Identification de facteurs de régulation du VIH-1 chez les
macrophages humains
Mémoire
Yann Breton
Maîtrise en microbiologie-immunologie
Maître ès sciences (M. Sc.)
Québec, Canada
© Yann Breton, 2016
Identification de facteurs de régulation du VIH-1 chez les
macrophages humains
Mémoire
Yann Breton
Sous la direction de :
iii
Résumé
Lors d'une exposition au VIH-1, bien qu'une seule petite proportion des macrophages soit infectée, il est proposé que ces cellules jouent un rôle important dans l'infection et la propagation du VIH-1. Pour approfondir nos connaissances dans ce domaine, des analyses transcriptomiques et protéomiques ont été effectuées afin de comparer les MDMs (Macrophages Dérivés de Monocytes) infectés aux non infectés. Ces analyses ont mené à la sélection de 50 gènes dont l'expression est modulée chez les cellules infectées pour effectuer un criblage par siRNA pour leurs rôles fonctionnels dans le cycle viral. Huit cibles ont été identifiées comme des régulateurs de l'infection chez les MDMs, mais seulement le gène MDM2 agissait comme un facteur de susceptibilité. Ce gène a donc été l'objet d'études plus approfondies. L'inhibition de l'expression de MDM2 induit une diminution de moitié de l'expression virale. Nos résultats indiquent que la résistance accrue au VIH-1 associée à l’interférence de MDM2 est maintenue même si le niveau d'ARNm est rétabli, suggérant que cette protéine serait impliquée indirectement dans l'infection par le VIH-1. L'identification des cofacteurs viraux régulés par MDM2 mènera à une compréhension des évènements signalétiques contrôlant la réplication du VIH-1 dans les macrophages.
iv
Abstract
Upon exposure to HIV-1, only a small proportion of macrophages are infected whereas most remain uninfected. It is proposed that these cells play an important role in the establishment and propagation of HIV-1 infection. To further our knowledge in this field, transcriptomic and proteomic comparative analyses of uninfected and HIV-1-infected MDMs (Monocyte-derived macrophages) were performed. These analyses led to the selection of 50 genes that were tested for their functional roles in HIV-1 replication by siRNA screen. Eight genes were identified as regulators of HIV-1 infection in MDMs, but only MDM2 acted as a susceptibility factor. The knockdown of MDM2 decreased HIV-1 expression by two folds. Our results indicate that the resistance to HIV-1 upon MDM2 silencing is maintained in MDMs even if MDM2 mRNA level is restored, thus suggesting that this protein might be indirectly involved in HIV-1 infection. Identification of viral cofactors regulated by MDM2 will bring a new understanding of signaling events controlling HIV-1 replication in macrophages.
v
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des tableaux ... viii
Liste des figures ... ix
Liste des abréviations ... x
Remerciements ... xiii
Chapitre 1: Introduction ... 1
1.1. Le virus de l'immunodéficience humaine de type 1 ... 1
1.1.1. Historique et état actuel de l'épidémie ... 1
1.1.2. Le génome ... 2
1.1.3. Structure du virion ... 3
1.1.4. Cycle de réplication du VIH-1 ... 4
1.1.4.1. Évènements précoces ... 4 1.1.4.2. Évènements de biosynthèse ... 5 1.1.4.3. Évènements tardifs ... 6 1.1.5. Immunopathogenèse du VIH-1 ... 8 1.1.5.1. Transmission du virus ... 8 1.1.5.2. Phases de l'infection ... 9
1.1.5.3. Les réservoirs viraux ... 11
1.1.6. Les antirétroviraux ... 12
1.2. Les macrophages ... 13
1.2.1. Les macrophages tissulaires ... 13
1.2.2. Les macrophages dérivés de monocytes ... 14
1.2.3. La polarisation des macrophages ... 15
1.3. L'infection des macrophages par le VIH-1 ... 17
1.3.1. Caractéristiques du cycle viral chez les monocytes et macrophages ... 17
1.3.2. Polarisation des macrophages et VIH-1 ... 19
1.3.3. Impacts du virus sur les fonctions cellulaires ... 20
1.3.4. Les facteurs de restriction virale ... 20
Chapitre 2: Hypothèse et objectifs de recherche ... 24
vi
3.1. Production de particules virales ... 25
3.1.1. Modèles viraux ... 25
3.1.2. Culture de la lignée cellulaire 293T ... 26
3.1.3. Transfection au calcium-phosphate ... 27
3.1.4. Récolte et conservation de la production virale... 27
3.1.5. Caractérisation de la production virale ... 28
3.1.5.1. ELISA de la protéine de capside p24 ... 28
3.1.5.2. Culture de la lignée cellulaire TZM-bl et TCID50 des particules virales ... 29
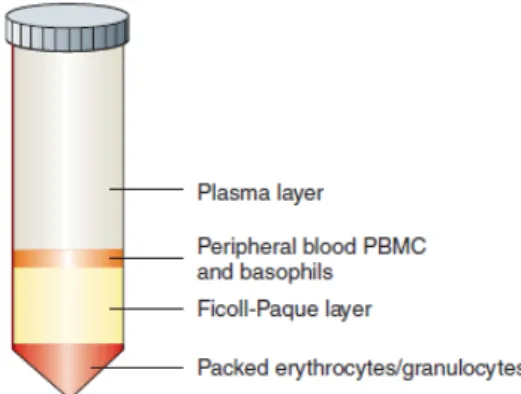
3.2. Isolement et culture de MDMs à partir de sang périphérique de donneurs sains 30 3.3. Criblage de petits ARN interférents chez les MDMs ... 32
3.3.1. Transfection des siRNAs ... 32
3.3.2. Infection des MDMs et analyse de l'infection ... 33
3.4. Validation des cibles potentielles ... 34
3.4.1. Cytométrie de flux ... 34
3.4.2. Extraction d'ARN et PCR quantitative ... 35
3.4.3. Immunobuvardage ... 36
3.4.4. Cinétique de production virale ... 37
3.5. Analyses statistiques ... 37
Chapitre 4: Résultats ... 38
4.1. Identification de protéines régulatrices du VIH-1 chez les macrophages humains ... 38
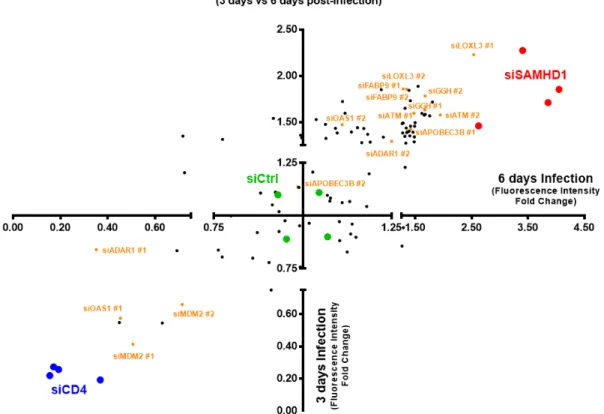
4.1.1. Criblage de petits ARN interférents ... 38
4.1.2. Efficacité des petits ARN interférents ... 40
4.2. Validation de MDM2 comme facteur de régulation du VIH-1 ... 42
4.2.1. Le gène MDM2 est surexprimé dans les macrophages infectés ... 42
4.2.2. La transfection des siRNAs contre MDM2 diminue l'infection par le virus NL4-3-Bal-IRES-HSA ... 43
4.2.3. La diminution du niveau de MDM2 n'induit pas de mort cellulaire ... 44
4.2.4. Vérification de l'expression de MDM2 suite à la transfection ... 45
4.2.5. Modulation de la production virale par les ARNs interférents ... 47
Chapitre 5: Discussion ... 50
5.1. Identification de facteurs de régulation potentiel du VIH-1 ... 50
5.2. Identification de MDM2 comme régulateur positif de l'infection ... 54
vii
5.2.2. Régulation induite par la voie p53-dépendante ... 56
5.2.3. Régulation induite par la voie p53-indépendante ... 57
Chapitre 6: Conclusion et perspectives ... 59
viii
Liste des tableaux
ix
Liste des figures
Figure 1: Prévalence du VIH chez les adultes (15-49 ans), 2014 par région du WHO ... 2
Figure 2: Le génome du VIH-1 et le rôle des protéines virales ... 3
Figure 3: Structure du VIH-1 ... 4
Figure 4: Cycle de réplication viral et les facteurs de restriction ... 8
Figure 5: Les stades de l'infection et leurs marqueurs immunologiques ... 9
Figure 6: Pathogénèse du VIH-1 ... 11
Figure 7: Classification actuelle des macrophages polarisés ... 16
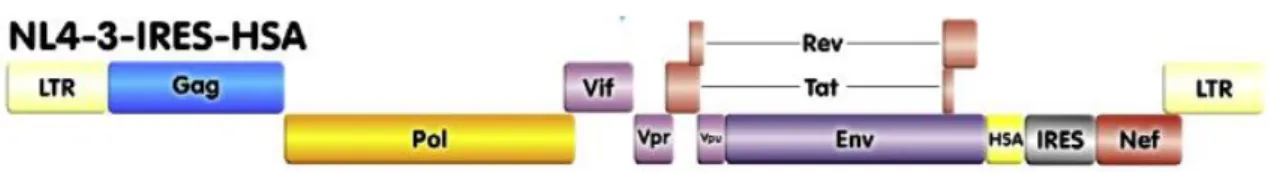
Figure 8: Représentation schématique du plasmide NL4-3-IRES-HSA ... 26
Figure 9: Séparation des cellules présentes dans le sang périphérique ... 30
Figure 10: Criblage de petits ARN interférents des gènes modulés par le VIH-1 révèle de nouveaux régulateurs de la réplication virale chez les macrophages ... 39
Figure 11: Efficacité des siRNAs sur le niveau d'ARNm ... 41
Figure 12: Modulation de l'infection par le siMDM2 ... 42
Figure 13: Le gène MDM2 est surexprimé chez les cellules infectées par le VIH-1 ... 43
Figure 14: La réduction de l'expression de MDM2 induit une diminution de l'infection ... 44
Figure 15: Des essais de viabilité ne montrent aucune toxicité apparente causée par l'interférence par ARN du gène MDM2 ... 45
