THÈSE
Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS École nationale supérieure d'ingénieurs (Poitiers) Institut de chimie des milieux et matériaux de Poitiers - IC2MP
(Diplôme National - Arrêté du 25 mai 2016)
École doctorale : Sciences pour l'environnement - Gay Lussac (La Rochelle) Secteur de recherche : Chimie appliquée
Présentée par :
Joakim Delaux
Activation de biopolymères par plasma
atmosphérique non thermique
Directeur(s) de Thèse : François Jérôme, Abdellatif Barakat Soutenue le 08 décembre 2016 devant le jury Jury :
Président Henri Cramail Professeur, LCPO, Université de Bordeaux
Rapporteur Patrick Da Costa Professeur, Université P. & M. Curie, Paris
Rapporteur Bernard Cathala Directeur de recherche, BIA, INRA, Nantes
Membre Abdellatif Barakat Chargé de recherches, IATE, INRA, Montpellier
Membre Jean-Michel Tatibouët Directeur de recherche CNRS, Université de Poitiers
Membre Henri Cramail Professeur, LCPO, Université de Bordeaux
Pour citer cette thèse :
Joakim Delaux. Activation de biopolymères par plasma atmosphérique non thermique [En ligne]. Thèse Chimie appliquée. Poitiers : Université de Poitiers, 2016. Disponible sur Internet <http://theses.univ-poitiers.fr>
THÈSE
Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS École nationale supérieure d'ingénieurs (Poitiers) Institut de chimie des milieux et matériaux de Poitiers - IC2MP
(Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l'environnement - Gay Lussac Secteur de recherche : Chimie appliquée
Présentée par :
Joakim Delaux
********************Activation de biopolymères par plasma atmosphérique non-thermique
********************
Directeur(s) de Thèse : François Jérôme Co-directeur de thèse : Abdellatif Barakat
Soutenance le 8 décembre 2016 devant le jury
Jury
Rapporteur Patrick DA COSTA Professeur, Université Pierre et Marie Curie Rapporteur Bernard CATHALA Directeur de recherche, INRA de Nantes
Membre Henri CRAMAIL Professeur, Université de Bordeaux
Membre François JEROME Directeur de recherche CNRS, Université de Poitiers Membre Abdellatif BARAKAT Chargé de recherche INRA, SupAggro de Montpellier Membre Jean-Michel TATIBOUET Directeur de recherche CNRS, Université de Poitiers
REMERCIMENTS
Ce travail a été réalisé au sein de l’institut des milieux et des matériaux de Poitiers (IC2MP) et le laboratoire des ingénieries des agro-polymères et technologie émergentes (IATE) de Montpellier. Ce projet a été financé par l’INRA et le CNRS, que je remercie grandement.
Je remercie Sabine Petit, directrice de l’IC2MP, ainsi que Hugo Vries, directeur de l’IATE de m’avoir permis de réaliser mes travaux de recherches dans les meilleures conditions.
Je remercie également Bernard Cathala, directeur de recherche à l’INRA de Nantes dans l’unité Biopolymères interactions assemblages (BIA) et Patrick Da Costa, professeur à l’université Pierre et Marie Curie à l’institut Jean Le Rond d’Alembert d’avoir accepté de rapporter ce travail. Je remercie également Henri Cramail, professeur à l’université de Bordeaux au sein laboratoire de chimie des polymères organiques à Pessac, Jean-Michel Tatibouët, directeur de recherche au sein de l’IC2MP pour avoir accepté de faire partie de ce jury.
Je remercie également François Jérôme, directeur de recherche à l’IC2MP, et Abdellatif Barakat, chargé de recherche à l’IATE, pour leur encadrement, leurs conseils, leur écoute et leur soutien lors de ces 3 années de thèse. Merci François pour tous tes conseils, pour le partage de tes connaissances et ton optimisme sans limite. Merci Abdellatif pour ton écoute, ta patience et ta compréhension.
Je souhaite remercier toute les personnes de l’IC2MP que j’ai côtoyées durant ces trois années. Je pense à Mehrad Tarighi qui m’a tout appris sur le MALDI TOF, Christine Canaff pour le temps qu’elle a passé pour m’expliquer l’XPS, Karine Doreau avec qui j’ai eu de longues conversations (chut faut pas trop le dire trop fort) et qui a toujours une solution à tout, à Jean Jacques pour le temps passé sur mes toutes petites plaques de verres, Claude pour avoir fait, refait et réparé mes réacteurs, Elodie Fourre pour son expertise plasma au labo, Philippe Ayrault pour le temps passé à faire mes analyses et pour ces récits de voyages à travers le monde, Jean Michel à nouveau pour nos conversations plus ou moins scientifiques (les bières belges ça compte dans la chimie) que j’ai vraiment appréciée, Sabine Valange pour toutes les histoires belges, les belgissismes (Oufti, je sais que je suis un pignouf !), le partage de chocolat et toutes les conversations que l’on a pu avoir au cours de ces 3 années.
Merci à mes collègues de labo, Florent, Caroline, Houcine, Ronan, Shi, Jialu, Ivan, Ayman et également les stagiaires et anciens thésards pour leur gentillesse et leur aide. Je tiens à remercier en particuliers deux collègues qui sont devenus des amis au cours du temps : Raluca et Maïté. Je n’oublie pas mon ami Clément que j’ai embarqué dans l’aventure de la thèse.
Merci à ma famille pour leur soutien au cours de ces 8 années d’études. Merci également à ma belle-famille pour leur support et leurs encouragements. Et enfin, et pas des moindres, je remercie la personne qui compte le plus au monde à mes yeux, qui m’a supporté, encouragé et conseillé durant toute cette aventure : Marie.
Introduction générale ... 1
Chapitre 1 : ... 5
Bibliographie ... 5
-I. La biomasse ... - 6 - A. L’hémicellulose ... 7 -B. La lignine ... 10 -C. La cellulose ... 13 -1. Liaisons hydrogènes ... 14-2. Polymorphisme et cristallinité de la cellulose ... 16
-3. Réactivité et activation de la cellulose ... 20
-a. Hydrolyse acide ... 21
-i. Homogène ... 21
-ii. Hétérogène ... 22
-b. Hydrolyse enzymatique ... 24
-c. Activation par voie chimique ... 25
-i. Dissolution dans une solution basique ... 26
-ii. Dissolution dans les solvants organiques ... 27
-iii. Dissolution dans les liquides ioniques ... 29
-a) Cations, anions et mécanisme de dissolution ... 31
-b) Caractéristiques après dissolution de la cellulose... 33
-d. Activation par voie physique ... 34
-iv. Le broyage ... 35
-a) Les broyeurs ... 35
-b) La cellulose après broyage ... 36
