG2, qui peut réguler négativement l’expression du gène CDKN1A. Ce sous-type présente également une expression de gènes du développement proneural, tels que les gènes SOX, ainsi que DCX, DLL3, ASCL1 et TCF4 (Verhaak et al., 2010).
Figu re 6 : L es alt ér ation s gé n étiq u es m aj eu re s d an s les GB M . A lt ér ati ons gé né ti que s comm une s et leur s pou rc ent age s re n contré es da ns le GBM. En moyenne , pl us de 60 alt éra ti ons g éné ti q ue s sont re trouv ée s d ans un se ul GBM. C er cles ro uge s : de s ampl ific ati ons le plus souve nt , ce rc le s bleus : de s dé lét ions homozygote s le plus souve nt . C erc les ve rt s : de s mut ati ons le plu s souve nt. C erc les ave c un ha lo coloré : alt éra ti ons gé n éti que s le s plus coura ntes d ans le s GBM. P I3K = phosph ati dyli nosi tol 3 -kinase , P IK 3R 1 = phospha ti dyli nosi tol 3 -kinase re gulator y subuni t, P IK 3C A = phospha ti dyli nosi tol 3 -kinase c atalyti c subuni t, P TEN = phospha tas e and ten -sin homol og, P 53 = prote in 53, C DK N2A = c y cli n -de pe nde nt kinase inhi bit or 2A, C DK 4 = c yc li n -d epe nde nt kinase 4, R b1 = r eti noblastom a 1, MDM2 = murine double mi nute 2, MDM4 = mur ine double mi nute 4, N F 1 = ne urof ib ro -matosi s 1, EG F R = e pider mal grow th fa ctor re ce ptor, P DG F R = plate let -de rive d gro wth fa ctor re ce ptor, I DH 1 = isocit ra te de hydro ge na se 1. (sc h ém a de ssi né via Ser vie r Me dica l Art, a da pt é de B eld en et al ., 2011) .
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
18
5. La biologie des GBM
Les caractéristiques cellulaires des GBM sont la conséquence de plusieurs altérations moléculaires. Un GBM typique présente plus de 60 altérations génétiques. Près de 90% de ces altérations sont des mutations (y compris les délétions), et les 10% restants sont des aberrations chromosomiques (amplifications) (Figure 6) (TCGA., 2008 ; Rao et al., 2010).
5.1. Perte de contrôle du cycle cellulaire 5.1.1. Les voies p53
Le gène p53, localisé sur le chromosome 17q13.1, code pour une protéine qui réagit à différents stress cellulaires : rayonnement UV, l’hypoxie, le stress oxydatif, etc. Cette protéine, qualifiée de suppresseur de tumeurs, régule l’expression de gènes cibles et joue un rôle important dans l’intégrité cellulaire en supprimant la transformation oncogénique par interruption du cycle cellulaire, laissant alors le temps pour réparer les dommages d’ADN ou induire l’apoptose si ces dommages sont trop sévères pour être réparés. La p53 peut aussi induire la différenciation cellulaire, la sénescence, ou la néovascularisation (Bogler et al., 1995).
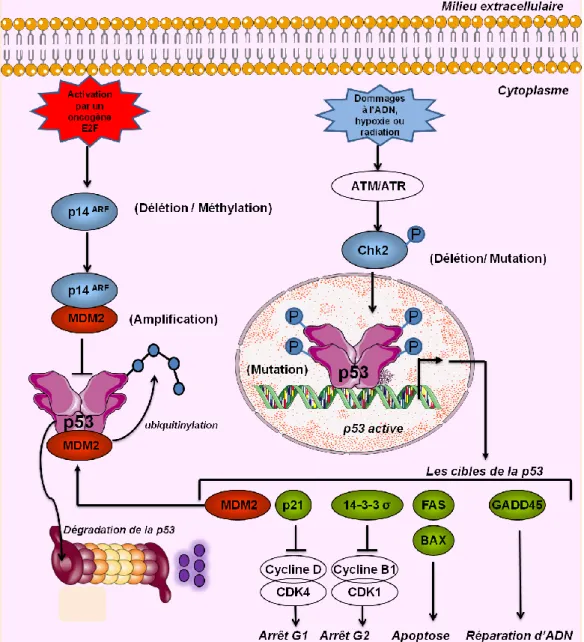
5.1.1.1. La voie p14ARF/MDM2/p53
Dans les cellules normales et en absence de stress, la protéine MDM2 inhibe l’activité transcriptionnelle de p53 en se fixant sur son domaine de transactivation N-terminal. En se fixant à p53, MDM2 va permettre l’ubiquitinylation de p53 et sa dégradation par le protéasome. Cependant, la transcription du gène MDM2 est induite par p53 sauvage (Barak et al., 1994, Zauberman et al., 1995). Cette boucle de rétroaction autorégulatrice permet de réguler l'expression de MDM2 et l'activité de p53. Suite à un stress cellulaire, tel que l’apparition de dommages à l’ADN, la protéine p14ARF (codée par un gène du locus CDKN2A localisé sur le chromosome 9p21) se lie directement à MDM2, inhibant ainsi l’association de ce dernier à la p53, ce qui empêche la dégradation et la répression transcriptionnelle de p53.