Figure 16: Les siRNAs MDM2 diminuent efficacement le niveau d'ARNm ... 46
Figure 17: L'interférence par ARN du gène MDM2 réduit son expression protéique ... 47
Figure 18: Modulation de la production virale et du métabolisme des MDMs par la transfection de petits ARN interférents ... 49
x
Liste des abréviations
AB-HS De l'anglais AB-human serum
ADN Acide désoxyribonucléique
ARN Acide ribonucléique
ARNm Acide ribonucléique messager
BSA De l'anglais Bovine Serum Albumin
CA Capside
CD Cluster de différenciation
DC De l'anglais Dendritic cell
DPBS De l'anglais Dulbecco's phosphate-buffered saline
dNTP Désoxyribonucléotides
dpi De l'anglais days post-infection
DSB De l'anglais Double strand break
ELISA De l'anglais Enzyme-linked immunosorbent assay
Env Enveloppe
FDA De l'anglais Food and Drug Administration F-Luc De l'anglais Firefly luciferase
Gag De l'anglais Group-specific antigen
GALT De l'anglais Gut-associated lymphoid tissue
GFP De l'anglais Green fluorescent protein HBS De l'anglais Hepes buffered saline
HBSS De l'anglais Hanks' Balanced Salt Solution hpi De l'anglais hours post-infection
xi
HRP De l'anglais Horseradish peroxydase HSA De l'anglais Heat stable antigen
IFN Interféron
IL Interleukine
IN Intégrase
LC De l'anglais Langerans cell
LPS Lipopolysaccharide
LTR De l'anglais Long terminal repeat
ly T CD4+ Lymphocytes T CD4+
M-CSF De l'anglais Macrophage colony-stimulating factor
MA Matrice
MDM Macrophage dérivé de monocyte
MOI De l'anglais Multiplicity of infection
NC Nucléocapside
Nef De l'anglais Negative factor
NTP Nucléoside triphosphate
p24 Protéine de capside virale
PBMC De l'anglais Peripheral blood mononuclear cells PBL De l'anglais Peripheral blood lymphocytes PBS De l'anglais Phosphate-buffered saline PCR De l'anglais Polymerase chain reaction
PE Phycoérythrine
PIC De l'anglais Pre-integration complex
xii
PR Protéase
Rev De l'anglais Regulator of viral gene expression RPMI De l'anglais Roswell Park Memorial Institute
RT De l'anglais Reverse Transcriptase
RT-PCR De l'anglais Reverse transcription polymerase chain reaction
SIDA Syndrome de l'immunodéficience acquise siRNA De l'anglais Small interfering ribonucleic acid Tat De l'anglais Transactivator of transcription TCID50 De l'anglais 50% Tissue culture Infective Dose
TLR De l'anglais Toll-like receptor TNF De l'anglais Tumor necrosis factor Vif De l'anglais Viral infectivity factor Vpu De l'anglais Viral protein U Vpr De l'anglais Viral protein R
VIH-1 Virus de l'immunodéficience humaine de type 1
xiii
Remerciements
Tout d'abord, je voudrais remercier le Dr Michel J. Tremblay d'avoir accepté de superviser le stage de recherche d'un jeune étudiant en microbiologie, puis ses études de maîtrise et prochainement ses études doctorales. Merci beaucoup Michel d'être un superviseur exemplaire, toujours prêt à écouter nos questions et nos inquiétudes, autant sur le plan personnel que professionnel et à nous encourager lors des bons moments, mais aussi dans les moins bons. J'aimerais aussi remercier mon comité d'évaluation pour le temps consacré à mon mémoire.
Merci à tous les membres de l'équipe MJT, passés et présents, d'être aussi fous tout en faisant « de la bonne science ». Vous rendez les longues journées dans le NC3 bien plus plaisantes que les bruits de l'autoclave! Un merci tout spécial à Alizé et Jean-François pour tous les petits moments de décompression. Je garderai de nombreux souvenirs de nos discussions parfois trop intenses. Merci aussi à Alexandre et Michel Ouellet pour le temps consacré à ma formation et à l'élaboration de ce projet.
Je n'aurai pas pu me rendre aussi loin dans mes études sans le soutien de ma famille. Malgré les quelque 800 km qui me séparent de mon Abitibi natal, sans oublier mes proches en Gaspésie (l'autre extrémité du Québec), j'ai toujours pu compter sur votre aide et votre support tout au long de mes études. Vous m'avez laissé partir en sachant bien que je ne pourrai pas travailler en région, mais vous m'avez soutenu dans toutes mes décisions et vous avez toujours cru en moi. Sachez que je vous en suis grandement reconnaissant! Merci au Consortium canadien de recherche sur la guérison du VIH (CanCURE) pour le financement. C'est d'ailleurs très encourageant et motivant de voir une collaboration si importante entre des équipes de recherche partageant le même but.
Finalement, un grand merci à mon conjoint Frédéric d'être présent dans ma vie. Je peux toujours compter sur toi pour me remettre dans le bon chemin pour atteindre mon but lorsque je commence à douter de moi-même. Merci pour ton écoute lorsque je te parle de ma journée et des multiples techniques et appareils qui ne te disent absolument rien! Tu as rendu ces deux dernières années beaucoup plus faciles, en espérant que tu es prêt pour m'accompagner dans celles à venir!
1
Chapitre 1: Introduction
1.1. Le virus de l'immunodéficience humaine de type 1 1.1.1. Historique et état actuel de l'épidémie
Entre 1979 et 1981, plusieurs cas de pneumonie causée par Pneumocystis carinii font surface dans une communauté homosexuelle, montrant tous les signes d'une immunosuppression [1]. En réponse aux cas de plus en plus fréquents dans plusieurs pays, le terme SIDA (syndrome d'immunodéficience acquise) fut adopté pour ce phénomène. C'est en France en 1983 que l'agent causal fut isolé pour la première fois. L'équipe du Dr Luc Montagnier, de l'institut Pasteur de Paris, a isolé un virus inconnu à partir d'un ganglion lymphatique d'un patient homosexuel, identifié comme un membre de la famille des HTLV (human T-cell leukemia virus). Cette particule virale a montré la capacité de se propager dans une culture de lymphocytes T provenant d'un donneur sain. Ce virus sera nommé LAV pour lymphadenopathy-associated virus [2]. En 1984, l'équipe du Dr Robert C. Gallot, située aux États-Unis, découvrit aussi un virus aux mêmes propriétés qu'ils nommèrent HTLV-III [3]. Des études génétiques ont montré qu'il s'agissant de deux variants d'un même virus, recevant officiellement en 1986 son nom actuel par une décision du International Committee on Taxonomy of Viruses: le virus de l'immunodéficience humaine (VIH) [4, 5].
Actuellement, selon le dernier rapport d'ONUSIDA de 2015, 36.9 millions de personnes vivaient avec le VIH. Le nombre de nouvelles infections est de 2 millions/année, alors que 1.2 million/an de personnes infectées au VIH-1 décèdent de maladies associées au SIDA. Cependant, le nombre de nouvelles infections a diminué de 35% depuis 2000 et le nombre de décès de 42%. C'est en Afrique que la prévalence est la plus forte (voir Figure 1 pour les données de 2014). Même si ces nombres sont toujours trop élevés, la tendance montre que la prévention et les thérapies antivirales sont efficaces. En juin 2015, il a d'ailleurs été calculé que 15.8 millions de personnes vivantes avec le VIH avaient accès aux thérapies antirétrovirales. Le but d'ONUSIDA est de mettre fin à l'épidémie d'ici 2030 et a élaboré le plan 90-90-90: que 90% des personnes infectées connaissent leur statut, que 90% des personnes séropositives aient accès au traitement antirétroviral et que 90% des personnes sous médication voient leur charge virale supprimée [6].