-v. Le broyage réactif ... 38
-II. Problématique, enjeux et verrous autour de la biomasse ...- 40 -
A. Avantages, inconvénients et applications industrielles ... 40
-B. Enjeux et verrous ... 41
-III. Plasma non thermique ...- 43 -
A. Définition ... 43
-B. Les différents types de plasma non thermique ... 44
-1. Formation du plasma ... 44
-2. Plasma sans diélectrique ... 45
-a. Décharge glissante 66 ... 46
-b. Couronne... 46
-c. Décharge à Barrière Diélectrique (DBD) ,, ... 48
-d. Plasma de surface 66 ... 48
-C. Espèces formées dans le plasma ... 49
-1. Réaction possible dans le plasma , ... 49
-2. Espèces formées en fonction du gaz utilisé , , ... 51
-D. Application du plasma non thermique ... 53
-3. Le traitement de l’air ... 54
-4. Le traitement de surface ... 55
-5. Application aux biopolymères ... 56
-IV. Conclusion ...- 58 -
Chapitre 2 : ... 61
Partie expérimentale ... 61
-I. Origine des produits utilisées ...- 62 -
A. Les solides ... 62
-1. Les polymères ... 62
-2. Les saccharides ... 63
-B. Les liquides ... 63
-C. Gaz ... 64
-II. Dispositif de traitement des composés ...- 64 -
A. Plasma DBD ... 64
-1. Générateur impulsionnel ... 64
-2. Réacteur DBD volumique « planplan » ... 65
-3. Mesure électrique et calcul de puissance ... 66
-B. Broyage : Ballmilling ... 67
-C. Microondes ... 68
-III. Caractérisation physico-chmique ...- 68 -
A. High pressure liquid chromatography (HPLC) ... 69
-B. Chromatographie gazeuse(GC) ... 71
-C. Matrix Assisted Laser Desorption Ionization/Time of Flight (MALDI TOF) ... 73
-D. Spectromètre de masse (analyses des gaz en sortie de réacteur) ... 74
-E. Spectrométrie infrarouge à transformée de Fourrier (FTIR) ... 74
-F. XRay Photoelectron Spectroscopy (XPS) ... 75
-G. Diffraction des rayons X ... 76
-1. Appareillage ... 76
-H. 13C CP/MAS NMR et RMN liquide (1H et 13C) ... 77
-1. RMN solide ... 77
-2. RMN liquide ... 77
-I. Mesure du degré de polymérisation ... 77
Chapitre 3 : ... 80
-Etude de la cellulose après traitement par plasma atmosphérique non-thermique ... -
80
-I. Introduction ...- 81 -II. Optimisation des paramètres plasma et de la nature de la cellulose par l’étude du degré de polymérisation ...- 81 -
A. Influence de la tension et de la puissance ... 82
-B. Influence de la masse introduite dans le réacteur ... 83
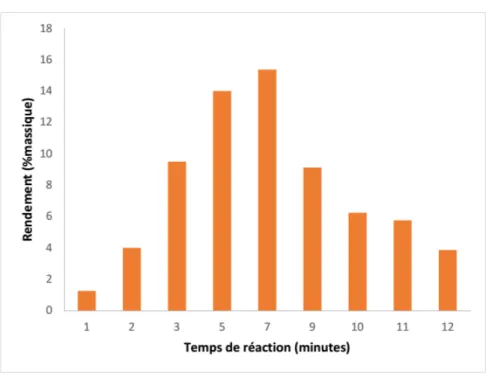
-C. Influence du temps de traitement... 84
-D. Influence de la nature de la cellulose ... 85
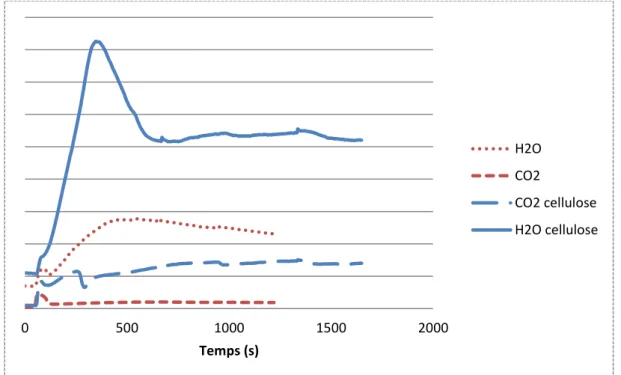
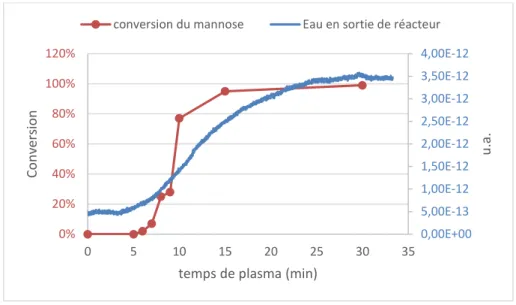
-E. Analyse des gaz en sortie du réacteur ... 87
-III. Dépolymérisation d’un autre biopolymère : l’inuline ...- 89 -
A. Généralité sur l’inuline ... 89
-B. Traitement plasma de l’inuline ... 90
-IV. Conclusion ...- 94 -
Chapitre 4 : ... 96
Polymérisation des monosaccharides assistée par plasma ... 96
-I. Introduction ...- 97 -
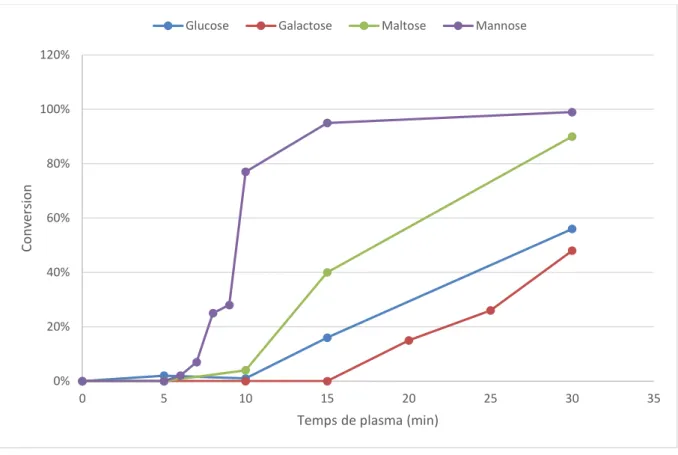
II. Cinétique et suivi de l’eau en sortie de réacteur pour quatre oligosaccharides - 97 - A. Structure du glucose, du galactose, du maltose et du mannose ... 98
-B. Analyse HPLC/SEC et spectromètre de masse des quatre saccharides ... 99
-C. Comparaison et conclusion ... 104
-III. Etude du mannose ... - 107 -
A. Influence du gaz ... 107
-B. Etude du CO2 en sortie de réacteur ... 109
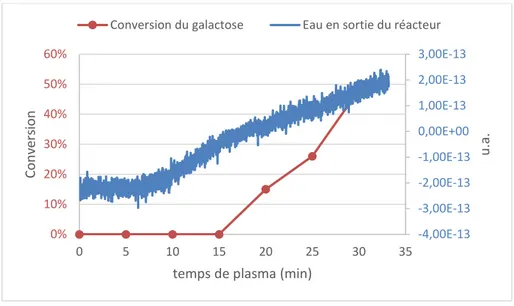
-C. Etude de la température initiale et pendant le traitement ... 110
-D. Evolution du pH du mannose après traitement plasma ... 112
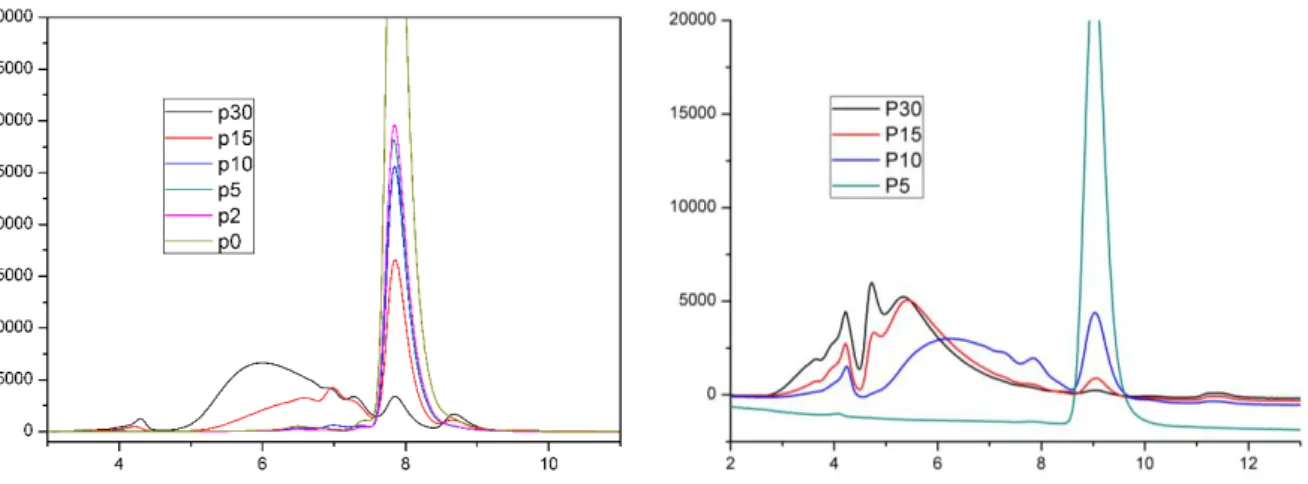
-E. Analyse du mannose polymérisé ... 113
-a. Analyse du spectre de masse pour l’échantillon P10 ... 116
-b. Analyse du spectre de masse pour l’échantillon P15 ... 117
-c. Analyse du spectre de masse pour l’échantillon P30 ... 118
-2. Analyse par Diffraction des rayons X du mannose traité par plasma ... 119
-3. Analyse par infrarouge du mannose traité par plasma ... 120
-4. Analyse par CP MAS RMN et par RMN liquide du mannose traité par plasma ... 122
-5. Analyse par XPS du mannose traité par plasma ... 124
-6. Analyse des dimères par chromatographie gazeuse après dérivatisation ... 125
-7. Analyse par chromatographie d’exclusion stérique ... 127
-8. Solubilité du mannose après traitement par plasma ... 129
-IV. Extrapolation à d’autres mono- et disaccharides ... - 129 -
Chapitre 5 : ... 133
-Plasma : tra
itement de surface ou de cœur ? ... 133
-I. Introduction ... - 134 -
II. Broyage, plasma et indice de cristallinité ... - 134 -
III. Analyses des modifications de la structure ... - 136 -
A. Impact du plasma sur le DP ... 136
-B. Etude de la solubilité ... 137
-C. Analyses XPS des celluloses broyées et traitées par plasma ... 138
-D. Analyse 13C CP/MAS RMN des celluloses ... 141
-E. Analyse par MALDI TOF de la cellulose ... 143
-F. Analyse par GCMS ... 145
-G. Hydrolyse acide par Microondes ... 147
-IV. Conclusion des analyses réalisées... - 148 -
V. Proposition de mécanisme ... - 149 -
VI. Etude du coût potentiel du procédé ... - 151 -
VII. Perspectives ... - 152 -
Conclusion ... 154
-- 1 --
- 2 -
Aujourd’hui, la société nous permet d’avoir accès à de nombreux biens et services qui impliquent une utilisation des ressources non renouvelables, que ce soit au niveau de l’énergie ou de la matière première. Une exploitation abusive de ces ressources entraine une augmentation de la pollution, une diminution irréversible de celles-ci et donc un appauvrissement des richesses de la Terre. La prise de conscience des problèmes de la surconsommation prend de plus en plus d’importance et de nouvelles alternatives commencent à voir le jour. Grâce à la recherche et à l’envie de sauver notre planète, il est possible d’offrir à nos enfants, nos petits-enfants et les générations futures une vie plus saine, plus respectueuse de l’environnement et avec un minimum de pertes et de déchets. L’utilisation de la biomasse peut en partie apporter des solutions aux problèmes liés à l’utilisation de matière première issue du pétrole. En effet, la biomasse est considérée comme une ressource renouvelable et peut être valorisée par voie énergétique ou pour en extraire des molécules d’intérêt médical ou encore pour en faire des matériaux innovants.
