Université de Montréal
î)
/1. 3 /
z
Mémoire présenté à la faculté des
Études
Supérieures
En vue de 1’ obtention du grade de
Maître ès Sciences (M.Sc.)
En Chimie
Synthesis of Heterocyclic Compounds of Medicinal Relevance
par
Jie Shi
Département de Chimie
Faculté des Arts et des Sciences
G
December 2003
fl—’ \
uH
Université
d11
de Montréal
Direction des bibliothèques
AVIS
L’auteur a autorisé l’Université de Montréal à reproduire et diffuser, en totalité ou en partie, par quelque moyen que ce soit et sur quelque support que ce soit, et exclusivement à des fins non lucratives d’enseignement et de recherche, des copies de ce mémoire ou de cette thèse.
L’auteur et les coauteurs le cas échéant conservent la propriété du droit d’auteur et des droits moraux qui protègent ce document. Ni la thèse ou le mémoire, ni des extraits substantiels de ce document, ne doivent être imprimés ou autrement reproduits sans l’autorisation de l’auteur.
Afin de se conformer à la Loi canadienne sur la protection des renseignements personnels, quelques formulaires secondaires, coordonnées ou signatures intégrées au texte ont pu être enlevés de ce document. Bien que cela ait pu affecter la pagination, il n’y a aucun contenu manquant.
NOTICE
The author of this thesis or dissertation has granted a nonexclusive license allowing Université de Montréal to reproduce and publish the document, in part or in whole, and in any format, solely for noncommercial educational and research purposes.
The author and co-authors if applicable retain copyright ownership and moral rights in this document. Neither the whole thesis or dissertation, nor substantial extracts from it, may be printed or otherwise reproduced without the author’s permission.
In compliance with the Canadian Privacy Act some supporting forms, contact information or signatures may have been removed from the document. While this may affect the document page count, it does not represent any loss of content from the document.
•
Université de Montréal
Faculté des Études Surpérieires
Ce Mémoire infitilé:
Synthesis ofHeterocycic Compounds ofMedicinal Relevance
Présenté par:
JieShi
A été évalué par un jury composé des personnes suivantes:
Dr. André Charrette
président-rapporteur
Dr. Yvan
Guindon
membre du
jury
Dr. Stephen
Hanessian
directeur de recherche
Mémoire
accepté le:
Abstract
My research was involved in the synthesis of heterocyclic compounds of medicinal relevance. f irstly, we designed and synthesized a novel 2-pyridone precursor related to ABT-719, a well-known antibacterial compound. An advanced intermediated was reached, but difficulties in the last steps precluded the synthesis of the intended bicyclic azaquinoline.
The second project focused on the synthesis of a small library of 28 compounds as Rho kinase inhibitors. The core structure was an amino piperidine, which was diversified as sulfonamides and amides. Modest activity was found with one ofthe compounds.
The third project was invoÏved in the synthesis of a series of monocyclic acylguanidines
as Na/H exchanger inhibitors. Biological testing identified four potent inhibitors.
Keywords: heterocvcle, 2-pyridone, DNA gyrase, piperidine, Rhokinase inhibitor, acylguanidine, Na/H exchanger (N}IE-1) inhibitor.
Résumé
Ma recherche décrit la synthèse de composés hétérocycliques d’importance biologique. En premier lieu, nous avons fait le design et la synthèse d’un nouvel analogue de type 2-pyridone basé sur la structure d’un composé antibactérien bien connu, le ABT-719. Un composé intermédiaire avancé a été atteint, mais des difficultés lors des dernières étapes ont mené à l’abandon de 1’ azaquinoline bicyclique désirée.
Le deuxième projet décrit la synthèse d’une petite librairie de 28 produits consistant en deux séries de dérivés sulfonamides et amides de pipéridines comme étant inhibiteurs de la Rho-kinase. Une activité modeste a été trouvée avec un des analogues.
Le troisième projet consiste en la synthèse d’acylguanidines monocycliques comme inhibiteurs potentiels de canaux NatH (Na/H échangeur, NFTE-1). Basé sur les structures connues d’inhibiteurs NHE, nous avons synthétisé une petite librairie de dérivés acylguanidines. Les analyses biologiques ont identifié quatre inhibiteurs potentiels.
Mots clefs: hétérocycle, 2-pyridone, ADN gyrase, pipéridine, Rhokinase inhibiteurs, acylguanidine, Na7W échangeur (NHE- 1) inhibiteur.
Table of Contents
Résumé I ListofSchemes W ListofFigures V Abbreviation VI Acknowledgment VIII CHAPTER 1 1Synthesis of a novel of 2-pyridone analogue 1
1.1 DNA gyrase inhibitors 2
1.2 The inhibition mechanism ofquinolones 3
1.3 Mode of action ofquinolones 3
1.4 2-Pyridone: A new quinolone analogue 5
1.5 Synthesis of 8-Chloro- 1 -cyclopropyl-7-ftuoro-9-methyl-4-oxo-4H-pyrido[ 1 ,2aJ
-pyrazine-3-carboxylic acid ethyl ester 6
1.6 Conclusion 10
1.7 General experimental notes 10
Attempted cyclization 25
1.8 References 26
CHAPTER2 29
Synthesis of Rho kinase inhibitors 29
2.1 The mechanism of action of Y-27632—an inhibitor of Ca2Lsensitizing enzyme... 30
2.2 Pipendine derivatives as potential inhibitors of Rho kinase 31
2.3 Synthesis ofintermediates 33
2.3.1 Synthesisoftrans4-isopropyl-cyclohexanecarboxylic acid 2.4 33
2.3.2 Synthesis oftrans-4-(trifluoromethyl)cyclohexanecarboxylic acid 2.8 34
2.3.3 Synthesis of 4-( 1 -tert-butoxycarbonylaminoethyl)benzoic acid 2.14 35
2.3.4 Synthesis of ti-ans (5)-( 1 -tert-butoxycarbonyÏaminoethyl) cyclohexanecarboxyÏic
acid 2.20 35
2.4.1 Michael addition and Beckmann regement .36
2.4.2. Synthesis ofpiperidine cores 38 2.4.2.1 Synthesis ofpiperidine cores 2.32 and 2.33 38 2.4.2.2 Synthesis ofpiperidine core 2.39 39 2.4.2.3 $ynthesis ofpiperidine cores 2.42 and 2.45 40 2.4.3 Synthesis of3-substituted piperidine derivatives 2.47, 2.49, 2.5 1, 2.54, and 2.57 40 2.4.4 Synthesis of4-substituted piperidine derivatives 2.60 and 2.63 42 2.5 Biological tests 43 2.6 Conclusion 43 2.7 Experimental notes (See Chapteri) 43 R-(+)-3 -(2-Nitropropane-2-yl)cyclopentanoxime (2.23) and 3 -epimer 50 General procedure for the preparation of sulfonamides (A) 62 General procedure for the preparation of amides (B) 62 Genereal pro cedure for reduction of nitro groups (C) 62 General procedure for N-Boc hydrolysis (D) 63
General procedure for the preparation ofhydrochloride saits (E) 63
2.8 References 94
CHPTER3 98
Synthesis of acylguanidines as Na/H antiporter inhibitors 98
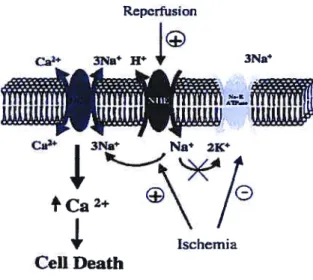
3.1 Na17H antiporters 99
3.1.1 NHE- 1 structure and cellular localization 100 3.1.2 Mechanistic basis for NHE involvement in myocardial ischemic and reperfusion
injury 101
3.1.3 NaiH antiporter inhibitors under clinical development 102
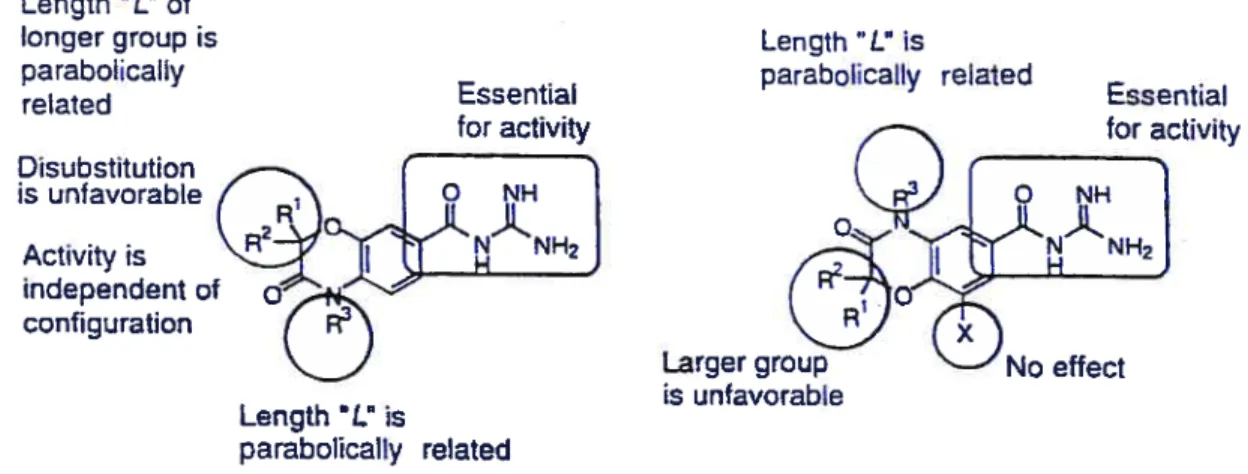
3.1.4 Functional requirements of acylguanidines 103 3.2 Monocyclic acylguanidines 104 3.3 Biological resuits 105 3.4 Experimental notes: (See Chapteri) 107 N-(4-Chloro-3 -trifluoromethyl-benzoyl)-guanidine hydrochloride (3.12) 112 3.5 References 115
List of Schemes
Scheme 1.1 Synthesis ofthe 2-pyridone analogue. 7
Scheme 1.2 Attempted Curtius rearrangement 7
Scheme 1.3 Alternative methods 2
Scheme 1.4 Attempts toward target compound 1.1 9
Scheme 1.5 Examples of cyclization 10
Scheme 2.1 Synthesis oftrans4-isopropyl-cyclohexanecarboxylic acid 2.4 33
Scheme 2.2 Synthesis oftrans-4-(trifluoromethyl)cyclohexanecarboxylic acid 2.8 34
Scheme 2.3 Synthesis of4-(1-tert-butoxycarbonylarnino-ethyl)-benzoic acid 2.14 35
Scheme 2.4 Synthesis of trans-(S)-4-(1 -tert-butoxycarbonylarninoethyl) cyclohexanecar
boxylic acid 2.20 35
Scheme 2.5 Synthesis of 3- and 4- substituted -1actarns 36
Scheme 2.6 Possible transition state model for the Michael addition 37
Scheme 2.7 Mechanism ofBeckmann rearrangement 37
Scheme 2.8 Synthesis ofpiperidine cores 2.32 and 2.33 38
Scheme 2.9 Synthesis ofpipendine core 2.39 39
Scheme 2.10 Synthesis ofpiperidirie cores 2.42 and 2.45 40
Scheme 2.11 Synthesis of 3-substituted piperidine derivatives 2.47, 2.49, and 2.51 40
Scheme 2.12 Synthesis of3-substituted piperidine derivatives 2.54 41
Scheme 2.13 Synthesis of 3-substituted piperidine derivatives 2.57 42
Scheme 2.14 Synthesis of4-substituted piperidine derivatives 2.60 and 2.63 42
Scheme 3.1 The synthesis of substitutedN-acylguanidines 3.1-3.13 104
List of Figures
Figure 1.1 Structures of core units of 4-quinolone and 2-pyridone 2
Figure 1.2 Schematic presentation of the catalytic firnction of DNA gyrase and the bactericidal effect of quinolone antibacterials 3 Figure 1.3 Head to tau association allows H-bonding to DNA 4
Figure 1.4 Inhibition 3D model for quinolone antibiotics 4
Figure 1.5 Structures ofABT-719 and atarget compound 5
Figure 1.6 Retrosynthesis ofthe target compound 6 Figure 2.1 Structure of Y-27632 30 Figure 2.2 The mechanism of action of Y-27632 27632 — an inhibition of the activity of
Ca2-sensitizing enzyme 31 Figure 2.3 The target compounds 32 Figure 2.4 Synthesis of five piperidine cores 32 Figure2.5 Retrosynthesis of substituted piperidine derivatives 33 Figure 2.6 Moderately active compound 43 Figure 3.1 Structure and cellular localization 100 Figure 3.2 NHE involvement in myocardial ischemic and reperfusion injury 102
Figure 3.3 Na/H antiporter inhibitors 103 Figure 3.4 Functional and structure requirements for activity 103
Figure 3.5 The structure ofmonoacylguanidines 104 Figure 3.6 1C50 values for inhibition 105 Figure 3.7 Change in pH with concentration 106
Abbreviation
Specific rotation Ac Acetyl Boc tert-Butoxycarbonyl Bp Boiling point Bu Butyl t-Bu tert-Butyl Chemical shifi in ppme Concentration in milligrams per milliliter
Calcd. Calculated
DCM Dichioromethane
DEAD Diethyl azodicarboxylate
DIPEA N,N-Diisopropylethylamine
DMAP 4-Dimethylaminopyridine
DMF N, N-Dimethylformamide
DMSO Dimethyl sulfoxide
DNA Deoxyribonucleic acid
DPPA DiphenylphosphoryÏ azide
EDC 1 -(3 -Dimethyllaminopropyl)-3 -ethylcarbodiimide hydrochioride
EtOAc Ethyl acetate
Et Ethyl
eq Equivalent
ether Diethyl ether
h Hours (s)
Hex Hexane
HOBt 1 -Hydroxybenzofflazole
HRMS High resolution mass spectrum
Hz Hertz
1C50 Concentraction of inhibition at 50%
R Infrared spectroscopy
LDA Lithium diisopropylamide (N Me Methyl mg Milligram min Minute mL Milliliter mmol Millimole Mp Melting point MS Mass spectrum
NMR Nuclear magnetic resonance
Ph Phenyl
PMB p-Methoxylbenzyl
ppm Parts per million
psi Pounds per square inch
rt Room temperature
$atd S aturated
SAR Structure activity relationship
TBM Tetrabutylammonium iodide
Tf Trifluoromethanesulfonyl
TNF Tetrahydrofiiran
TLC Thin layer chromatography
TMS Trimethylsilane
Ts 4-Toluenesulfonyl
Microliter
Wt Weight
Acknowledgment
I would like to take this opportunity to express my sincere gratitude to Professor Stephen Hanessian, for his guidance and inspiration as weII as giving me this training opportunity to improve my chemistry.