2
Figure 1: Prévalence du VIH chez les adultes (15-49 ans), 2014 par région du WHO Prévalence mondiale du VIH-1 chez les adultes en 2014. Figure issue du site internet du World Health Organisation (WHO) [7].
1.1.2. Le génome
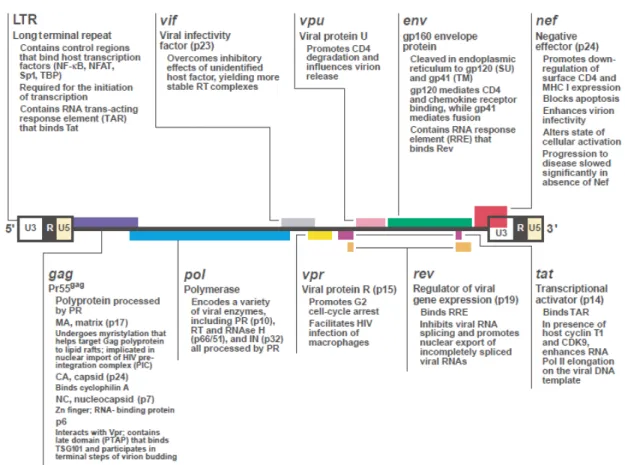
Le VIH-1 est un rétrovirus du genre Lentivirus ayant un génome petit, mais complexe. La particule virale renferme deux copies d'un même ARN simple brin de polarité positive et d'une longueur de 9200 nucléotides. Cet ARN code pour trois polyprotéines (gag, pol et env) et six protéines régulatrices et accessoires (voir Figure 2 pour le rôle de chacune). Le génome viral est flanqué de séquences non traduites (LTR) à chacune de ses extrémités. Ces séquences terminales longues répétées sont impliquées dans la régulation de la transcription des gènes viraux, dans la transcription inverse et dans l'intégration du génome viral dans celui de la cellule hôte [8, 9].
3
Figure 2: Le génome du VIH-1 et le rôle des protéines virales
Représentation du génome viral et description des diverses protéines virales codées par chacun des gènes. Figure issue de Greene, W.C. et Peterlin, B.M., 2002 [10].
1.1.3. Structure du virion
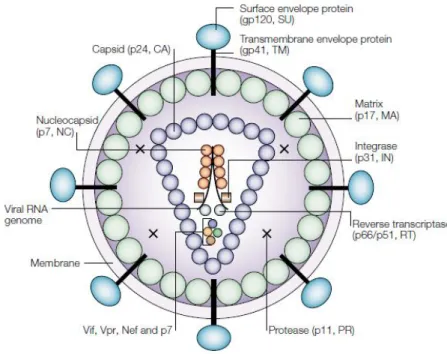
La particule virale est sphérique et son diamètre est d'environ 120 nm [9]. Elle est enveloppée d'une bicouche lipidique provenant de la cellule hôte lors du bourgeonnement du virion. Cette enveloppe comporte les trimères de gp120 et gp41 ainsi que plusieurs molécules de l'hôte telles que HLA-DR et ICAM-1, impliquées respectivement dans la présentation d'antigène et l'adhésion cellulaire [11]. Sous l'enveloppe virale se retrouve une structure formée par la protéine de matrice (MA, p17) qui confère la forme sphérique au virus (voir Figure 3). À l'intérieur du virion se trouve la capside virale, un assemblage conique d'environ 1500 copies de la protéine de capside (CA, p24). La capside est définie comme étant le cœur du virus. Elle contient les deux copies de l'ARN viral stabilisées par la nucléocapside (NC). On y retrouve aussi les enzymes nécessaires au cycle viral, soit la
4
transcriptase inverse (RT), l'intégrase (IN) et la protéase (PR), ainsi que les protéines régulatrices conférant le pouvoir infectieux du virus (Vif, Vpr et Nef) [12].
Figure 3: Structure du VIH-1
Coupe transversale d'un virion mature. Figure issue de Robinson, H.L., 2002 [13]. 1.1.4. Cycle de réplication du VIH-1
1.1.4.1. Évènements précoces
Le virus possède à sa surface des trimères de glycoprotéines. Ceux-ci sont composés des glycoprotéines gp120 et gp41 dont les noms proviennent de leur taille respective de 120 et 41 kilodaltons. Ces deux glycoprotéines résultent du clivage de la polyprotéine gp160, aussi nommée en fonction de sa taille, par des protéases cellulaires [14, 15]. Les deux sous-unités forment ensuite un hétéroduplexe où la gp41 est transmembranaire et la gp120 se trouve dans la matrice extracellulaire. Ces hétéroduplexes sont retrouvés sous forme de trimères et seront incorporés chez le virion lors du bourgeonnement [16, 17]. La gp120 est la glycoprotéine qui permet au virus de s'attacher à son récepteur cellulaire, le CD4. La gp120 contient 5 domaines variables (V1 à V5) et 5 domaines conservés (C1 à C5) [18]. Sa structure tridimensionnelle forme une cavité pouvant accueillir le CD4. Cette cavité possède une poche profonde qui reçoit un acide aminé du récepteur CD4 critique pour la
5
liaison: le phénylalanine-43 [19]. Cet attachement entraîne un changement de conformation de la gp120 et du CD4, rapprochant le virus de la membrane cellulaire pour permettre la liaison au corécepteur. Étant donné que le virus est fortement sujet aux mutations, les séquences des domaines variables risquent d'être modifiées [20]. La région V3, plus précisément, a un rôle à jouer dans le tropisme de la particule virale. Dépendamment de sa séquence, le virus aura un tropisme dit R5 ou X4, chacun reconnaissant respectivement le corécepteur CCR5 ou CXCR4 [21-23]. La liaison à ce corécepteur libère le peptide de fusion situé en position N-terminale de la gp41. Il s'agit d'une séquence d'acides aminés hautement hydrophobique qui s'associe aux lipides membranaires de la cellule et entraîne la fusion de l'enveloppe virale à celle-ci, libérant la capside virale dans le cytoplasme [24].
1.1.4.2. Évènements de biosynthèse
Une fois entrée dans le cytoplasme, la capside virale se désassemble (voir Figure 4). La décapsidation serait causée par la cyclophiline A, une molécule incorporée dans le virion lors de la formation de la capside. Cette molécule stabilise la capside virale et protégerait aussi le génome viral lors de sa migration dans le cytoplasme, permettant d'échapper au système immunitaire. Cependant, lors d'une nouvelle ronde d'infection, la p24 qui n'est pas associée à la cyclophiline A recrute la protéine Nup358, menant à la décapsidation et à la libération de son contenu dans le cytoplasme [25, 26]. L'ARN simple brin et les protéines virales sont libérés et participent à la formation du complexe de réplication. Celui-ci est composé de l'ARN viral, la protéine de matrice (MA), de nucléocapside (NC), la RT, l'intégrase, la protéase, Vpr, l'ARN de transfert Lys 3 ainsi que les histones H1, H2a, H2b, H3 et H4 [10, 27]. La protéine de matrice se retrouve sous forme phosphorylée, ce qui l'empêche d'être associée à la membrane cellulaire et l'amène plutôt à se lier au cytosquelette d'actine, permettant une rétrotranscription efficace de l'ARN viral en ADN double brin [28]. Cependant, la transcriptase inverse du VIH-1 permet plusieurs erreurs. Il a été calculé que celle-ci introduit une mutation par 6000 nucléotides, expliquant la grande variabilité génétique de ce virus [29]. L'ADN nouvellement synthétisé se retrouve sous forme linéaire et est flanqué de région LTR. Il formera ensuite le PIC (complexe de préintégration) avec l'intégrase, la RT, la MA, la Vpr et le HMGI(Y) (high-mobility group
6
DNA-binding protein) [10, 30]. Le PIC profite du réseau microtubulaire de la cellule pour
migrer vers le noyau et y entrer. Ce complexe protéique possède des propriétés caryophiles grâce à ses signaux de localisation nucléaire (NLS, nuclear localization signal) se trouvant sur la MA, la Vpr et l'intégrase. Il sera donc transporté activement au travers de la membrane nucléaire par les importines et karyophérines, expliquant pourquoi le VIH-1 peut infecter des cellules ne se divisant pas et y intégrer son génome [31-33]. Une fois à l'intérieur du noyau cellulaire, l'intégrase clive les extrémités de l'ADN viral pour y enlever deux nucléotides au bout 3' des brins, puis catalyse la réaction d'intégration dans l'ADN cellulaire [30]. Le virus est maintenant présent dans la cellule sous forme de provirus. Le provirus peut être présent sous deux états: latent ou réplicatif [34]. Le virus peut être intégré dans bien des locations sur les divers chromosomes, mais il aura tendance à choisir des régions transcriptionnellement actives, appelées hotspots, et évitera les régions répétées des centromères afin d'assurer sa transcription [35, 36]. Le LTR en position 5' du provirus recrute l'ARN polymérase de type II qui produira seulement de courts transcrits d'ARNm pour les gènes tat, rev et nef. Cependant, lorsque les facteurs de transcription tels que NF-κB et NFAT sont recrutés à un site amplificateur de la transcription en amont du LTR et que la protéine virale Tat est suffisamment présente pour se lier à la région TAR (transactivation response), l'ARN pol II peut effectuer une élongation efficace de l'ARN viral [37]. Des ARNm de diverses tailles seront produits, soit un ARN non épissé d'environ 9 kb (kilobases) codant pour Gag et Gag-Pol, des ARNm d'environ 4 kb produits par épissage unique codant pour Vif, Vpr, Vpu et Env et d'autres ARNm faisant près de 2 kb produits par épissages multiples, résultant en les protéines Tat, Rev et Nef. Les ARNm de 2 kb seront les premiers à quitter le noyau pour être traduits en protéines. Une fois qu'une certaine concentration de Rev est atteinte, cette dernière servira à l'exportation des autres transcrits viraux présents dans le noyau vers le cytoplasme [38].