L’expression de p14ARF est régulée négativement par p53 (Stott et al., 1998). Ainsi, l'inactivation de p14ARF/MDM2/p53 est causée par une expression modifiée de l'un des gènes p53, MDM2, ou p14ARF (Figure 7).
Figure 7 : La voie p53. Une boucle de régulation négative contrôle les niveaux cellulaires de la protéine p53. Dans les cellules normales, p53 induit l’activation de MDM2 qui favorise la dégradation de p53. En présence d’un stress cellulaire tel que l’activation d’un oncogène qui induit l’activation de
p14ARF, la protéine MDM2 est séquestrée. De plus, des dommages de l’ADN ou des agents
chimiothérapeutiques, activent des protéines kinases, tels que ATM et ATR, qui par l’intermédiaire d’une autre kinase CHK2, phosphorylent le domaine N-terminal de la p53 pour empêcher la liaison de MDM2. Le domaine Ct de la p53 est aussi phosphorylé pour augmenter les liaisons spécifiques à l’ADN. Ces événements augmentent les niveaux de p53 activant ainsi la transcription des gènes cibles de la p53. Les protéines p21 et 14-3-3 favorisent l’arrêt de la croissance en phase G1 et G2 en inhibant l’activité des CDK (cyclin-dependent protein kinase). FAS et BAX favorisent l’apoptose si la réparation des dommages d’ADN n’est pas possible, et GADD45 favorise la réparation de l’ADN. Les altérations génétiques induisant des « activations » sont représentées dans un cercle rouge. Les altérations génétiques qui conduisent à une perte de fonction sont représentées dans un cercle bleu
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
19
La voie p53 joue un rôle crucial dans le développement des gliomes. En effet, les mutations du gène p53 sont fréquentes : p53 est mutée dans deux tiers des astrocytomes diffus de bas grade. Cette fréquence est similaire à celle des astrocytomes anaplasiques (grade III) et des GBM (Watanabe et al., 1996 et 1997 ; Ohgaki et al., 2004). De plus, des anomalies moléculaires impliquant d'autres gènes de la voie, tels que p14ARF, MDM2, ou MDM4 ont également été décrites dans les GBM. L’amplification de MDM2 est observée dans environ 10% des GBM, exclusivement dans les GBM qui n'ont pas de mutation du gène p53 (Biernat et al., 1997 ; Reifenberger et al., 1993). La perte d'expression de p14ARF est souvent présente dans les GBM (76%) et semble être associée principalement à la méthylation aberrante de son promoteur ou à une délétion hémizygote, alors que l'inactivation par mutation est rare (Ichimura et al., 2000 ; Nakamura et al., 2001).
5.1.1.2. La voie ATM/Chk2/p53
Récemment, l’implication de la voie ATM/Chk2/p53 a été mise en évidence dans la gliomagénèse. Squatrito et ces collaborateurs ont démontré que la perte des composants de la voie ATM/Chk2/p53 accélère le développement des gliomes et contribue à la résistance aux radiations (Squatrito et al., 2010). En réponse à un rayonnement ionisant, les cellules activent des kinases « senseures » ATM, ATR et la DNA-PK. Ces kinases phosphorylent la p53 directement ou indirectement par l’intermédiaire d’autres kinases de point de contrôle appelées : Chk1 et Chk2 (Hirao et al., 2002) (Figure 7). Ces dernières induisent la phosphorylation de p53 ce qui permet d’empêcher son inhibition et sa dégradation par MDM2, conduisant ainsi à la stabilisation et l’activation de p53. Bien que des études antérieures n’aient montré qu’une faible fréquence, voire aucune, des mutations Chk2 (environ 6%) (Ino et al., 2000 ; Sallinen et al., 2005), 22% des patients atteints de gliome dans l'étude TCGA présentaient une perte d’une seule copie de la région chromosomique contenant Chk2, avec une réduction significative de l’ARNm Chk2. Ceci suggère que Chk2 pourrait représenter un important suppresseur de tumeur pour un sous-ensemble de patients atteints de gliome (TCGA, 2008 ; Squatrito et al., 2010). La perte de ce gène suppresseur de tumeurs ou de l’un des gènes impliqués dans la signalisation p53 est associée à une instabilité génomique, favorisant ainsi l’apparition de mutations génétiques successives entrainant une résistance à l’apoptose et la progression tumorale des GBM.
