Expression de la dystrophine humaine dans le Tibialis anterior
de souris Rag/mdx suite à une greffe de cellules myogéniques dérivées
d’hiPSCs dystrophiques et corrigées génétiquement.
Mémoire
William-Édouard Gravel
Maîtrise en biologie moléculaire et cellulaire
Maître ès sciences (M.Sc.)
Québec, Canada
III
Résumé
Les cellules souches embryonnaires humaines (hESCs) et les cellules souches pluripotentes induites humaines (hiPSCs) ont démontré leur capacité d'auto-renouvellement et peuvent potentiellement se différencier en tous les types de lignées cellulaires. Elles représentent donc une source illimitée de cellules pour le développement de thérapies curatives pour les maladies dégénératives, telles que la dystrophie musculaire de Duchenne (DMD). Cette maladie héréditaire est le résultat de diverses mutations dans le gène de la dystrophine. Ces mutations engendrent un changement dans le cadre de lecture du gène de la dystrophine, abolissant ainsi son expression. Elle se caractérise cliniquement par une progression rapide de la dégénérescence musculaire qui débute tôt dans la vie. Les hiPSCs dystrophiques ont été corrigées par notre collaborateur, le Dr. Hotta, en insérant une paire de bases dans l’exon 45 avec les Transcription Activator-Like Effector Nucleases (TALENs) pour rétablir le cadre de lecture du gène. Notre laboratoire a mis au point une procédure en deux étapes pour différencier des hiPSCs en cellules myogéniques. Nous avons d'abord utilisé un milieu de culture myogénique préparé spécialement dans le laboratoire (appelé MB1) pour promouvoir la différenciation des hiPSCs en cellules de type mésenchymateuses. Nous les avons ensuite transduites avec un lentivirus exprimant MyoD, un facteur de transcription myogénique sous le contrôle du promoteur synthétique CAG, afin d'induire leur différenciation en myoblastes. Ces myoblastes modifiés ont été greffés dans le muscle Tibialis anterior d’une souris Rag/mdx, un animal immunodéficient et dystrophique, et ont par la suite fusionné avec les fibres musculaires existantes. La présence de la protéine dystrophine humaine a été confirmée par immunohistofluorescence dans les muscles greffés avec les cellules corrigées génétiquement ainsi que dans le contrôle positif réalisé avec des myoblastes provenant d'un donneur sain. La thérapie cellulaire homotypique à partir de cellules corrigées génétiquement présente de grands avantages pour les patients souffrant de DMD, car elle permet l’expression d’un gène capable de produire une dystrophine fonctionnelle dans les fibres musculaires, de diminuer les risques de rejet de la greffe et d’accroitre la capacité de régénération du muscle et la force musculaire.
V
Abstract
Human embryonic stem cells (hESCs) and human-induced pluripotent stem cells (hiPSCs) have shown self-renewal capacity and can potentially differentiate into all types of cell lineages. They represent an unlimited source of cells for the therapy of degenerative diseases, such as Duchenne Muscular Dystrophy (DMD), a disease characterized by a rapid degeneration of muscles that starts early in life. Dystrophic hiPSCs have been corrected by our collaborator, Dr. Hotta, by inserting of a single base pair in the exon 45 with Transcription Activator-Like Effector Nucleases (TALENs) to restore the reading frame of the gene. Our laboratory has developed a two-step procedure to differentiate hiPSCs into myogenic cells. We first used a myogenic culture medium especially developped in the laboratory (called MB-1) to promote the differentiation of hiPSCs into mesenchymal-like precursor cells. We next transduced them with a lentivirus expressing the myogenic transcription factor MyoD under the control of the composite CAG promoter, in order to induce their differentiation into myoblasts. Transduced cells have been grafted in the Tibialis anterior muscle of Rag/mdx mice where they fused with existing muscle fibers. The presence of the human dystrophin protein has been confirmed by immunohistofluorescence in muscles grafted with the genetically corrected cells and in a control graft with myoblasts of a healthy donor. Cell therapy shows great promises for DMD patients since it allows the expression of a normal gene capable of producing a functional dystrophin in muscle fibers and increase the regenerative capacity of the muscle and the muscle strength.
VII
Tables des matières
Résumé ______________________________________________________________________ III Abstract ______________________________________________________________________ V Tables des matières ____________________________________________________________ VII Liste des Tableaux _____________________________________________________________ IX Liste des figures _______________________________________________________________ XI Liste des abréviations __________________________________________________________ XIII Remerciements ______________________________________________________________ XVII Introduction ___________________________________________________________________ 1
CHAPITRE 1 La myogenèse ______________________________________________________ 3
1.1 Mésoderme embryonnaire et tissu squelettique adulte _______________________________ 3 1.2 MyoD et les facteurs de régulations myogéniques ___________________________________ 5
CHAPITRE 2 La dystrophie musculaire de Duchenne (DMD) __________________________ 7
2.1 Description et diagnostic de la maladie ___________________________________________ 7 2.2 Le gène et la protéine dystrophine _______________________________________________ 8 2.3 La souris mdx, un modèle de la DMD ___________________________________________ 10
CHAPITRE 3 Traitements et stratégies thérapeutiques ______________________________ 13
3.1 Traitements pharmacologiques et essais cliniques __________________________________ 13 3.2 Édition du génome, thérapie cellulaire et génique __________________________________ 14 3.3 Les vecteurs viraux : le lentivirus basé sur le VIH de type 1 __________________________ 18
CHAPITRE 4 Les cellules souches ________________________________________________ 21
4.1 Les cellules souches embryonnaires humaines ____________________________________ 21 4.2 Cellules souches pluripotentes induites __________________________________________ 23
CHAPITRE 5 Expression de la dystrophine humaine dans les muscles de souris Rag/mdx suite à une greffe de cellules myogéniques dérivées d’hiPSCs dystrophiques génétiquement corrigées. __ 25 5.1 Introduction _______________________________________________________________ 25 5.2 Hypothèse et objectifs _______________________________________________________ 26 5.3 Matériel et Méthodes ________________________________________________________ 27 5.3.1 Construction du vecteur de transfert ___________________________________________ 27 5.3.2 Vecteur lentiviral (FUGW_CAG_MyoD_P2A_eGFP) ____________________________ 31 5.3.3 Culture cellulaire __________________________________________________________ 34 5.3.4 Greffes et immunohistochimies ______________________________________________ 37 5.4 Résultats __________________________________________________________________ 39 5.4.1 Construction du vecteur de transfert lentiviral ___________________________________ 39 5.4.2 Formation de myotubes _____________________________________________________ 43 5.4.3 Détection de la chaîne lourde de la myosine in vitro ______________________________ 46 5.4.4 Immunobuvardage de MyoD et de la dystrophine in vitro __________________________ 47 5.4.5 Greffe des cellules dans les muscles TA de souris Rag/mdx ________________________ 48 5.5 Discussion ________________________________________________________________ 51 5.6 Conclusion et perspectives ____________________________________________________ 53 Bibliographie _________________________________________________________________ 55
IX
Liste des Tableaux
Tableau 1 . Séquences des amorces utilisées pour la construction de l’insert à introduire dans le
vecteur FUGW. ... 28
XI
Liste des figures
Figure 1. Schéma d’une coupe transversale d’un embryon durant la myogenèse embryonnaire. .. 4
Figure 2. Formation de fibres musculaires. ... 5
Figure 3. Hiérarchie des facteurs régulateurs myogéniques principaux précoce dans l’embryogenèse et la formation du tissu squelettique adulte. ... 6
Figure 4. Le gène de la dystrophine. ... 8
Figure 5. Le complexe de la dystrophine. ... 9
Figure 6. Modèles murins expérimentaux pour la DMD.. ... 11
Figure 7. Génération d’une coupure dans l’ADN par une paire de TALENs. ... 15
Figure 8. Schématisation des étapes d’une greffe de myoblastes allogéniques. ... 16