1.1.4.3. Évènements tardifs
La dernière étape du cycle viral est la production de nouvelles particules virales infectieuses. Le précurseur de la polyprotéine Gag, nommé Pr55Gagen fonction de son poids
7
domaines correspondant aux protéines MA, CA, NC et p6 [39]. Les Pr55Gag synthétisés s'associent aux radeaux lipidiques présents dans la membrane plasmique via une interaction entre la séquence MA et les lipides membranaires [40-42]. Près de 2000 polyprotéines Gag vont se concentrer à la membrane cellulaire, en plus d'environ 200 polyprotéines Gag-Pol. Deux brins d'ARNs génomiques seront recrutés sous forme de dimère au site d'assemblage par le domaine NC de Gag, grâce à ses propriétés de protéine chaperonne d'acides nucléiques [43, 44] . La glycoprotéine d'enveloppe, quant à elle, est exprimée au niveau du réticulum endoplasmique rugueux (RER) sous forme de polyprotéine nommée gp160, puis transformée par l'appareil de Golgi en trimères de gp120 et gp41. Ces trimères sont transportés à la surface de la cellule et forment des spicules au travers de la membrane cellulaire. La partie cytosolique de gp41 s'associe aux domaines MA de Pr55Gag [45]. Le
virus utilise ensuite la machinerie cellulaire du sentier ESCRT (endosomal sorting
complexes required for transport) pour remodeler la membrane cellulaire et mener au
bourgeonnement du virus [46]. Le domaine p6 de Gag est aussi impliqué dans la production de particule virale, car des mutations dans sa séquence empêchent le bourgeonnement et mènent à une accumulation de particule virale à la membrane plasmique [47]. Lors de la sortie de la particule virale, celle-ci emporte une partie de la membrane plasmique contenant les protéines d'enveloppe et des molécules de l'hôte (voir section 1.1.3.). L'étape finale du cycle viral est la maturation du virus. La protéase virale clive les polyprotéines Gag et Pol, résultant en les protéines de structures (MA, CA, NC) et les enzymes virales (l'intégrase, la protéase et la transcriptase inverse), conférant au virus sa structure finale et son infectivité [48, 49].
8
Figure 4: Cycle de réplication viral et les facteurs de restriction
La réplication du VIH-1 peut être interrompue à plusieurs étapes du cycle par des protéines de la cellule hôte. Figure issue de Barré-Sinoussi, F. et al., 2013 [50].
1.1.5. Immunopathogenèse du VIH-1 1.1.5.1. Transmission du virus
Chez une personne infectée et non traitée, le virus peut être retrouvé dans ses fluides biologiques, dans ses tissus et ses muqueuses. Cependant, le sang, le lait maternel, le sperme et les sécrétions vaginales, contenant tous des particules virales, sont les causes de nouvelles infections via trois modes de transmission possibles. Le premier et le plus fréquent est par relations sexuelles non protégées, qu'elles soient vaginales ou anales. Le deuxième est par les produits sanguins alors que le troisième est la transmission de la mère à l'enfant, aussi appelée transmission verticale, lors de la grossesse, l'accouchement ou encore l'allaitement du nouveau-né [51]. Alors que la transmission sexuelle est la plus fréquente, elle n'est pas la plus efficace (8 infections sur 10 000 expositions chez le récepteur lors d'une relation vaginale et 138 infections sur 10 000 chez le récepteur d'une relation anale). C'est plutôt la transmission sanguine, spécifiquement lors de transfusion de produits sanguins (9250 infections sur 10 000 expositions), suivie de la transmission mère-enfant (2260 infections sur 10 000 expositions) [52].
9 1.1.5.2. Phases de l'infection
L'infection peut être séparée en 7 phases, dépendamment des marqueurs pouvant être détectés dans le plasma du patient (voir Figure 5). Il y a d'abord une phase éclipse suite à l'infection où le virus ne peut être détecté par la méthode la plus sensible, soit la PCR de l'ARN viral. Celui-ci est détectable à partir du 10e jour suivant l'infection et marque le
début de la phase 1. Le nombre de copies d'ARN viral grimpe de manière exponentielle à partir de cette phase. La phase 2 est marquée par la présence de l'antigène p24 et la phase 3 par la présence d'anticorps dirigés contre le VIH-1. La phase 3 représente donc la séroconversion du patient. Une fois la phase 4 débutée, la virémie commence à décliner et un certain patron de protéines est détectable par immunobuvardage, sans que celui-ci réponde aux critères de la Food and Drug Administration (FDA). L'étape 5 consiste en la fin de la phase aiguë de l'infection et la présence d'un patron spécifique pour les protéines virales, sauf pour la polyprotéine Pol (p31). Le début de la phase chronique de l'infection, la phase 6 représente une virémie stable et la présence de Pol [53].
Figure 5: Les stades de l'infection et leurs marqueurs immunologiques
Chaque stade de l'infection par le VIH-1 est accompagné d'antigènes ou d'anticorps spécifiques pouvant être détectés dans le plasma du patient. Figure issue de McMichael, A.J. et al., 2010 [54].
10
Lors de l'entrée des virions dans la lamina propria des muqueuses, ceux-ci se retrouvent devant un bassin de lymphocytes T CD4+ (lyT CD4+), macrophages et cellules dendritiques (DC), naturellement présents afin de prévenir les infections. Ici débute la séroconversion, c'est-à-dire l'activation de l'immunité humorale, où il y aura une production d'anticorps dirigés contre les protéines virales. Les premières cellules à être infectées sont les lyT CD4+
et les cellules de Langerhans (LC) [55]. DC-SIGN, une lectine de type C présente chez les DC, permet aussi à ces cellules de capter les particules virales en liant les gp120. DC-SIGN n'agit pas en tant que récepteur du VIH, mais permet plutôt de favoriser l'infection des lyT CD4+ ou d'autres cellules sujettes à l'infection [56]. Dans les 3 à 6 jours suivant l'infection,
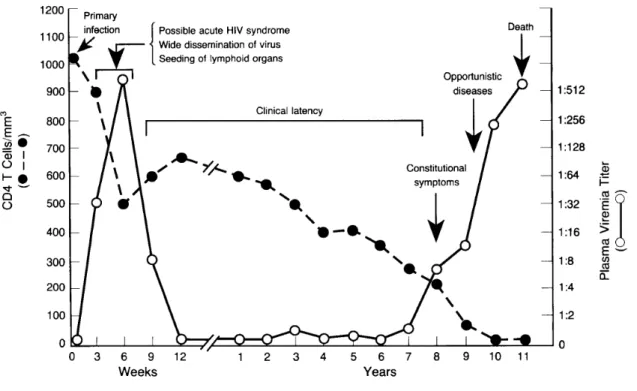
les cellules infectées et les DC ayant capté des virus, migrent vers les ganglions lymphatiques, principalement le GALT (gut-associated lymphoid tissue), où il y aura une réplication massive du virus. Cette expansion virale induira une déplétion dans le nombre de lyT CD4+ dans les ganglions lymphatiques, puis systémiques (voir Figure 6). [57] La
diminution des lyT CD4+ dans le GALT mène aussi à de la translocation bactérienne,
activant le système immunitaire et favorisant la réplication virale [58].
L'infection entraînera une réponse de l'immunité innée. Un phénomène appelé cytokine
storm se produit dans la phase aiguë de l'infection, où il y a une forte augmentation d'IFN-α
et d'IL-15 comme réponse antivirale et de la chimiokine IP-10. S'ensuit une production de TNF et de MCP-1 qui sera soutenue lors de l'établissement de l'infection. Cette vague de cytokine est produite principalement par les DC, mais les autres cellules de l'immunité innée (monocytes/macrophages, cellules NK) participent aussi à cette réponse [59, 60]. Ces cytokines activent constamment les lyT CD4+ et est l'un des facteurs menant à leur déplétion. Les cellules NK peuvent aussi éliminer les cellules infectées par le virus [61]. La réponse adaptative du système immunitaire, quant à elle, est inefficace pour contrôler l'infection. Les premiers anticorps, dirigés contre la gp41, sont produits par les lymphocytes B 8 jours après que le virus soit détectable dans le sang, retrouvés sous forme de complexes immuns avec les virions, et après 13 jours pour des anticorps libres. Les anticorps contre la gp120, eux, n'apparaissent qu'autour du 28e jour suivant la détection du virus. Les IgG et
IgM ainsi produits n'ont que peu d'impacts sur la réplication virale et l'établissement de l'infection [62]. Les premiers anticorps neutralisants apparaissent plus tard, soit à partir du 3e mois suivant l'infection. Ces anticorps sont spécifiques aux protéines d'enveloppe, mais
11
ceci mène à la sélection de virus pouvant évader la réponse adaptative grâce au fort taux de mutations lors de la réplication virale [63]. L'activité cytotoxique des lyT CD8+ débute lors du pic de virémie dans le plasma. Ces lymphocytes détruisent les cellules présentant les peptides dérivés de Nef, puis évoluent de manière à répondre à ceux provenant de Gag, Pol et Env [64]. Cette adaptation assure que le virus échappe de moins en moins au système immunitaire et finit par assurer un certain contrôle de la virémie.