I wish to thank the support of my husband and my lovely son as well as my friends and colleagues for their friendship and help, especially Dr. Vinh Pham. My thanks also go to madams Carol Major, Elaine foumelle and Lyne Laurin.
Finally, I would sincerely like to thank Professor Stephen Hanessian for revising the manuscript and Dr. Eric Therrien for preparing the Résumé.
CHAPTER 1
Synthesis of a novel of 2-pyridone analogue
Like the quinolones, the 2-pyridones are DNA gyrase inhibitors. The mechanism of
C
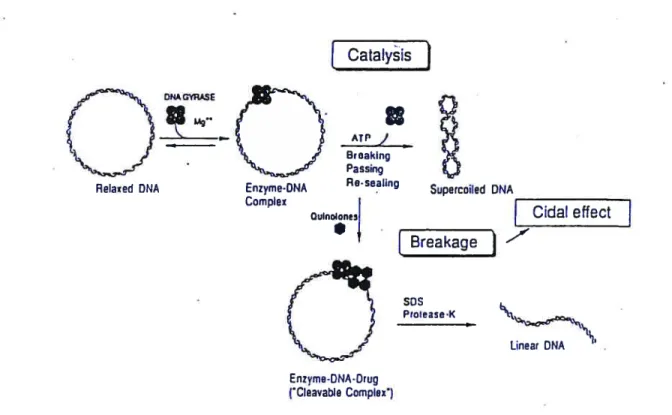
inhibiting bacteria by 2-pyridones is very similar to that ofthe quinolones.5 1.2 The inhibition mechanism ofquinolonesQuinolone-type dmgs have a unique capacity to trap the intennediate (DNA gate) by stabilizing the enzyme-DNA complex as illustrated in Figure 1.2. More importantly, such a process leads to the formation of a cleavable complex.
AtP} Broaking Passing Ro.seaiing
QuIn&aneJ
Cidal etfectj
s
Breakagj 7
figure 1.2 Schematic presentation of the catalytic ftmction of DNA gyrase and the bactericidal effect of quinolone antibacterials (Reproduced ftom Shen, L.L. Adv in Pharmacology, 1994, 29A, 285).
1.3 Mode of action of quinolones
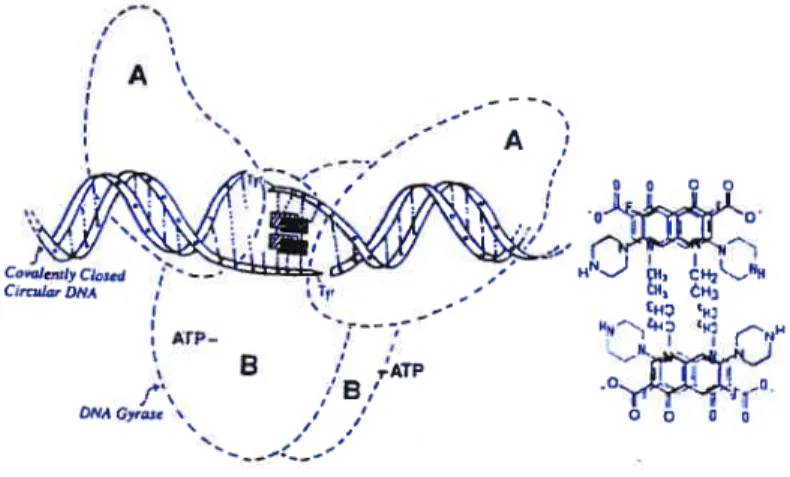
A model for the inhibition of ONA gyrase is shown in Figure 1.3. Quinolone molecules are
shown as solid and hatched rectangles that represent the dmg self-association, and binding to a gyrase-induced DNA site during the intermediate gate-opening step of DNA supercoiling process via hydrogen bonds to the unpaired bases indicated by doted fines. Gyrase A subunits
[
Catalysisflelaxed ONA Enzyme-DNA
Complex Supetcoiled DNA sos ProieaseK EnzymeDNA-Dtug (Cleavable Complox9 Lineat DNA
1.1 DNA gyrase inhïbitors
DNA gyrase1 is a bacterial motor protein in a class known as topoisomerases, which is responsible for controlling the topological properties of DNA (e.g. amount of supercoiling or catenation). Most topoisomerases can relax supercoiled DNA, which is an energetically favourable process. DNA gyrase is unique amongst this class enzyme, because it can introduce supercoils as well remove them. Quinolones were new generarion broad-spectrum antibacterial agents2 developed in 1980s’. The mechanism of action of quinolones is inhibition ofDNA gyrase.3
Four-generations of quinolone drugs have been developed.4 First-generation drugs (e.g.,
nalidixic acid) achieve minimal serum levels. Second-generation quinolones (e.g.,
ciprofloxacin) have increased Gram-negative and systemic activity. Third-generation dmgs (e.g., levofloxacin) have expanded activity against Gram-positive bacteria and atypical pathogens. Fourth-generation quinolone drugs (currently only trovafloxacin) add significant activity against anaerobes. We chose ciprofloxacin as a representative of quinolone and compared its structure with 2-pyridone. Figure 1.1 shows that changing the position and number ofnitrogen in ciprofloxacin can generate two structures related to 2-pyridones.
Quinolone
HN]
Ciprofloxacin
— N
4-H-4-oxoquinolizine
C
Figure 1.1 Structures of core units of 4-quinolone and 2-pyridone4-Pyridone 2-Pyridone
4-oxo-1 ,4-dihydro 6H-6-oxo-naphthyridine pyrido[ I .2-a]pyrimidine
form covalent bonds between Tyr-122 and the 5’ end of the DNA chain, and the subsequent
C
opening of the DNA chains along the 4-hp staggered cuts resuits in a locally denatured DNAbubble, which is an ideal site for the drug to bind. When a relaxed DNA is used, ATP is required for the induction of the drug-binding site. Dashed curves mimic the shape of the DNA gyrase, a tetramer of two A and two B subunits as revealed by the electron microscope image ofthe M Ïuteus enzyme. Figure 1.4 is a 3-dimensional presentation ofthe model.
00Go
cH ôi 04 4 HD
Figure 1.3 Head to tau association allows H-bonding to DNA (reproduced from Shen, L.L. Adv in Fharmacology, 1994, 29A, 285)
HbonU Acceptot Quinolone Ring -Hydtophobic Tau
o----H-bond Donot H-bond DNAFigure 1.4 Inhibition 3D model for quinolone antihiotics (reproduced from Shen, LL. Adv in Fharmacology, 1994, 29A, 285)
1.4 2-Pyridone: A new quinolone analogue
In an effort to discover nove! antibacterials related to the known fluoroquinolones6 sucli as ciprofloxacin,7 scientists at Abbott Laboratories explored the chemistry of new series. This involved transposition of the nitrogen of 4-quinolones to the bridgehead position at C5 quinolone membering) yielding two nove! heterocyclic nuclei related to a 2-pyridone, 6H-6-oxo-pyrido[ 1 ,2-a]pyrimidine and 4-H-4-oxoquinolizine (see figure 1.1), which had flot previously been evaluated as antibacterial agents and were found to be potent inhibitors of DNA gyrase. In addition, the so-called 2-pyridones also possess favorable physiochemical and pharmacokinetic properties.8’9
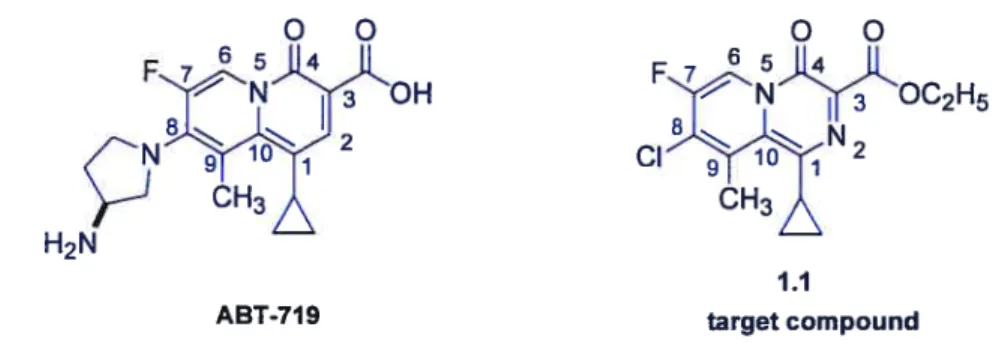
ABT-719 is a potential antibiotic compound,’° which was synthesized by scientists at Abbott Laboratories. The chemical structure of ABT-719 is similar to fluoroquinolone, the difference being transfer of the N atom to position 5. The target compound, 8-chloro-l-cyclopropyl-7-ftuoro-9-methyl-4-oxo-1, 4-dihydro-pyrido [1.2-a] pyrazine-3 -carboxylic acid ethyl ester is a nove! class of 2-pyridone core compared to ABT-719 (figure 1.5), The main difference is in the replacement of C-5. This leads to a 5-aza-isoquinolone-type structure.