Figure 9. Comparaison des génomes du lentivirus sauvage HIV-1 et du lentivirus recombinant de 3e génération. ... 18
Figure 10. Isolation des cellules souches embryonnaires et culture cellulaire. ... 22
Figure 11. Utilisation des hiPSCs en thérapie génique et en thérapie cellulaire.. ... 24
Figure 12. Cartographie du vecteur FUGW. ... 27
Figure 13. Construction du vecteur multicistronique. ... 29
Figure 14. Transfection par la méthode de phosphate de calcium. ... 33
Figure 15. Amplifications des gènes et analyse sur gel d’agarose.. ... 39
Figure 16. Analyse sur gel d’agarose après la digestion du vecteur original FUGW. ... 40
Figure 17. Analyse sur ge ; d’agarose après la digestion du vecteur original FUGW avec l’insertion ... 40
Figure 18. Analyse SDS-PAGE pour détecter la protéine MyoD humaine ... 41
Figure 19. Analyse SDS-PAGE pour détecter la protéine eGFP. ... 41
Figure 20. Analyse sur gel d’agarose et construction du vecteur de transfert lentiviral.. ... 42
Figure 21. Photographie au microscope optique de myotubes humains sains (BB 13M). ... 43
Figure 22. Photographie au microscope optique des MSCs-L DMD- transduites par le pseudo-lentivirus MyoD_eGFP. ... 44
Figure 23. Photographie au microscope optique des MSCs-L IFH30 transduites par le pseudo-lentivirus MyoD_eGFP.. ... 45
Figure 24. Analyse d’immunocytochimie de l’expression de la chaîne lourde de la myosine dans les différents types cellulaires. ... 46
Figure 25. Immunohistochimie sur des cryo-sections consécutives de muscles de souris greffés avec des myoblastes humains normaux.. ... 48
Figure 26. Immunomarquage de différentes cryo-sections consécutives des muscles greffés avec des MSCs-L DMD (-/-). ... 49
Figure 27. Immunomarquage de différentes cryo-sections consécutives de muscles greffés avec des MSCs-L IFH30.. ... 50
XIII
Liste des abréviations
ADN = acide déoxyribonucléique ADNc = ADN complémentaire ARN = acide ribonucléique ARNm = ARN messager ATP = adénosine triphosphate A.A = acide aminée
CAG = promoteur synthétique (cytomegalovirus early enhancer/chicken β-actin) CRISPR = clustered regularly interspaced short palindromic repeats
DMD = dystrophie musculaire de Duchenne DM = dermomyotome
ESCs = cellules souches embryonnaires
FUGW* = forme circulaire de FUGW (9941)
FUGW (eGFP) = Forme linéarisé de FUGW sans eGFP (9228 kb)
hESCs = ESCs humaines hiPSCs= iPSCs humaines
IGF = Facteur de croissance de l’insuline
iPSCs = cellules souches induites à la pluripotente HRP = peroxydase de raifort (horseradish peroxidase) Kb = kilo base
Mb = mégabase KDa = kilo Dalton
MCK = créatine kinase, liée au muscle
Mdx = dystrophie musculaire liée au chromosome X (muscular dystrophy X-linked) MEF = fibroblastes embryonnaire de souris
MY = myotome
MPCs = cellules myogéniques progénitrices mg = milligramme
mm = millimètre ml = millilitre Mm = Milli molaire
MI = multiplicité d’infection
MRFs = facteurs régulateurs de la myogenèse MSCs = cellules mésenchymateuses
MRF4 = facteur régulateur musculaire 4 Myf5 = facteur myogénique 5
MHC = chaine lourde de la myosine
MyoD = facteur de différenciation myogénique MyoG = myogénine
LTR = longue région répétitive ONA = oligonucléotide anti-sens
Pax 3/7 = facteur de transcription paired box 3 et 7 pb = paire de base
PBS = solution tampon de phosphate PCR = réaction en chaîne par polymérase
XIV
SDS-PAGE = sodium dodecyl sulfate polyacrylamide gel electrophoresis TA = Tibialis anterior
TALENs = Transcription activator-like effector nucleases Tris-HCl = Tris (hydroxymethyl) aminomethane hydrochloride µl = microlitre
µg = microgramme µm = micromètre V = volts
XV
XVII
Remerciements
Tout d’abord, je remercie ma femme Laurence, qui a su m’épauler durant toutes ces années de vie commune, et surtout durant mes deux années de maîtrise, qui ont été de loin les plus émouvantes et importantes de ma vie.
Je remercie du fond du cœur le Dr.Tremblay qui m’a accordé la chance de me faire valoir durant un stage en mai 2013 et qui a cru en moi en m’offrant l’opportunité de réaliser un programme de maîtrise. Il a vu en moi un potentiel, et ce malgré mes notes durant mon baccalauréat en biologie. De plus, je veux souligner le travail incroyable que Dominique Ouellet a accompli en me prenant sous son aile durant mon parcours académique. Elle m’a permis de me dépasser autant au niveau personnel, j’ai couru plus de 350 km en deux ans et accompli un premier demi-marathon grâce à sa motivation contagieuse, et au niveau professionnel par le partage de ses connaissances. Je remercie aussi Chantale Maltais qui m’a aidé à développer mes habiletés et ma confiance en matière de culture cellulaire. Grâce à elle, je suis maintenant un pilier de la de culture d’IPS dans le laboratoire. Sa minutie ainsi que sa méthodologie m’ont été fort inspirantes. Je remercie également Catherine Gérard pour avoir exécuté la lourde tâche de greffer mes cellules et d’avoir gardé le moral malgré les hauts et les bas des résultats des greffes. Je remercie aussi Véronique Dorval pour avoir réussi à me faire surmonter ma crainte des fautes d’orthographe et de m’avoir encouragé tout au long de la rédaction de ce mémoire. Finalement, je remercie l’homme qui à vue l’homme qui à vue l’ours, Joël Rousseau, de m’avoir si bien introduit dans l’équipe, et ce dès la première journée avec ses premières paroles si sages, en m’abordant avec une P1000 à la main. J’aimerais remercier mes égaux au sein du laboratoire. Arnaud ‘‘Immortal King’’ Perrin, Benjamin ‘‘Bridou’’ Duchenne, Daniel ‘‘Mexicain’’ Agudelo et Jean-Paul ‘‘Invendable’’ Iyombe. Sans vous les amis, mon cheminement ne serait pas aussi parfait.
1
Introduction
Ce mémoire met de l’avant le développement d’une thérapie cellulaire dans l’optique de produire un traitement curatif pour les patients atteints de la dystrophie musculaire de Duchenne (DMD). Cette thérapie se base sur l’utilisation de cellules souches pluripotentes induites humaines (hiPSCs) corrigées génétiquement par édition du génome. Certains thèmes comme la myogenèse, et le tissu musculaire squelettique, la dystrophie musculaire de Duchenne, ses traitements ainsi que le modèle animal expérimental et les cellules souches pluripotentes sont tout d’abord introduits pour permettre une compréhension globale de mon projet. Par la suite, une description approfondie du projet de recherche sera exposée et se résume par l’expression positive de la dystrophine humaine dans les tissus du Tibialis anterior de souris Rag/mdx suite à une greffe d’hiPSCs dystrophiques génétiquement corrigées.
3
CHAPITRE 1
La myogenèse
La myogenèse est le processus de formation du muscle et de sa réparation. Elle implique un cycle d’alternance de phases de différenciation et de quiescence cellulaires. La phase de différenciation débute dès l’embryogenèse et est active durant la réparation musculaire. Celle de la quiescence est liée à l’homéostasie du muscle et à son maintien durant son développement. Le tissu musculaire squelettique est généralement stable et son homéostasie est finement régulée. Lorsqu’il est sain, ce tissu accomplit l'ensemble de ses fonctions, soit la locomotion, la gestion de la posture et la respiration.
1.1 Mésoderme embryonnaire et tissu squelettique adulte
Lors de l’embryogenèse, la phase de gastrulation conduit à la formation des feuillets embryonnaires. Il existe trois types de feuillets distincts, soit l’ectoderme, le mésoderme et l’endoderme. Le feuillet externe, l’ectoderme, donne naissance à la peau et le feuillet interne, l’endoderme, assure la croissance du système digestif ainsi que des glandes connexes. Le mésoderme, feuillet central, produit une partie du système circulatoire en formant les vaisseaux sanguins et se développe en une partie du système locomoteur, soit les muscles squelettiques et le squelette [1].
Durant le développement du feuillet mésodermique, il y a formation d’une structure primitive, les somites, qui se différencient en divers axes géo-spatiaux pour engendrer la formation de structures diversifiées (figure 1) [2]. La section d’intérêt est la portion épithéliale dorsale du somite, car elle devient le dermomytome (DM). Une fine couche du DM se différencie en myotome (MY) tandis qu’une autre partie se désintègre pour faire place aux cellules myogéniques progénitrices (MPC), lesquelles formeront le bassin de cellules satellites postnatales du muscle [3].
4
Figure 1. Schéma d’une coupe transversale d’un embryon durant la myogenèse embryonnaire. Les structures du dermomyotome (DM) et du myotome (MY) sont à l’origine de
la formation du muscle squelettique adulte [3].
