Utilisation de la spectroscopie de fluorescence pour la
vérification du nettoyage d’un ingrédient
pharmaceutique actif sur les surfaces des équipements
de production
Mémoire
Marianne Guay
Maîtrise en génie chimique
Maître ès sciences (M. Sc.)
Québec, Canada
© Marianne Guay, 2017
Utilisation de la spectroscopie de fluorescence pour la
vérification du nettoyage d’un ingrédient
pharmaceutique actif sur les surfaces des équipements
de production
Mémoire
Marianne Guay
Sous la direction de :
Résumé
La vérification de nettoyage des équipements pharmaceutiques permet de déterminer si la concentration du résidu sur la surface est inférieure à une limite acceptable afin de limiter la contamination croisée. La façon de procéder est de prélever un échantillon sur la surface par écouvillon et de l’analyser par chromatographie liquide à haute performance. Le problème avec cette approche est qu’il peut s’écouler jusqu’à 2 jours avant que le résultat soit obtenu et, durant ce temps, l’équipement nettoyé ne peut être libéré pour produire un autre lot de médicaments. Il existe une opportunité pour le développement d’une nouvelle méthode analytique permettant la quantification en temps réel et sans prise d’échantillon.
La nouvelle méthode évaluée utilise la fluorescence ciblée pour la quantification directe de l’ibuprofène. Dans cette preuve de concept, les différents paramètres pouvant influencer le signal sont évalués. La façon de préparer les standards pour l’étalonnage de l’appareil (TraC) est déterminée ainsi que la façon de les analyser. Une première courbe d’étalonnage pour des surfaces en acier inoxydable est validée lors d’une collecte de données in situ et il est déterminé qu’il est impossible d’évaluer la propreté de certains équipements dû à la limite de quantification du TraC et que le TraC possède un biais positif par rapport à la méthode traditionnelle. Suite à cette étude, l’appareil a subi des modifications, une nouvelle courbe d’étalonnage est réalisée et la limite de quantification est réduite. Il est déterminé que la méthode est exacte (pourcentage de récupération situé entre 85 et 115%), précise (écart-type relatif inférieur à 5%) et que les excipients testés n’ont aucun impact sur le signal. L’étalonnage sur l’aluminium, le bronze, le polypropylène et le laiton est réalisé. La preuve de concept pour l’utilisation du TraC pour la quantification de l’ibuprofène est démontrée et il est recommandé d’implanter la méthode dans une première usine.
Abstract
Cleaning verification is used to assess the cleanliness of pharmaceutical process equipment. Cleanliness is established when the concentration of the residue of a pharmaceutical ingredient is lower than an acceptance limit. The method used to perform this verification consists of collecting a sample using a swab, and to analyze it using high performance liquid chromatography. The issue with this method is that it can take up to 2 days before the results are obtained, and before the equipment can be released for the production of another batch of drugs. Therefore, there is an opportunity to develop a new analytical method to quantify cleanliness in real-time without sampling (contactless).
The new method investigated in this thesis uses fluorescence for direct quantification of ibuprofen residue. In this proof of concept, the impact of various parameters on the fluorescence signal was assessed. The best way to prepare the standards and how to analyze them with the instrument (the TraC) were determined. Following the verification of a first calibration curve using in situ data collection, the equipment was modified to allow the evaluation of equipment with an acceptance limit lower than the quantification limit of the TraC. The plant tests also revealed that the TraC always gives a higher concentration than the traditional method. An upgraded unit was calibrated, and the limit of quantification was lowered. It was established that the method is accurate (recovery is between 85 and 115%) and precise (relative standard deviation is lower than 5%), and that the excipients studied have no impact on the signal. Calibration curves were also built for aluminum, bronze, polyethylene and brass. Finally, the proof of concept for the use of the TraC for the cleaning verification of ibuprofen was demonstrated, and it is recommended to implement the method at a first plant.
Table des matières
RÉSUMÉ ... III ABSTRACT ... IV TABLE DES MATIÈRES ... V LISTE DES TABLEAUX ... VIII LISTE DES FIGURES ... IX LISTE DES ABRÉVIATIONS ET DES SIGLES ... XI REMERCIEMENTS ... XII AVANT-PROPOS ... XIII
CHAPITRE 1 INTRODUCTION ... 1
1.1 VALIDATION ET VÉRIFICATION DES PROCÉDURES DE NETTOYAGE ... 2
1.2 MÉTHODE ACTUELLEMENT UTILISÉE POUR LA VÉRIFICATION DU NETTOYAGE ... 4
1.2.1 Méthode d’écouvillonnage ... 5
1.2.2 Chromatographie liquide à haute performance ... 6
1.3 PROBLÉMATIQUE ... 8
1.4 REVUE DE LITTÉRATURE ... 8
1.4.1 Technologie d’analyse de procédé ... 8
1.4.2 Méthodes non spécifiques ... 10
1.4.3 Méthodes spécifiques ... 10
1.4.3.1 Méthode avec prise d’échantillon par écouvillon ... 10
1.4.3.1.1 Spectrométrie de mobilité ionique ... 10
1.4.3.1.2 Spectroscopie Raman ... 11
1.4.3.1.3 Méthode capillaire ... 11
1.4.3.1.4 Fluorescence ... 11
1.4.3.1.5 Conclusion sur les méthodes demandant une prise d’échantillon ... 12