FioH FOCH
ÇN ‘à12 N2
H2N
1.1
A5T719 target compound
figure 1.5 Structures ofABT-719 and atarget compound
1.5 Synthesis of $-ChIoro-1-eycopropyJ-7-flnoro-9-methy1-4-oxo-4ll-pyrido[1 ,2a]
-pyrazïne-3-carboxylic acid ethyl ester
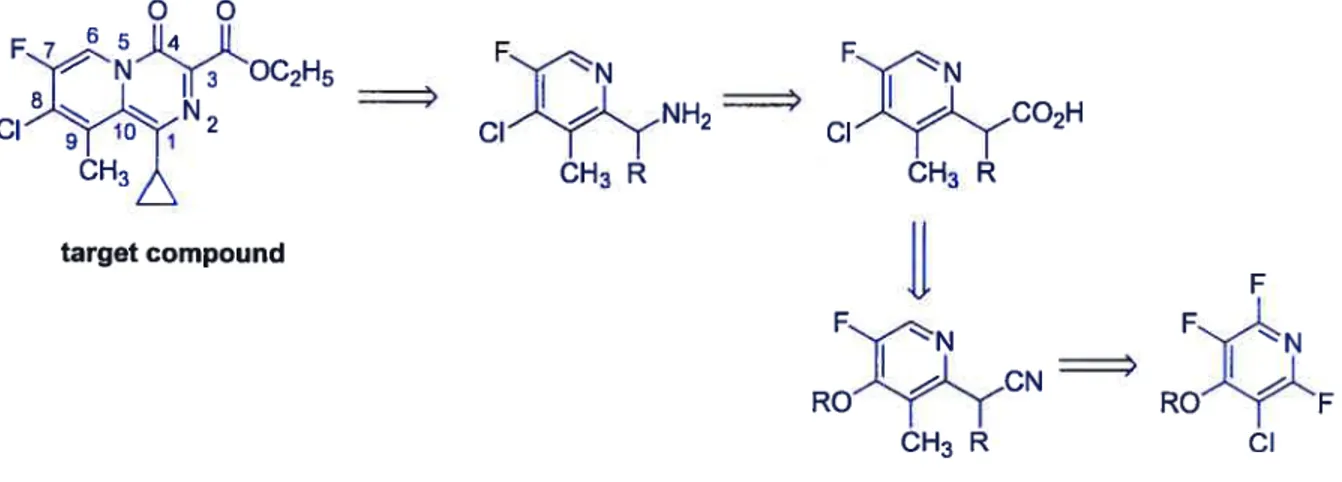
We envisaged the disconnections shown in Figure 1.6 for the synthesis of the bicyclic core. A series of aromatic substitutious would lead to the C-2 branched acid, which would be subjected to a Curtius rearrangement.
CiN2 > ciNH2 “ ciC02H
CH3 CH3 R CH3 R target compound F F FN ROCN ROLF CH3R CI
Figure 1.6 Retrosynthesis ofthe target compound
2,4,5,6-Tetrafluoro-3-chloro-pyridine was treated with lithium tert-butoxide to give two regioisomeric products 1.2 and 1.311 (Scheme 1.1). The next step involved metallation of aryl chloride by sec-butyl lithium followed by alkylation with methyl iodide to afford compound 1.4. The fluorine in the 6 position was substituted with hydrazine and the product was oxidized to yield compound 1.5. The carbanion formed by treatment of a substituted acetonitrile with LDA was then used to introduce carbon branching at position 2 to give 1.6. Treatment of 1.6 with POC13 effected the cleavage of the OtBu group and the replacement by chlorine to give 1.7. Hydrolysis of the nitrile group with ethanolic HC1 afforded the ester12 1.8, which was subsequently converted to the carboxylic acid 1.9.13
Scheme 1.1 Synthesis ofthe 2-pyridone analogue 1. NH.,NH, 2. 02, NaOH 66% FN t-Bu 0’f F 0H3 1.5 LDA, THF,-78°C RCH2CN 81 % (a) 83 % (b) FN tBuO(N CH3 R 1.6fa,b) POCI3, DMF CH2CI2, r.t 89 ¾ 95% FN J1&yCN CH3 R 1.7f a, b) HCI F-N O N-oC2H5 EtOH I CH3 R 89 % (a) 82%(b) 1.8(a,b)
The Curtius rearrangement 11, 14 was tried in the presence of DPPA and TEA. However, heating the compound 1.10 in tert-butyl alcohol didn’t lead to compound 1.11. Using the smaller methanol didn’t help to produce the carbamate (Scheme 1.2).
Unfortunately the standard conditions for a Curtius rearrangement (DPPA, Et3N) and heating in t-BuOH failed ta give the desired rearranged N-Boc derivative 1.11, Even though can obeserve the formation ofthe isocyanate by R spectraoscopy.
Scheme 1.2 Attempted Curtius rearrangement
0
9
DPPA, PhMe FN9
t-BUOHCIyoH Et3N CIf’N3
CH3R CH3R 1.9(a,b) 1.lOfa,b) t-BuOH F N O Cl NH-QBu-t CH3 R 1.11(a,b) F F Ot-Bu F F.LN tert-BuOLi F.N + R..._LN sec-BuLI Mel, -78°C -78°C t-BuO
I
F t-Bu01F CI 90%ci
ci
76% CH3 1.2 1.3 7:1 1.4 2N NaOH b MeOH 78 ¾ ta) 76 % (b) F-N O Cif’0H a R=Et CH3 R b,-< 1.9 (a, b)We therefore considered an alterative strategy as shown in Scheme 1.3. Compound 1.5 was reacted with 1 ,3-dithiane in the presence of n-BuLi15 to give 4-tert-butoxy-2-(2-ethyl-[1,3]dithian-2-yl)-5-fluoro-3-methyl-pyridine 1.12, which was alkylated by ethyl triflate afier deprotonation by n-BuLi to give compound 1.13.16 Removal of the 1.3-dithiane with BF3
OEt2 in MeCN and H20’7 gave 1.14. Reduction of the ketone with NaBH4 gave alcohol
1.15,18 which was transformed to the azide 1.16 under Mitsunobu condition.19
Triphenylphosphine was employed to reduce2° the azide to give amine 1.17.
Scheme 1.3 Alternative methods
BF3 CEt2 MeCN H20 41 ¾ NaBH4 MeOH 84 % target compound
e
We had effectively introduced the amino propyl group at C-2 of the pyridine nucleus by this method of branching. The remaining was to construct the pyrazine unit en route to intended target.
From compounds 1.14 and 1.17, we tried to prepare the six-membered ring using diethyl malonate derivatives2la, b
Unfortunately, we were flot successful in effecting the desired condensations. FN tBu0N CH3 1.5 S F N n-BuLiTHF F
()
_____ s _____________ t-Bu0S t BuO F n-BuLi, THF TfOEt, -78°C 61% CI-13 S - 6H3Ç
1.12 1.13 R CH3 1.14 F N DEAD, DPPA Ph3P, THF CH3 71% 1.15 00 F»N PhP CI N CH3 Ç 52 % - CH3 F NoEt H20 BuO2 CH3Ç
1.16 1.17 1.1Scheme 1.4 shows in one series of reactions where the ketone 1.14 was treated with diethyl 2-aminomalonate under acid catalysis. We envisaged formation of an imine followed by cyclization. In a second attempt, we reacted the aminopropyl pyridone with diethyl 2-oxo-malonate under acid catalysis, expecting to get the same imine. In both cases starting materials 1.14 and 1.17 were recovered with and without the loss of the tert-Bu group. The literature has hardly any precedence for the formation of the intended ring structure as shown
in Scheme 1.5.2223
Scheme 1.4 Attempts toward target compound 1.1
F 1. NaBH, MeOH 2.DEAD,DPPA t-BuO Ph3P, THF t-BuO’j 3. Ph3P, H20 CH3 O CH3 NH2 31 % 1.14 1.17 00 00
p-TsO (cat)
\
EtO0EtEtotOE,,,,4” (1) PhH, reflux (1) PhMe reflux
or NH2
/
(2) p-TsOH (cat) (2) DMSO, 140°C 0 PhMeEto
o
o
EtC O OEt F t-BuO(
t-SuO H3 x F NOEt/
t-BuON CH3 O O F NfOEt CI N 6H3Ç
1.1 targetcompoundThus, starting from compound 1.18, the reaction was conducted under varying conditions such as boiling glacial acetic acid or in polyphosphoric acid at 80 °C. hi no case compound 1.19 was formed. Aiso with diethyl oxalate oniy 1.20 was formed aithough the bicycle 1.19
is theoreticaily possible. (Scheme 1.5)
Scheme 1.5 Examples ofcyclization
÷ H3COC2H5 + EtOLOEt COOR \\ 1.19 COOR COCOOEt 1.20 1.6 Conclusion
Although we were successful in preparing suitably functionalized pyridines, the construction ofthe desired 6-azaisoquinoline nucleus could flot be achieved. It is possible that the pyridine nitrogen is too weakly basic to affect cyciization as shown in Scheme 1.4.
1.7 General experimental notes
Melting points (mp) were measured on a Fisher-Johns apparatus, and they are uncorrected. Unïess otherwise specified, ail non-aqueous reactions were carried out under a nitrogen atmosphere, using oven-dried giassware, and ail reaction soivents were removed by rotary evaporator. Ail solvents in dry reactions were distiiied over calcium hydride. Unless other stated, the reagents were purchased from Aidrich Chemical Co.
Analytical thin layer chromatography (TLC) was performed using EM Reagent 0.25 mm silica gel 60-F plates. Visualization of the deveioped chromatogram was performed by DV
J H
1.18
absorbance. Nuclear magnetic resonance of proton spectra (1H NMR) and carbon-13 (3C NMR) were recorded on a Bruker AMX-300, or Bruker AMX-400 spectrometer in a deuterated solvent as indicated using the signal from the residual non-deuterated solvent,
CHC13 (H, = 7.27 ppm; C, = 77.23 ppm) as internai reference. Chemical shifts
()
andcoupling constants (J) are expressed in ppm (part per million) and Hz (Hertz), respectiveiy. The abbreviations used for the description ofthe peaks are as follows: s, singiet; d, doublet; t, triplet; m, multiplet; br, broad; dd, doublet of doublet; doublet of triplets. DEPT-135 experiments were performed routinely, methyl (CH3) and methine (CH) give positive signal
(+), methylene (CH2) gives a negative signal (-), and tertracarbon give no signal (0). Ail chemical shifis are measured from the centre of the resolved peaks, the unresolved multiple and broad peaks are normally indicated as a range.
Low resolution mass spectra (MS) and high resolution mass spectra (HRMS) were
respectively determined on a VG Micro Mass 1212 and a kratos MS-50 TCTA mass
spectrometer by using methods of desorption chemical ionization (Cl) or fast atom bombardment (fAB).