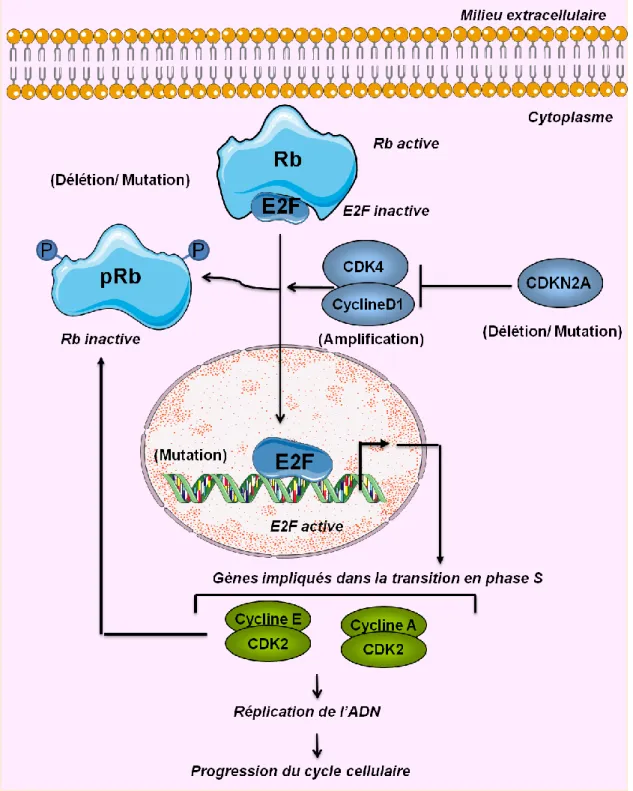
Figure 8 : La voie Rb. Le complexe Cycline D / Cdk4 se fixe sur la protéine Rb. Une fois activé, le complexe Cycline D / Cdk4 phosphoryle la protéine Rb. Ceci permet alors la libération par cette dernière du facteur de transcription E2F. Une fois libéré, E2F se fixe sur l'ADN, activant l'expression de gènes codant pour des protéines nécessaires au déroulement de la phase S. Les altérations génétiques qui conduisent à une perte de fonction sont indiquées en bleu (schéma dessiné via Servier Medical Art).
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
20 5.1.2. La voie RB (Rétinoblastome)
La voie RB empêche l'entrée du cycle cellulaire et sa progression, comme la voie p53. La protéine RB1 de 107 kDa codée par le gène RB1 (locus 13q14) contrôle la progression à travers la phase G1/S du cycle cellulaire (Serrano et al., 1993) (Figure 8). Cette voie est activée par la protéine p16INK4a, codée par un gène situé au même locus que celui de la protéine p14ARF, CDKN2A. La protéine p16 se lie à la protéine CDK4/6 et bloque la formation d’un complexe CDK4/6 actif avec la cycline D1. Cette liaison entre la p16 et la CDK4/6 rend cette dernière inactive et inhibe la phosphorylation de la protéine Rb, empêchant ainsi la libération du facteur E2F qui régule l’expression des gènes impliquées dans l’entrée en phase S. Ainsi, l’altération de RB1, CDK4, ou CDKN2A peut provoquer une dérégulation de la transition de phase G1-S.
Le projet pilote TCGA a révélé que la fréquence des altérations génétiques dans la voie de signalisation RB était de 77% dans les GBM. Ces altérations comprennent la délétion homozygote ou des mutations de CDKN2A (52%), la délétion homozygote de CDKN2B
(p15INK4b) (47%) ou de CDKN2C (p18INK4c) (2 %), l’amplification de CDK4 (18%), de
CCND2 (cycline D2) (2%) ou de CDK6 (1%), et la mutation ou la délétion homozygote de RB1 (11%) (ATCG, 2008).
5.2. Les facteurs de croissance et leur signalisation
Pour la croissance et la progression des GBM, la signalisation de la prolifération dépend largement de l'activité des récepteurs tyrosine kinases (RTK).
5.2.1. Les récepteurs tyrosine-kinases
Les RTK sont des glycoprotéines membranaires composées d’un site de fixation du ligand appartenant au domaine extracellulaire N-terminal relié au domaine cytoplasmique par une hélice α transmembranaire. Le domaine cytosolique C-terminal présente une activité enzymatique qui permet le transfert du phosphate γ de l’ATP vers l’hydroxyle des tyrosines des protéines cibles et/ou du récepteur lui-même. Dans ce dernier cas, on parle d’autophosphorylation (Hunter, 1998). Il existe environ 20 familles de RTK regroupées selon leur organisation structurale (Figure 9) (Blume-Jensen et al., 2001).