Figure 6: Pathogénèse du VIH-1
Évolution de la virémie lors de l'infection jusqu'à la phase SIDA et son impact sur le nombre de lyT CD4+. Figure issue de Pantaleo, G. et al., 1993 [65].
1.1.5.3. Les réservoirs viraux
Les patients sous traitement sont en mesure de conserver leur charge virale sous le seuil de détection. Malgré ce contrôle, des cellules infectées persistent de manière latente, c'est-à-dire qu'elles contiennent l'ADN viral, mais ne produisent pas de nouvelles particules infectieuses. Cette latence virale peut être soit pré ou post-intégration [66]. Ces cellules peuvent être réactivées afin de sortir le virus de sa phase latente et mener à la production de nouveaux virions [67]. L'établissement de ces réservoirs viraux semble se produire dès la phase éclipse de l'infection [68]. Lorsqu'un patient arrête sa thérapie, la charge virale se
12
rétablit dans le plasma dans les 3 semaines suivant l'interruption, l'obligeant à être sous médication à vie [69].
Les réservoirs viraux sont formés par plusieurs types cellulaires. Le plus étudié est le lyT CD4+ mémoire. Ces réservoirs sont maintenus lors de la thérapie par la prolifération des lymphocytes, mais la proportion de cellules portant le virus latent diminue avec le temps, si la thérapie est maintenue [70, 71]. Les lyT CD4+ effecteurs, soit les Th1 et Th17,
contribuent aussi au maintien de la persistance virale lors de la thérapie [72]. Les cellules de l'immunité innée forment d'autres réservoirs viraux. Les monocytes, microglies, astrocytes, macrophages intestinaux et macrophages alvéolaires sont tous rapportés pour contenir l'ADN viral intégré [73-76]. Ces cellules devront toutes être éliminées afin d'éradiquer le VIH-1 chez un patient.
Un problème dans l'élimination de ces cellules est qu'elles sont présentes dans des sanctuaires pharmacologiques, c'est-à-dire des tissus difficiles d'accès pour les drogues antivirales. Le GALT, le RALT (rectal-associated lymphoid tissue), les ganglions lymphatiques, le système nerveux central et le tractus génital (mâle et femelle) sont considérés comme des sanctuaires. Les macrophages sont aussi considérés comme des sanctuaires dus à leur expression de transporteurs à efflux, rendant difficile de garder une concentration intracellulaire efficace pour les drogues antivirales [77].
1.1.6. Les antirétroviraux
La découverte de molécules inhibitrices de la protéase virale et de la transcriptase inverse dans les années 90 a ouvert la porte à une monothérapie, qui rapidement a évolué en une thérapie combinatoire de 2 médicaments antirétroviraux pour finalement en ajouter un troisième, formant un cocktail mieux connu sous le nom de trithérapie. En combinant plusieurs antirétroviraux, les personnes séropositives sont en mesure de conserver leur charge virale sous le seuil de détection, étant maintenant de moins de 50 copies d'ARN viral par ml de sang [78, 79]. Les médicaments disponibles actuellement inhibent le cycle viral à diverses étapes et sont séparés en 4 catégories: (1) les inhibiteurs d'entrée, soit les inhibiteurs de fusion ou les antagonistes de CCR5 ou CXCR4; (2) les inhibiteurs de la transcriptase inverse, constitués par les NRTIs (nucleoside/nucleotide reverse transcriptase
13
inhibitors) et les NNRTIs (non-nucleoside reverse transcriptase inhibitors); (3) les
inhibiteurs d'intégrase et (4) les inhibiteurs de la protéase [79].
1.2. Les macrophages
Les phagocytes mononuclés, aussi appelés macrophages, sont des cellules ayant une grande capacité de phagocytose. Leur rôle est de patrouiller l'organisme et de se débarrasser des corps étrangers, tels que les bactéries, moisissures, ou encore les cellules endommagées. Les macrophages sont séparés en deux classes: (1) les macrophages tissulaires, résidents permanents des tissus sujets à des dommages et invasion par des microorganismes et (2) les macrophages dérivés de monocytes, principalement impliqués dans l'inflammation [80].
1.2.1. Les macrophages tissulaires
Les macrophages tissulaires forment une population hétérogène répartie dans la majorité des tissus, chacun ayant une niche spécifique. On y retrouve entre autres les macrophages alvéolaires dans les poumons, les cellules de Langerhans dans l'épiderme et les microglies du système nerveux central. Beaucoup de ces cellules ont longtemps été considérées comme des cellules dendritiques, mais l'importance du récepteur du M-CSF (macrophage
colony-stimulating factor) pour ces cellules fait d'elles des membres de la famille des
macrophages [81]. Le M-CSF est une cytokine permettant la différenciation des cellules myéloïdes en macrophages de phénotype M2, c'est-à-dire des macrophages anti-inflammatoires, bien que des études récentes ont montré que ces cellules ont une grande plasticité au niveau de leur polarisation [82]. Ces macrophages produisent donc du M-CSF constitutivement et il a été montré in vitro que cette cytokine améliore leur viabilité, aide à leur prolifération et leur permet de conserver un phénotype M2-like [83, 84]. Pour certains tissus, c'est plutôt l'interleukine-34 (IL-34) qui va lier le récepteur du M-CSF, le CSF1R, et entraîner la différenciation des macrophages [85].
Les macrophages résidents des tissus sont présents souvent dès le stade embryonnaire. Par exemple, les précurseurs des cellules de Langerhans proviennent tout d'abord de la vésicule vitelline, puis sont remplacés majoritairement par les monocytes du foie fœtal pendant
14
l'embryogenèse [86]. Les microglies, quant à elles, proviennent de macrophages primitifs s'étant différenciés dans la vésicule vitelline avant de migrer vers le tissu cérébral de l'embryon [87]. Ces cellules sont maintenues tout au long de la vie de l'organisme majoritairement par prolifération. Contrairement au modèle proposé par les Drs van Furth et Cohn, les macrophages tissulaires ne proviennent pas tous des monocytes circulants et ne sont pas maintenus dans le temps par une infiltration de monocytes dans le tissu, mais sont plutôt des macrophages présents dès le stade embryonnaire et sont maintenus par prolifération locale [80, 88].
Le rôle de ces macrophages tissulaires est de prévenir les infections microbiennes et de résoudre l'inflammation pour contrôler les dommages. Suite à la reconnaissance d'un pathogène ou d'un signal de danger, les macrophages tissulaires sécrètent des cytokines afin de déclencher l'inflammation et le recrutement de neutrophiles et de monocytes [89, 90]. Une fois le pathogène détruit, les macrophages résidents amorcent la résolution de l'inflammation. La phagocytose des cellules apoptotiques est restreinte aux cellules exprimant la 15-lipoxygénase, une enzyme présente chez les macrophages résidents et les cellules stimulées par l'IL-4, donc ayant un phénotype anti-inflammatoire [91].
1.2.2. Les macrophages dérivés de monocytes
Les monocytes représentent 10% des leucocytes chez l'homme. Ces cellules résultent de la différenciation de cellules souches hématopoïétique dans la moelle osseuse et patrouillent l'organisme grâce à la circulation sanguine, où ils y restent sur une période de 1 à 2 jours avant d'être éliminé ou de simplement mourir [92]. Les monocytes sont entreposés dans la rate et peuvent être déployés en cas d'inflammation [93]. Les monocytes sont classés en 3 sous-populations selon leurs clusters de différenciation (CD): (1) les classiques (85% des monocytes du sang), soit CD14++ et CD16-; (2) les intermédiaires (5% des monocytes)
CD14++ CD16+ et (3) les non-classiques (10% des monocytes) CD14+ CD16++ [92, 94]. Les
monocytes classiques sont principalement impliqués dans la phagocytose et la sécrétion de cytokines inflammatoires comme l'IL-6 et l'IL-8 tandis que la population intermédiaire sécrète fortement le TNF (tumor necrosis factor) en réponse aux lipopolysaccharides
15
(LPS). Les monocytes non classiques, eux, sont plutôt impliqués dans la réponse aux virus via leur TLR (toll-like receptor) 7 et 8 [95, 96].