L’engagement du mésoderme dans la voie myogénique débute quand des cellules exprimant divers facteurs transcriptionnels commencent à proliférer et à former une population de myoblastes. Une différenciation terminale en myocytes suivie par leur fusion engendre un syncytium multi-nucléés, soit les myotubes. Une fois matures, les myotubes deviennent des fibres musculaires contractiles (figure 2). Les myoblastes non différenciés font partie du bassin des cellules satellites adultes et résident à proximité des fibres [4]. Ces dernières ont été caractérisées comme étant une population myogénique, mononuclée, non différenciée, qui contribue à l’homéostasie du muscle [3, 4].
5 Figure 2. Formation de fibres musculaires. Les cellules du mésoderme se différencient en
myoblastes ou en cellule satellites. Les myoblastes deviennent des myocytes avant de fusionner en myotubes et ainsi former une fibre musculaire. Les cellules satellites peuvent se différencier en myoblastes et ensuite former des myotubes ou seulement migrer dans le muscle entre les myotubes où elles attendent le moment opportun pour proliférer et se différencier [4].
1.2 MyoD et les facteurs de régulations myogéniques
À la fin des années 1980, une expérience réalisée avec des fibroblastes de souris et un agent de déméthylation de l’ADN a démontré l’activation de l’expression des facteurs régulateurs de la myogenèse (MRF) et a permis le clonage du gène MyoD. De multiples expériences ont été réalisées en transfectant ce facteur transcriptionnel sur une grande variété de cellules, incluant des cellules nerveuses, des adipocytes et des cellules hépatiques. Toutes ont été converties en cellules musculaires squelettiques [5, 6], démontrant ainsi que ce seul gène peut initier une différenciation myogénique. MyoD est reconnu comme le gène maître de la différenciation cellulaire en cellules squelettiques musculaires [3]. Ce gène est composé de 963 paires de base (bp), et code pour une protéine de 320 acides aminés (aa), soit une taille moléculaire d’environ 37 kilo Dalton (kDa). MyoD, de concert avec le facteur myogénique 5 (Myf5), est impliqué dans la prolifération des myoblastes et ces facteurs de transcription activent le facteur de régulation myogénique 4 (Mrf4) et la myogénine (MYOG) qui sont impliqués dans la différenciation terminale des myotubes[7]. L’analyse détaillée de la myogenèse a permis de définir une échelle hiérarchique des facteurs de transcription responsables de la différenciation d’une lignée cellulaire en cellules musculaires squelettiques (figure 3).
6
L’expression de ces facteurs impliqués dans la progression d’une lignée myogénique n’est pas linéaire. Elle se caractérise par un réseau d’interactions positives et négatives complexes. Parmi cet ensemble de facteurs, Pax3 et Pax7 sont exprimés tôt dans le développement du muscle. Le rôle de Pax3 est relié à la croissance et la spécialisation des somites chez l’embryon, tandis que celui de Pax7 est indispensable pour le développement musculaire embryonnaire. De plus, Pax7 contribue à la formation des premières fibres musculaires et permet l’établissement du bassin de cellules satellites [8]. Des études réalisées chez des souris déficientes dans l’expression de Pax3 et/ou Pax7 démontrent un phénotype létal ou une défaillance musculaire [9].
Figure 3. Hiérarchie des facteurs régulateurs myogéniques principaux précoce dans l’embryogenèse et la formation du tissu squelettique adulte. L’expression des gènes Pax3 et
Pax7, observée dans les MPC, est suivie de l’expression de Myf5 et de MyoD, lesquels engagent les cellules dans la voie musculaire. Les gènes « tardifs » MyoG et MRF4 accomplissent la différenciation terminale en myotubes [3].
7
CHAPITRE 2
La dystrophie musculaire de Duchenne (DMD)
Les premiers cas de DMD ont été rapportés au début du 19e siècle. C’est en 1858 que le
physiologiste français Guillaume Benjamin Duchenne en fait la première description clinique. En 1868, Duchenne nomma cette maladie la « paralysie musculaire pseudo-hypertrophique » [10]. Il a été le premier à établir des critères diagnostiques bien précis.
2.1 Description et diagnostic de la maladie
La myopathie de Duchenne est une maladie récessive dont l'incidence est de 1 cas sur 3 500 naissances mâles, ce qui la classe dans la catégorie des maladies dites “orphelines” ou rares (fréquence <1/2000). Parmi les myopathies, c'est toutefois l'une des plus fréquentes. Elle est observée dans toutes les populations. Étant létale, il s'agit de la forme de dystrophie la plus sévère. Cliniquement, la maladie se caractérise par une diminution progressive de la force musculaire débutant en très bas âge, vers 2 à 3 ans. À ce stade de l’enfance, les symptômes sont peu apparents, mais les signes cliniques sont présents.
Progressivement, le jeune enfant développera une démarche pathologique et il est alors possible d’observer des pseudo-hypertrophies musculaires. L’apparition de la manœuvre de Gowers vers l’âge de 5 à 7 ans mettra en évidence la perte progressive musculaire. Ce phénotype indique que l’ensemble des muscles localisés dans les jambes et les hanches sont trop faibles pour soulever l’enfant du sol. Au stade juvénile, le patient est confiné dans un fauteuil roulant, ses muscles étant incapables de fournir le support et la force pour le soutenir.
Finalement, l’apparition de problèmes respiratoires oblige les personnes atteintes à utiliser un système de respiration assistée. L’espérance de vie des patients oscille entre 20 et 30 ans et les causes principales de décès sont en lien avec les infections respiratoires ainsi que des insuffisances au niveau respiratoire et cardiaque [11]. Dû au fait que la DMD est considérée comme une maladie « orpheline », il n’y aucun test néonatal exécuté, sauf s’il y a présence de la maladie dans le « pedigree » familial.
8
Dans ce cas, un diagnostic génétique prénatal peut être effectué par amniocentèse. Chez l’enfant, le dépistage de la maladie se fait par une analyse sanguine du taux de créatine kinase. Cette enzyme est en forte concentration dans le sang, car elle catalyse la libération de l’énergie (ATP) qui sert à différents organes, dont le cerveau et le muscle [12]. Par conséquent, une concentration sérique élevée de créatine kinase suggère que cette enzyme a été libérée dans le milieu extracellulaire suite à la rupture de la membrane des fibres musculaires durant les contractions.
2.2 Le gène et la protéine dystrophine
La DMD est due à plusieurs types de mutations dans le gène de la dystrophine localisé sur le chromosome X (Xp21)[13]. À ce jour, ce gène est reconnu comme étant le plus long gène codant dans le génome humain. Il est composé de 2,5 Mb d’ADN et contient 79 exons. L’ARNm pleine longueur transcrit à partir du site d’initiation de la transcription Dp427(M) fait plus de 14 kb et permet la synthèse de l’isoforme musculaire pleine longueur de la protéine dystrophine (figure 4). Cette isoforme est exprimée en prédominance dans les muscles squelettiques et cardiaque, le cerveau et les cellules de Purkinje localisées dans le cervelet. Des isoformes plus courtes sont présentes dans la rétine et les cellules de Schwann [14-16]. Environ 60% des mutations dans le gène de la dystrophine sont de grandes insertions ou délétions génétiques tandis que 40 % sont des mutations ponctuelles. Toutes ces mutations conduisent à un changement du cadre de lecture qui introduit un ou plusieurs codons « stop » prématurés, résultant en une absence de protéine fonctionnelle [17].
Figure 4.Le gène de la dystrophine. Le gène est composé de 2,5 Mb d’ADN et code pour sept isoformes
de la dystrophine dont l’expression localisée dans des tissus bien spécifiques [17]. L’ARNm de la dystrophine musculaire est transcrit à partir du site d’initiation Dp427(M).
9
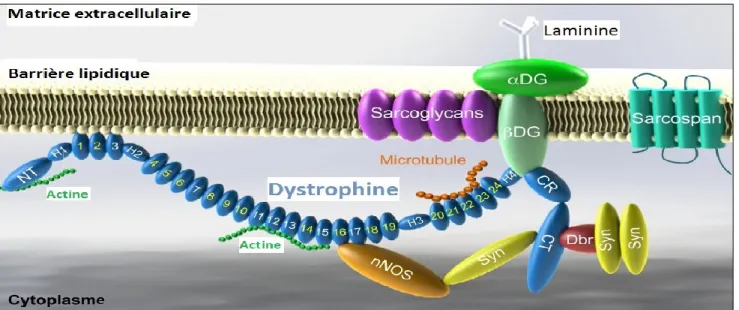
La protéine dystrophine musculaire a un poids moléculaire de 427 kDa et est constituée de 3685 aa. Elle est un membre de la famille des protéines -spectrine/-actinine. La structure de la dystrophine se divise en quatre domaines principaux bien distincts (figure 5). Le domaine de liaison à l’actine se trouve à l’extrémité N-terminale. Le domaine tige est composé de 24 répétitions de type spectrine en tandem, de style triple hélice, séparées par quatre domaines charnières qui contribue à augmenter la flexibilité de la protéine. Le domaine riche en cystéine et le domaine situé en C-terminal qui est spécifique à la dystrophine et permet le lien avec la dystrobrévine (Dbr). La dystrophine est localisée à la surface cytoplasmique du sarcolemme et permet l’ancrage du cytosquelette à la matrice extracellulaire via un complexe de glycoprotéines liées à la dystrophine (DGC). De plus, elle protège l’intégrité du sarcolemme et assure la transduction de la force tout au long de la fibre lors des contractions musculaires.