1.4.3.2 Méthode analysant le solvant du rinçage ... 12
1.4.3.3 Méthodes sans prise d’échantillon... 13
1.4.3.3.1 Technologie infrarouge ... 13
1.4.3.3.2 Spectroscopie de masse ... 16
1.4.3.3.3 Fluorescence ... 16
1.4.3.3.4 Conclusion sur les méthodes sans prise d’échantillon ... 17
1.4.4 Autres méthodes analytiques servant à la quantification de l’ibuprofène ... 17
1.4.4.1 Méthode de quantification en solution ... 17
1.4.4.1.1 Chromatographie liquide à haute performance ... 17
1.4.4.1.2 Méthode capillaire ... 18
1.4.4.1.3 Spectrophotométrie UV ... 18
1.4.4.1.4 Méthode infrarouge ... 19
1.4.4.1.5 Techniques utilisant la fluorescence ... 19
1.4.4.1.6 Conclusion sur les méthodes demandant une mise en solution ... 20
1.4.4.2 Méthode de quantification sous forme solide ... 20
1.4.5 Méthode de quantification des solides utilisant la fluorescence... 20
1.4.6 Méthode choisie pour effectuer la vérification de nettoyage ... 21
1.5 OBJECTIF DU PROJET ... 23
1.6 PLAN DU MÉMOIRE ... 23
CHAPITRE 2 FLUORESCENCE ET APPAREIL À L’ÉTUDE ... 24
2.1 PRINCIPE DE LA FLUORESCENCE ... 24
2.1.1 Phénomène de fluorescence ... 24
2.1.2 Instrumentation ... 27
2.2 FONCTIONNEMENT DE L’APPAREIL ... 28
CHAPITRE 3 MATÉRIEL ET MÉTHODES ... 30
3.1 MATÉRIEL ET CONSOMMABLES ... 30
3.1.1 Ingrédient pharmaceutique actif et solvant ... 30
3.1.2 Coupons ... 30
3.1.2.1 Acier inoxydable... 30
3.1.2.2 Autres matériaux de construction ... 31
3.2 PRÉPARATION DE STANDARDS EN ACIER INOXYDABLE ... 31
3.3 ÉTALONNAGE ET VÉRIFICATION DE LA PERFORMANCE DE L’APPAREIL ... 33
3.3.1 Étalonnage de l’appareil... 33
3.3.2 Collecte de données en production... 35
3.3.3 Calcul de la concentration avec le TraC ... 37
3.4 VÉRIFICATION DE LA MÉTHODE ANALYTIQUE ... 37
3.4.1 Linéarité et écart d’utilisation... 38
3.4.1.1 Description ... 38
3.4.1.2 Évaluation expérimentale ... 38
3.4.2 Limite de détection et de quantification ... 39
3.4.2.1 Description ... 39 3.4.2.2 Évaluation expérimentale ... 40 3.4.3 Exactitude ... 40 3.4.3.1 Description ... 40 3.4.3.2 Évaluation expérimentale ... 40 3.4.4 Précision ... 41 3.4.4.1 Description ... 41 3.4.4.2 Évaluation expérimentale ... 41 3.4.5 Spécificité ... 42 3.4.5.1 Description ... 42 3.4.5.2 Évaluation expérimentale ... 43 3.4.6 Robustesse ... 43 3.4.6.1 Description ... 43 3.4.6.2 Évaluation expérimentale ... 44
CHAPITRE 4 OPTIMISATION DE LA MÉTHODE... 45
4.1 ÉVALUATION DE L’INFLUENCE DES PARAMÈTRES SUR LE SIGNAL ... 45
4.1.1 Paramètres reliés à l’équipement... 46
4.1.1.1 Source lumineuse, intensité lumineuse et filtres ... 47
4.1.1.2 Fatigue ... 47
4.1.1.3 Bruit de fond... 47
4.1.1.4 Reproductibilité du signal dans le temps ... 48
4.1.2 Paramètres reliés à la méthode de préparation des standards ... 49
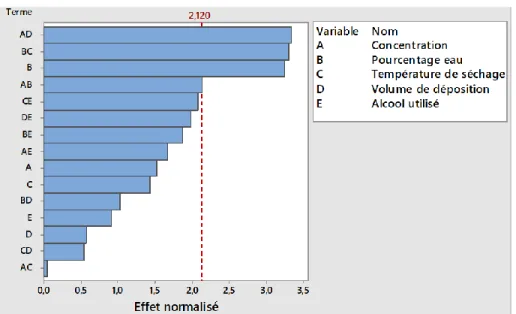
4.1.2.1 Plan d’expérience fractionné à 5 facteurs (2V5-1) ... 50
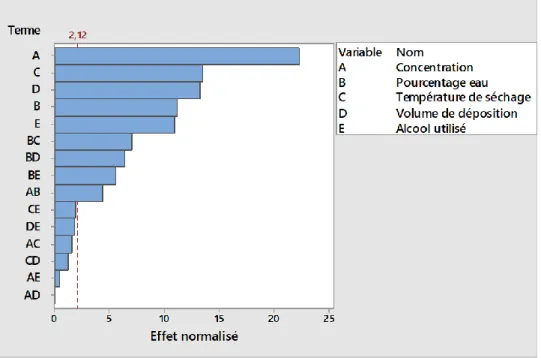
4.1.2.2 Plan d’expérience complet à 3 facteurs (23) ... 55
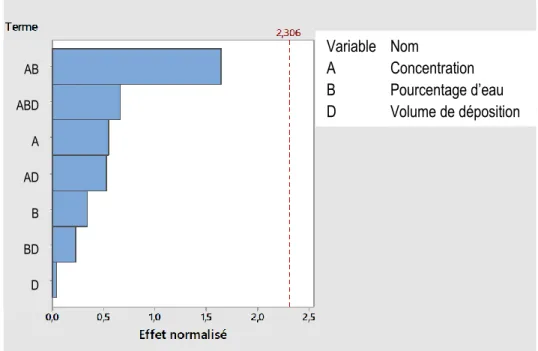
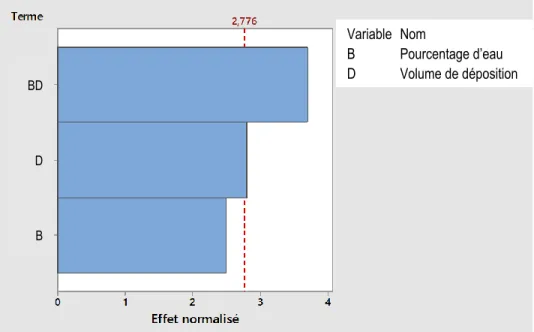
4.1.2.3 Plan d’expérience complet à 2 facteurs (22) ... 58
4.1.2.4 Conclusion des plans d’expérience ... 62
4.1.3 Paramètres reliés aux matériaux ... 62
4.1.3.1 IPA ... 63 4.1.3.1.1 Photoblanchissage ... 63 4.1.3.1.2 Fournisseur d’IPA ... 64 4.1.3.1.3 Effet de la température ... 65 4.1.3.2 Type de surface ... 67 4.1.3.2.1 Matériau de construction ... 67 4.1.3.2.2 Aspects de la surface ... 67 4.1.3.2.3 Réflexion lumineuse ... 67
4.1.4 Paramètres reliés à l’environnement... 68
4.1.4.1 Lumière ambiante ... 68
4.1.5 Paramètres reliés à la mesure des standards... 69
4.1.5.1 Section analysée... 69
4.1.5.2 Nombre de données collectées pour une section fixe ... 71
4.1.5.3 Nombre de mesures pour chaque donnée collectée ... 71
4.1.5.4 Distance entre le TraC et l’échantillon ... 72
4.1.5.5 Angle à lequel la mesure est prise... 74
4.1.6 Paramètres reliés à l’opérateur ... 74
4.2 FAÇON DE TRAVAILLER ... 74
CHAPITRE 5 ÉTALONNAGE ET VÉRIFICATION DES PERFORMANCES DE L’APPAREIL ... 76
5.1 PREMIÈRE COURBE D’ÉTALONNAGE ... 76
5.1.1 Construction de la première courbe d’étalonnage ... 76
5.1.2 Vérification de la première courbe d’étalonnage avec des données de laboratoire... 77
Vérification des performances de la première courbe d’étalonnage avec des données collectées in situ (en 5.1.3 production) ... 79
5.1.4 Optimisation des paramètres de l’appareil ... 84
5.2 COURBE D’ÉTALONNAGE CONSTRUITE SUITE À L’OPTIMISATION DE L’APPAREIL ... 84
5.2.1 Construction de la courbe d’étalonnage pour l’appareil optimisé... 84
5.2.2 Vérification de la courbe d’étalonnage pour l’appareil optimisé avec des données de laboratoire ... 85
5.2.2.1 Linéarité et intervalle de concentration ... 85
5.2.2.2 Limite de détection et de quantification ... 85
5.2.2.3 Exactitude et répétabilité ... 86
5.2.2.4 Précision intermédiaire ... 88
5.2.2.5 Spécificité ... 89
5.2.2.6 Robustesse ... 90
5.2.2.6.1 Détermination de la possibilité de quantifier sur chaque matériau... 91
5.2.2.6.2 Courbe d’étalonnage pour l’aluminium ... 93
5.2.2.6.3 Courbe d’étalonnage pour le laiton ... 93
5.2.2.6.4 Courbe d’étalonnage pour le polypropylène... 94
5.2.2.6.5 Courbe d’étalonnage pour le nickel ... 95
5.2.2.6.6 Conclusion sur l’impact des matériaux de construction ... 96
5.3 DÉTERMINATION DU NOMBRE DE POINTS À UTILISER POUR LA COLLECTE DE DONNÉES IN SITU ... 96