Parmi toutes les altérations des RTK dans les GBM, l'amplification du gène EGFR est l'altération la plus fréquente (environ 40%) (Libermann et al., 1985 ; Ohgaki et al., 2004).
F ig ure 9 : L a fam il le d es ré ce p te u rs tyr osi ne -k in ase (R T K ) con tient 20 sous -f am ill es , p rés ent ées ici de fa çon sc hé m at ique ave c le s m em br es de la fam ill e figur an t en d es sous de cha que réc ept eur . Les d o m ai nes st ruc tur el s da ns les ré gi ons ex tr ac el lu la ir es , id ent if iés par dét er m ina tion d e la st ru ct ur e o u de l'ana lys e de sé quenc e, son t rep rés ent és c onf or m ém ent aux m oti fs figur ant dan s l’en ca rt . Le s dom ai n es int rac el lul ai res son t pr és en tés sou s f o rm e de r ect angl es r ouge s. (D ’ap rès L em m on et Sch le ss inge r., 201 0) .
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
21
L'amplification du gène EGFR est également associée à des altérations structurelles. Sept variant majeurs mutés de l’EGFR ont été identifiés, mais le plus courant est le variant III appelé «EGFRvIII ». Ce dernier est un mutant qui peut envoyer des signaux de croissance constitutifs indépendamment du ligand (Wong et al., 1987 ; Yamazaki et al., 1988 ; Biernat et al., 2004). Cinquante pour cent des GBM présentant une amplification du gène EGFR expriment l’EGFRvIII, qui résulte de la délétion au niveau des exons 2-7 codant pour une partie du domaine extracellulaire. La surexpression de l‘EGFRvIII dans des lignées cellulaires de gliome induit une autophosphorylation constitutive conduisant à l'activation des voies de signalisation Ras et PI3K. Cela entraîne l’augmentation de la prolifération cellulaire, la résistance à l'apoptose induite par des agents endommageant l'ADN grâce à la modulation de l‘expression de Bcl-XL et le renforcement de la tumorigénicité (Huang et al., 1997 ; Nagane et al., 1998 ; Narita et al. 2002). Certaines études ont montré que les patients atteints de GBM surexprimant l'EGFR ou des mutants de ce récepteur ont une survie plus courte, ce qui suggère que des altérations de l'EGFR peuvent être corrélées à une agressivité accrue du GBM (Feldkamp et al., 1999 ; Shinojima et al., 2003).
5.2.2. La signalisation des RTK
5.2.2.1. PI3K/AKT/PTEN/mTOR
La transduction du signal en aval des RTK implique la signalisation PI3-kinase (Figure 10) qui joue un rôle central dans la survie de la cellule et dans la régulation de l'expression génique, du métabolisme cellulaire et du réarrangement du cytosquelette. La PI3-kinase catalyse la formation d’un phospholipide membranaire particulier, le phosphatidylinositol (3, 4,5)-triphosphate (PI (3, 4,5) P3 ou PIP3), à partir du phosphatidylinositol (4,5) biphosphate (PI(4,5) P2). Le PIP3 peut ainsi recruter et activer un grand nombre d'effecteurs cytoplasmiques, parmi eux des protéines sérine-thréonine kinases comme AKT et PDK1, des protéines tyrosine kinases telles que TEC ou encore des protéines adaptatrices comme GRB2-associated binding protein 1, ou GAB-1. La protéine Akt est aussi connue pour être activée directement par une sérine/thréonine kinase appelée mTOR (mammalian Target of Rapamycin). A l’inverse de ces effecteurs, la protéine PTEN (gène suppresseur de tumeur) intervient pour bloquer la voie PI3K/Akt et permet ainsi un arrêt en phase G1 du cycle cellulaire et une sensibilité accrue à l’apoptose. Cette protéine est dotée d’une activité phosphatase qui lui permet de déphosphoryler le PIP3 en PIP2. Elle agit ainsi à la fois sur la morphologie, le caractère invasif et la prolifération des cellules tumorales.
Figure 10 : La voie de signalisation RTK/PI3K/Akt. La liaison des RTKs, incluant l’EGFR et le PDGFR, avec la sous unité p85 de la protéine PI3K induit une activation de la sous unité catalytique p110, qui catalyse la phosphorylation de PIP2 (PI 3,4-bisphosphate) en PIP3 (3,4,5-triphosphate). Inversement PTEN convertit PIP3 en PIP2. PIP3 à son tour active la PDK1 (phosphoinositide-dependent kinase-1), qui phosphoryle Akt au niveau de la thréonine 308, tandis que la sérine 473 d’Akt est phosphorylée par mTORC2. AKT activée inactive le complexe suppresseur TSC1/TSC2, qui permet l’activation de mTORC1. Cette voie affecte de multiple processus cellulaires tels que la survie cellulaire, la prolifération et la motilité (schéma dessiné via Servier Medical Art, Adapté de Nakada et al., 2011).