La biomasse lignocellulosique est particulièrement intéressante car elle offre une grande disponibilité et peut provenir de la valorisation des déchets agricoles, par exemple.
La première étape de valorisation de la biomasse est son fractionnement en libérant la cellulose et la lignine grâce à l’hydrolyse des hémicelluloses. Bien qu’un intérêt croissant pour la lignine voie le jour au niveau de la recherche, actuellement, 98% de celle-ci est brulée pour fournir de l’énergie thermique. L’hydrolyse des hémicelluloses permet de récupérer des sucres qui sont ensuite fermentés afin d’obtenir des molécules plateformes (furfural par exemple) ou de l’éthanol.
La cellulose est quant à elle plus difficile à valoriser en raison de sa grande résistante aux traitements chimiques. L’utilisation de la cellulose à des fins énergétiques est peu rentable (prix cellulose > prix éthanol). Alors que la synthèse de molécules plateformes peut être intéressant pour des molécules à haute valeur ajoutée. Toutefois, l’obtention de ce genre de molécules reste relativement délicate dû à la nécessité de dépolymériser la cellulose et peut entrainer des dépenses énergétiques élevées et une gestion des déchets pouvant être compliquée. La cellulose peut également être utilisée dans le domaine des matériaux en offrant de nouvelles propriétés par rapport aux plastiques ou en renforçant ceux-ci par exemple.
Afin de rendre la cellulose moins réfractaire aux traitements, un prétraitement a été réalisé par application d’un plasma atmosphérique non thermique. Le plasma est une technologie de plus en plus employée dans le monde de la chimie notamment dans la dépollution de l’air et le traitement de l’air. Son emploi dans l’industrie textile est très développé pour rendre les tissus hydrophile ou hydrophobe suivant les conditions. Dans le cas du traitement de la cellulose par plasma, l’objectif est de « fragiliser » celle-ci et de la rendre plus réactive aux traitements chimiques (hydrolyse par exemple).
- 3 -
L’objectif de cette thèse est donc de traiter la cellulose par plasma non thermique afin de rendre celle-ci moins réfractaire aux traitements chimiques. Il est primordial de comprendre comment le plasma peut agir sur la cellulose pour en déduire un mécanisme réactionnel. Grâce à la connaissance du mécanisme, il sera possible de contrôler l’impact du plasma sur la cellulose et donc de contrôler la structure du produit obtenu en fonction des paramètres de réaction. Cette méthode d’activation de la cellulose rentre dans un cadre plus respectueux de l’environnement en évitant l’utilisation de solvant et de catalyseur.
Le premier chapitre de la thèse concerne l’étude bibliographique de la biomasse et du plasma l’accent étant mis sur les traitements de la cellulose (les traitements existants, la structure du polymère…) et sur l’utilisation du plasma au niveau chimique (différents types de plasma, espèces formées, applications…). Les verrous scientifiques liés aux traitements de la cellulose viendront conclure ce chapitre.
Le deuxième chapitre est consacré à la partie expérimentale de la thèse. Dans cette partie, nous développerons les différents produits utilisés, les méthodes de traitement employée et les méthodes analytiques utilisées au cours de cette étude.
Le troisième chapitre présente les premiers résultats qui nous ont permis de découvrir l’effet du plasma sur la cellulose. Dans cette partie, les premières hypothèses sont formulées sur le mécanisme réactionnel.
Le quatrième chapitre est axé sur l’étude des saccharides traités par plasma. Le mannose a été pris comme modèle pour les autres sucres. Les résultats obtenus ont ouvert la voie vers une nouvelle utilisation du plasma dans le domaine des sucres. De plus, ce chapitre nous a également permis de mieux comprendre les évolutions structurelles de la cellulose au cours d’un traitement plasma non thermique en apportant des réponses aux premières interrogations du chapitre trois.
Le chapitre cinq est orienté sur l’étude de la réactivité de la cellulose en fonction de ses caractéristiques physiques (cristallinité principalement). Il conclut cette étude et nous permet de proposer un mécanisme réactionnel potentiel de la transformation de la cellulose soumise à un plasma non thermique.
La dernière partie de ce manuscrit consiste en une discussion des résultats obtenus et dans la proposition de perspectives de recherche et d’éventuels développements industriels.
- 5 -
Chapitre 1 :
Bibliographie
- 6 -
I. La biomasse
La biomasse peut être séparée en deux grandes utilisations : l’énergie, les bioproduits et les matériaux. Dans le cadre de cette thèse, les biopolymères et plus particulièrement ceux issus de la cellulose seront étudiés afin de mieux comprendre les mécanismes réactionnels mis en jeu lors de traitements ou prétraitements physiques.
Depuis des millénaires, les matériaux issus de la biomasse, tels que le bois, sont utilisés dans différents domaines, tels que l’aéronautique, la construction, les vêtements, le papier, etc. Néanmoins, avec l’arrivée des ressources fossiles, les matériaux employés à travers le monde ont énormément évolué. Les plastiques, les caoutchoucs synthétiques ou les médicaments sont autant d’exemples de domaines qui peuvent être d’origine biosourcée.
La biomasse lignocellulosique est une des sources de substitution aux ressources fossiles, notamment grâce à son abondance sur Terre et à sa non-utilisation dans le domaine de l’alimentaire. En effet, selon l’Organisation des Nations Unies, la Terre serait recouverte d’environ 4 milliards d’hectares de forêt (principale source de la biomasse). Mais à ce chiffre, des tonnes de sous-produits agricoles non exploitées, ainsi que la biomasse marine peuvent être ajoutées.
La biomasse lignocellulosique est composée de 3 principaux polymères : la cellulose, l’hémicellulose et la lignine [Figure I-1]. Les proportions de ces trois composés varient en fonction de la source de la biomasse. [Tableau I-1]
Tableau I-1 Proportions cellulose/hémicellulose/lignine dans différentes sources de biomasse
Cellulose Hémicellulose Lignine
Coton 95 % 2 % 0 % Bois feuillus1 40-44 % 20-32 % 25-35 % Bois résineux1 40-44 % 15-35 % 18-25 % Herbe2 37 % 29 % 19 % Paille de blé3 38 % 29 % 15 % Bagasse3 49 % 31 % 19 % Agave 78 % 6 % 13 %
1Bryan F. Staley and Morton A. Barlaz, “Composition of Municipal Solid Waste in the United States and Implications for Carbon
Sequestration and Methane Yield,” Journal of Environmental Engineering 135, no. 10 (October 2009) : 901–9
2 A. Demirbaş, “Relationships between Lignin Contents and Heating Values of Biomass,” Energy Conversion and Management 42,
no. 2 (2001) : 183–88
- 7 -
Figure I-1 Schéma de la lignocellulose
A.
L’hémicellulose
Les hémicelluloses sont des polymères composés de différents saccharides et naturellement présents dans toutes les plantes. Elles sont composées de sucres tels que le glucose, le xylose, l’arabinose ou l’acide galacturonique [Figure I-2]. Insolubles dans l’eau, il est néanmoins possible de solubiliser ces polymères dans une solution alcaline. Ces chaînes, composées de xylose, glucomanane, xyloglucane ou encore d’arabinose, sont généralement assez courtes. De l’ordre de 500-3000 unités de répétition ou également appelé degré de polymérisation (DP). Elles sont structurées par un squelette de résidus β-(1,4)-D-Pyranose avec l’O4 en position équatoriale.4
Xylose Glucose Arabinose
Mannose Acide galacturonique Fructose
Figure I-2 Exemples de saccharides présents dans les hémicelluloses
Les hémicelluloses peuvent être classées en quatre grandes familles :
- 8 - Les Xylanes qui englobent :
o Les Glucuronoxylanes qui sont composés d’un squelette de xylose avec des ramifications d’acide glucuronique et/ou son dérivé O-méthylé.
o Les Arabinoxylanes qui sont composés d’un squelette de xylose avec des ramifications d’arabinose.
o Les Glucuronoarabinoxylanes qui sont composés d’un squelette de xylose avec des ramifications d’arabinose et d’acide glucuronique.
o Les Homoxylanes qui sont composés d’une chaîne linéaire de xylose.