5.3.1 Nombre de données à récolter calculé à partir des données in situ ... 97
5.3.2 Nombre de données à récolter calculé à partir de données de laboratoire ... 98
5.4 MÉTHODE D’UTILISATION PROPOSÉE POUR L’APPAREIL ... 102
CHAPITRE 6 CONCLUSION ET RECOMMANDATIONS ... 104
BIBLIOGRAPHIE ... 108
ANNEXES ... 114
PROCÉDURE DE PRÉPARATION DES STANDARDS ET SÉLECTION DES COUPONS ... 114
ANNEXE A ANALYSE DES PLANS D’EXPÉRIENCE ... 116
ANNEXE B CODE MATLAB POUR LA DÉTERMINATION DU NOMBRE DE DONNÉES À RÉCOLTER ... 122
Liste des tableaux
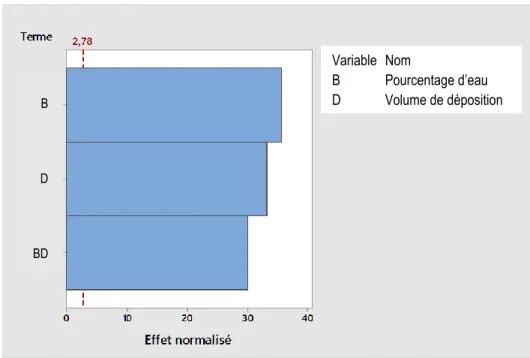
Tableau 3.1. Pourcentage d’eau dans la solution d’IPA ... 33
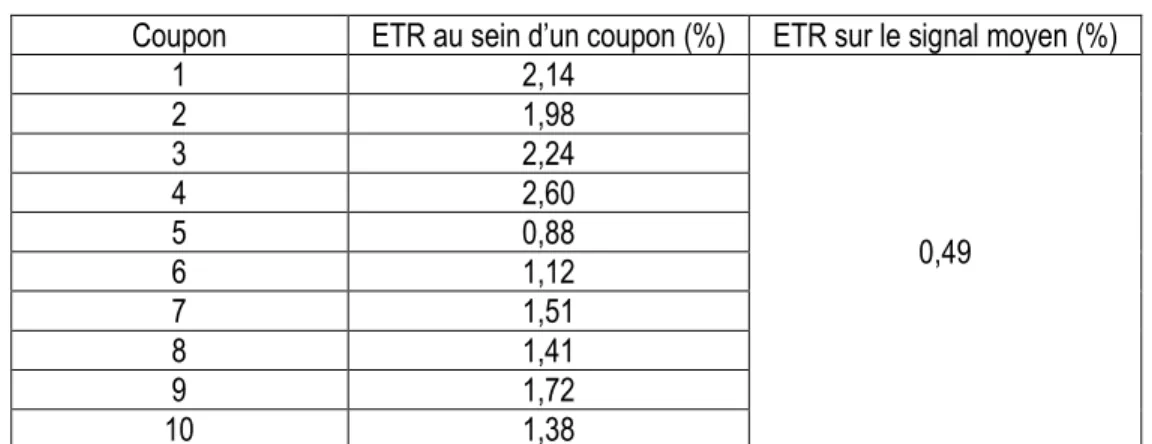
Tableau 4.1. Valeur de l’ETR des blancs pour l’unité non optimisée... 48
Tableau 4.2. Valeur de l’ETR des blancs pour l’unité optimisée ... 48
Tableau 4.3. Résultats du test-t à deux échantillons lorsque les données sont comparées avec le temps t0 ... 49
Tableau 4.4. Plan d'expérience fractionné à 5 facteurs (2V5-1) ... 50
Tableau 4.5. Plan d'expérience complet à 3 facteurs (23) ... 56
Tableau 4.6. Plan d'expérience complet à 2 facteurs (22) ... 59
Tableau 4.7. Paramètres recommandés pour la première courbe d’étalonnage ... 62
Tableau 4.8. Résultats sur l’évaluation de l’impact de la température... 66
Tableau 4.9. Valeur P pour le test-t à deux échantillons pour l’impact de la lumière ... 69
Tableau 4.10. Valeur P du test-t à deux échantillons pour déterminer l'effet de la section d'analyse ... 70
Tableau 4.11. Valeur P du test-t à deux échantillons pour le nombre de lectures prises pour une section fixe ... 71
Tableau 4.12. Valeur P du test-t à deux échantillons pour le nombre de mesures prises à chaque point ... 72
Tableau 4.13. Évaluation de l’impact de la distance pour l’appareil optimisé ... 74
Tableau 5.1. Évaluation de la précision et de l’exactitude pour la première courbe d’étalonnage à partir des données de vérification ... 78
Tableau 5.2. Données obtenues lors de l’évaluation de l’utilisation du TraC en production ... 82
Tableau 5.3. Évaluation de l’exactitude et de la précision basée sur la LQ... 86
Tableau 5.4. Résultats pour l’exactitude et la répétabilité pour l’appareil optimisé ... 87
Tableau 5.5. Évaluation de la variation de la réponse dans le temps pour le TraC optimisé ... 88
Tableau 5.6. Évaluation de l’impact de l’opérateur sur la réponse lorsqu’une méthode de collecte de données utilisant 20 points aléatoires est utilisée pour le TraC optimisé ... 89
Tableau 5.7. Résultats de l’étude de spécificité pour les standards contenant de l’IPA ... 89
Tableau 5.8. Variation du signal entre les blancs et les coupons placebo ... 90
Tableau 5.9. Évaluation de l’impact des différents matériaux échantillonnés sur les mesures du TraC optimisé .. 92
Tableau 5.10. Taille minimale de l’échantillon respectant les cibles d’incertitudes pour les données in situ ... 98
Tableau 5.11. Taille minimale de l’échantillon respectant les cibles d’incertitudes pour les données de laboratoire ... 100
Tableau 5.12. Comparaison des deux modes de collecte de données in situ ... 102
Tableau B.1. Résultats du plan d’expérience pour l’intensité du signal pour le plan d'expérience à 5 facteurs (2V5-1) ... 116
Tableau B.2. ANOVA pour l’intensité du signal transformée pour le plan d'expérience à 5 facteurs (2V5-1) ... 117
Tableau B.3. ANOVA pour l’ETR transformé pour le plan d'expérience à 5 facteurs (2V5-1) ... 117
Tableau B.4. ANOVA pour l’ETR pour le plan d'expérience à 3 facteurs (23) ... 117
Tableau B.5. ANOVA pour l’intensité du signal transformée pour le plan d'expérience à 3 facteurs (23) ... 118
Tableau B.6. ANOVA pour l’ETR pour le plan d'expérience à 3 facteurs (22) ... 118
Liste des figures
Figure 1.1. Schéma des étapes à suivre pour effectuer le nettoyage d'un équipement de production ... 1
Figure 1.2. Méthode d’échantillonnage par écouvillon (Cole-Parmer, 2016) ... 5
Figure 2.1. Diagramme de Jablonski (Valeur & Berberan-Santos, 2012) ... 25
Figure 2.2. Exemple (a) d’un grand et (b) d’un petit déplacement de Stokes (ThermoFisher Scientific, Sans date) ... 25
Figure 2.3. TraC et plateforme « XY » fabriqué par Photon Systems Inc. ... 29
Figure 3.1. Coupon servant à la préparation de standard ... 31
Figure 3.2. Modèle à suivre pour la préparation des standards ... 32
Figure 3.3. Surface d’analyse et diamètre du faisceau (cercle bleu) ... 33
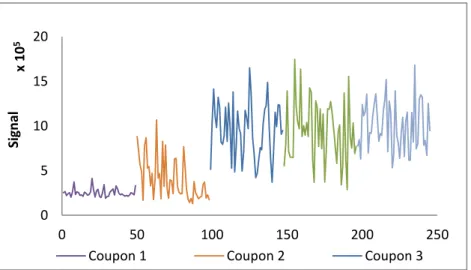
Figure 3.4. Variation du signal pour différents coupons d’une même concentration d’IBU ... 34
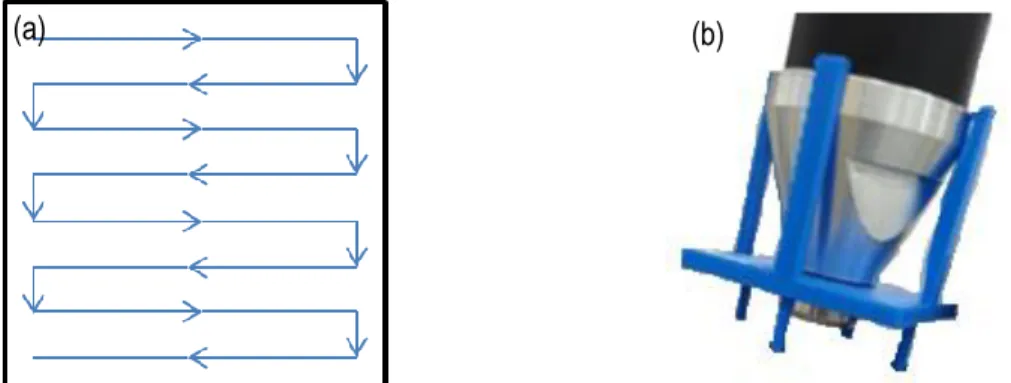
Figure 3.5. (a) Patron et (b) embout utilisé pour la collecte de données in situ ... 36
Figure 3.6. Schéma utilisé pour effectuer la collecte de données in situ ... 36
Figure 4.1. Diagramme d’Ishikawa présentant les facteurs pouvant influencer le signal en fluorescence ... 46
Figure 4.2. Diagramme de Pareto pour l’ETR transformé pour le plan d'expérience à 5 facteurs (2V5-1) ... 52
Figure 4.3. Effet (a) des facteurs principaux et (b) des interactions sur l’ETR transformé pour le plan d'expérience à 5 facteurs (2V5-1) ... 52
Figure 4.4. Apparence général d’un coupon avec (a) des points concentrés et pour (b) une déposition homogène ... 53
Figure 4.5. Diagramme de Pareto pour l’intensité du signal transformée pour le plan d'expérience à 5 facteurs (2V5-1) ... 54