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
22
Le gène PTEN est souvent altéré dans les GBM. Il subit une perte génomique, une mutation ou une inactivation épigénétique dans 40% à 50% des gliomes, causant ainsi des niveaux élevés d'activité de PI3K et de la signalisation en aval (Koul 2008). Comme la voie PI3K est une force motrice dans les gliomes, même des changements mineurs dans l'expression ou la fonction de son régulateur négatif critique, PTEN, ont de profondes répercussions sur le comportement des cellules tumorales. La perte de PTEN est fortement corrélée à l’activation de la protéine mTOR via Akt. Des mutations oncogéniques d’Akt n'ont pas été détectées dans les GBM (Bleeker et al., 2009). Cependant, l’élévation de la phosphorylation d’Akt a été rapportée dans plus de 85% des lignées cellulaires de GBM et dans des échantillons de tumeurs (Holland et al., 2000 ; Haa-Kogan et al., 1998 ; Wang et al., 2004), ce qui est corrèlé à l’observation que la signalisation RTK/PI3K/Akt est altérée dans 88% des GBM (TCGA, 2008). Quant à la protéine mTOR, l’amplification de son gène est fréquemment observée dans le cas des GBM.
5.2.2.2. RAS/MAPK
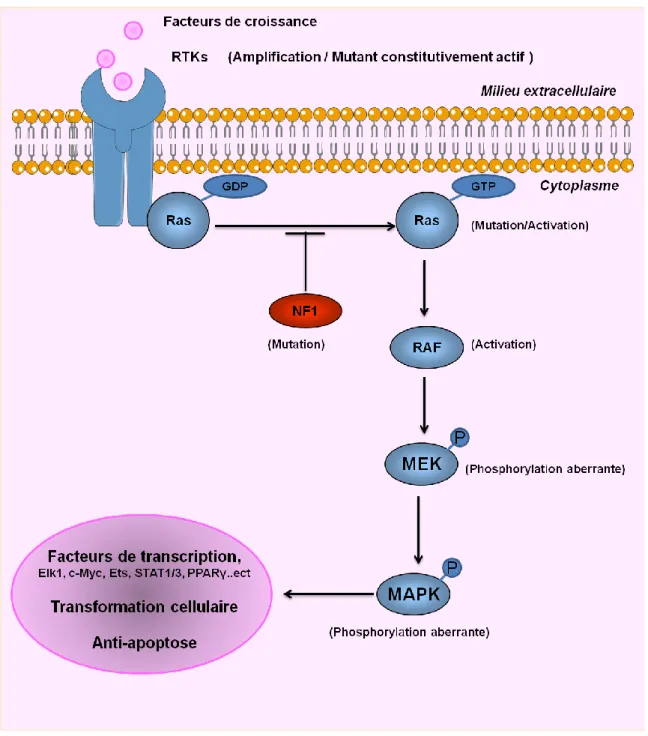
La transduction du signal en aval des RTK peut aussi activer la voie RAS/MAPK. RAS active les sérine-thréonine kinases (STKs) Raf, MAPK (ERK1 et ERK2), PI3K, et un certain nombre de protéines qui sont transloquées vers le noyau pour promouvoir la prolifération cellulaire, la différenciation et la survie (Figure 11). La kinase RAF activée phosphoryle et active la kinase MAPK (MAPKK), également appelée MEK. Une fois activée, MEK phosphoryle à son tour et active MAPK (Adjei et al., 2005). MAPK activée se transloque dans le noyau pour activer plusieurs facteurs de transcription qui induisent la progression du cycle cellulaire et la transcription de gènes anti-apoptotiques (Kapoor et al., 2003 ; Kapoor et al., 2003).
L’activation combinée de RAS et Akt dans les progéniteurs neuronaux induit la formation de GBM dans un modèle de souris (Holland et al., 2000). Bien que les mutations activatrices de RAS soient observées chez environ 30% des cancers humains (Bos et al., 1989), les mutations de RAS sont rares dans les GBM humains (2%, selon l'étude TCGA). Par conséquent, la dérégulation observée de la voie de signalisation Ras/RAF/MAPK dans les GBM est attribuée à ses régulateurs positifs en amont, dont l'EGFR et le PDGFR, qui sont hautement actifs dans la majorité des gliomes malins (Feldkamp et al., 1999 ; Guha et al., 1997).