Figure 5. Le complexe de la dystrophine. La dystrophine est localisée au niveau du sarcolemme
et est associée à un complexe transmembranaire qui, ensemble, est relié au cytosquelette au niveau de la lame basale. Figure adaptée de Joe W. McGreevy et al. 2015 [18].
Chez le patient DMD, l’absence de dystrophine font que les contractions musculaires provoquent des ruptures membranaires entrainant un cycle de destruction et réparation des myofibrilles. Ces cycles épuisent le réservoir de cellules satellites au point où elles deviennent sénescentes, ce qui entraîne la dégénération musculaire progressive. De plus, l’absence de la dystrophine mène à de multiples micro-ruptures du sarcolemme et cause des fuites d’ions qui augmentent la nécrose. Finalement, le décès survient lorsque les tissus musculaires cardiaques et respiratoires sont complètement détériorés [19-21].
10
2.3 La souris mdx, un modèle de la DMD
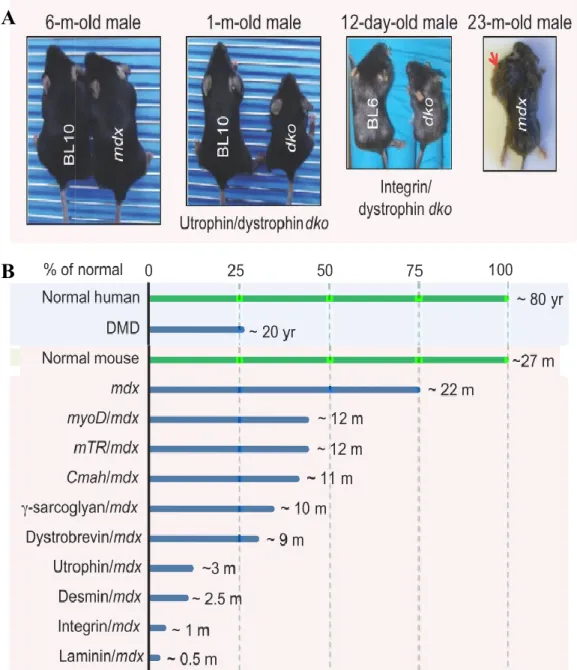
Plus de 60 modèles animaux, incluant des modèles canins, félins ou même primates non humain, ont été développés à des fins expérimentales pour la pathologie de la DMD [18]. Le modèle murin le plus fréquemment utilisé pour l’étude de la DMD est la souris mdx. Ce modèle provient de l’analyse d’une colonie de souris de type C57BL/10ScSn qui démontraient des taux élevés de créatine kinase ainsi que des évidences d’une myopathie observable sur des coupes de muscles [18]. Il a été mis en évidence que les souris possédaient une mutation ponctuelle non-sens dans l’exon 23 de la dystrophine, résultant en un codon stop prématuré et, par conséquent, produit une protéine tronquée . Bien que ce modèle murin mime la pathologie chez l’humain, il demeure que le phénotype de la longévité observé chez la souris diffère largement de celui observé chez l’homme.
En effet, une souris dystrophique perd seulement environ 25% de sa durée de vie et ce, même si elle n’exprime aucune dystrophine, comparativement à l’homme qui voit sa durée de vie raccourcie de 75% [22, 23]. Une raison possible pour expliquer cette différence est la surexpression des gènes codant pour l’utrophine et l’α7β1-intégrine chez la souris et pas chez l’humain. Ces protéines ont des fonctions similaires à celle de la dystrophine au niveau du cytosquelette [23]. Ceci a été mis en évidence lors de la délétion génique de ces fonctions [24]. De plus, il est possible de modifier le phénotype des souris mdx par mutagenèse dans divers gènes, tels les gènes spécifiques aux muscles squelettiques comme ceux qui codent pour les facteurs de régulation myogénique (MRF) ou encore les gènes codant pour des protéines qui interagissent avec le complexe associé à la dystrophine (figure 6).
Dans l’optique de développer de futures thérapies cellulaires, diverses souches de souris mdx ont été croisées avec des souris provenant de lignées immuno-déficientes pour limiter les réponses du système immunitaire et éviter le rejet de xénogreffes [18, 23, 25].
La souris Rag est très populaire comme modèle immunodéficient car elle possède une mutation dans le gène RAG-1 ce qui permet l’arrêt de la production de lymphocytes B et la différenciation des lymphocytes T mature [26]. Le modèle de souris Rag/mdx est celui que nous utilisons dans le cadre du présent projet de recherche puisse qu’elle mime la maladie et est immunodéficiente.
11 Figure 6. Modèles murins expérimentaux pour la DMD. A) Comparaison phénotypique entre
une souris sauvage (BL10), le modèle mdx et certains dérivés. B) Représentation de la durée de vie d’un individu sain comparée à celle d’un patient DMD, ainsi que celles de différents modèles murins expérimentaux pour la DMD comparées à une souris normale. Figure adaptée de Joe W. McGreevy et al. 2015 [18].
A
13
CHAPITRE 3
Traitements et stratégies thérapeutiques
Les traitements et les soins offerts pour les patients DMD ne permettent que de retarder et de réduire certains symptômes de la maladie. L’utilisation d’orthèses ou d’un fauteuil roulant aide à prévenir la dégénération des muscles. La chirurgie vise la réparation des scolioses causées par l’affaiblissement des muscles dorsaux . L’emploi de la ventilation artificielle aux stades tardifs de la maladie sont des exemples de soins palliatifs, auxquels peuvent s’ajouter des traitements pharmacologiques, dont deux ont été étudiés en essais cliniques [27].
3.1 Traitements pharmacologiques et essais cliniques
Bien qu’ils ne soient pas curatifs, les traitements avec des glucocorticoïdes permettent le renforcement de la force musculaire, la réduction des risques de scoliose ainsi que le ralentissement du développement des complications respiratoires et cardiaques. La Prednisolone ou encore le Deflazacort sont administrés dès que le diagnostic est confirmé avant la détérioration musculaire. Les traitements à long terme aux glucocorticoïdes engendrent des effets secondaires tels le gain de poids, un déséquilibre hormonal et l’ostéoporose [28].
Les essais cliniques en cours utilisent l’administration de molécules ayant pour but de produire une protéine tronquée fonctionnelle soit en ignorant un codon d’arrêt pour les patients qui ont une mutation stop ponctuelle, ou soit en restaurant le cadre de lecture du gène de la dystrophine par le saut d’un ou plusieurs exons.
Dans la première étude, les petites molécules de gentamicine ou d’ataluren ont été administrées dans le but de négliger le codon stop prématuré. Bien que les résultats avec la gentamicine sont très variables et parfois nuls, l’ataluren offre de meilleurs résultats en ne démontrant pas de sensibilité aux codons stop prématurés UAG et UAA [29]. Les résultats de phase IIa ont démontré que 61% des patients DMD traités avec l’ataluren montrent une augmentation de l’expression de la dystrophine [30].
14
La seconde stratégie est le saut d’exon, lequel est basé sur le retrait du codon stop prématuré. L’utilisation d’oligonucléotides antisens (ONA) complémentaires à une séquence cible de l’ARN pré-messager promeut l’épissage de l’exon muté pour produire une protéine plus courte, mais fonctionnelle. Les résultats obtenus lors des premiers essais cliniques s’avèrent variables, allant jusqu’à 55 % de fibres positives pour la dystrophine chez un patient [31]. Malgré l’avenir prometteur de cette méthode, les essais cliniques ont été arrêtés durant la phase III, car les traitements ne démontraient aucune amélioration significative en comparaison à l’administration d’un placebo [32].
3.2 Édition du génome, thérapie cellulaire et génique
Un désavantage des traitements pharmacologiques est qu’ils demandent l’administration continue des molécules thérapeutiques. L’avancement des connaissances et des technologies en génétique humaine permet maintenant de modifier le génome de cellules malades afin de rétablir leurs fonctions, et ce de façon permanente.