Figure 4.6. Effet (a) des facteurs principaux et (b) des interactions sur l’intensité du signal transformée pour le plan d'expérience à 5 facteurs (2V5-1) ... 55
Figure 4.7. Diagramme de Pareto pour l’ETR pour le plan d'expérience complet à 3 facteurs (23) ... 57
Figure 4.8. Diagramme de Pareto pour l’intensité du signal transformée pour le plan d'expérience complet à 3 facteurs (23) ... 57
Figure 4.9. Effet (a) des facteurs principaux et (b) des interactions sur l’intensité du signal transformée pour le plan d'expérience complet à 3 facteurs (23) ... 58
Figure 4.10. Diagramme de Pareto pour l’ETR pour le plan d'expérience complet à 2 facteurs (22) ... 59
Figure 4.11. Effet (a) des facteurs principaux et (b) des interactions sur l’ETR pour le plan d'expérience complet à 2 facteurs (22) ... 60
Figure 4.12. Diagramme de Pareto pour l’intensité du signal transformée pour le plan d'expérience complet à 2 facteurs (22) ... 61
Figure 4.13. Effet (a) des facteurs principaux et (b) des interactions sur l’intensité du signal transformée pour le plan d'expérience complet à 2 facteurs (22) ... 61
Figure 4.14. Différence absolue entre la moyenne de quatre mesures consécutives et la moyenne des quatre premières mesures en fonction du nombre d’exposition pour deux concentrations ... 64
Figure 4.15. Distribution du signal pour chaque fournisseur ... 65
Figure 4.16.Variation de l’intensité du signal pour le test de réflexion de la lumière ... 68
Figure 4.17. Section d'analyse (a) partielle et (b) complète ... 70
Figure 4.18. Coupon pour l'échantillon 9 ... 70
Figure 5.1. Première courbe d’étalonnage sur de l’acier inoxydable pour un intervalle de (a) 0 à 667% et de (b) 0 à 250% ... 77 Figure 5.2. Première courbe d’étalonnage pour un intervalle de 0 à 250% avec les données de vérification sur de
l’acier inoxydable ... 78 Figure 5.3. Trois scénarios possibles lors d’une collecte de données in situ avec le TraC ... 80 Figure 5.4. Courbe d’étalonnage sur de l’acier inoxydable suite aux modifications de l’appareil TraC ... 84 Figure 5.5. Courbe d’étalonnage sur de l’acier inoxydable suite aux modifications de l’appareil pour un intervalle
de concentration allant de la LQ à 167% ... 86 Figure 5.6. Courbe d’étalonnage avec les données de vérification sur de l’acier inoxydable pour l’unité optimisée
... 87 Figure 5.7. Courbe d’étalonnage de l’IPA sur de l’aluminium ... 93 Figure 5.8. Courbes d’étalonnage de l’IPA sur (a) le laiton et sur (b) le laiton et l’acier inoxydable ... 94 Figure 5.9. (a) Courbe d’étalonnage de l’IPA sur du polypropylène et (b) courbe d’étalonnage normalisée pour
l’acier inoxydable et le polypropylène ... 94 Figure 5.10. Courbe d’étalonnage (a) sur du nickel pour une plage de 17 à 167%, (b) sur le nickel pour une plage
de 51 à 167% et (c) sur l’acier inoxydable et sur le nickel ... 95 Figure 5.11. Distribution des données brutes récoltées pour (a) l’échantillon #2 et (b) l’échantillon #11 ... 98 Figure 5.12. Distribution des données brutes pour le coupon #2 pour une concentration à 83% ... 101 Figure 5.13. Organigramme décisionnel pour la détermination du résultat de la vérification de nettoyage avec le
TraC ... 103 Figure A.1. Procédure à suivre pour la préparation des standards ... 114 Figure A.2. Procédure de sélection des coupons pour l’étalonnage ... 115 Figure B.1. Analyse des résidus pour (a) l’ETR transformé et pour (b) le signal transformé pour le plan
d'expérience à 5 facteurs (2V5-1)... 119 Figure B.2. Analyse des résidus pour (a) l’ETR et pour (b) le signal transformé pour le plan d'expérience complet à 3 facteurs (23) ... 120 Figure B.3. Analyse des résidus pour (a) l’ETR et pour (b) le signal transformé pour le plan d'expérience complet à 2 facteurs (22) ... 121
Liste des abréviations et des sigles
Abréviation DéfinitionACP Analyse en composantes principales ANOVA Analyse de la variance
BPF Bonne pratique de fabrication
CCD Caméra avec dispositif à transfert de charges CIH Conférence internationale sur l'harmonisation COT Carbone organique total
DEL Diode électroluminescente EQM Erreur quadratique moyenne ETR Écart-type relatif
FDA Food and Drug Administration
HPLC Chromatographie liquide à haute performance
IBU Ibuprofène
IC-PIR Imagerie chimique par proche infrarouge IPA Ingrédient pharmaceutique actif
IR Infrarouge
IRRAS Spectroscopie d’absorption infrarouge en réflexion LD Limite de détection
LIF Fluorescence induite par laser LQ Limite de quantification PEHD Polyéthylène haute densité PIR Proche infrarouge
PLS Régression par moindres carrés partiels SMI Spectrométrie de mobilité ionique TAP Technologie d’analyse de procédés TF-IR Transformation de Fourier en infrarouge
TraC Instrument portatif pour la vérification de nettoyage
Remerciements
Je tiens à exprimer mes sincères remerciement à mon directeur de recherche, monsieur Carl Duchesne, qui par ses conseils et son soutien a permis de mener à bien cette recherche. Je tiens également à remercier ma superviseure au sein de Pfizer Montréal Inc., madame Joanny Salvas, pour son temps ainsi que pour son expertise qui ont permis à ce projet de se concrétiser. J’aimerais aussi remercier monsieur Jean-Sébastien Simard pour m’avoir offert l’opportunité d’effectuer ma maîtrise au sein d’une entreprise ainsi que pour l’apport financier sans lequel le projet n’aurait pu être réalisé.