Figure 11: la voie de signalisation Ras/MAPK. Les protéines RAS sont des petites protéines G et oscillent entre leurs deux conformation on/off (RAS-GTP/RAS-GDP). Cette conversion est contrôlée par les RTK et le gène suppresseur de tumeur NF-1. Ras activée (Ras-GTP) active la sérine-thréonine kinase RAF. Cette dernière active une MAPKK (mitogen-activated protein kinase kinase), appelée aussi MEK, qui à son tour active la MAPK. MAPK activée induit l’activation de nombreux facteurs de transcription, tels que Elk1, c-myc, Ets et STAT1/3 qui provoquent la transformation cellulaire et bloquent l’apoptose (schéma dessiné via Servier Medical Art, Adapté de Nakada et al., 2011).
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
23
Le gène suppresseur de tumeur NF-1 codant pour la neurofibromine fonctionne principalement comme un régulateur négatif de RAS et joue un rôle dans les voies de signalisation médiées par l'adénylyl cyclase et Akt-mTOR (Lee et al., 2007 ; Gottfried et al., 2010) (Figure 11). Dans l'étude pilote TCGA, des mutations/délétions homozygotes de NF-1 ont été identifiées dans 18% des GBM. Il semble que la perte de fonction du gène NF-1 par mutation entraîne une augmentation de la signalisation RAS qui est impliquée dans la tumorigenèse. De plus, une perte d’expression de NF-1 en absence d’altération du gène NF-1 a été observée dans les GBM (TCGA., 2008).
La signalisation aberrante par cette voie RAS/MAPK mène à la transformation cellulaire et à la résistance à l'apoptose. Ainsi, cette voie est une cible de choix pour le traitement du gliome malin (Krakstad et al., 2010).
5.3. La migration et l’invasion
Par nature, le GBM est fortement envahissant, mais ne métastase pas vers d'autres organes comme d’autres cancers. Les cellules de GBM ont tendance à migrer le long des vaisseaux, des dendrites et des fibres dans la substance blanche. Le caractère très infiltrant des gliomes humains est similaire au comportement migratoire des progéniteurs gliaux au cours du développement du système nerveux central, ce qui suggère que les activateurs, les récepteurs et les protéines de signalisation qui contribuent à la migration des cellules de la crête neurale sont des acteurs clés dans l'invasion des gliomes.
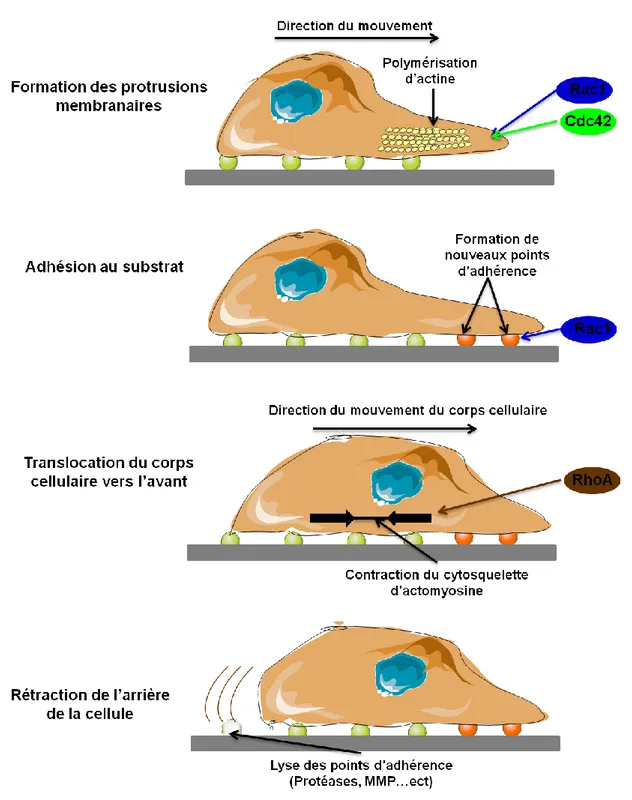
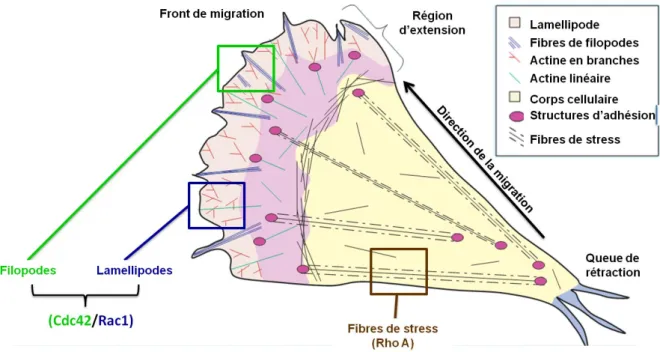
L’invasion des gliomes est un processus complexe impliquant : (1) le détachement du site d'origine, (2) l'adhérence à la MEC, (3) le remodelage de la MEC, et (4) la migration cellulaire (Nakada et al., 2007 ; Onishi et al., 2011). De nombreuses molécules sont impliquées dans chaque étape. Les mécanismes de ces différentes étapes sont détaillés ci-dessous et résumés dans la Figure 12.
