Study of imprinted genes in bovine embryos produced by assisted reproductive technologies
Texte intégral
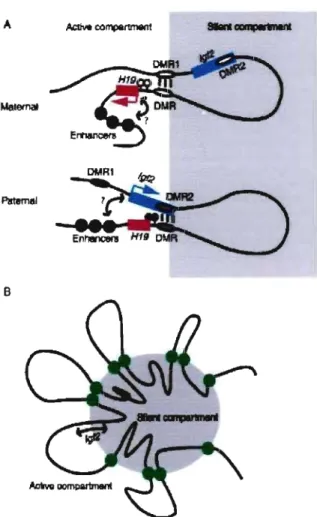
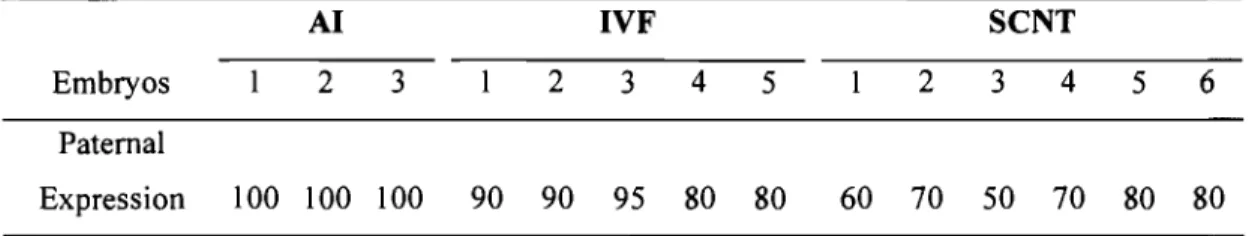
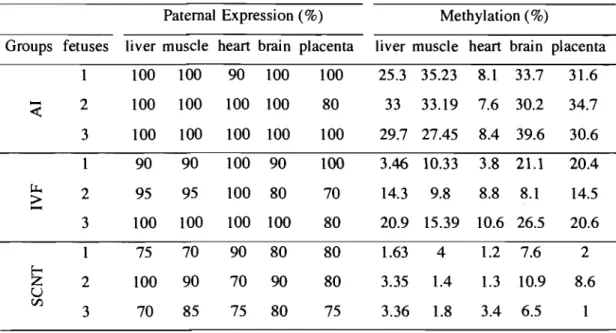
Figure
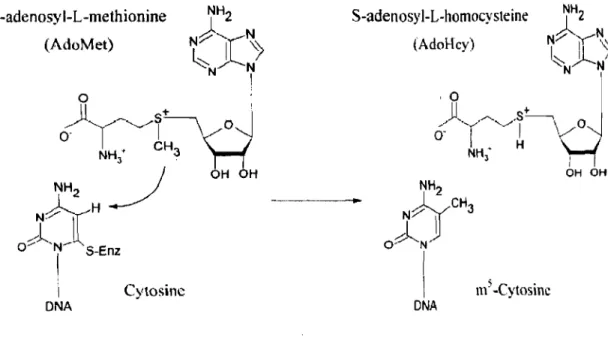
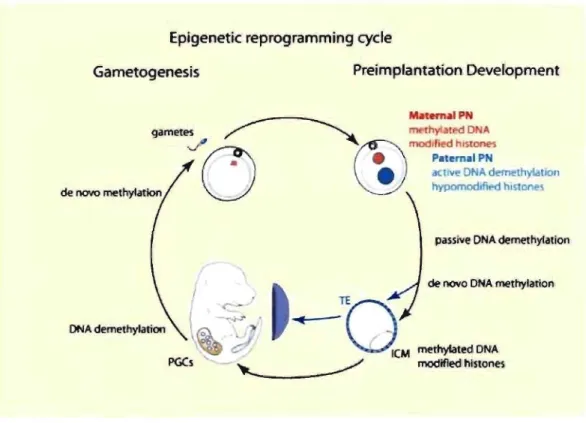
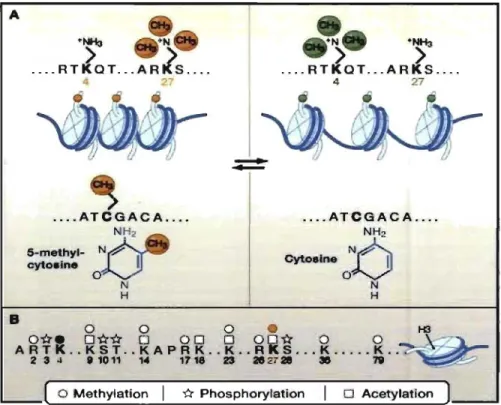
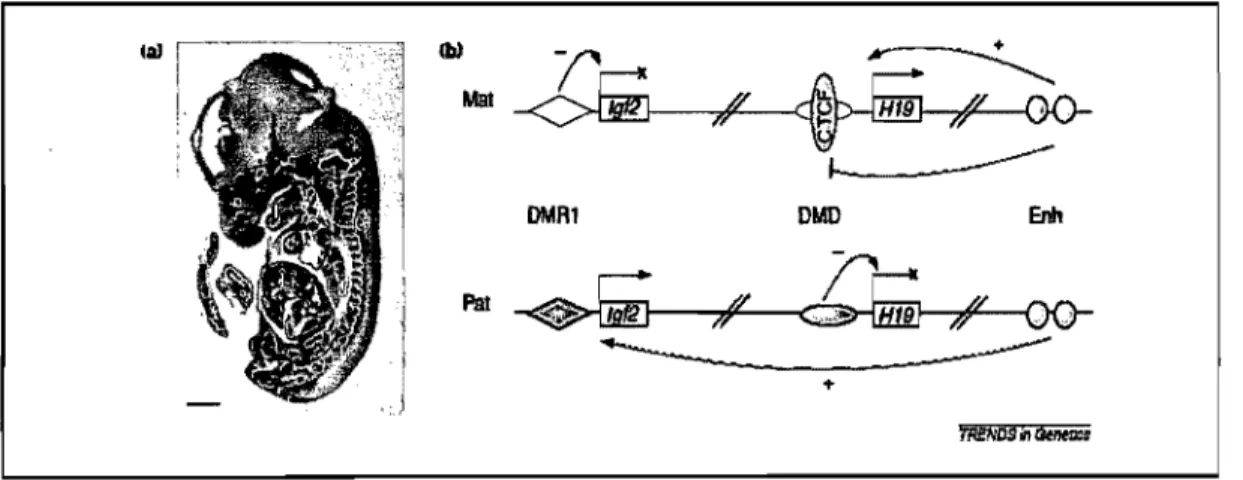
Documents relatifs
Les poutres sont des éléments structuraux horizontaux chargés de la transmission des charges verticales et horizontales aux éléments porteurs (poteaux et voiles). Elles
Sander, A Simple Method for Determining the Heating Energy Requirement of Direct-Gain Passive Solar Houses, National Research Council Canada, Division of Building Research,
l’art. 221 al. 1 let. d et e CPC est de permettre au juge de déterminer sur quels faits
The Algerian war was a great event of the 20 century. It had a deep impact on the society. In order to go straight on freedom of Algeria, among several methods which
One can evaluate that product iter- atively, keeping the real and imaginary parts of the partial product of all already considered terms as double-word num- bers, and just using
Les résultats que nous avons générés suggèrent donc qu’ e n p hotopério de lo ngue, l’ horm one juvénile est synthétisée e n qua ntité suffisante pour
N'abandonnons tout de même pas, ils nous assassinent avec leur haine, nous les violerons d'amour, nous reprendrons notre souffle et nous taperons à chaque battement de cœur.
- La maîtresse reste dans la même classe, et moi, je monte dans la