Je tiens à souligner l’aide de monsieur Ben Lyons pour ses suggestions concernant le projet et pour la formation qu’il m’a donné sur le fonctionnement du TraC ainsi que madame Naimah Majeed et monsieur Alan Rhoden. Je tiens à remercier madame Lourdes Lopez-Caban, messieurs Louis Hodges et Jose Montenegro pour leur aide lors de la collecte de données en production sans qui celle-ci n’aurait pas été réalisée. Je remercie également les gens du groupe PASG qui ont participé de près ou de loin au projet.
J’aimerais également remercier les gens de Photon Systems pour leur support lors de l’évaluation de l’appareil, plus particulièrement messieurs Ray Reid et Mike Reid pour leur temps et leur expertise.
Finalement, je remercie ma famille, ma mère Suzanne, mon père Michel et ma sœur Katherine pour leur support durant mes études de maîtrise.
Avant-Propos
Le présent projet de maîtrise est le fruit d’une collaboration entre l’Université Laval et le groupe PASG de Pfizer Montréal Inc. Le projet consiste à évaluer le potentiel d’un prototype pour la vérification de nettoyage, fabriqué par un partenaire industriel de Pfizer, soit la compagnie Photon Systems Inc. Le projet s’inscrit dans un cadre précis et les conditions de nettoyage des équipements ainsi que les processus de fabrication des médicaments contenant l’ingrédient pharmaceutique actif (IPA) sont fixes. L’étude concerne seulement la vérification de nettoyage de l’ibuprofène sur la surface des équipements de production. Le prototype a été fabriqué pour effectuer uniquement la vérification de l’ibuprofène en sélectionnant les longueurs d’onde d’émission et d’excitation optimale pour la spectroscopie de fluorescence.
Dans le domaine pharmaceutique, les méthodes analytiques doivent respecter des critères bien précis avant de pouvoir être utilisées lors des opérations de l’usine. Les méthodes analytiques sont validées en utilisant les critères définis dans les lignes directrices de la Conférence internationale sur l’harmonisation. La méthodologie appliquée dans ce mémoire pour valider la méthode est donc prédéfini par les normes.
Chapitre 1 Introduction
L’industrie pharmaceutique est régulée par les organismes gouvernementaux tels Santé Canada et la « Food and Drug Administration » (FDA) aux États-Unis. Ces organismes permettent d’assurer que les médicaments sont fabriqués de manière à réduire les dangers pour la population. Les bonnes pratiques de fabrication (BPF) sont un point important des réglementations implantées par ces organismes gouvernementaux. Les BPF permettent d’assurer « la fabrication de drogues de haute qualité » (Santé Canada, 2009). Dans ces règlementations, le nettoyage des équipements de production est notamment abordé.
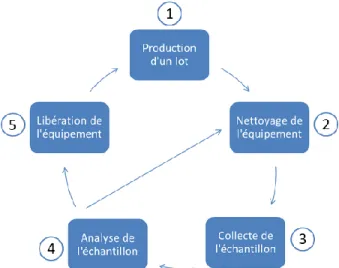
La fabrication de médicaments demande l’utilisation de plusieurs types d’équipement, comme les mélangeurs, les presses à comprimés, etc. Le même équipement peut être utilisé pour la production de plusieurs médicaments possédant des ingrédients actifs différents ou des doses différentes. Par exemple, un médicament ayant un ingrédient pharmaceutique actif (IPA) « B » est manufacturé sur le même équipement qu’un médicament utilisant un IPA « A ». Afin d’éviter que des résidus de l’IPA « A » se retrouvent dans le produit « B » et qu’il y ait contamination croisée, un nettoyage de l’équipement est effectué entre les lots. Ce nettoyage s’effectue selon des procédures établies et validées. La Figure 1.1 présente un résumé des étapes à suivre pour effectuer le nettoyage des équipements de production. La première étape consiste à fabriquer un lot avec un équipement qui est par la suite nettoyé (étape 2). Après le nettoyage, il faut vérifier que le résidu présent sur la surface ne dépasse pas une certaine limite, appelée limite acceptable. Pour effectuer cette vérification, il faut collecter un échantillon sur la surface (étape 3) et l’analyser (étape 4). Si la concentration de résidu est supérieure à la limite acceptable, l’équipement est nettoyé de nouveau (retour à l’étape 2), sinon il est libéré (étape 5) pour la préparation d’un autre lot de médicaments (étape 1). Le présent projet de maîtrise se concentre sur la vérification des procédures de nettoyage dans le domaine pharmaceutique pour des résidus d’ibuprofène (IBU).
Ce chapitre aborde la validation et la vérification des procédures de nettoyage, la méthode utilisée actuellement dans l’industrie pharmaceutique pour effectuer la vérification, la problématique reliée à cette méthode, une revue de littérature sur ce qui a été fait jusqu’à présent pour répondre au problème, la méthode choisie afin de répondre à la problématique dans le cadre de cette maîtrise ainsi que les objectifs du projet et finalement l’organisation du mémoire.
1.1 Validation et vérification des procédures de nettoyage
Le nettoyage des équipements servant à la fabrication de médicaments est une pratique qui fut incluse dans la réglementation sur les BPF de la FDA en 1963. Dans cette réglementation, il était stipulé que les équipements doivent être maintenus dans un état propre (FDA, 1993). Le but premier de cette étape d’un procédé de fabrication de médicaments est de prévenir la contamination et d’assurer la qualité du produit (FDA, 1993). Avant 1993, aucun guide spécifique portant sur les procédés de nettoyage n’avait été publié afin de faciliter la compréhension de la réglementation antérieure à 1993 (Hall, 2007), c’est pourquoi la FDA publia le guide « Validation of Cleaning Processes (7/93): Guide to Inspections Validation of Cleaning Processes » (FDA, 1993). Ce guide explique comment effectuer la vérification des procédures de nettoyage. La procédure de nettoyage à suivre est établie selon l’utilisation de l’équipement, c’est-à-dire si l’équipement est dédié à une substance en particulier ou non et si le mode d’opération est en batch ou en continu (ICH, 2005a) ainsi que selon le type d’équipement et son design. Le procédé de nettoyage des équipements de production demande donc des procédures écrites et validées pour chaque type d’équipement et de catégorie de substances à nettoyer (FDA, 1993).