5.3.1. Détachement du site d’origine
Afin d’envahir le stroma environnant, les cellules tumorales doivent d'abord se détacher de la masse tumorale naissante (primaire). Cette étape implique plusieurs événements (Figure 12) :
- la déstabilisation et la désorganisation des jonctions assurées par les molécules d'adhérence, cadhérine et NCAM,
Figu re 12 : Le s di ff ére nts méc anism es mol éc ulair es im pli qué s da ns l ’inva sion d es GBM.
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
24
- une diminution de l'expression de la connexine 43 (Cx43), ce qui conduit à une réduction de la formation des jonctions communicantes gap,
- le clivage de la protéine CD44, qui ancre la masse tumorale primaire à la MEC.
5.3.1.1. Les cadhérines
Un groupe important de molécules d’adhérence liées à l'invasion des gliomes est la superfamille des cadhérines. Les cadhérines sont des molécules d’adhérence cellulaire calcium-dépendantes, qui interviennent dans l'adhérence homophilique (cadhérine-cadhérine) entre deux cellules, et jouent un rôle important dans la construction des tissus et de la morphogenèse dans les organismes multicellulaires. Il existe une trentaine de membres de cadhérines, dont l’expression est spécifique du tissu : E-cadhérine (épithéliale), N-cadhérine (nerveuse), T-(ou H-) cadhérine et VE-cadhérine. Les E- et N-cadhérines, qui sont les plus étudiées à ce jour, se lient à une protéine cytosolique, la β-caténine (Gumbiner, 2005). Cette dernière se lie à une α-caténine reliant ainsi le complexe « cadhérine-caténine » au cytosquelette d'actine par une interaction avec les fibres de stress d'actine via l’α-actinine, une protéine liant l'actine. Une perte d'expression de l’E-cadhérine dans les tumeurs épithéliales est associée à un phénotype plus invasif et métastatique (Cavallaro and Christofori, 2004).
Dans tous les grades de malignité des gliomes, une faible expression de l’E-cadhérine a été rapportée, alors que des résultats contradictoires ont été obtenus concernant la participation de la N-cadhérine dans l'acquisition de propriétés invasives. Dans les lignées cellulaires de GBM, aucune corrélation n'a été trouvée entre l'expression de N-cadhérine et le comportement invasif ou la migration cellulaire (Shinoura, 1995), mais l'activité de l'invasion des cellules de GBM (U87 et U373) est supprimée par un inhibiteur de la N-Cadhérine (Takino et al., 2003 ; Kotelevets et al., 2001). Dans des biopsies chez des patients présentant un GBM, l'expression de la N-cadhérine est plus importante que dans le tissu normal de cerveau, mais est inversement corrélée à l'invasion (Asano, 2004). La T-cadhérine, qui est exprimée dans les astrocytes normaux mais pas dans les gliomes de haut grade, diminue la motilité cellulaire quand elle est transfectée de façon stable dans les cellules de gliomes (Huang et al., 2003). Ces résultats contradictoires ont conduit à l'hypothèse que l'instabilité et la désorganisation des jonctions médiées par les cadhérines, plutôt que la réduction de l'expression des composants du système cadhérine-caténine, favorisent la migration et le caractère invasif dans des lignées cellulaires de GBM (Perego et al., 2002).
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
25 5.3.1.2. Les NCAM
La NCAM est un membre de la superfamille des immunoglobulines. Cette protéine établit généralement des liaisons homophiliques. Il existe quatre isoformes de molécules NCAM (180, 170, 140 et 120 kDa) dans les neurones et les astrocytes du système nerveux central. Dans les tumeurs astrocytaires, l’expression de chaque isoforme NCAM diminue proportionnellement avec le grade histologique de malignité. L’expression des isoformes de 140 et 180 kDa de NCAM provoque une réduction significative de la motilité cellulaire et de la prolifération in vitro et in vivo (Sasaki et al., 1998 ; Owens et al., 1998 ; Prag et al., 2002).