Inftared (W) spectra were recorded on Perkin-Elmer fTR Paragon 1000 spectrophotometer in a chloroform solution with a sodium chloride ceil, or mixture film with KBr. Only characteristic peaks are reported.
Opticai rotations ([Œ]) were measured at room temperature using a Perkin-Eimer polarimeter,
modele 241 apparatus with a sodium lamp (wavelength of 589 nm) at ambient temperature using a 10 cm-lenghtceil containing 1 mL of a solution prepared at the indicated concentration (c, g /100 mL).
Chromatography
f lasli cbromatography was done by general procedure using Kieselgel (Merck 9385, 230-400 mesh) silica gel. Thin Layer Chromatography (TLC) was performed using commercial availabie glass plates coated with Silica Gel 60 f254 with 0.25 mm tbickness (Merck, Kieselgel 60F254).
TLC visualization
C
DV 254 lamp was used to observe the DV visible compounds and to evaluate the
advancement of the reaction. Chemical visualization was donc using one of the following solutions:
ta) Molybdate/Ceric sulfate solution.
Ammonium molybdate (VI) tetrahydrate: (NH4)6Mo7O24.4H20 50g
Ammonium cerium(W) sulfate dihydrate: (NH4)4Ce($04).2H20 20g
Concentratedd suiphuric acid: H2S04 200 mL
Distilled water: H20 1200 mL (b) Ninhydrin solution. Ninhydrin dihydrate 2 g Butanol 600 mL Acetic acid 18 mL (c) KMnO4 solution.
A 10% aqueous solution ofKMnO4 was used with olefin compounds. Reagents
CommerciaÏÏy available were purchased from Aldrich, Sigma or Lancaster and used without further purification. Ail commercially unavailable reagents were prepared.
Solvents
EtOAc, Hexane, and dichloromethane (DCM) were distilled prior to chromatography and general use.
Toluene, THF, DCM and ether were dried using the dry alumina column. Triethylamine, diisopropylamine benzene and methanol were distilled over calcium hydride.
Ail reaction were carried out under argon. The entire flasks were flame-dried under vacuum. Ail needles and syringes were dried under vacuum before to use. Yieids refer to chromatographically pure products.
F FN t-BuO F
CI
4-tert-Butoxy-3-chloro-2, 5, 6-trïfluoropyridine (1.2)
A solution of tert-BuOLi (7.5 g, 93.7 mmol) in TifF (75m!) was cooied to —78 oc in a dry ice/acetone bath. A solution of 2,4,5,6-tetrafluoro-3-chloropyridine (22.5 g, 84.9 mmol) (Aldrich, 70% pure) mixed with 2,4,5,6-tetrafluoro-3-chloropyridine 30% in THF (45 mL) was added dropwise. The reaction mixture was stirred for 2 h at —78 °C, then at ambient temperature ovemight. The reddish brown mixture was concentrated at —30 °c, hexane (lOOmL) and celite (-5 g) were added and the mixture was stirred for 30 min. The solid was removed by filtration the solvent was removed under reduced pressure to yieid a colorless liquid (18.4 g, 90%) as a mixture of 1.2 (4-butoxy, desired) and 1.3 (6-butoxy, undesired) in ratios of 7:1. The two compounds couid be separated by flash coÏumn chromatography (ethyl acetate:hexane 3:97) to get pure compound 1.2 (15.lg, 71%)
‘H NMR (400 MHz, CDC13) 6: 1.2 1.52 (s, 9H); 1.3: 1.61 (s, 9H);
‘3c
NMR (400 MHz, CDcl3) 6: 163.9, 159.1, 150.7, 132.1, 104.3, 73.1, 28.4. ‘9F NMR (400 MHz, cDcl3) 6 (ppm): 1.2: -73.75 (dd, J = 14.2, 23.2 Hz, 1F), -89.71 (dd, J = 14.2, 21.9 Hz, 1F), -152.42 (t, J= 22 Hz, 1F); 1.3: -74.95 (dd, J 9.0, 24.5 Hz, 1F), -121.69 (dd, 1=9.0, 18.1 Hz, 1F), -162.47 (dd, 1= 18.1,24.5Hz, 1F). MS (M): 239.03F
FyÇ
t-BuO F CH,
4-tert-Butoxy-3-methyl-2, 5, 6-trifluoropyridine (1.4)
A 250mL three-necked flask equipped with a mechanical stiner, a graduated addition funnel and a digital thermometer was charged with compound 1.2 (6.06 g, 0.0252 moi) and THF (23 mL). The internai temperature ofthe mixture was cooled to —70 oc using a dry ice / acetone bath. A solution of sec-BuLi (24 ml, 1.3 M in cyclohexane, 0.03 13 mol) was added via syringe to this above stirred solution over a period of 1.0 h. The speed of addition was adjusted as to maintain an internai temperature between —61 to —70 °C. Afier the addition was completed, stirring was continued for an additional 1 h in a dry ice / acetone bath. MeT (2.38 ml, 0.0383 mol) was added over -15 mm, the lithium sait dissolved and the internai temperature rose quickly to —39 °c. The mixture was stirred for 1 h at ambient temperature. The reaction was quenched with saturated aqueous NRC1 (7 mL) and extracted with 100 mL of ether. The extract was washed with water (1 x 25 mL), brine (2 x 15 mL), dried over MgS04, and concentrated to give the crude product (7.15 g). This material was distilled under reduced pressure to give 1.4 (4.2 1 g, 76%) as a pale yellow liquid, bp. 75-81 °C (7.5 mmHg). It was used without fiirther purification.
‘H NMR (400 MHz, CDC13) 6 2.12 (s, 3H), 1.47 (s, 9H). ‘3C NMR (400 MHz, CDCI3) 6 164.3, 161.0, 149.8, 130.6, 105.1, 71.1, 28.5. 11.5. 19F NMR) (400 MHz, CDC13) 6 -75.91 (dd, J= 15.0, 22.1 Hz, 1F), -93.17 (dd, J= 15.0, 22.1 Hz, 1F), -156.54 (m, 1F). HRMS: c10H12F3N0 (M); calcd.: 219.0897; found: 219.0881. FN t-BuOF CH,
e
4-tert-Butoxy-2, 5-difluoro-3-methylpyridine (1.5)
.
A solution of 1.4 (4.2 g, 19.2 mmol) and hydrazine monohydrate (>98%, 2.33 ml, 0.048 mol) in methanol (7.5 mL) was refluxed for 9 h. The methanol was removed and the residue was dissolved in methylene chloride (10 mL) and washed with water (2 x 5 mL). Solvent was distilled under reduced pressure, leading an orange ou. It was redissolved in methanol (21.3 mL). To this was added aqueous sodium hydroxide (20%, 11.3 mL), and air was passed through the solution for 6 days with vigorous stirring. The methanol was removed under vacuum at 30-35 °C, the residue was dissolved in ether (38 mL), washed with water (1 x 15 mL), 10% HCi (1 x 10 mL), saturated bnne (1 x 20 mL), and dried over MgSO4. The solvent was removed and the residue was purified by flash chromatography (ethyl acetate:hexane 5:95) to afford 2.56 g of 1.5 (66.5%) as a colorless liquid.
‘H NMR (400 MHz, CDC13) 6 7.85 (br, H), 2.18 (d, J= 1.5 Hz, 3H), 1.43 (d, 1= 1.5 Hz, 9H).
13c
NMR (400 MHz, CDCI3) 6 162.5, 158.2, 143.7, 135.4, 106.6, 75.1, 28.5, 11.3. 19F NMR (400 MHz, CDCY3) 6 -73.37 (d, J= 24.5, 1F), -142.17 (d, J= 24.5 Hz, 1F) MS: 201.10. HRMS: C10H13F7N0 (M); Calcd.: 201.1072; found: 201.108$. F t-Bu 2-(4-tert-Butoxy-5-ftuoro-3-methyl-2-pyridyl)-2-cyclopropylacetonitrile (1.6 a)LDA xvas formed by adding n-BuLi (2.5 M in hexanes, 15 mL, 37.5 mmol), dropwise to a stirred solution of diisopropylamine (5.15 mL, 73.5 mmol) in THF (15 mL) at —78 °C. The reaction was allowed to stir at O °C for 15 min and then cooled to —7$ °C with a dry ice/acetone bath. Cyclopropylacetonitrile (3.0 g, 37.0 mmol) in anhydrous 111F (7.5 mL) was added over a period of 15 min to the above solution of LDA, keeping the internai
temperature between —51 and —67 °C. The mixture was stirred for an additional 35 min at the
O
same temperature. To the above solution, 1.5 (3.0 g, 14.9 mmol) in T (7.5 mL) was addedover 20 min maintaining an internai temperature of —65 to —71 °C. The cooling bath was removed and stirring was continued for 30 min. When the temperature reached —30 °C, an exothermic reaction was observed and the temperature rose quickly to 17 °c. The reaction was quenched with saturated aqueous NH4ci (10 mL) and was extracted with ether (50 mL). The extract was washed with saturated brine, dried over Mg$04 and concentrated. The excess cyclopropylacetonitrile was removed at 40-45 oc at 0.2 mmllg. The residue was purified by flash chromatography (ethyl acetate:hexane 5:95) to give 1.6a (3.25 g, 83%) as a colorless liquid, which solidified on standing.
Mp. 52-54 °c. ‘II NMR (400 MHz, cDcl3) 8.29 (s, 1H), 3.75 (d, J= 7.16 Hz, 1H), 2.28 (s, 3H), 1.4$ (m, 1H), 1.42 (s, 9H), 0.73 (m, 1H), 0.63 (m, 1H), 0.50 (m, 2H).
13c
NMR (400 MHz, CDC13) 157.1, 154.7, 153.4, 150.7, 149.9, 137.2, 85.3, 38.9, 29.6 HRMS: C15H20N20f (M+1); Calcd.: 263.1560; found: 263.1565. F t-BuoCN 2-(4-tert-Butoxy-5-lluoro-3-methyl-2-pyrïdyl)-butyronitrïle (1 .6b)The procedure was same as 1.6a to give 1.6b as a colorless solid (0.$g, 81%)
1H NMR (400 MHz, Dl3) (ppm): 8.12 (s, 1H), 3.91 (t, 1=7.1, 1H), 2.13 (s, 3H), 1.85
(m, 2H), 1.30 (s, 9H), 0.82 (t, 1= 7.1 Hz, 3H).