Les procédures écrites font en sorte que le nettoyage est toujours effectué de la même manière par les différents opérateurs (ICH, 2000). Elles permettent de savoir quand le nettoyage doit être fait, la méthode à suivre pour effectuer le nettoyage, y compris les détergents utilisés, ainsi que la méthode pour effectuer la vérification du nettoyage, comment entreposer les équipements propres et la personne qui s’est chargée des différentes étapes de la procédure (FDA, 2016). La première étape de l’élaboration d’une procédure de nettoyage est de déterminer les caractéristiques du nettoyage, par exemple, si le procédé est automatique ou non, la cible du nettoyage (IPA, détergent, etc.), le type d’équipement à nettoyer, le matériau de fabrication de l’équipement, etc. (PDA, 1998). Elle doit également identifier les endroits les plus à risque d’occasionner de la contamination, le produit occasionnant le pire scénario (cible du nettoyage) et les équipements les plus difficiles à nettoyer (Hall, 2007). Une des étapes cruciales de l’élaboration d’une procédure de nettoyage est de déterminer la limite acceptable du résidu sur la surface. Cette limite correspond à la quantité maximale d’un résidu par unité de surface qui peut être toléré après le nettoyage. Il n’existe aucune valeur cible pour la limite acceptable suggérée par les organismes gouvernementaux, dû à la grande variété de produits pharmaceutiques. Cette limite doit, par contre, être logique, fondée sur un raisonnement scientifique et doit
être vérifiable et atteignable (FDA, 1998). De plus, elle ne doit pas occasionner de risque pour la santé, ne doit pas occasionner de perte de qualité du produit et, à cette valeur, aucune trace ne devrait être visible sur la surface de l’équipement (FDA, 2015). Cette limite peut être calculée à partir des données toxicologiques, comme la quantité maximale d’exposition quotidienne, en fonction de la dose quotidienne thérapeutique ce qui correspond à 1/1000 de la dose thérapeutique normale (Satinder, Shashikant, & Bharat, 2012). Finalement, cette limite peut correspondre à une valeur maximale de 0,1% dans le prochain produit fabriqué avec l’équipement (Satinder et al., 2012). La limite acceptable est souvent exprimée en masse de résidus par surface nettoyée (µg/cm2). Cette valeur correspond donc à la quantité maximale de résidu qui peut se retrouver dans un autre lot lorsque la surface totale de l’équipement est considérée (APIC, 2014). La procédure indique également les points de collecte d’échantillons pour effectuer la vérification et la validation du nettoyage. Les points d’échantillonnage correspondent aux endroits les plus difficiles à nettoyer et doivent être justifiés par un raisonnement scientifique avant d’être utilisés pour effectuer la vérification de nettoyage (Pluta & Sharnez, 2011). La collecte d’échantillon s’effectue soit par rinçage, à l’aide d’un écouvillon ou par toutes autres méthodes qui permettent la collecte d’échantillons (ICH, 2000). La méthode analytique utilisée pour la détermination de la quantité de résidu sur la surface est également détaillée dans la procédure. La détermination de la quantité résiduelle sur la surface s’effectue à l’aide d’une méthode analytique. Il faut que cette méthode permette de quantifier le résidu en deçà de la limite acceptable, sinon une autre méthode doit être utilisée. Les éléments contenus dans une procédure de nettoyage ne se limitent pas à ceux mentionnés précédemment. L’application de la procédure de nettoyage doit donc être documentée afin de respecter les BPF (FDA, 2016). Il faut également que ces procédures traitent de l’élimination des produits servant au nettoyage comme les solvants et détergents (FDA, 1998).
Avant qu’une procédure de nettoyage ne soit appliquée pour des opérations de routine, celle-ci doit être validée. La validation s’effectue à l’aide d’un protocole de validation. La validation des procédures de nettoyage permet de prouver l’efficacité et la reproductibilité du nettoyage ainsi que de corriger certains problèmes non envisagés lors de l’élaboration de la procédure (Lakshmana Prabu & Suriyaprakash, 2010). La validation de nettoyage permet aussi de vérifier que la procédure de nettoyage contrôle la contamination par les microorganismes (Satinder et al., 2012). La procédure sujette à la validation doit permettre d’atteindre la limite acceptable du résidu sur la surface sinon une nouvelle approche doit être envisagée. La méthode analytique utilisée pour déterminer la quantité résiduelle sur la surface doit être également validée. La validation d’une méthode analytique pour effectuer les validations et vérifications de nettoyage est abordée au Chapitre 3. Le protocole de validation comprend donc la façon dont la méthode analytique est appliquée ainsi que la manière de prélever les échantillons (ICH, 2000). Au cours de la validation de la procédure, le pourcentage de recouvrement de la méthode d’échantillonnage doit également être évalué (Pluta & Sharnez, 2011). Finalement, pour qu’un procédé de nettoyage soit validé, il faut que celui-ci passe avec succès le
protocole de validation un minimum de trois fois (PDA, 1998) afin de déterminer si le procédé à l’étude permet de nettoyer de façon adéquate et reproductible l’équipement. Les résultats de la validation d’une procédure de nettoyage sont présentés dans un rapport de validation où la conclusion de la validation est décrite (FDA, 1998). Une fois la procédure de nettoyage validée et le rapport de validation complété, la procédure peut être appliquée pour les opérations de routine en limitant le nombre de vérification de nettoyage (Pharmaceutical Inspection Convention, 2007). Une revalidation des procédures de nettoyage peut être nécessaire si la méthode de nettoyage est modifiée pour un équipement, s’il y a un changement au niveau des produits, du procédé et de l’équipement, après la maintenance ou une fermeture (« shutdown »), etc. (Agalloco, 1992). En conclusion, la validation des procédures de nettoyage permet de déterminer si la façon de nettoyer l’équipement décrite par la procédure assure que la quantité du résidu présent sur la surface est inférieure à la limite acceptable préalablement établie.
Contrairement à la validation de nettoyage qui sert à déterminer si une procédure permet d’atteindre les objectifs visés, la vérification de nettoyage, quant à elle, s’effectue lors des opérations normales de l’usine et permet de déterminer si la procédure de nettoyage en place a effectivement permis de réduire le résidu à une valeur inférieure ou égale à la limite acceptable (Nash & Berry, 2003). La différence entre la validation et la vérification est donc minime. La vérification du nettoyage utilise donc la même méthode analytique que la validation pour quantifier le résidu sur la surface des équipements de production qui ont été nettoyés selon les procédures établies.
Une méthode analytique est donc utilisée lors de l’élaboration des procédures de nettoyage, lors de la validation de la procédure de nettoyage, lors des opérations routinières de vérification de nettoyage et lors de la revalidation. La section suivante décrit la méthode utilisée actuellement pour effectuer la vérification du nettoyage dans le domaine pharmaceutique.
1.2 Méthode actuellement utilisée pour la vérification du nettoyage
La méthode conventionnelle pour effectuer les vérifications de nettoyage est le prélèvement d’un échantillon sur la surface à l’aide d’un écouvillon, suivi de l’analyse de l’échantillon par chromatographie liquide à haute performance (HPLC). La méthode d’échantillonnage par écouvillon (« swab » en anglais) est la méthode favorisée par la FDA pour réaliser les vérifications des procédures de nettoyage (Dubey, Mandhanya, & Kumar Jain, 2012). La seconde méthode décrite dans les lignes directrices de la FDA est le rinçage qui consiste à analyser le solvant du dernier rinçage, ce qui signifie que l’IPA est détecté directement dans le solvant (Santé Canada, 2008). Pour utiliser cette technique, il faut donc que l’IPA soit soluble dans le solvant et qu’il ne doit pas être dissimulé dans l’équipement (Lakshmana Prabu & Suriyaprakash, 2010). La vérification de nettoyage n’est pas limitée aux ingrédients pharmaceutiques actifs. Elle s’applique également
aux résidus laissés par les produits utilisés lors du nettoyage. Par contre, ces travaux de maîtrise se concentrent uniquement sur la vérification de nettoyage d’un IPA spécifique.