Les Xyloglucanes qui sont composés d’un squelette de glucose avec des ramifications de xylose, de galactose et de fructose.
Les Mannanes qui regroupent :
o Les Galactomannanes qui sont composés d’un squelette de mannose avec des ramifications de galactose.
o Les Galactoglucomannanes qui sont composés d’un squelette de mannose et de glucose avec des ramifications de galactose.
Les β-1, 3 ;1,4 -glucanes qui sont composés de différents glucanes avec plus ou moins de ramifications.
Ces structures différentes ne sont pas présentes dans toutes les plantes. Certains de ces biopolymères ne sont présents que dans des familles de plantes spécifiques telles que les herbacées ou les conifères et dans certaines parties de la cellule (paroi primaire, paroi secondaire). La synthèse des hémicelluloses dans les plantes est réalisée grâce une réaction enzymocatalytique. Chaque polymère a besoin d’un cocktail d’enzymes bien spécifiques incluant la synthèse de la chaîne principale du squelette et la synthèse de la chaîne latérale ou ramification.5,6 la présence de groupements hydroxyles
sur les hémicelluloses et la cellulose favorise les interactions entre les chaînes saccharidiques.
À l’instar de leur synthèse, la dégradation des hémicelluloses peut se faire par voie enzymatique. Ce sont les micro-organismes qui dégradent les hémicelluloses pour en extraire les sucres (dégradation enzymatique). Certaines enzymes spécifiques peuvent également être extraites de micro-organismes et utilisées une à une pour dégrader les hémicelluloses de manière sélective et ainsi éviter la dégradation complète du polymère afin de récupérer des oligosaccharides précis (dimères, oligosaccharides solubles dans l’eau). C’est une des valorisations possibles des chaînes de polymères hémicellulosiques.
5 Chris Somerville et al., “Toward a Systems Approach to Understanding Plant Cell Walls,” Science 306, no. 5705 (December 23,
2004): 2206–11
6 Karthikeyan, “Pathway: Xyloglucan Biosynthesis,” 2008,
- 9 -
Comme ressources renouvelables, les hémicelluloses peuvent être employées en tant qu’additif alimentaire, plastique, cosmétique ou produits pharmaceutiques.7
Exemple d’une hémicellulose :
Gomme de Guar [Figure I-3] : Polymère de type Galactomannane, il peut être utilisé dans l’industrie alimentaire en tant qu’épaississant. Tandis que dans l’industrie non alimentaire, il est utilisé dans le domaine du textile, du papier, de la cosmétique, de l’industrie pharmaceutique, du forage pétrolier ou des explosifs.8
Figure I-3 Structure de la gomme de guar
Le Tableau I-2 qui suit représente une partie des applications possibles des hémicelluloses en fonction de leur structure.
Tableau I-2 Exemples d'applications de différentes hémicelluloses
Type Applications possibles
Hémicelluloses en général
Film et revêtements
Produit chimique par hydrolyse et conversion des sucres
Biotechnologie et fermentation des sucres
Xyloglucanes9 Papier, alimentaire, produits cosmétiques et
pharmaceutiques, textiles
7 Paul Gatenholm and Maija Tenkanen, eds., Hemicelluloses: Science and Technology, vol. 864, ACS Symposium Series
(Washington, DC: American Chemical Society, 2003)
8 “Guar Gum,” accessed May 17, 2016, http://en.wikipedia.org/wiki/Guar_gum.
9 S.V.Patil and S.C.Dhawale, “Tamarind Gum: A Pharmaceutical Overview,” 2008,
- 10 -
Galactomannanes
Fibres diététiques solubles dans l’eau, Textiles, papier, explosifs, produits cosmétiques
et pharmaceutiques, industrie alimentaire, Exploitation minière, hydro-ensemencement
B. La lignine
La lignine compose 15 à 35 % de la biomasse et représente la teneur en énergie spécifique la plus élevée des trois fractions. Ce polymère amorphe tridimensionnel [Figure I-4] est issu de la polymérisation de trois principaux monomères : l’alcool ρ-coumarylique, l’alcool coniférylique et l’alcool sinapylique [Figure I-5]. La réaction de polymérisation est de nature radicalaire, avec des couplages radicalaires combinatoires. Sa structure composée de cycles aromatiques lui confère des propriétés hydrophobes, ce qui permet une imperméabilité des parois cellulaires. Par ailleurs, des liaisons covalentes de type éther entre la lignine et les hémicelluloses peuvent être créées, renforçant encore plus les liens entre les parois de la cellule végétale.10 Elle est l’une des sources des carbones
aromatiques les plus abondantes dans la nature.
Figure I-4 Schéma de la lignine
- 11 - Alcool
courmarylique Alcool coniférylique Alcool synapylique
Figure I-5 Les trois composés principaux de la lignine
Il existe plusieurs méthodes d’extraction de la lignine,11 suivant la méthode utilisée et de la
nature de la biomasse (Tableau I-3). Il est possible d’obtenir des structures de polymères différentes afin d’obtenir des caractéristiques particulières. Le Tableau I-3 présente les lignines extraites par le procédé de traitement acide des pâtes pour une lignine organo-soluble. Une fois extraites, les bioraffineries utilisent principalement la lignine en tant que combustible pour les chaudières, pour produire de l’énergie (électrique et/ou chaleur). Néanmoins, une partie peut servir à l’extraction d’aromatiques tel que le benzène, le toluène ou encore le xylène (BTX). Pour se faire, il est nécessaire de faire dans un premier temps une pyrolyse de lignine afin de rompre les liaisons C-C et C-O, puis d’effectuer une hydrodésoxygénation catalytique. Enfin, les BTX peuvent être récupérés.12
Tableau I-3 Composition de la lignine en fonction de l'origine de la biomasse
Alcool Coumarylique Alcool Coniférylique Alcool Sinapylique
Bois feuillus 0 % 90-95 % 5-10 %
Bois résineux 0 % 50 % 50 %
Herbe 5 % 75 % 25 %
En fonction de l’origine de la biomasse, les lignines récupérées possèdent des masses molaires en poids (Mw = masse molaire moyenne du produit) ou en nombres variables (Mn = masse molaire
médiane du produit). Le rapport Mw/Mn correspond à la polydispersité du produit final. Plus ce rapport est proche de 1 et plus la polydispersité est faible. [Tableau I-4]
11 Pooya Azadi et al., “Liquid Fuels, Hydrogen and Chemicals from Lignin: A Critical Review,” Renewable and Sustainable Energy
Reviews 21 (May 2013): 506–23
12M. P. Pandey and C. S. Kim, “Lignin Depolymerization and Conversion: A Review of Thermochemical Methods,” Chemical
- 12 -
Tableau I-4 Caractéristiques des lignines en fonction de l'origine de la biomasse
Type de biomasse % acide
acétique % HCl T (°C) Mw Mn Mw/Mn
Bois résineux13 85 - 185 3200 - -
Bagasse14 90 0,1 110 1710 990 1,7
Paille de blé15 90 0,1 85 3960 2330 1,7
Selon les procédés utilisés, il est possible d’utiliser la lignine dans diverses applications. Le Tableau I-5 montre quelques exemples d’application en fonction du procédé employé.
Tableau I-5 Applications possibles de la lignine suivant le procédé utilisé
Procédés Application actuelle Application possible Application à long terme Procédé d’extraction
Kraft pulping Énergie et chaleur
Production de dihydrogène, production d’alcool Procédé Fischer-Tropsch pour du carburant
Sulfite pulping Macromolécules à
faible valeur ajoutée
Macromolécules à haute valeur ajoutée
Procédé de transformation
Pyrolyse Carburant solide
Production de dihydrogène, production d’aromatiques Dépolymérisation Carburant solide, production d’aromatiques
De nos jours, la communauté scientifique s’intéresse de plus en plus à la lignine et la Figure I-6 nous montre que les publications traitant du sujet sont de plus en plus nombreuses. L’abondance
13 Xiaoxiang Jiang, Naoko Ellis, and Zhaoping Zhong, “Characterization of Pyrolytic Lignin Extracted from Bio-Oil,” Chinese Journal
of Chemical Engineering 18, no. 6 (December 2010): 1018–22
14 DJ Hayes et al., “The Biofine Process –production of Levulinic Acid, Furfural, and Formic Acid from Lignocellulosic Feedstocks”
(Germany, 2006), http://www.carbolea.ul.ie/files/HFHR_Chapter%204_FINAL.pdf.
15 A. G. Gayubo et al., “Pyrolytic Lignin Removal for the Valorization of Biomass Pyrolysis Crude Bio-Oil by Catalytic
- 13 -
des aromatiques dans la lignine en fait un excellent candidat pour leurs extractions et, ainsi, fournir une autre source que celle des produits pétroliers.