Édition du génome
Il est possible de cibler spécifiquement une séquence d’ADN mutée causant une maladie et de la corriger directement à l’aide de complexes protéiques exogènes [33]. Le mécanisme implique la génération de coupures dans une région précise de l’ADN génomique, ce qui enclenche la réparation des cassures, l’introduction ou l’élimination de quelques nucléotides, ce qui permet dans un cas sur trois de rétablir le cadre de lecture et la poursuite de la transcription normale du gène. Les Zinc-Finger Nucleases (ZFN), les Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) et les Transcription Activator-Like Effector-Nucleases (TALENs) [34] sont parmi ces systèmes de nucléases. Ces dernières dérivent des Transcription Activator-Like Effectors (TALEs), lesquels ont été découverts chez une bactérie pathogène exclusive au groupe de plantes du genre Xanthomonas. L’étude de cette classe de protéines a permis de caractériser un nouveau site de liaison à l’ADN, soit le domaine de répétition TALE. Le phénotype sauvage TALE reconnait une séquence cible dans l’ADN génomique de la cellule hôte et active la transcription de gènes promouvant l’infection [34, 35].
15
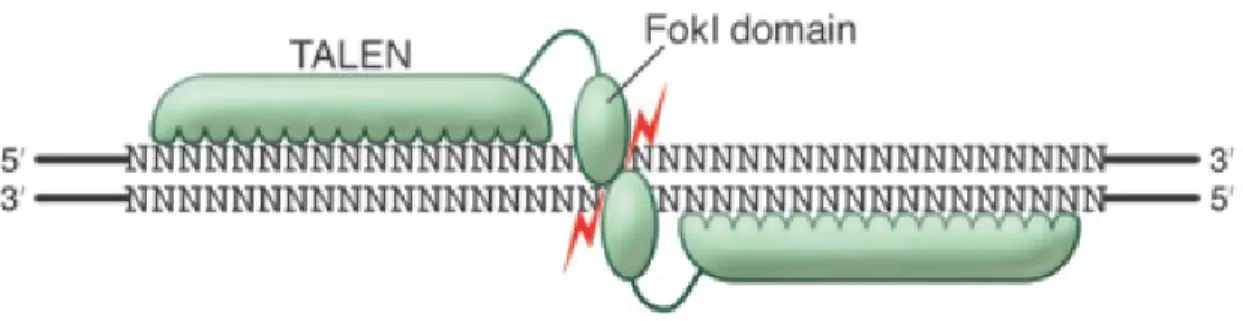
Par ingénierie moléculaire, il a été possible de créer les TALENs en modifiant les TALEs pour cibler les régions mutées du gène de la dystrophine et en les combinant à l’endonucléase FokI. Les TALENs reconnaissent non seulement une séquence spécifique de nucléotides mais induisent aussi une coupure double brin dans l’ADN (figure 7) [36]. Après la coupure, des mécanismes de réparation de l’ADN s’activent. La réparation peut se faire par recombinaison homologue ou jonction d’extrémités non homologues (NHEJ) [37]. Les TALENs peuvent cibler théoriquement n’importe quelle séquence d’ADN qui est précédée par une thymidine [33, 38]. C’est le potentiel de cette technologie qui a été utilisé pour corriger le gène de la dystrophine dans notre étude.
Figure 7. Génération d’une coupure dans l’ADN par une paire de TALENs. Les TALENs se
lient spécifiquement à une séquence de nucléotides et l’endonucléase FokI génère une coupure double brin dans l’ADN. Adapté de Rajat M. Gupta et al, 2015 [36].
Thérapie cellulaire
Le concept de thérapie cellulaire consiste à greffer des cellules saines à un patient. La greffe peut être de type allogénique dans le cas où les cellules proviennent d’un donneur sain (figure 8), ou de type autologue lorsque les cellules sont propres au patient. Lors d’une greffe autologue on doit préalablement corriger génétiquement les cellules à transplanter ex vivo. Plusieurs essais cliniques ont été réalisés avec des greffes de myoblastes sains, avec des résultats variables. Aux cours des premiers essais cliniques de 1990 à 1995, plusieurs résultats négatifs ont été obtenus à cause de la mort des cellules greffées, du faible taux de migration des cellules à travers le muscle de l’hôte et du rejet immunitaire [18]. Par la suite, notre laboratoire a réalisé en 2005 un essai clinique de Phase I au cours duquel une greffe de myoblastes provenant d’un donneur sain haplocompatible a été faite chez neuf patients DMD immunosupprimés avec le Tacrolimus [39, 40]. Cet essai clinique a permis de détecter dans les muscles greffés la présence du gène normal de la dystrophine, l’expression de son ARNm et la présence de la protéine dystrophine normale.
16
Dans le but d’améliorer les résultats suite à une greffe myoblastes sains, il est possible d’augmenter l’aire de la zone greffé en promouvant la migration des myoblastes grâce à une co-injection de facteurs de croissance comme ‘‘insulin-like growth factor-1 (IGF)’’ ou encore ‘‘basic fibroblast growth factor (bFGF)’’ [41]. Une autre alternative pour prévenir le rejet de greffe est l’emploi d’agents immunosuppresseurs comme le tacrolimus [42]. L’augmentation de la quantité de cellules greffées et une forte densité d’injections sur l’ensemble du muscle augmentent aussi le succès des greffes [43].
Figure 8. Schématisation des étapes d’une greffe de myoblastes allogéniques. (i) Prélèvement
d’une biopsie de tissu musculaire d’un donneur sain afin d’obtenir des fibres musculaires. (ii-iii) Libération des cellules satellites du muscle donneur pour la mise en culture. (iv) Prolifération et isolation des myoblastes. (v) Les myoblastes obtenus sont injectés dans les muscles d'un patient DMD. Adaptée de Tremblay et al. 2001[44].
17
Thérapie génique
En 1990, le concept théorique de transférer chez un pateint une copie fonctionnelle d’un gène est devenu réalité. Le gène fonctionnel de l’adénosine déaminase (ADA) a été introduit dans des lymphocytes d’un patient ayant une mutation de cette enzyme. Cet essai clinique a démontré des résultats encourageants, et par le fait même engendré un intérêt pour la thérapie génique. Dans le
contexte d’un patient DMD, il faut introduire une copie fonctionnelle du gène de la dystrophine dans les fibres musculaires squelettiques et cardiaques. Cependant, il est difficle de livrer dans les noyaux des cellules un gène d’une taille de 2,4 Mb. De plus, le gène doit être délivré dans tous les muscles squelettiques et dans chaque fibre musculaire, lesquelles sont entourées de tissus conjonctifs.
Actuellement, les principaux vecteurs utilisés dans la livraison de gènes sont les vecteurs plasmidiques et les vecteurs viraux. Les vecteurs plasmidiques sont introduits généralement par électroporation. Par contre, l’expression du transgène est d’une durée limitée. Les vecteurs viraux sont un outil de prédilection car certains de ces vecteurs sont capables d’infecter des cellules en division on non. Malgré les succès de certains traitements de la DMD par thérapie génique dans des modèles animaux, les problèmes comme le rejet immunitaire, la courte durée d’expression du transgène et la toxicité doivent être résolus [45].
18
3.3 Les vecteurs viraux : le lentivirus basé sur le VIH de type 1
Les vecteurs viraux se démarquent généralement par leur grande efficacité dans le transfert de gènes. Les premiers vecteurs viraux ont été développés au milieu des années 80 [46]. Il existe un inventaire très large de virus utilisés en thérapie génique, incluant les adénovirus, les virus adéno-associé (AAVs) et les lentivirus. Le virus de l'immunodéficience humaine de type 1 (HIV-1) est responsable d’engendrer le syndrome d’immunodéficience acquise (SIDA) chez l’humain. Les interactions et les mécanismes de réplication ainsi que la structure virale sont bien connus [47]. Les lentivirus sont des dérivés du HIV-1 et possèdent donc de grandes similarités. Les ressemblances sont au niveau de leur structure génomique, leur cycle de réplication et leur habilité à infecté un large inventaire de cellules, lesquelles peuvent être ou non dans un cycle de division [48, 49]. Ces vecteurs sont dépourvus de plusieurs séquences génomiques pour limiter leur réplication et diminuer leur virulence (figure 9). Le lentivirus est un outil moléculaire important utilisé dans le cadre de notre étude.
Figure 9. Comparaison des génomes du lentivirus sauvage HIV-1 et du lentivirus recombinant de 3e génération. A) Génome du lentivirus sauvage HIV-1 B) Les quatre vecteurs utilisés pour produire un lentivirus recombinants de 3e génération. L’ensemble des protéines
impliquées dans la virulence du type sauvage sont retirées et seulement les éléments essentiels sont conservées et divisé sur trois plasmides différents. Ceci permet de maximiser la sécurité lors de la production d’un lentivirus de 3e génération [48].