1.2.1 Méthode d’écouvillonnage
Le prélèvement d’un échantillon par écouvillon est réalisé en suivant les procédures établies et validées. Pour utiliser cette technique, il faut que la substance à échantillonner soit extractible par l’écouvillon (Kohli, 2012). Les endroits à échantillonner ont été évalués lors de l’élaboration de la procédure de nettoyage afin de déterminer les endroits représentant le pire scénario, c’est-à-dire les endroits les plus difficiles à nettoyer (APIC, 2014). Il faut, par contre, que ces endroits soient relativement accessibles pour que l’échantillonnage puisse s’effectuer (Satinder et al., 2012). Le type d’écouvillon, le solvant ainsi que la technique à utiliser pour effectuer le prélèvement sont décrits dans les procédures de nettoyage (APIC, 2014). Le solvant choisi pour le prélèvement ne doit pas entraîner la dégradation du résidu (Lodhi, Padamwar, & Patel, 2014). Il doit permettre de recouvrer le résidu et d’éviter d’endommager l’équipement. La plupart des solvants utilisés sont aqueux ou organiques (Nassani, 2005). La limite acceptable est représentée sous la forme de masse de résidu par unité de surface lorsque l’échantillonnage par écouvillon est utilisé (Santé Canada, 2008). La collecte d’échantillon par écouvillon consiste à mouiller l’écouvillon, sans le saturer de solvant (Cole-Parmer, 2016) et à passer celui-ci sur la surface en suivant un certain patron comme illustré à la Figure 1.2. Dans cette figure, deux écouvillons sont utilisés pour échantillonner la surface. L’utilisation d’un ou de plusieurs écouvillons dépend du résidu. Par la suite, la tête de l’écouvillon est placée dans un contenant fermé puis le résidu est extrait de l’écouvillon et analysé par une méthode analytique validée (Cole-Parmer, 2016). L’échantillon est généralement récolté sur une section de 25 cm2 (Forsyth, 2016).
Figure 1.2. Méthode d’échantillonnage par écouvillon (Cole-Parmer, 2016)
Les avantages de cette méthode sont qu’elle permet de faire une collecte physique du résidu. Donc la méthode permet de collecter les résidus qui ont séché sur la surface ainsi que les résidus insolubles (Carlson, 2010). Elle permet également d’échantillonner une section précise, est applicable sur un grand nombre de surfaces et peut être utilisée pour la collecte d’IPA, d’agents nettoyants et de microorganisme (PDA, 1998).
Finalement, cette méthode d’échantillonnage permet la détermination de la quantité du résidu par unité de surface (FDA, 1998) et cette quantité peut être convertie en masse totale pour l’équipement (APIC, 2014). La méthode d’échantillonnage par écouvillon comprend également des inconvénients. Un des premiers inconvénients est que l’écouvillon peut causer des interférences lors de la quantification du résidu (Lakshmana Prabu & Suriyaprakash, 2010). Il faut donc que la réponse de l’écouvillon lui-même soit minimale (Corrigan, Salton, Preston, & Piletsky, 2010). De plus, l’utilisation d’un écouvillon peut introduire des fibres sur la surface nettoyée (Kumar, Sanjeev, & Sharma, 2012). Comme l’échantillonnage par écouvillon sélectionne les endroits les plus critiques qui sont accessibles pour la collecte, il ne permet pas de faire l’échantillonnage complet de la surface et de certains endroits non accessibles (Carlson, 2010). De plus, cette technique dépend de la personne qui réalise la collecte des échantillons (Carlson, 2010). Il faut donc que le personnel qui s’occupe de la vérification soit formé et suive une procédure pour réduire la variabilité lors de la prise d’échantillon (Pluta & Sharnez, 2011). Cette méthode ne permet pas une récupération totale du résidu sur la surface échantillonnée, donc une étude de récupération est nécessaire pour déterminer le pourcentage de résidus récupérés par l’écouvillon (Cole-Parmer, 2016). Finalement, cette technique demande beaucoup de temps pour être réalisée (Corrigan et al., 2010).
Une fois que la collecte de l’échantillon par écouvillon est terminée, l’échantillon doit être extrait de l’écouvillon et analysé par une méthode analytique validée. La chromatographie liquide à haute performance est une des méthodes les plus utilisées pour effectuer la quantification des résidus sur la surface des équipements pharmaceutiques (Fekete, Fekete, & Ganzler, 2009).
1.2.2 Chromatographie liquide à haute performance
La chromatographie liquide à haute performance compte trois sections, soit le système de livraison du solvant, la colonne de séparation et le détecteur (Hansen, Pedersen-Bjergaard, & Rasmussen, 2012). Une HPLC repose sur la séparation des composés qui sont injectés dans la phase mobile avant le passage de celle-ci à travers une colonne garnie (Hansen et al., 2012). La phase mobile est alimenté dans la colonne par la pression exercée par une pompe (Agilent Technologies, Sans date) et passe à travers la phase stationnaire. Certains composés possèdent une plus grande affinité pour la phase stationnaire. Ceux-ci seront retenus par la phase stationnaire avant de retourner dans la phase mobile et d’être analysés (Ohannesian & Streeter, 2001). Le temps de rétention correspond au temps entre l’injection du soluté et son passage au détecteur (Ohannesian & Streeter, 2001). Plus un composé possède un temps de rétention élevé, plus il possède d’affinité avec la phase stationnaire. À la sortie de la colonne, les composés sont identifiés et/ou quantifiés (Agilent Technologies, Sans date).
Afin d’obtenir une bonne séparation, il faut que le couple phase mobile et phase stationnaire choisi permette la plus grande différence d’affinité pour la phase stationnaire entre les composants de l’échantillon (Ohannesian & Streeter, 2001). Le gradient d’élution est utilisé pour les composés ayant de grandes différences de rétention et repose sur le fait que la composition change dans la phase mobile pendant l’analyse (Hansen et al., 2012). Cette méthode permet de réduire le temps d’analyse qui peut être grand lorsque l’élution isocratique est utilisée. De plus, avec cette méthode, seuls les composés qui ont un temps de rétention moyen sont bien caractérisés tandis qu’avec le gradient d’élution, tous les composés sont considérés. (Hansen et al., 2012). Le temps d’analyse par HPLC est aux alentours de 10 à 30 minutes par échantillon et peut aller jusqu’à 2 heures (McMaster, 2007).
Le détecteur sert à déterminer la concentration d’un analyte en fonction de sa réponse qui est, la plupart du temps, calculée par l’aire sous la courbe d’un pic ou la hauteur du pic pour le composant à l’étude (Hansen et al., 2012). Il existe deux types de détecteurs, soient ceux qui permettent de voir des changements dans la phase mobile ou ceux qui sont spécifiques à des composés selon leurs propriétés (Hansen et al., 2012). Le détecteur le plus utilisé dans le domaine pharmaceutique est le détecteur utilisant l’ultraviolet (UV) (Hansen et al., 2012). L’utilisation d’un détecteur UV consiste en l’absorption de la lumière à une longueur d’onde précise par les chromophores d’une molécule, donc ce type de détecteur ne peut pas être appliqué pour toutes les molécules (Hansen et al., 2012). Parmi les autres types de détecteur, on retrouve ceux qui utilisent la fluorescence, l’indice de réfraction, l’électrochimie, etc. (Hansen et al., 2012). Il est également possible de combiner différents détecteurs, mais il faut qu’un certain ordre soit suivi (Hansen et al., 2012). L’avantage d’avoir un détecteur sélectif est qu’il n’est pas toujours nécessaire de séparer préalablement tous les analytes puisque le détecteur identifie uniquement la substance ciblée (Ohannesian & Streeter, 2001).