Figure I-6 Nombre de publications sur la lignine en fonction des années
C. La cellulose
La cellulose est le polymère naturel le plus abondant sur Terre. Un français, Anselme PAYEN découvrit la cellulose en 1838 en plongeant des fibres végétales dans une solution d’acide puis une solution d’ammoniaque. Le composé fibreux et insoluble qui en résultait fut nommé cellulose et Anselme PAYEN lui donna la formule de C6H10O5. Ce n’est qu’en 1922 que Hermann
STAUDINGER proposa une structure à ce polymère, en affirmant que les fibres étaient en réalité des petites molécules liées entre elles pour former des chaînes moléculaires beaucoup plus grandes. Il proposa donc la molécule représentée en [Figure I-7]. Aujourd’hui, c’est cette représentation qui est utilisée. La cellulose est constituée de D-anhydroglucose (AGU) reliés entre eux par une liaison covalente glycosidique β-(1-4) en conformation chaise. Le degré de polymérisation (DP) correspond au nombre de répétitions de l’AGU.
0 500 1000 1500 2000 2500 3000 3500 4000 1985 1987 1989 1991 1993 1995 1997 1999 2001 2003 2005 2007 2009 2011 2013 2015 N o m b re d e p ap ie rs sur la li g n in e
- 14 -
Figure I-7 Structure de la cellulose
Chaque AGU possède trois groupements hydroxyles sur les carbones C2, C3 et C6. Grâce à ces fonctions, il est possible de faire diverses réactions sur la cellulose. Les extrémités de la cellulose peuvent également être sujettes à des réactions spécifiques. L’extrémité réductrice possède une fonction aldéhyde offrant de nouvelles possibilités de réactions tandis que l’extrémité non réductrice offre une nouvelle fonction hydroxyle en C4. Ces multiples fonctions permettent aux chercheurs de modifier ce polymère naturel par des réactions telles que l’oxydation, la méthylation ou la dépolymérisation par voie acide.
1. Liaisons hydrogènes
La cellulose peut former des liaisons hydrogènes entre les groupements hydroxyles C2, C3 et C6. Ces interactions peuvent se faire de manière intermoléculaire et intramoléculaire [Figure I-8]
- 15 - Intramoléculaire :
Selon Liang et Marchessault16, l’hydrogène du groupement C3 serait capable de
former une liaison avec l’oxygène hétérocyclique et selon Balckwell et al17, l’oxygène sur
le C6 serait capable de former une liaison avec l’hydrogène de la fonction hydroxyle en C2. Néanmoins, c’est deux siècles après sa découverte que la structure cristalline de la cellulose a été élucidée par synchrotron de rayon X et diffraction de neutron par Nishiyama et al.1819
Intermoléculaire :
Entre deux chaînes de celluloses proches, des liaisons entre les groupements hydroxyles sur le C6 de la première chaîne et les groupements hydroxyles sur le C2 et le C3 peuvent se créer. Mais il existe d’autres arrangements en fonctions du type de cellulose.
Figure I-9 Exemples de liaisons hydrogènes au sein de la cellulose
À l’échelle macromoléculaire, les chaînes d’AGU sont organisées sous forme de feuillet. Les liaisons hydrogènes intra et intermoléculaires se font au sein du polymère, créant ainsi une structure en trois dimensions sous forme de feuillet. [Figure I-10]
16 C. Y. Liang and R. H. Marchessault, “Infrared Spectra of Crystalline Polysaccharides. I. Hydrogen Bonds in Native Celluloses,”
Journal of Polymer Science 37, no. 132 (June 1959): 385–95
17 J Blackwell, FJ Kolpak, and KH Gardner, In Cellulose Chemistry and Technology, J.C. Arthur, vol. 48, ACS Symposium Series
(Washington, DC, 1977).
18 Yoshiharu Nishiyama, Paul Langan, and Henri Chanzy, “Crystal Structure and Hydrogen-Bonding System in Cellulose Ibeta
from Synchrotron X-Ray and Neutron Fiber Diffraction,” Journal of the American Chemical Society 124, no. 31 (August 7, 2002): 9074–82.
19 Yoshiharu Nishiyama et al., “Crystal Structure and Hydrogen Bonding System in Cellulose I(alpha) from Synchrotron X-Ray
- 16 -
Figure I-10 Structure en feuillet de la cellulose
2. Polymorphisme et cristallinité de la cellulose
La conformation chaise des AGU, les liaisons hydrogènes, les interactions Van Der Waals, les interactions électroniques et la structure en feuillet sont responsables de la cohésion de la cellulose, induisant une structure cristalline importante et un taux de cristallinité élevé. Grâce à ces caractéristiques, la cellulose est un polymère peu réactif et difficile à faire réagir avec une chimie dite « douce ». Néanmoins, il est possible de modifier la cristallinité de la cellulose. C’est communément appelé le polymorphisme de la cellulose. La Figure I-11 montre les différentes possibilités de cristallinité de cellulose en fonction du traitement chimique effectué.
- 17 -
Figure I-11 Polymorphisme de la cellulose
Les mailles cristallines sont déterminées par six paramètres a, b et c les arêtes de la maille et α, β et γ les angles de la maille. La Figure I-12 permet de visualiser ces paramètres en trois dimensions dans la maille.
Figure I-12 Paramètre de la maille cristalline
La cellulose I :
La cellulose I correspond à la cellulose native, celle retrouvée dans la nature. Néanmoins, il existe 2 types de cellulose I : la Iα et la Iβ.20 Les deux types de cellulose possèdent la même orientation parallèle des chaînes, mais leur maille cristalline diffère.21 [
20 R. H. Atalla and D. L. Vanderhart, “Native Cellulose: A Composite of Two Distinct Crystalline Forms,” Science (New York, N.Y.)
223, no. 4633 (January 20, 1984): 283–285
21 Mike Jarvis, “Chemistry: Cellulose Stacks up,” Nature 426, no. 6967 (December 11, 2003): 611–12
Ammo niac <80°C
- 18 -
Figure I-13]. Les paramètres de mailles de la cellulose Iα et de la cellulose Iβ 22
sont représentés dans le Tableau I-6. L’orientation des chaînes est régie par le parallélisme ou l’anti parallélisme des liaisons β-(1-4) des AGU tandis que la maille cristalline est régie par les liaisons hydrogène formées en inter et intramoléculaire.
Suivant la provenance de la cellulose, la cellulose I possède des ratios Iβ/Iα
variables. Par exemple, les bactéries et les algues possèdent majoritairement de la cellulose Iα tandis que les plantes (bois, coton, paille de blé…) possèdent majoritairement
de la cellulose Iβ.
Figure I-13 Représentations schématiques de la maille cristalline de la cellulose Iα et Iβ
Tableau I-6 Paramètres de la maille cristalline de la cellulose Iα et Iβ
Paramètre cristallin Iα Iβ Symétrie de la maille P1 P21 a (Å) 6,717 7,784 b (Å) 5,962 8,201 c (Å) 10,4 10,38 α (°) 118,08 90 β (°) 8,201 90 γ (°) 80,7 96,5 La cellulose II :
La cellulose II est obtenue à partir de la cellulose I par traitement de celle-ci par une solution de NaOH. En 1844, John Mercer découvre les propriétés d’une cellulose traitée par la soude : augmentation de la force de la fibre et augmentation de la tenue des colorants. Ce matériau aux propriétés renforcées est très utilisé dans le domaine textile. D’un point de vue chimique, la cellulose passe d’un arrangement parallèle [Figure I-14] à un arrangement antiparallèle des chaînes [Figure I-15]. Ce changement d’orientation
22 Masahisa Wada et al., “The Structure of Celluloses,” Powder Diffraction 23, no. 2 (June 2008): 92–95
- 19 -
induit un renforcement du réseau des liaisons hydrogènes en diminuant la distance et en augmentant le nombre des liaisons.23 La maille cristalline de la cellulose II 22 est comparé
à la cellulose Iβ dans le Tableau I-7. Avec les différents changements, la cellulose II
possède une plus grande stabilité thermodynamique.
Figure I-14 Cellulose Iβ
Figure I-15 Cellulose II
Tableau I-7 Paramètres de la maille cristalline de la cellulose Iβ et II
Paramètre cristallin Iβ II Symétrie de la maille P21 P21 a (Å) 7,784 8,10 b (Å) 8,201 9,03 c (Å) 10,38 10,31 α (°) 90 90 β (°) 90 90 γ (°) 96,5 117,10
La cellulose IIII et la cellulose IIIII :
La cellulose IIII et IIIII sont obtenues par traitement de la cellulose I et de la
cellulose II, respectivement, par l’ammoniac liquide à une température inférieure à 30 ° C. La maille cristalline de la cellulose IIII [Figure I-16] est de type monoclinic P21 avec pour
valeur a = 4, 450 Å ; b = 7, 850 Å ; c = 10,31 Å ; α= β= 90 ° ; γ= 105,10 °. Tandis que la maille cristalline de la cellulose IIIIIest encore inconnue à l’heure actuelle. La cellulose
IIII est particulièrement étudiée dans la littérature. En effet la transition de la cellulose I
vers la cellulose IIII permet d’obtenir de meilleurs rendements et une meilleure cinétique
23 P. Langan, Y. Nishiyama, and H. Chanzy, “A Revised Structure and Hydrogen-Bonding System in Cellulose II from a Neutron
- 20 -
par voie enzymatique lors de l’hydrolyse de la biomasse.24 Contrairement à l’irréversibilité
de la réaction de mercerisation, la cellulose III (IIII et IIIII) peut revenir à la cellulose
initiale (cellulose I et cellulose II).