Vecteur Encapsidation
REV Enveloppe
19
Brièvement, le génome du lentivirus sauvage HIV-1 contient trois régions majeures qui permettent l’expression des protéines responsables de l’encapsidation et de l’intégration dans le génome (gag/pol), ainsi que les protéines de l’enveloppe (env). De plus, on peut dénombrer plusieurs protéines accessoires impliquées dans la réplication et par le fait même dans sa virulence. L’ensemble du génome est flanqué de longues régions répétitives terminales (LTR). La production des vecteurs lentivirus recombinants de 3e génération requièrent la transfection de
quatre plasmides distincts dans la cellule. Ceux-ci codent pour les éléments essentiels permettant d’obtenir une grande quantité de pseudovirus. Par contre, les séquences codantes pour les protéines accessoires Vif et Vpu impliquées dans la virulence du lentivirus sauvage sont absentes. Le vecteur plasmidique contient le transgène ainsi que les éléments essentiels à l’intégration et la réplication du lentivirus. Le vecteur gag/pol assure la synthèse des protéines liées à la capside, à la réplication, grâce à la transcriptase inverse, et à son intégration par l'intermédiaire de l’intégrase. Le troisième plasmide code pour une protéine très importante, la protéine Rev, qui est impliquée dans l’importation des ARNm et de l’ARN viral au noyau de la cellule infectée. Ce transport assure une protection contre la dégradation ou l’épissage non-désiré. Le dernier plasmide code pour la glycoprotéine G du virus de la stomatite vésiculaire (VSV-G) qui produit l’enveloppe virale.
Le lentivirus a la capacité de s’intégrer de façon stable dans le génome, de permettre une expression du transgène à long terme et de posséder une capacité de clonage relativement grande. Un atout crucial du lentivirus est son efficacité de transduire des cellules ayant un faible taux de division cellulaire comme les myocytes.
21
CHAPITRE 4
Les cellules souches
Les cellules souches sont des cellules indifférenciées qui ont la capacité d’engendrer des cellules spécialisées et de se maintenir indéfiniment par prolifération in vitro ou dans l’organisme [50]. Elles ont le potentiel de régénérer des tissus, voire des organes, et sont donc un atout important dans la thérapie cellulaire et en médecine régénérative (figure 10).
4.1 Les cellules souches embryonnaires humaines
Ce sont des chercheurs canadiens qui ont découvert l’existence des cellules souches. Une vingtaine d’années plus tard, la première lignée de cellules souches embryonnaires murines (mESCs) a été isolée et cultivée [51]. La différenciation de ces cellules a commencé à être étudiée. Il a été démontré que ces cellules peuvent former des corps embryonnaires (EB) qui sont des agrégats cellulaires en différenciation, finement régulés et très organisés [52].
En 1998, aux États-Unis, la première lignée d’hESCs a été établie à partir d’un embryon préimplantatoire. Les cellules souches embryonnaires humaines (hESCs) sont totipotentes. Elles ont la capacité de se différencier en cellules spécialisées issues d’un des trois feuillets embryonnaires.
Actuellement, il existe diverses lignées de cellules souches embryonnaires (ESCs) bien établies qui peuvent être différenciées pour le développement de traitements thérapeutiques. Cependant, les hESCs sont des cellules allogéniques. Le rejet immunitaire ainsi que les problèmes éthiques sont les limitations principales de leur utilisation thérapeutique. Un comité de Surveillance des Cellules Souches a été mis en place au Canada [53].
22
Figure 10. Isolation des cellules souches embryonnaires et culture cellulaire. Schématisation
de la production de divers types cellulaires originaires d’une culture de cellules souches embryonnaires. Stem Cell Transportation (www.asiancancer.com)
23
4.2 Cellules souches pluripotentes induites
Les cellules souches pluripotentes induites (iPSCs) sont dérivées de cellules adultes, contournant ainsi les problèmes éthiques reliés à l’utilisation des hESCs. La pluripotence est obtenue en reprogrammant une cellule différenciée en cellule embryonnaire à l’aide d’un ensemble de facteurs transcriptionnels. En 2006, la première lignée d’iPSCs a été produite à partir de fibroblastes embryonnaires de souris (MEFs) et de quatre facteurs de transcription : Oct4, Klf4, Sox2 et c-Myc [53]. Il s’est avéré que ces cellules sont similaires aux ESCs car elles peuvent se différencier en cellules spécialisées issues des trois feuillets embryonnaires et possèdent la capacité d’auto-renouvellement [54, 55].
En 2007, les cellules souches pluripotentes induites humaines (hiPSCs) ont été produites à partir de cellules somatiques humaines. Des lentivirus codant pour Oct4, Klf4, Sox2 et c-Myc ont été utilisés pour reprogrammer des fibroblastes de peau humaine en hiPSCs [53]. Il a été démontré qu’il était possible d’utiliser la même approche pour obtenir la pluripotence, mais utilisant les facteurs de transcription différents, soit Nanog et Lin28 au lieu de KFL4 et c-myc, sans altération du résultat final [56].
La technique la plus usuelle pour délivrer les facteurs de transcription utilise le virus Sendaï, un virus à ARN non intégratif et capable d’une expression prolongée [57]. Actuellement, on retrouve plusieurs lignées d’hiPSCs et certaines d’entre elles sont produites afin de modéliser diverses maladies ou encore afin de découvrir de nouvelles drogues [58].
En plus de se comporter comme des ESCs, les hiPSCs peuvent être dérivés du patient malade. Ceci est un avancement majeur pour la médecine régénérative et les thérapies cellulaires car les risques de réactions immunitaires lors d’une greffe autologue sont largement réduits (figure 11).
24
Figure 11. Utilisation des hiPSCs en thérapie génique et en thérapie cellulaire. Production
des hiPSCs et leurs applications multiples. Des fibroblastes prélevés chez le patient sont mis en culture. Par la suite, ils sont reprogrammés en cellules souches induites pluripotentes. Les hiPSCs peuvent être corrigées génétiquement et différenciées. Finalement, une transplantation de ces cellules corrigées génétiquement et modifiées peut être exécutée [59].
25
CHAPITRE 5
Expression de la dystrophine humaine dans les muscles de souris
Rag/mdx suite à une greffe de cellules myogéniques dérivées
d’hiPSCs dystrophiques génétiquement corrigées.
5.1 Introduction
La DMD est une myopathie sévère et létale causée par l’absence de dystrophine fonctionnelle dans les tissus musculaires, résultat d’une mutation génétique dans le gène codant pour la protéine. L’une des thérapies cellulaires considérées pour le traitement de patients atteints de la DMD est la transplantation allogénique de cellules myogéniques saines. Cependant, certaines limitations sont à considérer, incluant une prolifération cellulaire variable, une mortalité prématurée des cellules injectées et un faible niveau de migration vers les fibres musculaires non endommagées. Outre les défis en culture cellulaire, la prise d’immunosuppresseurs est inévitable pour le patient afin de prévenir une réaction immunitaire contre les cellules du donneur [60]. L’utilisation des hiPSCs est une stratégie alternative face à tous ces obstacles. Leur capacité d’auto-renouvellement et leur potentiel de différenciation en une multitude de types cellulaires sont des aptitudes à exploiter. Les hiPSCs utilisées expérimentalement sont généralement des fibroblastes provenant du patient puis sont reprogrammés à l’aide des facteurs transcriptionnels tels Oct4, Sox2, KLF4 et l’oncogène c-myc [61].
La correction génétique s’effectue avec une paire de ciseaux moléculaires, les TALENs, qui introduisent une coupure autour de la séquence d’ADN comprenant un codon « Stop » prématuré pour rétablir le cadre de lecture du gène de la dystrophine [62]. Les hiPSCs corrigés subissent ensuite une transduction avec des gènes viraux pour devenir des « myoblastes corrigés ». En plus d’être efficaces pour une transplantation autologue, les myoblastes corrigés sont capables de fusionner avec les myotubes du patient et d’exprimer une dystrophine fonctionnelle dans le muscle.
26
5.2
Hypothèse et objectifs
Dans le cadre de notre étude, la stratégie thérapeutique développée s’inspire de certains aspects de la médecine régénérative et de la thérapie cellulaire. L’hypothèse de notre étude est que les myoblastes dérivés d’hiPSCs corrigés génétiquement fusionneront les fibres musculaires de la souris Rag/mdx in vivo, et exprimeront la dystrophine humaine.
L’objectif principal est d’obtenir l’expression de la dystrophine humaine suite à une xénogreffe de myoblastes dérivés d’hiPSCs dystrophiques corrigées génétiquement par les protéines TALENs. La différenciation des hiPSCs corrigés sera faite en deux étapes : les hiPSCs seront d’abord différenciés en cellules de type mésenchymateuses (MSCs-L) et seront ensuite transduites avec un lentivirus exprimant MyoD, produisant ainsi une population de myoblastes modifiés.