5.3.1.3. La connexine 43 (Cx43)
La Cx43 est la largement exprimée dans le SNC et principalement dans les astrocytes (Dermietzel and Spray, 1993). En 1999, deux équipes ont mis en évidence l’implication des Cx43 dans l’invasion des GBM. En effet, l’équipe de McDonough et ses collaborateurs ont montré que la réduction de la formation des jonctions communicantes gap est corrélée avec une augmentation de la motilité des cellules de gliomes in vitro (McDonough et al., 1999). De plus, Zhang et ses collaborateurs ont montré l’existence de jonctions communicantes entre les astrocytes et les cellules de gliomes via la Cx43. Ceci induit une transformation phénotypique des astrocytes qui pourrait rendre le parenchyme cérébral permissif à l’invasion des cellules tumorales de GBM (Zhang et al., 1999 ; Sin et al., 2012).
5.3.1.4. La protéine CD44
CD44 est une glycoprotéine transmembranaire de la superfamille des immunoglobulines qui fonctionne comme une molécule d'adhérence et qui a pour ligand l'acide hyaluronique (HA). Ce dernier représente une fraction essentielle de la MEC du cerveau et est impliqué dans une grande variété de processus physiologiques et pathologiques. La forme standard de CD44 se compose d'une queue cytoplasmique, d'une région transmembranaire, et d'un large domaine extracellulaire (90 acides aminés). Dans des cellules tumorales, le CD44 est clivé par des protéases telles que les MMP, associées à la membrane. Cette protéolyse du CD44 joue un rôle essentiel dans le détachement efficace des cellules tumorales du substrat HA et favorise la migration des cellules. Des anticorps monoclonaux dirigés contre le CD44 entraînent une diminution de l’invasion intracérébrale des cellules de gliome in vivo et de l’invasion en Matrigel in vitro (Gunia et al., 1999 ; Merzak et al.,1994). Dans les GBM, la protéine CD44 peut être clivée par les protéases ADAM (A Disintegrin And
Introduction / Chapitre I. Les glioblastomes et leurs traitements Partie I : Les glioblastomes
26
Metalloproteinase) 10 et 17 ou par la MMP 9, ce qui favorise la migration cellulaire (Okamoto et al., 1999 ; Nagano et al., 2004 ; Chetty et al., 2012). Le produit de clivage de CD44 est détecté dans 60% des gliomes, mais pas dans le cerveau normal (Okamoto et al., 1999).
5.3.2. L'adhérence à la MEC
La migration cellulaire requiert la coordination parfaite entre l’adhérence et la désadhérence de la cellule à la matrice attenante. Les molécules les plus connues qui permettent l’adhérence des cellules de GBM à la MEC sont les intégrines.
Les intégrines sont des glycoprotéines transmembranaires qui forment des dimères entre 14 différentes sous unités α et 8 sous unités β. Les intégrines interagissent avec 2 classes majeures de ligands : (i) les protéines de la MEC telles que la fibronectine, la vitronectine et le fibrinogène, (ii) les membres de la famille des immunoglobulines tels que les molécules d’adhérence intracellulaire (ICAM-1, ICAM-2) et la vascular cell adhesion molecule (VCAM-1). Les intégrines agissent comme des ancreurs mécaniques à la MEC et comme des médiateurs de signaux via les protéines qui leur sont associées telles que CD47, la tétraspanine et les récepteurs des facteurs de croissance (Hemler., 1998).
Les intégrines ont été proposées pour jouer un rôle clé dans la biologie des gliomes incluant la migration cellulaire (D’Abaco and Kaye 2007). Plusieurs études ont montré la surexpression de la sous unité β1 dans les GBM en comparaison avec le tissu cérébral normal. Le blocage de la β1 par des anticorps entraine une diminution de la migration et de l’invasion des cellules de gliomes in vitro (Paulus et al. 1993 ; Rooprai et al. 1999 ; Tysnes et al. 1996). La surexpression expérimentale de la sous-unité α6 dans des cellules U87 de GBM humain augmente la migration et l’invasion cellulaire in vitro, et favorise l’infiltration et la diffusion des cellules U87 dans le cerveau des souris nude (Delmarre et al., 2009). De plus, des études immunohistochimiques ont démontré que les intégrines αvβ3 et αvβ5 sont surexprimées dans les cellules de gliome et que leur expression est corrélée au grade de la tumeur (Stupp et Ruegg., 2007). La stimulation des cellules de GBM humain avec les cytokines TGF-β1 ou TGF-β2 permet d’augmenter le taux d’intégrine αvβ3 à la surface cellulaire ce qui induit la migration des cellules des gliomes (Platten et al., 2000). Ces différents travaux indiquent donc que l’expression de diverses intégrines favorise la migration et l’invasion des cellules de GBM.