Figure I-16 Cellulose IIII
La cellulose IVI et IVII :
La cellulose IV peut être obtenue par traitement de la cellulose III par le glycérol à 260 °C pendant 20 min. La cellulose IVI provient de la cellulose IIII et conserve
l’arrangement des chaînes parallèles alors que la cellulose IVII provient de la cellulose IIIII
et conserve l’arrangement des chaînes antiparallèles.25
3. Réactivité et activation de la cellulose
La quantité de la cellulose sur Terre en fait un polymère naturel très abondant. Il est donc très intéressant à valoriser et en particulier dans le domaine des matériaux. Actuellement, l’industrie papetière consomme environ 200 000 tonnes de cellulose par an, mais ce n’est qu’une petite partie des 100 milliards de tonnes de production de cellulose par la nature par an. L’industrie textile utilise également la cellulose en grande quantité qui est majoritairement issue du coton. Les constructions en bois sont également une source d’utilisation de la cellulose. Dans une moindre mesure, les matériaux composites à base de fibre de bois commencent également à voir le jour.
C’est dans ce domaine des matériaux que le sujet sera orienté. En modifiant la cellulose de manière chimique ou physique, il est possible d’obtenir de nouvelles propriétés. La cellulose est un polymère de glucose et par conséquent une source importante de ce saccharide. Les hydrolyses acide et enzymatique sont deux techniques très employées à ce jour. Toutefois, la production de glucose à partir de la cellulose n’est pas intéressante, car la cellulose est plus chère que le glucose. De plus, pour conserver certaines propriétés de la cellulose, une dépolymérisation totale du polymère n’est pas
24 Kiyohiko Igarashi, Masahisa Wada, and Masahiro Samejima, “Activation of Crystalline Cellulose to Cellulose III(I) Results in
Efficient Hydrolysis by Cellobiohydrolase,” The FEBS Journal 274, no. 7 (April 2007): 1785–92
- 21 -
forcément intéressante. L’activation par voie chimique, biochimique ou physique peut s’avérer plus judicieuse pour une application orientée vers les matériaux.
a. Hydrolyse acide
Deux grandes réactions sont possibles pour réaliser une hydrolyse acide : par voie homogène et par voie hétérogène. Dans le cas d’une hydrolyse homogène, le catalyseur acide est plus difficile à récupérer que dans le cas d’une hydrolyse hétérogène où il est possible de le récupérer en fin de réaction par filtration par exemple. Nous allons traiter les deux cas.
i. Homogène
L’hydrolyse acide a été découverte en 1834 par Henri Braconnot en utilisant de l’acide sulfurique concentré.26 Suite à cette découverte, deux stratégies ont été étudiées :
Acide dilué et haute température
Cette méthode a été développée par Scholler en 1923. En mettant en contact la cellulose avec l’acide sulfurique à 0,5 % à 170 °C, Scholler a obtenu un rendement en glucose soluble d’environ 50 %. Le résidu solide obtenu est principalement composé d’oligosaccharides.27 En 2011, Fardim et al.
ont mis au point un protocole avec l’acide chlorhydrique dilué dans l’éthanol. À 79 °C, température d’ébullition de l’éthanol, et en 5 h, le degré de polymérisation de la cellulose issue du bois passe de 800 à 150. Grâce au reflux de l’alcool, la pénétration du solvant dans le réseau cristallin est facilitée par l’interaction hydrophobe de l’alcool et permet donc une attaque de l’acide de manière plus efficace.28
Acide concentré et basse température
Cette méthode a été développée par Berguis en 1937. Dans une solution d’acide chlorhydrique 40 % et à température ambiante, la cellulose est mise en contact pendant 2 h avec l’acide. La dépolymérisation est effective et les produits obtenus sont principalement du glucose et des oligosaccharides. Néanmoins cette technique induit des contraintes financières. Il est nécessaire de
26 Colonna, La Chimie Verte.
27 Roberto Rinaldi and Ferdi Schüth, “Acid Hydrolysis of Cellulose as the Entry Point into Biorefinery Schemes,”
ChemSusChem 2, no. 12 (December 21, 2009): 1096–1107
28 Jani Trygg and Pedro Fardim, “Enhancement of Cellulose Dissolution in Water-Based Solvent via Ethanol–hydrochloric Acid
- 22 -
faire la réaction dans un matériau inerte aux acides (très couteux) ; la neutralisation de la solution à la fin de la réaction et le traitement des déchets rendent ce procédé difficilement exploitable à l’échelle industrielle.27
ii. Hétérogène
En suspension et sous forme solide, le catalyseur acide permet de réaliser la réaction d’hydrolyse avec de multiples avantages. Le premier, et le plus important, est la récupération du catalyseur à la fin de la réaction tandis que les oligosaccharides restent dans la phase liquide. La dangerosité et l’aspect corrosif de l’acide sont nettement diminués par rapport aux acides minéraux. Afin d’augmenter l’interaction entre le catalyseur et la cellulose, les paramètres tels que la nature du support ou l’acidité du matériau sont particulièrement étudiés par la communauté scientifique. Il existe plusieurs familles de catalyseurs, dans cette étude, nous parlerons des résines d’échangeuses d’ions et des charbons sulfonés.
Les résines échangeuses d’ions
Hartler et al furent les premiers à étudier l’hydrolyse acide de la cellulose par des résines échangeuses d’ions.29 Sur une base de polystyrène réticulé avec du divinylbenzène, des groupements
sulfoniques -SO3H sont greffés lui apportant une forte acidité. À 100 °C, les rendements de
l’hydrolyse de la cellulose en glucose sont < 2 %. Cependant, avec la résine A15 DRY, Rinaldi et al ont pu atteindre les 3 % en mono et disaccharide après 5 h de traitement à 100 ° C dans un liquide ionique grâce à la formation de HCl dans le milieu.30 Ces faibles rendements sont principalement
causés par l’encombrement stérique du support polymérique et la force du réseau des liaisons hydrogène qui diminue fortement l’interaction entre les sites acides et la cellulose et plus particulièrement les groupements SO3H et les liaisons glycosidique β-(1-4). Néanmoins, si la molécule
est un disaccharide tel que le saccharose, il est possible d’obtenir des conversions élevées en glucose et fructose (97 %).31 Ces résultats confirment que la coupure, et donc la dépolymérisation, est possible,
mais que trop de paramètres limitent les interactions entre la cellulose et le catalyseur (l’encombrement stérique du support, la cristallinité de la cellulose, la température supportée par le catalyseur). De plus, l’exemple du saccharose n’est pas représentatif des liaisons présentes dans la cellulose.
29 Nils Hartler and Kari Hyllengren, “Heterogeneous Hydrolysis of Cellulose with High Polymer Acids. Part 3. The Acid
Hydrolysis of Cellulose with Finely Divided Cation-Exchange Resin in the Hydrogen Form,” Journal of Polymer Science 56, no. 164 (February 1962): 425–34
30 Roberto Rinaldi, Regina Palkovits, and Ferdi Schüth, “Depolymerization of Cellulose Using Solid Catalysts in Ionic Liquids,”
Angewandte Chemie International Edition 47, no. 42 (October 6, 2008): 8047–50
31 Karine De Oliveira Vigier and François Jérôme, “Heterogeneously-Catalyzed Conversion of Carbohydrates,” in Carbohydrates
in Sustainable Development II, ed. Amélia P. Rauter, Pierre Vogel, and Yves Queneau, vol. 295 (Berlin, Heidelberg: Springer Berlin
- 23 - Les charbons sulfonés
Les charbons sulfonés sont issus de la pyrolyse de la cellulose, de l’amidon ou de saccharides et l’acidification de ceux-ci par une réaction de sulfonation. Les groupements formés en surface du matériau possèdent un caractère acide grâce aux fonctions sulfoniques, carboxyliques et hydroxyles (dans une moindre mesure). Suganuma et al ont fait réagir la cellulose avec un charbon sulfoné pendant 3 h à 100 °C et ont obtenu une conversion de 68 % avec 4 % de glucose et 64 % d’oligosaccharides solubles dans l’eau.32 L’interaction forte entre les groupements OH et COOH avec
les groupements de la cellulose facilite grandement le contact entre les liaisons glycosidiques et l’acide sulfonique à la surface du catalyseur. Ainsi l’hydrolyse acide est facilitée par ce rapprochement entre le catalyseur et la cellulose. L’eau, le solvant, est un facteur clef dans la réaction d’hydrolyse. Elle permet de contrôler la sélectivité de la réaction en glucose et oligosaccharides ; plus il y a d’eau dans le milieu moins il y aura de glucose. En effet, plus il y a d’eau moins l’acide est concentré et donc l’hydrolyse des oligosaccharides et de la cellulose devient plus difficile, car le contact entre le catalyseur et la cellulose diminue.