Pour atteindre l’objectif principal, il faut : - Construire un lentivirus codant pour MyoD.
- Différencier les hiPSCs corrigées en cellules de type mésenchymateuses (MSC). - Transformer, par transduction, les MSCs en myoblastes.
- Faire fusionner in vitro ces myoblastes (détection l’expression de chaine lourde de la myosine) - Greffer les myoblastes corrigés et les faire fusionner in vivo dans la souris Rag/mdx.
- Évaluer l’expression des protéines dystrophine et lamine A/C humaines dans les muscles greffés.
27
5.3 Matériel et Méthodes
5.3.1 Construction du vecteur de transfert
Le squelette d’origine du lentivirus, le FUGW, a été offert gracieusement par le Dr David Baltimore (Addgene plasmid #14883) (figure 12) [63]. Ce plasmide est l’élément principal du projet de recherche. Ce vecteur a été modifié pour contenir le gène MyoD humain relié par un peptide 2A à un gène rapporteur, le eGFP. Le tout est flanqué de longues répétitions terminales (LTR) pour faciliter l’intégration dans le génome de l’hôte. De plus, le vecteur contient dans sa séquence originale un promoteur ubiquitine-C humain (hUbC) en amont d’une séquence exprimant la protéine eGFP. Ce promoteur entraîne une expression du transgène caractérisée comme étant modérée.
Figure 12. Cartographie du vecteur FUGW. Le plasmide FUGW contient le promoteur
28
Construction du vecteur multicistronique MyoD-P2A-eGFP
Le gène MyoD humain a été amplifié à partir du plasmide fourni par le Dr. Rénald Gilbert, tandis que le gène eGFP a été amplifié à partir du vecteur FUGW. Le protocole d’amplification est celui du kit PCR Protocol for Phusion® High-Fidelity DNA Polymerase (New England Bioloabs, Ipswich, Ma, USA). Les paires d’amorces FORWARD sont construites pour permettre l’amplification des gènes MyoD et eGFP. De plus, ces amorces contiennent des séquences supplémentaires en aval du gène MyoD (séquence REVERSE) et en amont du gène eGFP (séquence FORWARD) pour permettre leur liaison grâce à la formation d’un peptide 2A entre les deux gènes (Tableau 1) [64]. La validation du produit PCR complet a été faite par électrophorèse sur gel d’agarose 0,7%.
Tableau 1. Séquences des amorces utilisées pour la construction des transgènes à introduire dans le vecteur FUGW. En vert sont les sites de restriction, en bleu le codon « start » et en
rouge le codon « stop ».
À l’aide d’une lumière ultraviolette, il a été possible de visualiser sur le gel les fragments PCR amplifiés, de les récupérer avec une lame de scalpel et de procéder à leur extraction. Les gènes MyoD humain et eGFP ont été extraits en suivant le protocole du manufacturier (Thermo Scientific GeneJET Gel Extraction Kit #K0691). Finalement, le séquençage a validé la nouvelle insertion MyoD_P2A_eGFP (figure 13).
29 Figure 13. Construction du vecteur multicistronique. Schématisation de la stratégie utilisée
pour l’amplification et le clonage des gènes MyoD et GFP liés par un peptide 2A (P2A). Adapté d’Andrea et al. 2014 [64].
Assemblage du vecteur de transfert
Le vecteur FUGW et l’insert ont été digérés par les enzymes de restriction EcoRI et BamHI. Cette linéarisation du plasmide permet l’extraction du gène eGFP. Le vecteur résultant est de 9228 pb. Les extrémités cohésives engendrées par les coupures enzymatiques permettent la ligation du vecteur multicistronique dans le vecteur FUGW linéarisé. Après la ligation exécutée selon le protocole du kit Quick ligation M2200 (New England Bioloabs, Ipswich, Ma, USA), le vecteur recombinant a été transfecté dans les bactéries TOP 10 et étalées sur pétri contenant une concentration d’ampicilline de 50 g/mL. Ces bactéries possèdent une mutation dans le gène recA, diminuant ainsi les mécanismes de recombinaison de l’ADN avec nos constructions génomiques.
Le séquençage a permis de confirmer certains clones. Un clone positif a été cultivé en grande quantité et l’ADN plasmidique a été extrait. À cette étape, le vecteur original FUGW contient l’insertion MyoD_P2A_eGFP sous le contrôle du promoteur ubiquitine-C humain.
30
Transfection du vecteur FUGW_hUbC_MyoD_P2A_eGFP
La transfection d’une lignée de cellules embryonnaires humaines de rein immortalisées (HEK293T) avec le vecteur exprimant les transgènes a été réalisée avec l’agent de transfection lipidique Lipofectamine 2000, selon la procédure du fabricant (Lipofectamine® 2000 Transfection Reagent, #11668-019), en utilisant un ratio ADN : lipofectamine de 1 : 2. Après 24 heures d’incubation, les cellules ont été lysées sur glace (137 mM de NaCl, 50 mM Tris-HCl pH 8.0 et 1% de Triton X-100) et les protéines recueillies par centrifugation et dosées avec la méthode de Bradford. La production des protéines humaines MyoD et eGFP a été confirmée par immunobuvardage.
Détection des protéines par immunobuvardage
20 µg de protéines totales ont été séparées par électrophorèse sur un gel dénaturant de 10% polyacrylamide (SDS-PAGE). La migration a débuté à 70 V pendant 15 minutes et s’est poursuivie pendant 90 min à 100 V dans un tampon de migration composé de Tris 50 mM pH 8.0, 0.2 M Glycine et 0.1 % SDS. Par la suite les protéines ont été électrotransférées sur une membrane de nitrocellulose 0.2µm (Bio-Rad), à 200 mA, à 4°C, pour 1h (Tris 50 mM pH 6.8, 0.2 M Glycine, 0.1 % SDS et 20 % méthanol). Les membranes ont été incubées dans une solution de blocage contenant 0.1 % PBS 1X, 0.05% Tween™ 20 et 5 % de lait écrémé en poudre pendant une heure à température de la pièce, sous agitation.
Les membranes ont ensuite été incubées pendant 1h à 4°C, sous agitation, avec une solution d’anticorps primaire dilués dans la solution de blocage. Pour la détection de la MyoD humaine, la solution d’anticorps monoclonal de souris a été utilisée à une concentration de 1 : 250. Dans le cas de la détection de eGFP, l’anticorps polyclonal de lapin a été diluée 1 : 2000. Après 1h, les membranes ont été lavées trois fois, pendant 5 min, avec la solution de lavage (0,1 % PBS 1X, 0,05% Tween™ 20). Puis, les membranes ont été incubées avec les anticorps secondaires respectifs (anti-souris ou anti-lapin) pendant 1h à température de la pièce. Les anticorps secondaires, couplés à l’enzyme HorseRadish Peroxidase (HRP), ont été dilués 1 : 15 000 dans la solution de blocage. Finalement, trois lavages ont été effectués avant la détection des protéines positives par chemiluminescence (Clarity Western ECL substrate). Les résultats ont été visualisés en exposant un film autoradiographique HyBlot ® CL (Denville Scientific).
31
Amplification et substitution du promoteur CAG
Le promoteur hUbC du vecteur FUGW_hUbC_MyoD_P2A_eGFP a été substitué par le ‘‘cytomegalovirus early enhancer/chicken β-actin (CAG)’’. L’avantage de ce promoteur synthétique est qu’il permet une expression plus forte des 2 transgènes liés par le peptide 2A. Le promoteur ubiquitine du vecteur a été éliminé par digestion avec les endonucléases PacI et XbaI. Puis, le promoteur CAG a été amplifié avec une amorce qui introduit les paires de bases complémentaires au vecteur digéré (tableau 2).
Tableau I. Séquence d’oligonucléotide utilisée pour amplifier et cloner le promoteur CAG dans FUGW. Le site de restriction est indiqué en vert.
La ligation du promoteur CAG dans le vecteur a été réalisée à l’aide du kit In-Fusion Cloning technology (Clonetech Laboratories), et la transformation du plasmide a été effectuée dans les bactéries TOP 10. La nouvelle construction FUGW_CAG_MyoD_P2A_eGFP a été confirmée par séquençage et par digestion enzymatique visualisée par électrophorèse sur gel d’agarose 0,7%.