‘3C-NMR (400 MHz, D13) (ppm): 154.9, 152.4, 150.9, 150.9, 149.6, 136.2, 84.6, 38.3,
29.3, 26.6, 12.7, 12.0.
o
2-(4-Chloro-5-fluoro-3-methy)-2-pyridyl)-2-cyclopropylacetonitrile (1 .7a)
b a solution of 1.6a (2.25 g, 8.65 mmol) and DMF (3.4 mL, 43.9 mmol) in anhydrous
methylene chloride (19 mL), POCI3 (3.17 mL, 34.0 mmol) was added slowly with an ambient temperature bath cooling since there was a delayed exothermic reaction. The solution was stirred ovemight before being poured into crushed ice. (Caution: make sure POC13 is consumed before doing the extraction!). The mixture was extracted with methylene chloride (2 x 30 mL). The combined extracts were washed with water (lx 15 mL), saturated aqueous NaHCO3 (lx 15 mL), water (2x 10 mL), dried over MgSO4, and concentrated. The product was purifled by flash chromatography (ethyl acetate:hexane 1:4) to yield 1.7a as a pale yellow solid (1.83 g, 95%). Mp. 43—44°C ‘H NMR (400 MHz, CDC13) 8.39 (s, 1H), 3.80 (d, J= 8.0 Hz, 1H), 2.49 (s, 3H), 1.50 (m, 1H). 0.77 (m, 1H), 0.66 (m, 1H), 0.58 (m, 1H), 0.48 (m, 1H). ‘3C NMR (400 MHz, CDCY3) & 161.1, 158.2, 136.6, 133.1, 128.8, 117.7, 34.1, 29.3, 18.7, 13.7, 11.2. HRMS: C1,H10C1FN2 (W); Calcd.: 224.0484; found: 224.0489. F CN 2-(4-Chloro-5-fluoro-3-methyl-2-pyridyl)-butyronitrïie (1 .7b) The procedurewas same as 1.6b to give 1.7b (0.72 g, $9
%)
III NMR (400 MHz, CDCÎ3) (ppm): 8.39 (s, 1H). 4.06 (t, J 7 Hz, 2H), 2.45 (s, 3H), 2.1 (m, 1H), 1.1 (t,J=6.5Hz,3H). ‘3C-NMR (400 IV[Hz, CDC13) (ppm): 156.7, 154.1, 150.3, 135.9, 131.8, 119.8, 39.1, 26.7, 15.6, 12.8. HRMS: C10H10C1FN2 (M); Calcd.: 213.0542; found: 213.0529. F CI OC2H5
Ethyl 2-(4-chloro-5-lluoro-3-methyl-2-pyridyl)-2-cyclopropylacetate (1 .8a)
A solution of 1.7a (1.36 g, 6.0 mmol) in ethanol (0.9 mL) was added to a solution ofethanol (10 mL) saturated with HC1 gas (—4 g) at O °C, which was prepared by the dropwise addition
of concentrate H2S04 onto CaC12 The reaction was stirred for 3 h at O °C. To this solution
was added F120 (0.9 mL). The reaction was heated at 80 °C for 2 h. The mixture was poured over ice to give a total volume of 40 mL. This solution was neutralized with 50% NaOH to pH 8 while maintaining a temperature less than O °C. The solid was filtered, dissolved in
CH2l2, and the residual water layer removed. The organic layer was dried over MgSO4 and evaporated and purified by flash chromatography (ethyl acetate:hexane 2:8) to provide 1.$a as a pure tan solid (1.34 g, 82%).
H NMR (400 MHz, CDC13) & 8.36 (s, 1H), 3.23 (d, J= 9 Hz, 1H), 0.12 2.40 (s, 3H), 1.67 (m, 1H), 1.20 (t,J=z7Hz, 3H), 0.0.76 (m, 1H), 0.53 (m, 1H), 0.38 (m, 1H), 0.12 (m, 1H). ‘3CNMR (400 MHz, CDC13) : 172.0, 156.1, 154.1, 153.5, 136.2, 132.7, 66.4, 45.9, 24.3, 18.9 15.8, 12.9. HRMS: C13H15C1FN02 (M); Calcd.: 271.0871; found: 271.0904. c CI OEt
o
2-(4-Chloro-5-fluoro-3-methyl-2-pyridyl)-butyric acid ethyl ester (1 .8b)
The procedure was same as 1.8a to give 1.$b (0.72 g, 89%)
1fl NMR (400 MHz, CDC13) 6: 8.35 (s, 1H). 4.10 (q, J= 6.3 Hz, 2H), 3.82 (d, J= 8.7 Hz, 1H), 2.44 (s, 3H), 2.09 (m, 1H), 1.95 (m, 1H), 1.21 (t, J= 6.5 Hz, 3H), 1.85 (t, J= 6.1 Hz) 13C NMR (400 MHz, CDCI3) 6: 172.62, 155.9, 154.4, 153.4, 135.4, 132.6. 61.3, 52.1, 24.8, 15.8, 14.5, 12.5. RRMS: C12H15C1FN02 (M); Calcd.: 259.0834; found: 259.0849. )H
2-(4-Chloro-5-fluoro-3-methyl-2-pyridyl)-2-cyclopropylacetic acid (1 .9a)
A solution of 1.Sa (1.34 g, 5.8 mmol) in 10% NaOH (10 mL) was heated to 90 oc for 2 h. Afier cooling the residue was removed by filtration. The solution was adjusted to pH 5 with 18% HC1 at O °c and a white solid precipitated. The solid was collected by filtration and dried under vacuum to give (0.98 g, 76%) ofpurecompound 1.9a.
‘H NMR (400 MHz, D20) 6: 8.12 (s, 1H), 2.94 (d, J= 9.9 Hz, 1H), 2.21 (s, 3H), 1.32(m, 1H), 0.59 (m, 1H), 0.36 (m, 1H), 0.34 (m, 1H), 0.06 (m, 1H).
‘3C-NMR (400 MHz, D70) 6: 172.5, 156.1, 154.3, 153.5, 135.4, 133.6, 23.7, 18.3, 15.0, 11.7.
HRMS: c11H11clFNo2 (M); Calcd.: 243.0594; found: 243.0613.
F N O C OH O CL c’-13
2-(4-Chloro-5-fluoro-3-methyl-2-pyridyl)-butyric acïd (1 .9b) The procedure was same as 1.9a to give 1.9b (0.68 g, 78%)
‘H NMR (400 MHz, CDC13) 6 8.35 (s, 1H), 4.06 (q, J= 7 Hz, 2H), 2.57 (s, 3H), 2.1 (m, 1H), 1.95 (m, 1H), 0.90 (t,J 6.2 Hz, 3H).
‘3C-NMR (400 MHz, CDCY3) 6 175.9, 156.2, 153.9, 153.7, 134.5, 134.3, 50.9, 26.6, 15.7, 12.2.
HRIVIS: C10H,,C1FNO2 (M); Calcd.: 231.0462; found: 231.0465.
F
CI N3
(4-Chloro-5-fluoro-3-methyl-2-pyridyl)-cyclopropyl-acetyl azide (1.1 Oa)
To a solution of acid 1.9a (243 mg, 1.0 mmol) in toluene (10 mL) at O °C was added triethylamine (396 pL, 3.0 mmol) follow by diphenyÏphosphoryl azide (430 j.iL, 2.0 mmol). The ice bath was removed and afier 1.5 h of stirring at the room temperature, the reaction was diluted with ether and H20 solution 45 mL (5:1). The layers were separated, and the aqueous layer was extracted with ether (2 x 15 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (10 mL) and saturated aqueous NaC1 (10 mL). The organic layer was dried over MgSO4 and concentrated under reduced pressure to give crude product, which was purified by a very short column (ethyl acetate:hexane 5:95) to give 1.lOa. (174 mg, 65%) IR:i = 2139.1, 1725.9 cm’ ‘II NMR (400 MHz, D20) 6: 8.4 (s, 1H), 3.2 (d, 2H), 2.4 (s, 3H), 1.7 (m, 2H), 1.24 (2 H), 0.8 (m, 1H), 0.27 (m, 1H). ‘3C-NMR (400 MHz, D20) 6: 172.5, 156.1, 154.3, 153.5, 135.4, 133.6, 15.0, 12.9, 5.2, 3.8.
.
tBUOXS7
CH3 H
4-tert-Butoxy-2- [1,31 dithian-2-yl-5-fluoro-3-methyl-pyridine (1.12)
A solution of 1,3-dithiane (0.7$ g, 6.5 mmol) in TRF (degassed with argon, 6.5 mL) was cooled to —45°C and n-butyl lithium (hexane, 2.62 mL, 6.5 mmol) was added dropwise over
15 min. the reaction mixture was stirred for 2 h at —40 °C and then for 2 h at O °C. The
solution was cooled to —40 °C and the pyridine 1.5 (0.41 g, 2.2 mmol) was added dropwise, then stirred for 2 h at —40 °C. The reaction mixture was quenched by the addition of aqueous
NH4C1 and extracted with CH2C12 (50 mL x 3). The combined organic layers were dried over
Na2S04, concentrated under reduced pressure, and then purified by flash column chromatography (ethyl acetate:hexane 15:85) to give 2-dithianyl pyridine 1.12 as a white solid (0.43 g, 6 1%). Mp. 125 oc 1H NMR (400 MHz, CDC13) 6: 6 8.27 (s, 1H), 5.32 (s, 1H), 3.02 (m, 4H), 2.36 (s, 3H), 2.15 (m, 1H), 2.01 (s, 1H), 1.39 (d,J 1.1 Hz, 9H). 13C-NMR (400 MHz, CDC13) 6 (ppm): 6 155.2, 153.35 149.5, 136.5, 129.4, 84.5, 51.7. 32.1, 29.47, 25.9, 12.8. MS (M): 301.1, 245.0, 212.1, 160.0, 147.1, 106.0. HRMS: C14H20FN0S2 (M); Calcd.: 301.097036; found: 301.097521. 4-tert-Butoxy-2-(2-ethyl- [1,3] dithian-2-yl)-5-fluoro-3-methyl-pyridine (1.13)
A solution of 2-dithianyl pyridine 1.12 (301 mg, 1 mmol) in THF (degassed with argon, 2.5 mL) was cooled to -78 °C, and n-butyl lithium (hexane, 550 iiL, 1.1 mmol) was added
dropwise over 15 min. The resultant solution was stirred at -78 °C for 30 min and then TfOEt (155 iiL, 1.2 mmol) was added. The mixture was stirred in —40 °C for 1h, the cold bath was removed, the temperature was raised to O °C for 1h, when a dark red color was appeared. Quenched the reaction by the addition of saturated NII4C1 solution, extracted with CH2CÏ2 (3
x 15 mL), washed with NaHCO3 and H20, dried over Na2SO4. Concentration in vacuo and
purification by flash chromatography (ethyl acetate:hexane 10:90) afforded 1.13 (148 mg, 45%) as a yellowish white solid.
Mp. 114°C ‘H NMR (400 MHz, CDC13) : 8.39 (s, 1H). 4.06 (q, J= 7 Hz, 2H), 3.23 (d, J= 9 Hz, 1H), 2.43 (s, 3H), 2.1 (m, 1H), 1.05 (t, J= 7 Hz, 3H) ‘3C NMR (400 MHz, CDC13) & 156.5, 153.9, 149.4, 137.2, 129.9, 85.2, 51.3, 41.2, 33.4, 29.7, 26.1, 13.0, 9.8. MS (M): 329.1, 296.1, 273.1, 240.1, 216.1, 160.0, 1487.1, 106.0. HRMS: C16H24FN0S2 (M); Calcd.: 329.1283; found: 329.1299. t-Bi 1 -(4-tert-Butoxy-5-fluoro-3-methyl-2-pyrïdyl)-propan-1-mie (1.14)
Red mercuric oxide (432 mg, 2.0 mmol), boron trifluoride diethyl etherate (252 tL, 2.0 mmoÏ) and 15% aqueous tetrahydrofuran (10 mL/g of dithiane) were stirred vigorously in a three-neck flask equipped with a dropping funnel and a nitrogen inlet tube. Compound 1.13
(330 mg, 1.0 mmol) was dissolved in the minimum ofTHF and was added via the dropping
funnel in the course of 10-15 min under nitrogen. Stin-ing was maintained for 10-20 min afler addition was complete. The red mercuric oxide gradually dissolved and a white precipitate appeared. Ethyl ether (5 mL) was then added, the precipitated saits were filtered, and the ether was washed to pH 10 with saturated Na2CO3, and to neutrality with satd. NaC1, afler drying over Na2SO4, the ether was evaporated under vacuum and purified by flash chromatography (ethyl acetate hexane 7 93) to yield compound 114 (98 mg, 41%)
‘H NMR (400 MHz, CDC13) : 8.2$ (s, 1H), 3.12 (dd, J= 6.4 Hz, 2H), 2.4, 3H), 1.40 (d, J 1.4 Hz, 9H), 1.14 (t, J= 6.3 Hz, 3H). ‘3C-NMR (400 MHz, CDC13) & 204.5, 155.8, 154.2, 150.9, 135.6, 133.6, 84.9, 33.77, 29.5, 13.8, 8.5. HRMS: C13H18FN02 (M); Calcd.: 239.1349; found: 239.1357. H t-Bu( 1 -(4-tert-Butoxy-5-fluoro-3-methyl-2-pyridyl)-propan-1 -ol (1.15)
Sodium borohydnde (113.5 mg, 3.0 mmol) was added portionwise over 30 min to a cooled (ice bath) stirring suspension of ketone 1.14 (120 mg, 0.5 mmol) in anhydrous MeOH (3
mL). Complete dissolution was obtained at the end of the addition. The ice bath was
removed, and stirring was continued for 8 h. Monitoring by TLC (20% acetone-hexane on silica gel) conflrmed that the reaction had gone to completion. The reaction mixture was concentrated to a residue that was diluted with H70 and extracted with CH2C12 (3 x 15 mL). The organic extract were combined, dried over anhydrous Na2SO4, and concentrated to colourless ou. The crude product was purified by flash chromatography (ethyl acetate: hexane 1:4) to affordthe alcohol 1.15 (109 mg, 91%).