La récupération du catalyseur est également étudiée. Grâce aux particules de fer incorporées dans les pores de la silice, Lai et al ont élaboré un matériau paramagnétique.33 Grâce à ce nouveau
procédé, il est possible de séparer la cellulose et le catalyseur par magnétisme. Liu et al ont développé un charbon sulfoné contenant des nanoparticules de Fe3O4 pouvant servir à l’hydrolyse de la
cellulose.34
Pour conclure, l’hydrolyse acide est un moyen efficace d’obtenir des oligosaccharides. Les deux méthodes ont chacune leurs avantages. Le meilleur contact entre le catalyseur et la cellulose est en faveur de l’hydrolyse homogène et cela conduit donc souvent des rendements plus élevés. Les grandes quantités de matière utilisées pour l’hydrolyse acide homogène à haute température et faible concentration impliquent des frais de traitement des déchets conséquents. Dans le cas de l’hydrolyse acide homogène à basse température et forte concentration, le matériel utilisé doit être particulièrement résistant aux acides entrainant un coût qui doit être pris en compte pour une application industrielle. Les hydrolyses hétérogènes offrent la meilleure alternative pour récupérer les produits à la fin de la réaction. Néanmoins, le contact entre le catalyseur et la cellulose est plutôt difficile dans le cas des résines échangeuses d’ions. Dans le cas des charbons sulfonés, les rendements se rapprochent de ceux de l’hydrolyse homogène, mais la quantité de charbon utilisée pour obtenir ces résultats est supérieure à 100 % massique par rapport à la cellulose. La stabilité du catalyseur et la faible concentration en cellulose dans le milieu sont deux autres problèmes pour réaliser une grande
32 Satoshi Suganuma et al., “Hydrolysis of Cellulose by Amorphous Carbon Bearing SO3H, COOH, and OH Groups,” Journal of
the American Chemical Society 130, no. 38 (September 24, 2008): 12787–93
33 Da-ming Lai et al., “Hydrolysis of Cellulose into Glucose by Magnetic Solid Acid,” ChemSusChem 4, no. 1 (January 17, 2011):
55–58
34 Ning Liu et al., “Preparation and Characterization of Magenetically Core-Shell Structure Carbon Based Solid Sulfonic Acid,”
- 24 -
production en oligosaccharides. Pour conclure, il existe diverses réactions d’hydrolyse possible et, suivant le but recherché, une technique pourrait être préférable à une autre.
b. Hydrolyse enzymatique
Contrairement à l’hydrolyse acide, l’hydrolyse enzymatique nécessite des conditions plus douces. L’enzyme est une association de trois molécules agissant chacune à un stade de la réaction. Elles sont présentes chez la majorité des herbivores, mais pas chez les humains. Dans un premier temps l’endo glucanase diminue la cristallinité de la cellulose puis les cellobiohydrolases déconstruisent la cellulose en cellobiose et enfin les B-glucosidases transforment la cellobiose en glucose [Figure I-17]. Ces trois réactions se déroulent simultanément et en couche par couche de cellulose.35
Figure I-17 : Mécanisme de l'hydrolyse enzymatique de la cellulose par la cellulase36
L’activité des enzymes dépend de nombreux paramètres. La nature du substrat est un paramètre clef de la réaction. Dans le cas d’une cellulose issue du coton possédant un DP de 15 000, seulement 20 % de la cellulose est hydrolysée en oligosaccharides solubles. Les 80 % restant sont composés d’une cellulose possédant un DP d’environ 15 000. En augmentant la concentration en enzymes, il est possible d’améliorer les rendements, mais cela pose problème au niveau du coût du procédé.37
En 2011, Ferreira et al. ont démontré qu’en prétraitant la cellulose par « haute pression » (300 à 400 MPa) il a été possible d’augmenter fortement l’activité enzymatique de la cellulase et ainsi
35 D. Klemm et al., Comprehensive Cellulose Chemistry, vol. 1 (Weinheim, FRG: Wiley-VCH Verlag GmbH & Co. KGaA, 1998) 36 Daniela Alonso Bocchini Martins et al., “Agroindustrial Wastes as Substrates for Microbial Enzymes Production and Source of
Sugar for Bioethanol Production,” in Integrated Waste Management - Volume II, ed. Sunil Kumar (InTech, 2011)
37 H Pala, M Mota, and F Gama, “Enzymatic Depolymerisation of Cellulose,” Carbohydrate Polymers 68, no. 1 (March 1, 2007):
- 25 -
augmenter les oligosaccharides solubles [Figure I-18]. À haute pression, la cellulose est plus accessible pour les enzymes et le contact est donc favorisé.38 Il est également possible d’utiliser l’eau à
l’état supercritique afin de dépolymériser la cellulose.39
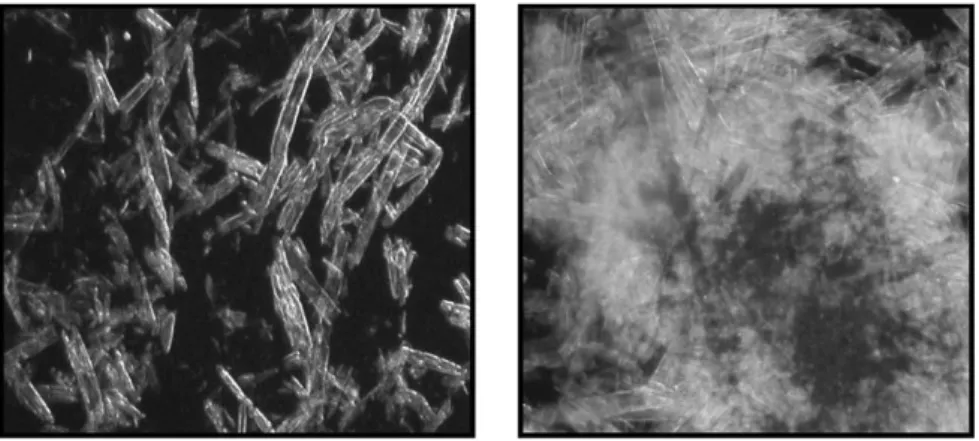
Figure I-18 : À gauche la cellulose non traitée / à droite la cellulose traitée haute pression
Le procédé d’hydrolyse enzymatique peut être respectueux de l’environnement et peut donc entrer dans les procédés durables. Les conditions de réaction sont généralement fixées à basse température (environ 40 °C) et à un pH de 5. C’est le principal avantage de cette réaction, mais son plus grand désavantage est la concentration en cellulose pouvant être traitée. En effet, pour une meilleure efficacité, il est nécessaire d’être dans un milieu avec une faible quantité de cellulose (entre 1 % et 10 %) malgré une faible proportion d’enzyme dans la solution. De plus, le coût de la cellulase (10 $ à 30 $/kg) relativement élevé, ainsi qu’un temps de réaction conséquent (48 H à 72 H) sont des facteurs limitants pour une exploitation à très grande échelle.
c. Activation par voie chimique
L’activation de la cellulose par voie chimique permet de rendre celle-ci plus accessible aux différents traitements en diminuant notamment la cristallinité et les liaisons hydrogène. Mais il également possible de diminuer le DP de la cellulose pour atteindre des chaînes plus courtes et facilitant le contact cellulose et réactifs.
Il existe diverses solutions pour activer la cellulose par voie chimique. Les solutions basiques, les solvants organiques et les liquides ioniques seront présentés dans ce manuscrit.
38Ana R. F. C. Ferreira et al., “High Pressure Pre-Treatments Promote Higher Rate and Degree of Enzymatic Hydrolysis of
Cellulose,” Green Chemistry 13, no. 10 (2011): 2764
39 Danilo A. Cantero, M. Dolores Bermejo, and M. José Cocero, “Kinetic Analysis of Cellulose Depolymerization Reactions in
- 26 - i. Dissolution dans une solution basique
Utilisé depuis des dizaines d’années dans le monde du textile, le procédé de mercerisation fait appel au traitement de la cellulose par NaOH. C’est en mettant le polymère en contact avec une solution de soude concentrée que le procédé est encore employé à ce jour. Les cations Na+ et les
anions OH- s’introduisent dans le réseau cristallin de la cellulose40 et rendent celle-ci plus brillante et
permettent la pénétration de molécules, comme les colorants, de manière plus efficace.
La dissolution de la cellulose par la soude dépend de la nature de la cellulose, de la température et de la concentration de soude. À -35 °C et dans une solution NaOH à 20 %, un eutectique se forme de composition NaOH-5 H2O ; 4 H2O. Dans ces conditions précises, la cellulose
est soluble.41
Roy et al. ont étudié le système cellulose/NaOH/eau et ont pu mettre en évidence la dissolution de la cellulose dans une solution 9 % NaOH à -6 °C et en seulement 3 minutes. Si la température est augmentée à 20 °C, un gel très stable se forme.