Lors de l'inflammation, les monocytes seront attirés dans le tissu par la présence de chimiokines de la famille des MCPs (monocyte chemoattractant protein) dont MCP-1 (aussi connu sous le nom de CCL2), des ligands du récepteur CCR2 [97]. Une fois dans l'environnement tissulaire, les monocytes seront en présence de multiples cytokines et deviendront des macrophages dérivés de monocytes (MDMs).
1.2.3. La polarisation des macrophages
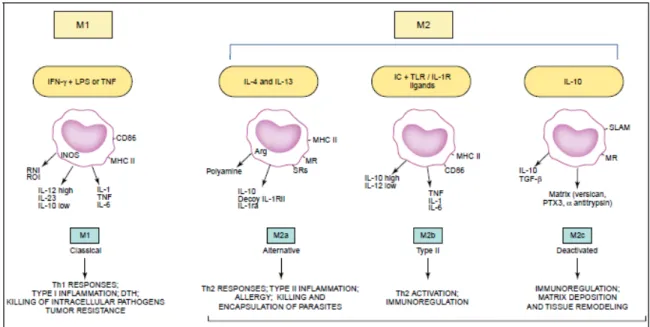
Le système actuel pour classer les macrophages est celui des phénotypes M1 (pro-inflammatoire) et M2 (anti-(pro-inflammatoire) (voir Figure 7). Cette nomenclature, à la base, servait à reproduire celle des lymphocytes T auxiliaires (helper), soit les Th1 et les Th2, car les macrophages M1 semblent induits par les cytokines des Th1, tandis que les M2 par celles des Th2 [98]. En réponse à l'interféron-γ (IFN-γ), une cytokine produite en réponse aux microorganismes, les macrophages adoptent un phénotype M1, pouvant être renforcé par d'autres cytokines comme le TNF et le LPS. Pour les monocytes, l'exposition au GM-CSF (granulocyte-macrophage colony-stimulating factor) les tend vers un phénotype M1 lors de la différenciation en macrophages. Ces cellules produisent fortement des dérivés réactifs d'oxygène (ROS) et des cytokines inflammatoires comme le TNF, l'IL-6 et l'IL-12 pour agir dans la réponse inflammatoire [98]. Les macrophages stimulés par les cytokines produites par les Th2 et les Treg adoptent un phénotype différent, appelé alternatif ou M2. Ces macrophages ont un niveau beaucoup plus élevé de récepteurs de mannose que les M1, une lectine de type C servant à reconnaître certains pathogènes et l'exposition au M-CSF oriente les monocytes/macrophages vers ce phénotype. L'état alternatif des macrophages peut être représenté en 3 sous-groupes: (1) les M2a, suite à une stimulation par l'IL-4 ou l'IL-13; (2) les M2b suite à la reconnaissance d'un complexe immun et (3) les M2c, aussi appelés macrophages désactivés, lorsqu'ils sont exposés à l'IL-10 [99-102]. Ces sous-populations, à l'exception des M2b, produisent moins de cytokines pro-inflammatoires et ont peu d'activités microbicides. Les M2a ont une activité phagocytaire nettement moindre que les macrophages non stimulés, tandis que les M2c phagocytent beaucoup plus, sans
16
toutefois tuer les pathogènes [103, 104]. Les M2a sécrètent de l'Il-10 et l'antagoniste du récepteur de l'IL-1 (Il-1ra) tandis que les M2c produisent de l'Il-10 et du TGF-β (Transforming growth factor beta). Les M2b, eux, conservent leur sécrétion élevée de cytokines pro-inflammatoires, mais participent à la différenciation des lymphocytes T en Th2 [105].
Figure 7: Classification actuelle des macrophages polarisés
Spectre d'activation des macrophages en réponse aux stimuli. Figure issue de Mantovani, A. et al., 2004 [105].
Bien que la classification des macrophages soit bien établie, de récentes études ont montré que les phénotypes ne sont pas fixés à la dualité M1 ou M2. L'exposition des monocytes au GM-CSF ou au M-CSF est connue pour induire leur différenciation en macrophages de type M1 ou M2 respectivement. Or, il a été montré que des macrophages dérivés de monocytes via le M-CSF qui sont ensuite exposés à du LPS et de l'IFN-γ se retrouve avec un profil transcriptomique semblable à celui des monocytes différenciés avec le GM-CSF, puis stimulés avec le LPS et l'IFN-γ [106]. La différenciation préparerait plutôt les macrophages à adopter un état et la stimulation serait responsable du changement de phénotype.
17
Une deuxième étude montre que la polarisation est beaucoup plus large que seulement deux phénotypes. Des MDMs différenciés avec du GM-CSF ou M-CSF ont été mis en présence de 28 combinaisons de stimuli différents pour en définir leur profil transcriptomique. Suite à des analyses bio-informatiques, cette étude révèle que la polarisation des macrophages est représentée par un spectre d'activation plutôt que le système M1/M2 [82]. Il y a donc une grande plasticité au sein de l'activation des macrophages qui doit être prise en compte lors de l'étude de leur réponse face à un pathogène.
1.3. L'infection des macrophages par le VIH-1
Plusieurs types cellulaires de l'immunité peuvent être infectés par le VIH-1, le plus étudié étant les lymphocytes T CD4+. Cependant, l'importance des autres lignées cellulaires dans
l'établissement de l'infection et la formation de réservoirs viraux ne doit pas être sous-estimée. Parmi ces cellules, les monocytes et les macrophages sont permissifs au VIH-1 et sont des réservoirs viraux importants, en raison de leur présence dans les tissus où les antirétroviraux ont difficilement accès. L'ADN viral peut être retrouvé dans les monocytes et les macrophages en plus des microglies et des macrophages périvasculaires du système nerveux central chez une personne infectée, même lorsque sous thérapie [73, 76, 107]. Les macrophages ont une longue durée de vie et sont grandement résistants à l'effet cytopathogène du virus. Ceci fait d'eux de grands producteurs de particules virales dont il est primordial de se débarrasser pour éventuellement guérir une personne du VIH-1.
1.3.1. Caractéristiques du cycle viral chez les monocytes et macrophages
Les monocytes peuvent être classés en trois groupes selon leur expression du CD16 (voir section 1.1.2.). Or, le taux d'infection est différent selon le type de monocytes. Les monocytes CD16+ sont plus permissifs au VIH-1 R5-tropique que la population CD16-.
Deux raisons expliquent ce phénomène. Tout d'abord, l'expression de CD16 corrèle avec une expression plus élevée du corécepteur CCR5. Ensuite, les monocytes CD16+ expriment
la forme inactive d'APOBEC3G, un facteur de restriction du VIH-1, tandis que les CD16
18
l'infection des monocytes CD16+ résultant en une réplication virale accrue une fois l'infection établie [108].
Les macrophages expriment à la fois le corécepteur CXCR4 et CCR5. La plupart des souches X4 et R5 sont capables d'infecter les macrophages, mais l'utilisation de CCR5 semble privilégiée et plus efficace. Lors d'une faible présence de CD4 à la surface d'une cellule, ce qui est le cas chez les macrophages, l'utilisation de CCR5 au lieu de CXCR4 faciliterait l'entrée du virus [109]. L'utilisation de virus R5 facilite donc l'infection des macrophages lors d'études in vitro. Bien que l'entrée du virus par attachement à la surface soit la voie typique d'infection, la grande activité phagocytaire des macrophages permet une autre voie d'entrée. La macropinocytose permet l'internalisation de molécules indépendamment des récepteurs cellulaires. Ainsi, les macrophages peuvent internaliser le virus dans des vésicules intracellulaires. Certaines particules résisteront à la dégradation et seront en mesure de fusionner à la membrane vésiculaire, libérant la capside dans le cytosol [110].
Ensuite, le VIH-1 peut infecter productivement les macrophages alors que ceux-ci sont des cellules différenciées ne se divisant pas, tandis que les lymphocytes T nécessitent une activation et une prolifération. L'intégration du génome est possible sans que le cycle cellulaire se produise [111]. Le mécanisme précis derrière cette capacité spécifique aux Lentivirus reste à élucider pour mieux comprendre comment le PIC peut traverser une membrane nucléaire intacte.
Une autre caractéristique du cycle viral chez le macrophage est l'assemblage des particules virales. Alors que les virions s'assemblent à la membrane cellulaire chez la plupart des types cellulaires, cette étape se passe différemment chez les macrophages. Les virions se forment majoritairement à la membrane des endosomes tardifs ou des corps multivésiculaires (MVB, multivesicular body) et bourgeonnent à l'intérieur de ceux-ci. Le virus acquiert dans son enveloppe certaines molécules de l'hôte spécifiques aux endosomes comme le CD63 et peu de molécules de surface comme ceux produits chez le lymphocyte T CD4+. Les virus sont ensuite relâchés dans l'espace extracellulaire par la fusion de la
19
dans les endosomes tardifs au lieu de la surface cellulaire chez le macrophage reste à élucider.