5.3.2 Vecteur lentiviral (FUGW_CAG_MyoD_P2A_eGFP)
Pour produire un pseudo-lentivirus, il faut co-transfecter quatre vecteurs d’expression. Outre le vecteur de transfert, on retrouve les trois vecteurs plasmidiques servant à produire la transcriptase inverse, les protéines permettant l’encapsidation de l’ADN viral (GAG/POL et REV) et les protéines de l’enveloppe (VSVG). Ces vecteurs ont été introduits par transfection avec la méthode du phosphate de calcium.
32
Transfection des vecteurs viraux
Des cellules de la lignée HEK293T ont été cultivées jusqu’à 80 % de confluence dans du High glucose Dulbecco's Modified Eagle medium (DMEM ; Wisent, Saint-Jean-Baptiste, Québec, Canada) contenant de la L-glutamine, glucose, sodium pyruvate, phénol rouge et supplémenté avec 10 % de sérum fœtal bovin (FBS ; Gibco, Burlington, Ontario), à 37oC sous une atmosphère
de 5% CO2. La transfection au phosphate de calcium se fait en deux temps (figure 14). Pour un
pétri de 10 cm, la solution A contient 15 µg des vecteurs FUGW_CAG_MyoD_P2A_eGFP et GAG/POL ainsi que 5 µg des vecteurs REV et VSVG. De plus, 50 µL de chlorure de calcium (2.5 M CaCl2) sont ajoutés et le volume est complété à 500 µL avec de l’eau. Cette solution a été déposée goutte-à-goutte dans 500 µL d’une solution tampon B (280 mM NaCl, 50 mM HEPES, 1,5 mM Na2HPO4 pH 7.1), pour un volume final de 1 ml, lequel a été incubé 20 min à
température ambiante.
Pour un bon taux de transfection, l’ajustement du pH est critique. Le phosphate de sodium monobasique (Na2HPO4) et le chlorure de sodium (NaCl) provenaient du Laboratoire MAT Inc
(Québec, Canada). Le tampon HEPES 1M (C8H18N2O4S) et le chlorure de calcium (CaCl2)
provenaient de Thermo Fisher Scientific (Waltham, MA USA). Le mélange a été par la suite réparti uniformément dans le milieu de culture des cellules, pour une durée de 16 h. Puis, le milieu de culture a été changé et les cellules ont poursuivi leur croissance ainsi que la production du lentivirus pendant 48 h.
Purification du lentivirus
Le milieu de culture des cellules transfectées a été récolté et centrifugé à 2000 rpm pendant 10 min pour éliminer les cellules et les résidus récupérés lors du prélèvement. Le surnageant a été ensuite filtré à travers une membrane de 0.22 µm de ‘‘molecular weight cut off (MWCO)’’. Le filtrat a été transféré délicatement sur une solution de sucrose 20%, dans un tube Beckman stérile pour ultracentrifugeuse, et le volume a été ajusté à 36 mL avec du tampon phosphate salin 1X (PBS) provenant de Wisent Inc. L’ultracentrifugation (OPTIMA™ L-80 XP, rotor SW 28) a été effectuée à 4°C, pendant 90 min, à une vitesse de 24 500 rpm. Le surnageant a été éliminé et le culot, qui contient les particules pseudovirales, a été resuspendu dans 100 µL de PBS 1X et entreposé à 4°C pendant 60 min. Finalement, le pseudo-lentivirus a été conservé à -80°C.
33 Figure 14. Transfection par la méthode de phosphate de calcium. Récapitulatif des étapes de
transfection des quatre vecteurs pour la production du lentivirus. Adapté de www.mirusbio.com/transfectopedia/methods
34
Titrage du lentivirus par cytométrie en flux
Des cellules HEK293T ont été cultivées dans une plaque de 24 puits, incluant un puits témoin avant la transduction, laquelle a été effectuée 24h après l’ensemencement des cellules, à une confluence d’environ 60 %. À ce moment, les cellules ont été incubées pendant 48h dans 500 µL de milieu de culture. Celui-ci contenait du DMEM, une quantité de particules pseudo-virales diluées en série et une concentration finale de 4 g/mL de Polybrène, lequel sert à augmenter l’efficacité d’absorption des virions lentiviraux [65]. Après 48h, les cellules ont été rincées au PBS 1X et remises en suspension. Elles ont été centrifugées pendant 10 min à 1000 rpm et fixées dans une solution de 3,75 % formaldéhyde/PBS. La quantité de cellules fluorescentes, c’est-à-dire transduites par le pseudo-lentivirus qui exprime la protéine eGFP, a été déterminée par cytométrie en flux. Le titre viral du pseudo-lentivirus FUGW_CAG_MyoD_P2A_eGFP était de 2 x 108 unités/mL.
5.3.3 Culture cellulaire
Culture des cellules myogéniquesDes myoblastes sains ont été isolés d’une biopsie post-mortem d’un enfant âgé de 13 mois (BB13M). Ils ont été cultivés dans le milieu MB-1 commercial (Hyclone, Mississauga, Canada). Ce milieu spécifique aux cellules humaines a été complémenté de 15% de sérum fœtal bovin (FBS) (Gibco, Burlington, Ontario) et de 10 ug/L de « basic Fibroblast Growth Factor » (bFGF) (Feldan, Québec, Canada). Les cellules ont été maintenues à faible passage et à une confluence approximative de 80%. Des analyses par cytométrie de flux du marqueur CD56+ dans cette
population de cellules myogéniques ont indiqué une présence majoritaire, soit 85% et plus, de myoblastes. Ces cellules ont été utilisées comme contrôle positif in vitro pour valider la détection de la chaine lourde de la myosine après fusion. De plus, elles ont permis de confirmer la présence de la dystrophine humaine ainsi que la lamine A/C humaine, après que les cellules aient fusionné avec les fibres du muscle de la souris Rag/mdx.
35
Culture des cellules mésenchymateuses
Les lignées d’hiPSCs dystrophiques (DMD -/-) et le clone corrigé (DMD-IFH30) sont une
gracieuseté du laboratoire du Dr Hotta (CiRA Kyoto University, Japan). Ces cellules proviennent de fibroblastes d’un patient DMD possédant une délétion de 75 484 bp, incluant l’exon 44. Ces cellules ont été reprogrammées par des vecteurs plasmidiques épisomaux sans intégration [66]. Les hiPSCs sélectionnées possédaient un caryotype normal, exprimaient des marqueurs de pluripotence comme Nanog et engendraient la formation de tératomes in vivo. Les hiPSCs IFH30
ont été corrigées par une paire de Platinum-TALEN. L’insertion d’une paire de base ‘‘A’’ dans l’exon 45 a permis de rétablir le cadre de lecture, et cette correction a été validée par séquençage de Sanger de l’ADNc des clones [62].
Dans la présente étude, ces cellules hiPSCs dystrophique et corrigées ont été cultivées dans notre laboratoire dans des plaques de 6 puits (Starstedt, Newton, USA) à l’aide de cellules nourricières (MSTO), en suivant les indications du Dr. Hotta. Les zones de différenciation spontanée de nos colonies ont été minutieusement retirées afin de préserver des colonies au contour lisse et bien défini et ayant un faible ratio cytoplasme/noyau. Lorsque les colonies d’hiPSCs ont été stables et que leur aptitude d’auto-renouvellement a été atteinte, les cellules nourricières ont été retirées avec une solution CTK [1 mg/mL de collagénase IV (Sigma), 2.5% de trypsine (Invitrogen), 0.1 mM CaCl2 (Laboratoire MAT) et 20% knockout sérum replacement (KSR)] et les hiPSCs ont été
cultivées dans le milieu mTeSR1 (Stem Cell Technologies, Vancouver, Colombie-Britannique, Canada). Une fois la confluence de 80% obtenue, les colonies ont été rincées au DMEM/F12 KnockOut™ (Gibco).
Les colonies ont été incubées pendant 7 minutes dans une solution 1 U/mL de Dispase (Stem Cell Technologies), une protéase capable de faire décoller doucement les cellules souches. Lorsque le contour des colonies commence à décoller, les cellules ont été rincées avec du DMEM/F12 KnockOut™ à deux reprises, et 2 mL de ce milieu ont été ajoutés dans chaque puits pour faciliter leur détachement mécanique à l’aide d’un grattoir à cellule. La suspension a été centrifugée à 1000 rpm pendant 5 min et le culot a été remis en suspension dans 10 mL de MB-1 et transféré dans un flacon de culture cellulaire, à 37oC.


![Figure 4. Le gène de la dystrophine. Le gène est composé de 2,5 Mb d’ADN et code pour sept isoformes de la dystrophine dont l’expression localisée dans des tissus bien spécifiques [17]](https://thumb-eu.123doks.com/thumbv2/123doknet/6411463.169835/26.918.109.808.727.901/figure-dystrophine-composé-isoformes-dystrophine-expression-localisée-spécifiques.webp)