D’après ces différents exemples de dissolution de cellulose dans le système NaOH/eau, il est préférable d’être à faible température pour améliorer la solubilité de la cellulose. Le DP de la cellulose initiale a été également étudié et il s’avère que les faibles DP se dissolvent mieux.42
Malgré de bons résultats dans la dissolution de la cellulose, le système cellulose/NaOH/eau reste complexe, principalement pour le mécanisme de gélification et les propriétés associées. Le gonflement et la dissolution des fibres sont les deux principales réactions du système NaOH/H2O et
ceci, sans dépolymérisation notable. En 2011, en combinant le prétraitement à la soude et l’hydrolyse enzymatique, Wan et al. ont obtenu 50 % de sucres solubles. La solution obtenue est principalement composée de xylose, de glucose et d’oligosaccharides.43
L’ammoniac liquide à basse température (-30 °C) peut également être utilisé. En effet, il est possible de venir former des liaisons N—HO en remplacement des liaisons O—HO et ainsi fragiliser la structure cellulosique. Li et al ont utilisé la paille de blé et l’ammoniac pour réaliser une hydrolyse enzymatique avec de meilleurs rendements. En mettant en contact la paille et l’ammoniac pendant 4
40 Cédric Roy, Tatiana Budtova, and Patrick Navard, “Rheological Properties and Gelation of Aqueous Cellulose-NaOH
Solutions,” Biomacromolecules 4, no. 2 (April 2003): 259–64
41 Magali Egal, Tatiana Budtova, and Patrick Navard, “Structure of Aqueous Solutions of Microcrystalline Cellulose/Sodium
Hydroxide below 0 °C and the Limit of Cellulose Dissolution,” Biomacromolecules 8, no. 7 (July 2007): 2282–87
42 Magali Egal, Tatiana Budtova, and Patrick Navard, “The Dissolution of Microcrystalline Cellulose in Sodium Hydroxide-Urea
Aqueous Solutions,” Cellulose 15, no. 3 (2008): 361–70
43Caixia Wan, Yuguang Zhou, and Yebo Li, “Liquid Hot Water and Alkaline Pretreatment of Soybean Straw for Improving
- 27 -
semaines à 30 °C, ils ont obtenu des oligosaccharides et des hexoses qui fermentent plus rapidement pour former de l’éthanol.44
ii. Dissolution dans les solvants organiques
Les solvants organiques pouvant solubiliser la cellulose ne sont pas nombreux. Généralement constitués de groupements N-oxydes ou en combinaison avec LiCl, les solvants possèdent une forte bipolarité. Grâce aux ions Cl- ou RO-, le solvant s’imprègne dans la cellulose, dans les parties
cristallines, et dissout le polymère. De plus, l’arrangement spatial des solvants va permettre de stabiliser les interactions avec la cellulose et faciliter la dissolution. Dans cette étude 2 systèmes de solvant seront étudiés.
Diméthyle-Acétamide (DMAc)/Chlorure de Lithium (LiCl)
L’utilisation du DMAc/LiCl pour dissoudre la cellulose a été développée par Mc Cormick45 et Turbak46 dans les années 1980. Le lithium et le groupement carbonyle du DMAc
sont fortement liés et ce qui a pour conséquence d’augmenter la nucléophilie du Cl -. C’est par
cet ion chlorure que la déstructuration de la partie cristalline et donc la solubilisation de cellulose peut se faire. Le Cl- forme des liaisons hydrogène avec les protons des groupements
alcools de la cellulose et induit une forte diminution des interactions entre les chaînes. Tandis que les oxygènes des groupements hydroxyles interagissent avec le complexe cationique DMAc/Li+.47 [Figure I-19]
Figure I-19 : Complxe cellulose/DMAc/LiCl 48
44 Xuan Li and Tae Hyun Kim, “Low-Liquid Pretreatment of Corn Stover with Aqueous Ammonia,” Bioresource
Technology 102, no. 7 (April 2011): 4779–86
45 C.L. McCormick, Novel Cellulose Solutions (Google Patents, 1981) US4278790. 46 A.F. Turbak et al., “Solvent System for Cellulose,” November 24, 1981 US4302252.
47 Anne-Laurence Dupont, “Cellulose in Lithium chloride/N,N-Dimethylacetamide, Optimisation of a Dissolution Method Using
Paper Substrates and Stability of the Solutions,” Polymer 44, no. 15 (July 2003): 4117–26
48 Biranchinarayan Tosh, Chowdhury N. Saikia, and Narendra N. Dass, “Homogeneous Esterification of Cellulose in the Lithium
chloride–N,N-Dimethylacetamide Solvent System: Effect of Temperature and Catalyst,” Carbohydrate Research 327, no. 3 (July 2000): 345–52
- 28 -
Une étape d’activation est nécessaire pour amorcer la dissolution. En chauffant la solution (<80 °C), la cellulose et des vapeurs de DMAc entrent en contact et amorcent la réaction de décristallisation. La quantité de LiCl par rapport à la cellulose joue un rôle important. Plus le degré de polymérisation est élevé plus la teneur en LiCl doit être importante (entre 2 % et 12 %). Néanmoins en maintenant la solution à une température de 80/85 °C il est possible de diminuer la quantité de LiCl nécessaire à la dissolution. Mais une température supérieure à 85 °C entrainerait une forte dégradation de la cellulose.49
Diméthyle Sulfoxide (DMSO)/Fluorure de tétrabutyl-ammonuim (TBAF) [Figure I-20]
Figure I-20 DMSO/TBAF
En mélangeant le TBAF et le DMSO, la nucléophilie de l’ion F- va augmenter et
faciliter l’interaction entre les hydroxyles de la cellulose et le solvant. Cela induit une diminution des liaisons hydrogène et favorise la solubilisation du polymère. Le mécanisme est similaire au système DMAc/LiCl toutefois l’ion F- étant plus petit et plus nucléophile que
l’ion Cl -, il est possible de dissoudre des DP de l’ordre de 10 000. Avec une solution de 10 %
de TBAF/DMSO à 25 °C et en agitant 15min, la cellulose se solubilise dans une solution limpide. En augmentant la température et le pourcentage de TBAF, une fonctionnalisation est possible par réaction des hydroxyles et du DMSO.50
En conclusion sur les solvants organiques, il est possible de solubiliser la cellulose dans un milieu très polaire. Ces solubilisations n’entrainent pas de dépolymérisation, mais peuvent conduire à certaines fonctionnalisations et surtout permettent une forte diminution de la cristallinité. Grâce à ce procédé, la cellulose est moins réfractaire au traitement. Néanmoins, l’utilisation de solvants organiques implique un traitement des déchets conséquent et la récupération des produits formés peut s’avérer délicate.
49 Antje Potthast et al., “Degradation of Cellulosic Materials by Heating in DMAc/LiCl,” Tetrahedron Letters 43, no. 43 (October
2002): 7757–59
50 Sarah Köhler and Thomas Heinze, “New Solvents for Cellulose: Dimethyl Sulfoxide/Ammonium Fluorides,” Macromolecular
- 29 - iii. Dissolution dans les liquides ioniques
Un liquide ionique est défini par un sel liquide pour une température inférieure à 100 °C. Composé d’un cation organique et d’un anion organique ou inorganique, il a les propriétés suivantes (cas pour les liquides ioniques à base imidazolium notamment) :51
- Basse tension de vapeur - Non inflammable
- Bonne stabilité thermique - Bon électrolyte
- Pouvoir de solvatation élevé
Découvert en 1914 par Walden et al., ce n’est que dans les années 30 que les liquides ioniques (LIs) sont devenus attractifs. Les premières applications ont été développées par l’US Air Force Academy en tant qu’électrolyte pour des batteries.52 Les LIs peuvent être facilement modifiables en
changeant l’anion ou le cation. Cette facilité de changer les ions permet, à ce jour, de réaliser 1012
combinaisons cations/anions.48
Les LIs possédant un cation imidazolium sont les plus synthétisés. La Figure I-21 représente le schéma réactionnel général de la synthèse d’un LI avec une base d’imidazolium. La première étape est la réaction de Menchoutkine et consiste à faire réagir l’imidazole (ou une amine tertiaire) et un halogénoalcane. Ensuite, en faisant réagir soit un acide soit un sel, il est possible d’échanger l’anion initial (halogène).
Figure I-21 Schéma réactionnel de la synthèse d'un LI imidazolium
51 H. Olivier-Bourbigou, L. Magna, and D. Morvan, “Ionic Liquids and Catalysis: Recent Progress from Knowledge to
Applications,” Applied Catalysis A: General 373, no. 1–2 (January 31, 2010): 1–56
52 André Pinkert et al., “Ionic Liquids and Their Interaction with Cellulose,” Chemical Reviews 109, no. 12 (December 2009):