1.3.2. Polarisation des macrophages et VIH-1
La polarisation des macrophages induit des changements dans l'activité cellulaire (voir section 1.2.3.) et modifie ainsi la susceptibilité des cellules à l'infection. Suite à la stimulation de MDMs avec du TNF et de l'IFN-γ (M1) ou de l'IL-4 (M2a), ces cellules deviennent réfractaires au VIH-1. La réplication virale dans les cellules M1 et M2a, mesurée par l'activité de la RT, est diminuée de plus de 50% pour les M1 et d'au moins 25% chez les macrophages M2a. Cette résistance à l'infection n'est cependant pas permanente et perd de l'efficacité dans le temps, surtout pour les M1. Cet effet s'explique par la modulation de l'expression des récepteurs et corécepteurs à la surface de la cellule. Le nombre de cellules exprimant le CD4 diminue significativement chez les macrophages M1 avec un effet moindre chez les M2a. Le corécepteur CXCR4 diminue aussi, mais pas le CCR5. La polarisation joue donc un rôle à des étapes différentes, soit aux étapes préintégration pour les M1 et les évènements tardifs pour les M2a. Finalement, les cytokines sécrétées diffèrent aussi selon le phénotype. Les M1 sécrètent fortement CXCL10, une chimiokine augmentant la réplication virale chez les cellules infectées, mais ne semblant pas avoir d'impact chez les M1 à cause de la restriction au niveau de l'intégration de virus. Les M2a, eux, produisent beaucoup de CCL22, une chimiokine inhibant la réplication virale au niveau des étapes tardives [113-116].
Pour les macrophages non stimulés, l'infection par le VIH-1 induit une polarisation. Ceux-ci se dirigent vers un phénotype pro-inflammatoire suite à l'infection. Il y a une diminution du récepteur éboueur (scavenger receptor) pour l'hémoglobine et l'haptoglobine (CD163) et le récepteur du mannose (CD206), deux récepteurs utilisés pour décrire le phénotype M2 des macrophages. Les cellules adoptent donc une polarisation M1 suite à la rencontre du virus [116].
20 1.3.3. Impacts du virus sur les fonctions cellulaires
Lorsqu'un macrophage est infecté par le VIH-1, ses fonctions cellulaires sont affectées par le virus. Tout d'abord, la fonction principale des macrophages, la phagocytose, est altérée par l'infection virale. Les macrophages infectés se retrouvent avec un niveau élevé d'AMP cycliques (AMPc), ce qui diminue leur capacité à phagocyter les pathogènes opsonisés par les molécules du complément [117]. La phagocytose induite par la liaison de pathogènes aux récepteurs FcγR est aussi plus faible [118]. Dans les deux cas, le VIH-1 n'induit pas une diminution du nombre de récepteurs de surface, mais interfère plutôt avec la transduction du signal suite à la reconnaissance d'un pathogène, menant habituellement à la phagocytose de celui-ci. Cependant, l'infection peut aussi mener à une augmentation de l'internalisation de certains pathogènes, comme le parasite de la Leishmania qui infectera le macrophage à son tour [119]. Ces facteurs influencent donc l'élimination des microorganismes par les macrophages menant à des infections opportunistes, souvent présentes chez les personnes atteintes du SIDA.
L'infection par le VIH influence aussi les cytokines sécrétées par les macrophages. Pour la famille des interleukines, plusieurs sont régulés à la hausse suite à l'infection. Les MDMs infectés sécrètent plus d'IL-1, d'IL-6, d'IL-8, d'IL-10 et d'IL-12. La production d'IL-1, IL-6 et IL-8 devient même constitutive. Pour les interférons, il y a une baisse de production d'IFN-α chez les macrophages infectés, alors que la sécrétion d'IFN-β est augmentée. Du côté des colony-stimulating factor, le VIH-1 induit une hausse de production pour le M-CSF et une diminution pour le GM-M-CSF. Finalement, la production des chimiokines MIP-1α et MIP-1β est régulée à la hausse, induisant la chimiotaxie des lymphocytes T non-activés, ainsi que CCL5 (RANTES), un autre chimioattractant pour les lymphocytes T [120].
1.3.4. Les facteurs de restriction virale
Le macrophage, comme d'autres cellules ciblées par le VIH-1, possède des protéines capables de lutter contre l'infection. Ces protéines sont appelées des facteurs de restriction virale et agissent contre des propriétés du virus ne pouvant être sujet à des mutations, telles que les lipides de l'enveloppe ou la structure des bases nucléiques. Afin de mériter ce titre,
21
une protéine doit répondre à 4 critères. Premièrement, son expression doit montrer une activité antivirale qui doit être causée directement par la protéine et dont la restriction doit être causée majoritairement par celle-ci. Une protéine régulant un autre peptide qui interagit avec le VIH-1 ne peut être considérée comme un facteur de restriction. Deuxièmement, le virus doit posséder un mécanisme de défense contre l'activité antivirale de la protéine cellulaire. Une souche actuelle doit contenir ce mécanisme qu'elle a obtenu au cours de son évolution. Troisièmement, le facteur de restriction doit avoir été sujet à une sélection positive lors de l'évolution de l'homme. Cette pression mène à des substitutions d'acides aminés qui ont été conservés et ont mené à une évolution rapide. En comparant la séquence de la protéine avec celle d'une autre espèce animale proche de l'homme, il est possible de voir cette évolution rapide de la protéine. Cependant, la sélection positive des mutations ne doit pas être obligatoirement causée par le VIH-1 ou une de ses souches ancestrales. Elle peut être le résultat d'une exposition à un pathogène autre que le virus. Finalement, le quatrième critère est l'induction de l'expression du facteur de restriction. La protéine doit être fortement exprimée suite à la réponse immunitaire innée. Après la rencontre d'un pathogène, les cellules de l'immunité innée sécrètent de l'interféron. La liaison de l'interféron à son récepteur chez une cellule doit mener à l'expression de la protéine ayant la propriété antivirale. Donc, en résumé, les 4 critères pour qu'une protéine cellulaire soit considérée comme un facteur de restriction sont: (1) la forte activité antivirale; (2) un mécanisme viral pour la contrer la restriction; (3) une sélection positive lors de l'évolution et (4) une induction par la réponse interféron [121].
Un facteur de restriction bien connu dans le contexte du VIH-1 est SAMHD1 (SAM domain
and HD domain-containing protein 1). Cette protéine est fortement exprimée chez les
cellules ne se divisant pas, soit les monocytes, les macrophages, les cellules dendritiques et les lymphocytes T CD4+ non activés, mais est aussi retrouvée à un niveau basal dans les
cellules de divers tissus [122-125]. Le gène codant pour SAMHD1 est induit préférentiellement par l'interféron de type 1 (IFN-α ou IFN-β) [126]. Cette enzyme réduit grandement l'infectivité du virus lors de la rétrotranscription de l'ARN viral en ADN. SAMHD1 est un déoxynucléoside triphosphate triphosphohydrolase, c'est-à-dire qu'elle clive le groupement triphosphate du nucléotide. Le bassin de dNTPs (désoxyribonucléotides) cytoplasmique se retrouve grandement diminué, impactant
22
directement la synthèse d'ADN [127]. Par contre, une protéine virale peut prévenir l'action de SAMHD1. Le gène de la protéine accessoire Vpx est présent dans le génome du VIH-2 et du VIS, mais pas dans celui du VIH-1, permettant à ces souches virales d'infecter plus efficacement les cellules qui sont habituellement réfractaires à l'infection. Lors du cycle viral, Vpx est incorporé dans le virion en interagissant avec le domaine p6 de la protéine Gag et sera relâché dans le cytoplasme lors d'un nouveau cycle viral [128]. Vpx s'associe à l'E3 ubiquitin ligase CRL4DCAF1 pour ubiquitiner SAMHD1 et mener à sa dégradation par
le protéasome [122].
D'autres facteurs de restriction sont les protéines de la famille des APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like), des cytidine déaminases. Cette famille comporte 11 membres dont certains sont connus pour posséder une activité antivirale contre le VIH-1 [129]. L'emphase sera mise ici sur le membre le plus étudié, APOBEC3G (A3G). Cette protéine est exprimée dans une variété de cellules, dont les cellules myéloïdes, les lymphocytes B et les lymphocytes T. La stimulation des cellules myéloïdes avec l'IFN-α induit l'expression de ce facteur de restriction [130]. Cette protéine possède une activité cytidine déaminase. Lors de la synthèse du premier brin d'ADN complémentaire à l'ARN viral, A3G désamine le groupement cytidine, le transformant en uridine. Cette modification entraîne des hypermutations de guanoside (G) en adénosine (A) dans l'ADN viral, diminuant ainsi le pouvoir infectieux des virions qui seront produits [131, 132]. Lors de la formation de nouveaux virions, A3G peut être incorporé par des interactions avec la protéine virale NC et avec l'ARN [133]. Cependant, le VIH-1 possède une protéine accessoire pour prévenir l'action des protéines APOBEC. La protéine virale Vif, codée par le gène vif, est exprimée tardivement dans le cycle viral et est incorporée dans les virions produits [134]. Lorsque présente, Vif peut interagir avec A3G soit en recrutant une E3 ubiquitin ligase, entraînant la dégradation d'A3G par le protéasome, soit en formant un complexe empêchant l'incorporation d'A3G dans les virions produits [135, 136].