1fl NMR (400 MHz, CDCJ3) E,: 8.22 (s, 1H), 4.71 (s, 1H), 4.45 (m, 1H), 2.1$ (s, 3H), 1.72 (m, 1H), 1.53 (m, 1H), 1.41 (d, J= 1.3 Hz, 9H). 0.95 (t,J 6.2 Hz, 3H) ‘3C-NMR (400 MHz, CDC13) E,: 157.7, 154.9, 149.7, 134.8 127.5, 84.4, 71.4, 30.9, 29.5, 12.1, 10.1. MS (M): 241.1, 212.1, 185.1, 156.0, 147.1 HRMS: C13H70FN07 (M); Calcd.: 241.147$; found: 241.1472
F
tBuON3
2-(1 -Azido-propyl)-4-tert-butoxy-5-ftuoro-3-methyl-pyridine (1.16)
b a stirred solution of 1.15 (24 mg, 0.1 mmol), triphenylphosphine (52.4 mg, 0.2 mmol) and diisopropyl azodicarboxylate (42 tL, 0.2 mmol) in dry THF, a solution of diphenylphos phoryl azide (44 pi, 0.2 mmol) was added over a period of 15 minutes and stirring continued for about 24 h. after which when the solvent was removed from the reaction mixture on a
rotary evaporator under reduced pressure. The thick oily liquid was purified by flash
chromatography (ethyl acetate:hexane 1:9) to afford 1.16 as a colourless liquid (37.5 mg, 71%).
1H NMR (400 MHz, CDC13) : 8.32 (s, 1H), 4.38 (t, J= 7.1 Hz, 1H), 2.28 (s, 3H), 2.04 (m, 2H), 1.43 (s, 9H). 0.97 (t, J 6.9 Hz, 3H)
13C-NMR (400 MHz, CDC13) : 157.2, 152.3, 149.7, 131.9, 128.4, 83.1, 71.3, 30.2, 29.1, 12.4, 11.4
HRI’IS: C13H19FN40 (M); Calcd.: 266.154$; found: 266.1521
F
t-BuOH2
1 -(4-tert-Butoxy-5-fluoro-3-methyt-2-pyrîdyl)-propylamïne (1.17)
A mixture ofazide 1.16 (113.15 mg, 0.5 mmol), triphenylphosphine (262 mg, 1.0 mmol), and water (1$ pi, 1.0 mmol) was stirred in THF (15 mL) for 24h. The mixture was concentrated and the residual ou was purified by flash chromatography (CHC12:MeOH:NH4OH 85:14:1) to give pyridine amine 1.17.
1H NMR (400 MHz, CDC13) & 8.25 (s, 1H), 4.12 (m, 1H), 2.25 (s, 3H), 1.73 (m, 2H), 1.42 (d,J= 1.0 Hz, 9H). 0.89. ‘3C-NMR (400 IVIHz, CDC13) & 156.1, 154.7, 148.4, 131.3, 127.7, 83.2, 70.1, 30.9, 29.4, 11.7, 10.0. HRMS: C13H21FN20 (M); Calcd.: 240.1627; found: 240.1639. Attempted cyclization A. From ketone 1.14
Commercial diethyl aminomalonate hydrochioride was converted to its free amineby stirring
in ethanol with excess potassium carbonate for about 1 h. The solids were then flltered and the ethanol removed in vacuo. Diethyl aminomalonate was subsequently distilled at reduced pressure (10 Torr) using a Kugelrohr apparatus. This material was stored in a reffigerator, and it maintained its integrity for several days as determined by 1H N1VIR.
The aminomalonate (263 mg, 1.5 mmol) was dissolved in toluene (7.5 mL), the ketone 1.14
(583 mg, 1.5 mmcl) was added to the mixture, the reaction flask was fifted with a Dean-Stark
apparatus and heated to reflux. Afier 13 h, the mixture was cooled, and the toluene was
removed in vacuo. Afler column chromatography, starting material was recovered. B. From amine 1.17
To a solution of compound 1.17 (264 mg, 1.1 mmcl) in benzene (3 OmL) were added diethyl 2-oxomalonate (161 jiL, 1.0 mmcl) andp-toluenesulfonic acid (9.5 mg, 0.05 mmcl) under an argon atmosphere. The reaction mixture was heated at reflux for 20 h with azeotropic removal. The solvent was evaporated, and the residue was purified with Kugelrohr distillation to give 4-hydroxyl starting material derivative of 1.17 (loss of t-Bu).
O
1.8 References1. Isaacson, R. E. “Novel targets for antibiotics” Expert Opin Investig Drugs. 1994, 3, 83-91. 2. Bail, P. “Quinolone generations: natural history or natural selection?” J Antimicrob
Chernother. 2000, SuppÏ T], 17-24.
3. Shen, L. L. “Molecular mechanism ofDNA gyrase inhibition by quinolone antibacterials”
Adv in Fharmacology, 1994, 29A, 285-303.
4. King, D. E.; Malone, R.; Lilley, S. H. “New classification and update on the quinolone antibiotics” Am. Fam. Physician, 2000, 6], 2741-8.
5. Hooper, D. C. “From fluoroquinolones to 2-pyridones” The Lancet, 1995, 345, 1192-1193. 6. Zhanel, G. G.; Ennis, K.; Vercaigne, L.; Walkty, A.; Gin, A. S.; Embu, J.; Smith, H.;
Hoban, D. J. “A critical review ofthe fluoroquinolones: Focus on respiratory tract infections” Drugs, 2002, 62, 13-59.
7. Shah, P. M. “Ciprofloxacin” Int. J Antimicrob. Agents, 1991, 1, 75-96.
8. Li,
Q,
Mitscher, L. A; Shen, L. L. “The 2-pyridone antibacteriai agents: bacterialtopoisomerase inhibitors” Med. Res. Rev. 2000, 20, 23 1-93.
9. Saiki, A. Y.; Shen, L. L.; Chen, C. M.; Baranowski, J. Lemer, C. G. “DNA cleavage
activities cf Staphylococcus aureus gyrase and topoisomerase IV stimulated by quinolones and 2-pyridones” Antirnicrob. Agents Chemother. 1999, 7, 1574-7.
10. Aider, J.; Clement, J.; Meulbroek, J.; Shipkowitz, N.; Mitten, M.; Janris, K.; Oleksijew,
A.; Hutch, T. S.; Paige, L.; Flamm, B. “Efficacies of ABT-719 and related 2-pyridones, members of a new class of antibacterial agents, against experimental bacterial infections” Antirnicrob. Agents Chemother. 1995, 39, 971-5.
11. Li,
Q.;
Chu, D.; Claibome, A.; Cooper, C. S.; Lee, C. M.; Raye, K.; Berst, K. B.; Donner,Baranowski, J.; Nilius, A.; Aider, J.; Meuibroek, J.; Marsh, K.; Croweil, D.; Hui, Y.; Seif, L.; Meicher, L. M.; Henry, R.; Spanton, S.; Faghih, R.; Klein, L. L.; Tanaka, S. K.; Plattner. J. “Synthesis and structure-activity relationships of 2-pyridones: A novel series of potent DNA gyrase inhibitors as antibacteriai agents” I Med. Chem. 1996, 39, 3070-3088.
12. Li.
Q.;
Sowin, T.; Chaibome, A.; Lijewski, L.; Zhang, X.; Raye, K.; Mazdiyasni, H.;Asmold, W.; Meicher, L. M.; Wang, W.; Hasvoid, L.; fung, A.; Chu, D. T. W.; Plattner, J. “Practical synthesis of 2-pyridone core: Ethyi 8-chioro-1-cyciopropyl-7-fluoro-9-methyl-4-oxo-4H-quinoione-3-carboxylate” Heterocycles, 1999, 51, 1345-1353.
13. Baggaley, K. H.; Fears, R.; Ferres, H.; Geen, G. R.; Hatton, I. K.; Jennings, L. J.; Tyrreli,
A. W. “N-Substituted amino acid derivatives with hyperalphalipoproteinaemic activity” Eur.
I Med. Chem. 1988, 23, 523-3 1.
14. Charette, A. B.; Côté, B. “ Stereoselective synthesis of ail four isomers of coronamic
acid: A General approach to 3-methanoamino acids” I Am. Chem. Soc. 1995, 117, 12721-12732.
15. $mith, A. B.; Rano, T. A.; Chida, N.; Sulikowski, G. A.; Wood, J. L. “Total synthesis of
the cytotoxic macrocycle (+)-Hitachimycin” I Am. Chem. Soc. 1992, 114, 8008-8022.
16. Nishiyama, Y.; Katoh, T.; Deguchi, K.; Morimoto, Y.; Itoh, K. “$tereoseiective synthesis
of 2,2,5-trisubstituted tetrahydrofurans via the Lewis acid-assisted reaction of cyciic hemiketals with nucleophiles” I Org. Chem. 1997, 62, 9339-9341.
17. Vedejs, E.; Fuchs, P. L. “An improved aidehyde synthesis from 1,3-dithianes” I Org.
Chem. 1971, 36, 366.
18. Efange, S. M. N.; Tu, Z.; Hohenberg, K.; Francesconi, L.; Howeli, R.C.; Rampersad,
R.V.; Todaro, L. J.; Papke, R. L.; Kung, M. P “2-(2-Piperidyl)- and 2-(2-pyrrolidyl)
chromans as nicotine agonists: synthesis and preliminary pharmacological characterization”
19. Lai, B.; Pramanik, B. N.; Manhas, M. S.; Bose, A. G.; “A novel reagent for the
C
stereospecific sthesis ofazides from alcohols” Tetra lett. 1977, 23, 1977-1980.20. Uenishi, J. I.; Hiraoka, T.; Yuyama, K.; Yonemitsu, O. “Synthesis ofoptically pure 1-(2-pyridinyl)ethylamine and 4-(2-pyridinyl)- 1 ,3-oxazolin-2-one” Heterocyctes, 2000, 2, 719-732.