Un troisième facteur de restriction bien étudié est la tetherin, aussi appelée BST2, désignée comme étant le CD317 et codée par le gène BST2. La tetherin est une protéine transmembranaire pouvant être exprimée à la surface des cellules et dans les compartiments intracellulaires. Elle est exprimée chez plusieurs cellules immunitaires dont les
23
macrophages et son expression est induite par la réponse à l'interféron de type 1 ou 2 [137, 138]. Cette protéine forme un lien entre la membrane cellulaire et l'enveloppe de la particule virale, empêchant le relâchement des nouveaux virus [139]. Chez les macrophages, où les virus bourgeonnent dans des compartiments intracellulaires, une forte concentration de theterin est retrouvée à la surface de ces vésicules et serait nécessaire à la formation de ces compartiments [138]. Pour inhiber l'effet de la tetherin, le VIH-1 exprime la protéine accessoire Vpu. Cette protéine virale est aussi transmembranaire et se retrouve colocalisée à la tetherin [140]. Vpu interagit avec la tetherin nouvellement synthétisée grâce à leur domaine transmembranaire, l'internalise et entraîne sa dégradation, nuisant ainsi au renouvellement de la protéine à la surface cellulaire [141]. Certaines études montrent que la voie des protéasomes est utilisée, tandis que d'autres montrent que c'est plutôt celle des lysosomes [142, 143].
La protéine TRIM5α (Tripartite motif-containing 5 isoform-a) montre aussi une restriction de l'infection par le VIH-1. Cette restriction n'est cependant possible que par le TRIM5α exprimé chez le singe rhésus, alors que l'orthologue humain ne montre aucune inhibition de l'infection [144]. Cette protéine interagit avec la capside et perturbe la décapsidation du virus suite à son entrée dans le cytoplasme [145].
Finalement, des protéines découvertes récemment, SERINC3 et SERINC5 (serine
incorporator) sont des facteurs de restriction potentiels. Ces protéines transmembranaires
sont exprimées à la surface des cellules susceptibles à l'infection et sont incorporées lors du bourgeonnement des virions. L'incorporation de ces protéines dans les particules virales interfère avec la fusion de l'enveloppe virale à celle d'une cellule lors d'une nouvelle infection. La protéine virale Nef empêche l'incorporation des protéines SERINC lors de la formation des nouveaux virus [146, 147].
24
Chapitre 2: Hypothèse et objectifs de recherche
Les médicaments antirétroviraux ont grandement évolué depuis leur découverte. Les personnes infectées par le VIH-1 ont maintenant l'opportunité d'avoir accès à divers médicaments dépendamment de leur état et peuvent aussi être suivies par PCR afin d'adapter le traitement en fonction de l'évolution du virus. Cependant, même s'il est possible de contrôler la charge virale chez un individu, nous devons trouver le moyen d'éliminer le virus d'une personne infectée afin de parvenir à une guérison complète. Pour y arriver, les réservoirs viraux devront être éradiqués. Ces cellules, dont les macrophages font partie, rétabliront la charge virale chez un patient dès que celui-ci arrêtera son traitement. Le rôle des macrophages dans le contexte d'infection par le VIH-1 est de plus en plus étudié. Avec leur grande résistance à l'effet cytopathogène du virus et leur large dissémination dans l'organisme, l'impact de ces cellules dans la pathogenèse et l'établissement des réservoirs n'est pas à sous-estimer. Il est donc important de mieux comprendre les interactions du virus avec les molécules de l'hôte qui pourraient agir comme facteur de régulation de l'infection au sein des macrophages. Plusieurs protéines sont déjà connues pour jouer un rôle dans l'infection, mais nous estimons qu'il en reste une panoplie à découvrir. Une analyse transcriptomique a d'ailleurs été effectuée pour comparer le profil des cellules infectées avec celui de la population bystander, c'est à dire qui ne montre pas d'infection productive. Comme prévu, environ 5000 gènes sont régulés à la hausse ou à la baisse chez les cellules infectées (résultats en cours de publication).
Avec ces résultats, nous avons émis comme hypothèse que le taux d'infection des macrophages in vitro sera modulé par l'absence de ces protéines. Ceci mettra en lumière de nouveaux sentiers signalétiques qui pourront être la cible de nouvelles thérapies pour venir à l'élimination du virus chez les macrophages et ainsi éliminer l'un des réservoirs viraux. Les objectifs de ce projet sont donc les suivants:
1. Réaliser un criblage moléculaire sur les MDMs en utilisant des ARN interférents dirigés contre des gènes sous/surexprimés chez les macrophages infectés.
2. Identifier et valider de nouveaux facteurs de restriction ou de susceptibilité potentiels chez le macrophage humain.
25
Chapitre 3: Matériels et Méthodes
Pour atteindre nos objectifs, le projet s'est déroulé en deux étapes. Tout d'abord, un criblage d'ARN interférents a été effectué sur les MDMs pour tester l'importance des gènes correspondants sur l'infection par le VIH-1. Ceci nous permettra de rapidement mettre en évidence plusieurs gènes potentiellement impliqués dans le cycle viral. Cependant, un criblage n'est pas la méthode la plus précise. Il sera fait sur un petit nombre de cellules et nécessitera beaucoup de pipetages en chaîne. Le taux d'erreur est donc un peu plus élevé. C'est pourquoi la deuxième étape consiste à étudier les cibles dont les résultats seront plus significatifs avec des techniques plus précises, dans le but de confirmer la fiabilité du criblage et ainsi identifier de nouveaux régulateurs du VIH-1.
3.1. Production de particules virales
Ce projet utilisera plusieurs clones moléculaires du VIH-1. L'utilisation de clones au lieu de souches retrouvés chez les patients fait en sorte que nous contrôlons l'accumulation de mutations, contrairement à une souche cumulant plusieurs années d'évolution. En utilisant un plasmide, nos productions virales possèdent le même génome, évitant que les résultats diffèrent avec l'utilisation d'un nouveau stock viral. Il est évident que le virus va quand même muter suite à l'infection lors des expérimentations, mais le nombre de mutations sera limité.
3.1.1. Modèles viraux
Plusieurs virus ont été utilisés dans ce projet. Le premier est le NL4-3-Bal-IRES-HSA. Le plasmide NL4-3 est un clone moléculaire du VIH-1, contenant tous ses gènes et est pleinement infectieux [5]. Ce clone a ensuite été modifié afin d'y ajouter le gène rapporteur HSA (Heat Stable Antigen) murin, qui va être exprimé à la surface des cellules productivement infectées (voir Figure 8) [148]. Cependant, ce virus est X4-tropique et a donc été modifié afin d'y ajouter l'enveloppe de la souche Bal, de tropisme R5, afin d'infecter les macrophages.
26
Figure 8: Représentation schématique du plasmide NL4-3-IRES-HSA
Le plasmide NL4-3-IRES-HSA est de tropisme X4 et contient le gène rapporteur HSA. L'enveloppe a été remplacée par l'enveloppe de tropisme R5 Bal, provenant du plasmide NL4-3balenv [149]. Figure issue de Imbeault, M. et al., 2009 [148].
Le plasmide 3-Bal-IRES-HSA a ensuite été dérivé en deux autres plasmides, le NL4-3-Bal-IRES-eGFP contenant le gène rapporteur de la enhanced green fluorescent protein (eGFP) et le NL4-3-Bal-IRES-FLuc contenant la firefly luciferase (FLuc) au lieu du gène HSA.
Finalement, une production appelée « Mock » est préparée pour le contrôle de cellules non infectées. Le mock est le surnageant ultracentrifugé de cellules transfectées avec un plasmide «vide», donc sans production de particules virales.
3.1.2. Culture de la lignée cellulaire 293T
La lignée cellulaire 293T (aimablement fourni par le Dr. Warner C. Greene du J. Gladstone Institutes, San Francisco, CA) est dérivée des cellules embryonnaires de rein HEK293. Ces cellules ont un taux de transfection très élevé et permettent d'avoir des productions virales efficaces.
Les 293T sont conservées dans l'azote liquide et doivent tout d'abord être décongelées. Ils sont ensuite maintenus dans le milieu de culture Dulbecco's Modification of Eagle's
Medium (DMEM) (Corning, New York, NY) supplémenté de 10% de Fetal Bovine Serum
(FBS) (Wisent, St-Bruno, QC) préalablement inactivé à la chaleur. Les 293T sont placés dans un flacon de 75 cc (Corning) et incubées à 37°C sous 5% de CO2. Les cellules sont en
culture jusqu'à l'atteinte d'environ 75% de confluence. Lorsque ceci est atteint, le milieu est retiré et le flacon est lavé avec 10 ml de PBS (Phosphate-Buffered Saline) 1X, puis 1 ml de trypsine 1X contenant 0.5% d'EDTA (acide éthylène diamine tétracétique) (Thermo Fisher