21. ta) Blazey, C. M.; Heathcock, C. H. “Regiochemistry in 1,3-dipolar cycloadditions ofthe azomethine ylide formed from diethyl aminomalonate and paraformaidehyde” I Org. Chem. 2002, 67, 298-300; (b) Niwa, Y.; Takayama, K.; Shimizu, M. “Iminomalonate as a convenient electrophilic amination reagent for Grignard reagents” Buli. Chem. Soc. Jpn. 2002, 75, 1819—1825.
22. Kolar, P.; Tiier, M. “Heterocyciic amino acids as synthons: Reactions with dicarbonyl compounds” I Heterocyclic Chem. 1993, 30, 1253-1260.
23. Kolar, P.; Pizzioli, A.; Tiier, M. “Transformations of the pyrido[1,2-Œ]pyrazine ring system into imidazo [1 ,2-Œ]pyridines, imidazo[ 1 ,2-Œ]pyrimidines and 2-oxo-6a, 1 Oc-diaza aceanthrylenes” I Heterocyclic Chem. 1996, 33, 639- 642.
o
CHAPTER 2
Synthesis of Rho kinase ïnhibîtors
2.1 The mechanism of action of Y-27632—an inhibitor of Ca2-sensitizing enzyme
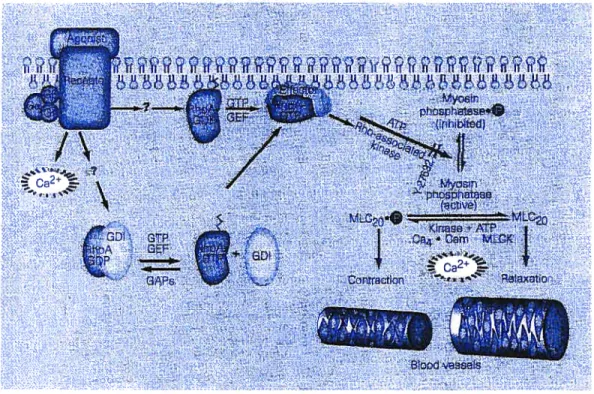
Abnormal smooth muscle contractility may be a major cause of disease states such as hypertension, and a smooth-muscle relaxant that modulates this process would be useful therapeuticalÏy. Smooth-muscle contraction is regulated by the cytosolic Ca2 concentration and by the Ca2 sensitivity of myofilaments. The former activates myosin light-chain kinase and the latter is achieved partly by inhibition of myosin phosphatase. Calcium sensitization of smooth muscle is mediated by a Rho-associated protein kinase in hypertension.1
N-_NH--Y-27632
Figure 2.1 Structure of Y-27632
Narumiya and colleagues1 have identified a drug (Y-27632) that inhibits the activity of a
Ca2-sensitizing enzyme (Rho-associated kinase) leading to a reduction of high blood
pressure in experimental animals (figure 2.1) Activation of receptors coupled to certain guanine-nucleotide-binding proteins G releases intracellular Ca2 that binds to calmodulin (Cam), and this complex activates myosin light-chain kinase (MLCK). By phosphorylating the regulatory light chain of myosin (MLC50) in smooth muscle, MLCK causes vascular smooth muscle to contract and the lumen of blood vessels to narrow. Many of the same receptors also activate RhoA and, with the help of guanine-nucleotide exchange factors (GEFs), dissociate cytosolic RhoA-GDP from guanine-nucleotide dissociation inhibitor (GDI). which allows the exchange of GTP for GDP on RhoA. The active RhoA-GTP activates Rho-associated kinase, wbich phosphorylates and so inhibits-myosin phosphatase. Myosin phosphatase dephosphorylates smooth-muscle myosin, causing the smooth muscle to relax and blood vessels to dilate. Y-27632 inhibits Rho-associated kinase, thereby blocking the inhibition of smooth muscle myosin phosphatase2 and Ca2 sensitization.3 Although Ca2 is the main activator of smooth-muscle contraction (through MLCK), the level of force can
C
be modulated independently of it.4 figure 2.2 shows the mechanism of inhibition.5 Wewished to test the activity of a small library of substituted piperidines as Rho-kinase inhibitors (figure 2.3).2.2 Piperidïne derïvatives as potential inhibitors of Rho kinase
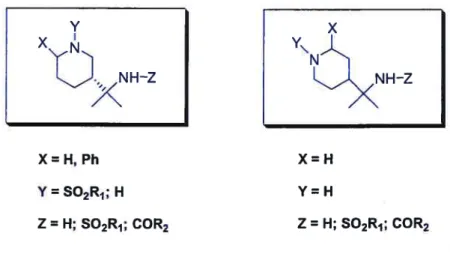
Based on the structure of Y-27632, we postulated that 1,3- and 1,4-substituted piperidine derivatives might have inhibitory activity against Rho Kinase. The intended derivatives and their provenance are shown in figure 2.3.
Figure 2.2 The mechanism of action of Y-27632 27632 — an inhibition of the activity of
2+ . .
X= H, Ph Y=S02R1; H Z= H; S02R1; COR2 X=H Y=H Z= H; S02R1; COR2
The common starting material was cyclopentenone, which would undergo a proline catalyzed Michael addition (sce 2.4) to give the corresponding adduct. Beckmann rearrangement would then lead to the corresponding lactams. Using this synthetic strategy we prepared five different piperidine cores as shown in figure 2.4. The sites of diversification are shown in Figure 2.5. oc Ph..û core-1 Boc. N NH2 cote-5 Boc 3XNH2 core-4 H core-2 core-3
e
Figure 2.4 Synthesis offive piperidine coresY H-Z X Y\ N NH-Z
Figure 2.3 The target compounds
.
H9
Ç-J’,,,NH—S—R1 /\o
H ‘yR2 HÛ%
,,,,NH—S—R3 /\o
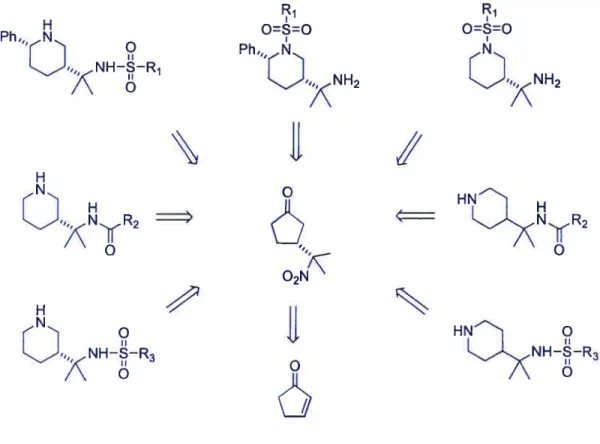
Figure 2.5 Retrosynthesis of substituted piperidine derivatives
2.3 Synthesis of intermedïates
2.3.1 Synthesïs oftraits 4-isopropy]-cyclohexanecarboxylic acid 2.4
Isopropyl benzoic acid was hydrogenated with platinum oxide to give the cis-4-isopropyl-cyclohexanecarboxylic acid as the major product. Esterification, isomerization and hydrolysis gave the desiredtrans carboxylic acid 2.46as the major diasteremer
Scheme 2.1 Synthesis oftrans 4-isopropyl-cyclohexanecarboxylic acid 2.4
NH2
.11
o==o
/
HNLH HNL9‘E
1.Pt02/H2,AcOH + 1. NaH, 160°C
L] 2.p-TsOH, MeOH Ç(J 2.IN NaOH, MeOH
CO2H 78 % CO2CH3 CO2CH3 64 %
5 1
2.1 2.2
CO2H CO2H separable
1 : 6
2.3 2.4
2.3.2 Synthesis of trans-4-(trffluoromethyl)cyclohexanecarboxy]ic acid 2.8
Methyl 4-trifluoromethylbenzoate was reduced and hydrolyzed to afford the cis-4-(trifluoro-methyl)cyclohexanecarboxylic acid 2.7 as a major product following a proceduce in the patant literature.7 Reaction with thionyl chloride and treatment with sodium hydroxide resulted in theii-ans acid 2.8 as a major product.
Scheme 2.2 Synthesis of trans-4-(trifluoromethyl)cyclohexanecarboxylic acid 2.8
CF3 CF3 CF3 CF3 CF3
RhC/H, 5 bar
+
I NNaOH
+
d
CO2CH3 CO2CH3 CO2CH3 CO2H CO2H
2.5 2.6 2.7 2.8 CF3 CF3 CF3 CF3
__
+ 3O%NaOH +Ç
coci coci CO2H CO2H
29 210 1 5
2.7 2.8 separabIe
2.3.3 Synthesis of 4-(1-tert-butoxycarbonylaminoethyl)benzoic acid 2.14
Using optically pure 1 $-(4-bromophenyl) ethylamine, three steps8 were necessary to obtain optically pure compound 2.13, which was protected9 with Boc2O to give compound 2.14. Scheme 2.3 Synthesis of 4-(1-tert-butoxycarbonylamino-ethyl)-benzoic acid 2.14
Br Br CN
(L
CH3CoCi(L
CuCN, DMF(L
6M EtO,-lO°C ÇJ 180°C,48h 11O°C,75h 81 %_L.
quant. - NH2 NHAc NHAc 2.11 2.12 COOH COOH(L1
Boc2O(L1
NaHCO3, MeOH I HCI f NH2 83 % NHBoc 2.13 2.142.3.4 Synthesis of trans (S)-(1-tert-butoxycarbonylaminoethyl) cyc]ohexanecarboxylic
acid 2.20
Compound 2.13 was reduced,’° isomerized and protected to give optically pure 2.20. (Scheme 2.4)
Scheme 2.4 Synthesis of trans-(5)-4-(1-tert-butoxycarbonylaminoethyl) cyclohexanecar boxylic acid 2.20
CO2H CO2H CO H CO2Na CO2Na
PtO/H
+
à
1NNaOH +
HCI HCI HCI
NH2 NH2 NH2 NH2 NH2
2.13 major minor minor major
2.4 Synthesis of piperïdine derivatives
2.4.1 Michael addition and Beckmann rearrangement.
2-Nitropropane was introduced by Michael addition to 2-cyclopentenone catalyzed by L proline to give enantioenriched 2.21,” which reacted with hydroxylamine to afford the oxime
2.23.12 Unfortunately the enantioselectivity of this reaction is mediocre compared to that
using cyclohexenone. Nevertheless, we proceeded with the modestly enriched mixture
towards the intended mini-library. Compound 2.23 was protected by p-TsC1,’2 and then subjected to a Beckmann rearrangement’3 in the presence of A1203 to give a regioisomeric mixture 2.25 and 2.26 in a proportion of 3:2 (Scheme 2.1). The stereochemistry indicated relates to the enriched isomer (75: 25 RIS).
Scheme 2.5 Synthesis of 3- and 4- substituted 5-lactams
H
N NO2
O H N
2.23 2.26
It is not possible to derive clear a mechanistic pathway,” but it is known that the proline catalyzed addition in presence of 2,5-dimethylpiperazine shows a complex non-linear effect.
CO2Na CO2Na CO2H CO2H
H2 + H2
H20/Dioxane
HBoc HBoc
minor major (21 % for 3 steps) : 5
2.17 2.18 2.19 2.20 separable + O CHCI3 L-proline, r.t 86% NO
‘X2
2.21 (75:25) NH2OH, HCI 2.22 95% NOH NOTs Hà
NO2 TsCI 6NO2 A1203 O wPyridine, O °C toluene
No2
÷‘ 92% ‘ 54%
ee 50 %
e
L-Proline first reacts with 2-cyclopentenone to give an iminium ion, which is attacked by nitropropane anion to give the R-product 2.21 as ffie major isomer. Although several secondary and tertiary bases were used as additives, only trans-2,5-dimethylpiperiazine gave a good ratio.Scheme 2.6 Possible transition state model for the Michael addition
O
II
CHCI3(
3 mol% proline \_JI cat. HN NH C,L
>=
H°1
O6
2.21 75 25 2.21 (ee=50 %)Beckmann rearrangement in the presence of dry alumina’3 afforded two constitutional isomeric 3-substituted and 4-substituted lactams because of the existing of Z and E two conformations in the oxime. A plausible mechanism is shown in Scheme 2.7. The mechanism involves conversion of the oxime hydroxyl group to a leaving group. Ionization and migration then occur as a concerted process, with the group, which is anti to the leaving group migrating. This resuits in formation of an iminium ion, which captures water. Eventually, hydrolysis leads to the lactams 2.25 and 2.26.
Scheme 2.7 Mechanism of Beckmann rearrangement
TsO\ Ts,AI2O3 Ts.AI2O3
r 0f I N N A1203 i’) ,N02 NO NO2 2.24(E) H HO,N 0N 2 NO2 XN02 2.25
C
OTs / NO2X
2.4(Z) Ts,AI2O3 /) NO2X
Ts..5AI2O3 N NO2X
OH O + H2O[NÏ
HNN 2.26The structural arrangement was made from analysis of their ‘H NMR spectura. Thus the major isomer 2.25 shows a doublet of doublets for the C-6 methylene hydrogens next to the lactam NH. The minor isomer 2.26 showed a multiplet for the C-6 methylene hydrogens 2.4.2. Synthesis of piperidine cores.
2.4.2.1 Synthesis ofpiperidine cores 2.32 and 2.33
The mixture of 2.25 and 2.26 was derivatized with Boc2O to give separable N-protected
lactams’4 2.27 and 2.2$. The desired compound 2.27 was treated with a Grignard reagent’ to
open the ring to give compound 2.29. Treatment with TFA16 gave the imine 2.30, followed
by a 2-step reaction sequence protocol17’ to give the amino N-Boc piperidine core
compound 2.32. Reduction’7 in the presence of Pd-C afforded the nitropiperidine core
compound 2.33. The relative stereochemistry of the 2-phenyl substituent was flot determined, but it can be assumed to be cisby analyzing the coupling constants ofbenzylic proton by ‘H NMR,
Scheme 2.8 Synthesis ofpiperidine cores 2.32 and 2.33
+ O Boc\ N +
X2
o
2.27 PhMgBr THF 70 ¾ NHBoc IFA Ph CH2CI2 84 % 2.29 Ph N Pd-C, H2/\
NO2 EtOAc 94 ¾ 2.30C
2.25 Boc2O DMAP 88% oc00
2.26 2.27 separable 2.28 3 2 core 2Boc Boc
Q
2.30 Pu-c, H2 NaBH4,MeOHPh
EtOAc, fBoc)O NiCI6H2O
91 % 2.31 84 % 2.32
core 1
2.4.2.2 Synthesis of pïperidine core 2.39
The mixture of lactams 2.25 and 2.26 was protected19 with PMBCI to yield two separable compounds 2.34 and 2.35. BH3•SMe22° followed by NaBH4 and NiC122’ reduced the major product to amine 2.37. Protection of 2.37 with Boc2022 and deprotection23 of PMB lead to compound 2.39. ($cheme 2.9)
$cheme 2.9 Synthesis ofpiperidine core 2.39
H
o
PMBo
HN NaH,THF PMBN) iKN02÷ LJ,kNo2 PM:cL O OC LJN c2 2 2.25 2.26 2.34 2.35 PMB PMB2 BH3.SMe2 NaBH4, NICI2 Boc2O
THF, 70 °C /N02 MeOH ÇJ, /NH2 CH2CI2, r.t
81% 78% 93%
2.36 2.37
PMB H
PU-c, H2
NHBoc MeOH
LJ
NHBoc68%
2.38 2.39
2.4.2.3 Synthesis of piperidine cores 2.42 and 2.45
Compounds 2.42 and 2.45 were prepared using a 4-step sequence from 2.27 and 2.28 as
shown in Scheme 2.10. In this sequence the lactam was reduced with the borane dimethylsulfide complex,2° the piperidine protected as the N-Boc derivative, and the nitro group reduced to the corresponding amine in the presence ofPd-C and hydrogen.24
Scheme 2.10 Synthesis ofpiperidine cores 2.42 and 2.45
oc 1.HCO2H ÇJ., NO2 2. BH3•SMe2
/K
THF 2.27 H BOC Boc2O Pd-C, H2L.J.,
NO2 CH2CI2L.J.,
NO2 60 psi (quant) 81% 2.40 2.41 O Boc “N 1. HCO2H -, NO2 2. BH3SMe2/K
THF 75% 2.28 HN Boc2O Ç_J>No2 CH2CI2 2.43 81 % Boc Pd-C, H2 60 psi/\
(quant) 2.442.4.3 Synthesis of 3-substituted piperïdine derivatives 2.47, 2.49, 2.51, 2.54, and 2.57
Piperidine cores 2.32, 2.33, and 2.39 reacted with substituted benzenesulfonyl chlorides to yield sulfonylamides25 2.46, 2.48, and 2.50, which were deprotected26 or reduced2’ to Ïead to the desired 3-substituted piperidine derivatives 2.47 a-e, 2.49 a-e, and 2.51 a-e respectively (Scheme 2.11).
Scheme 2.11 Synthesis of3-substituted piperidine derivatives 2.47, 2.49, and 2.51
Ph,, N
‘t
‘ TEA, CH2CI2 R2SO2CI H Ph, N HCO2H ‘t 1 O Ç>NH--R, core 4 cote 5 BocG
oC Ph, N‘C
/\ O 2.32 2.46 a-e 2.47a-eH
o=s=o
O=s=o Ph N Ph,, NaBH4, MeOH Ph,,/
TEA, CH2CI NO2 R2SO2CI C><\N02 N1C12.6H20 2.33 2.48a-e 2.49a-e H F1o=s=o
NC
/K
TEA, CH2CI2 N HCO2H N
R2s02c1 NHBoc NHBoc 2.39 2.50a-e 2.51a-e a b c d e CF3 R1 Ci —(J-CH3 —(J-OCH3
Core compound 2.42 was coupled27 with substituted benzoic acids and saturated carboxylic acids to give compounds 2.52 f-k, which were deprotected and treated with iN HC128 to give hydrochioride saits 2.54 f-k. (Scheme 2.12)
Scheme 2.12 Synthesis of3-substituted piperidine derivatives 2.54
Boc Boc H
N
DIEA, EDC, HOBt, N HCO2H N
o
DMF, R2CO2H, r.t XNHR2 cote 2.42 2.52 f-k 1N HCI 2.53 f-k 2.54 f-k HCi sait f g h ï
j
k R2—KD—<
Q<NH2 CF3e
• Core compounds 2.42 was reacted with sulfonyl chlorides to give compounds 2.55 1, m,
Deprotection and conversion to the hydrochioride sait gave 2.57 1, m (Scheme 2.13). Scheme 2.13 Synthesis of3-substituted piperidine derivatives 2.57
Boc OC H
Et3N, CH2CI2 HCOH
R3SO2CI J><\NH_s_Ra r.t
r 2.56 I m
cote 2.42 2.55 I,m iN HC1 I
L_e..2.57 I,m HC1sait
I m
2.4.4 Synthesis of 4-substïtuted piperïdine derivatives 2.60 and 2.63
Applying the same procedure that was used to prepare as 2.54 and 2.57 gave the 4-substituted compounds 2.60 g-k and 2.63 1, m as hydrochioride salis (Scheme 2.14).
Scheme 2.14 Synthesis of 4-substituted piperidine derivatives 2.60 and 2.63
Boc\ Boc\
o
DIEA, EDO, HOBt,_______ ii HCO2H H Nt H-—R2 NCNH DMF, R2CO2H, r.t NL<HCR2 82 ¾ 81% cote2.45 2.58 g-k iN HCI 2.59 g-k
quant. 2.6O g-k HC1 sait
Et3N, CH2Ci2 N HCO2H
Boc o HNL><\ R3SO2CI NH-R3 r.t NH-—R3
o
/ 0 80% 79 ¾ cote 2.45 2.61 m iN HCI — 2.62 (I, m) quant. L_,... 2.63 (I, m) HCI sait2.5 Biologïcal tests
C
The set of substituted piperidine derivatives was tested for inhibition of the Rho Kinase.29’30 Unfortunately only moderate inhibition was observed with 4-tert-butyl-N-[1-methyl-1-(6S-phenyl-piperidin-3R-yl)-ethyl] -benzenesulfonamide, 2.47a.
H Ph,
30 % inhibition at 10 tM Figure 2.6 Moderately active compound
2.6 Conclusion
We have prepared a small library of substituted piperidines as N-acyl and N-acylsulfonyl derivatives, starting with cyclopentenone using a recently developed Michael addition with nitroalkances. These compounds were obtained as enantiomerically partly enriched isomers
(--50% ee colTesponding to a 75:25 ratio of enantiomers in the 3-substituted piperidine series). Biological testing reveraled that only one analogue (2.47a) was moderately active as a Rho Kinase inhibitor.
2.7 Experimental notes (See Chapterl)
For some compounds the carbon resonances do flot match the formulae due to signal overlap.
and
CO2CH3 CO2CH3
CIS trans
2.1 2.2
Q
platinum oxide (50 mg) under 60 psi ofhydrogen at room temperature. The reaction mixturesCuminic acid (1 g, 6.1 mmol) was hydrogenated in acetic acid (5 mL) in the presence ofwere continuously stirred for 2 h. The acetic acid was distilled off from the reaction mixture under reduced pressure, and 0.95 g of the mixture of cis- and trans-4-isopropylcyclohexane-carboxylic acid was obtained by distillation (113-116 °C, 1 mmHg). b a solution of this acid and methanol (15 mL), a catalytic amount ofTsOH (10% wt) was added and the mixture was refluxed until starting material disappeared. Evaporation of methanol gave an ou which was purified by flash chromatography (ethyl acetate:hexane 5:95) to afford a mixture of 2.1 and 2.2 in aratio of 3:1 (0.88 g, 79%). ‘U NMR (400 MHz, CDC13): 3.60 (s, 3H), 2.58-2.14(m, 1H), 2.08-0.83 (m, 10H) ‘3C-NMR (400 MHz, cDcl3): 177.07(d), 51.84(d), 43.72(d), 43.50(d), 32.4(d), 29.40(d), 27.02(d), 20.29(d) Ô02H trans-4-(Isopropyl)cyctohexanecarboxylic acîd (2.4)
The mixture of esters 2.1 and 2.2 (0.8 5 g, 4.6 mmol) was isomerized in the presence of 60% sodium hydride (18.5 mg, 0.46 mmol) at 150 °C without solvent for 2 h. Water (3 mL) was carefully added to quench the reaction, extraction with CH2CI2 (3x 20 mL), drying over anhydrous Na2S04 gave 0.78 g ofthe trans methyl ester 2.2 and cis methyl ester 2.1 in a ratio of 6:1 afier distillation (64 °C, 0.7 mmHg). The methyl ester was dissolved in 4.2 mL of methanol and hydrolyzed by 4.2 mL of 2 N NaOH for 10 min. The solution was acidified with iN HCi to pH 2, and the powdery precipitate was fiÏtered. The crude product 2.3 and 2.4 was recrystallized from 80% MeOH aqueous to afford the trans acid 2.4 (0.49g, 64 %) as a white solid.