L’analyse par HPLC comporte certains avantages comme sa spécificité, sa sensibilité ainsi que la possibilité de quantifier les résidus (Peles, Ely, Crowder, & Ponstingl, 2013). Une HPLC est une méthode précise et reproductible pour la quantification. La méthode est également robuste (Dong, 2013). Un des inconvénients de la quantification par HPLC est que la méthode est coûteuse puisque les colonnes utilisées pour effectuer la séparation sont dispendieuses et leur durée de vie est courte (McMaster, 2007). De plus, les solvants utilisés peuvent être coûteux et il est parfois difficile d’en disposer. Également, la méthode demande une grande quantité de solvant et de phase mobile (Peles et al., 2013). Un autre inconvénient est le temps d’analyse par échantillon qui est long. L’analyse par HPLC demande généralement la préparation de standards, des échantillons et de la phase mobile, la configuration de la colonne, un test de compétence du système, un étalonnage de l’appareil avec les standards, l’analyse des échantillons, la quantification des échantillons ainsi que le rapport sur les résultats (Dong, 2013). Finalement, cette méthode demande que le personnel soit formé pour l’opérer (Ahuja & Rasmussen, 2007).
1.3 Problématique
La méthode traditionnelle utilisée pour effectuer les vérifications de nettoyage emploie l’échantillonnage par écouvillon suivi d’une quantification par HPLC. L’inconvénient principal de cette méthode est qu’elle demande une prise d’échantillon suivi d’une analyse, ce qui prend un certain temps. Cette méthode requiert que la personne qui collecte l’échantillon prépare le matériel nécessaire, récolte l’échantillon puis le place dans un flacon qu’il envoie au laboratoire après l’avoir identifié. Par la suite, le personnel de laboratoire documente l’échantillon, prépare le matériel nécessaire à l’analyse, effectue une extraction de l’échantillon, détermine la quantité de résidu obtenue par HPLC et finalement rapporte le résultat. Donc, tant que le résultat de l’analyse n’est pas obtenu, l’équipement de production ne peut être utilisé pour produire un autre lot de médicaments (Peles et al., 2013). Le délai entre le nettoyage de l’équipement et la libération de celui-ci peut aller jusqu’à 2 jours (Peles et al., 2013). Afin de réduire le temps d’attente entre le nettoyage d’un lot antérieur et un lot subséquent, il pourrait être possible et intéressant d’utiliser une méthode de quantification directe. La quantification directe correspond à une analyse de la surface sans collecte physique d’échantillon. Un autre problème de la méthode traditionnelle est qu’elle permet seulement d’échantillonner une portion de la surface, ce problème ne sera pas considéré puisque les mêmes sections que la méthode traditionnelle sont analysées. Une revue de la littérature a été réalisée afin de voir ce qui a été fait jusqu’à présent pour résoudre le problème présenté ainsi que pour déterminer ce que la méthode choisie pourrait apporter de plus en vérification de nettoyage dans le domaine pharmaceutique.
1.4 Revue de littérature
Cette section présente ce qu’est une technologie d’analyse de procédé (TAP). Les méthodes développées jusqu’à maintenant pour effectuer la vérification de nettoyage avec prise d’échantillons plus rapide que la méthode traditionnelle sont présentées. Les méthodes sans prise d’échantillon ainsi que les méthodes développées jusqu’à présent pour quantifier l’ibuprofène, qui est la cible de la vérification de nettoyage étudiée dans ce mémoire, sont également passées en revue. Finalement, la méthode choisie est décrite avec ces avantages par rapport aux autres méthodes développées jusqu’à présent.
1.4.1 Technologie d’analyse de procédé
Dans un guide pour l’industrie, la FDA encourage l’implantation de technologies d’analyse de procédé (« Process Analytical Technologies (PAT) ») par les entreprises pour mesurer en temps réel les différentes variables de procédés (FDA, 2004). Le but des TAP est d’obtenir une meilleure compréhension des procédés ainsi qu’un meilleur contrôle de ceux-ci afin de maintenir un certain niveau de qualité. Les TAP se séparent en quatre grandes catégories, soit les techniques pour la planification d’expérience, l’acquisition de données et l’analyse, les analyseurs de procédés, les instruments pour le contrôle des procédés et les instruments
servant à l’amélioration en continu et à la gestion des connaissances. La première catégorie de TAP sert lors du développement de procédés et permet d’évaluer quelles variables pourraient avoir un impact critique sur le procédé. Les analyseurs de procédé permettent la collecte de données au sein de celui-ci. Les analyseurs comprennent les sondes de pH, de température, de pression, l’utilisation de l’infrarouge, la spectroscopie Raman, etc. Les instruments de contrôle permettent de garder le procédé à un niveau préétabli.
Les TAP sont souvent accompagnés de méthodes statistiques multivariées qui permettent, à partir des données récoltées tout au long du procédé, de prédire la qualité du produit (Bondi & Drennen, 2011). Les méthodes multivariées les plus utilisées sont l’analyse en composantes principales (ACP) (« Principal component analysis (PCA) ») et la régression par moindres carrés partiels (« Partial least square (PLS) »). L’avantage de l’utilisation des TAP est qu’ils permettent de prédire la qualité du produit final en observant les différentes variations dans le procédé. Le fait d’observer les variations tout au long du procédé de fabrication permet d’apporter des modifications à celui-ci afin d’obtenir un produit final avec le bon niveau de qualité. Cela permet donc de réduire le nombre de lots rejetés en raison du non-respect des critères de qualité (Bondi & Drennen, 2011).
Les TAP ont pour objectif d’augmenter notamment la productivité ainsi que de réduire la consommation de certaines ressources coûteuses comme les solvants (Kandelbauer, Rahe, & Kessler, 2013). Ces objectifs peuvent être atteints en remplaçant une méthode hors ligne (« off-line ») par une méthode « at-line », « in-line » ou « on-in-line ». Une méthode analytique hors ligne demande une prise d’échantillon suivi d’une analyse en laboratoire de l’échantillon, ce qui cause des délais entre la prise d’échantillon et l’obtention du résultat de l’analyse (Kandelbauer et al., 2013). La méthode traditionnelle utilisée pour effectuer la vérification de nettoyage correspond à ce type de méthode analytique. Une méthode « at-line » se caractérise par une prise d’échantillon suivi de son analyse dans la même pièce que le procédé à analyser. Une méthode « on-line » demande que l’échantillon soit séparé de façon temporaire du procédé puis analysé avant d’être retourné au procédé. Finalement, une méthode analytique « in-line » correspond à un instrument qui est directement en contact avec le produit (Kandelbauer et al., 2013).
Les TAP offrent donc une alternative intéressante à la méthode traditionnelle de collecte d’échantillon par écouvillon suivi d’une analyse par HPLC pour la vérification de nettoyage. Dans les sections suivantes, différentes méthodes analytiques développées pour la vérification de nettoyage en utilisant des TAP sont abordées. Il existe deux types de méthodes analytiques, soit les méthodes spécifiques et les méthodes non spécifiques. Une méthode analytique spécifique permet de détecter un composé unique lorsque celui-ci est en présence d’autres substances (Kumar et al., 2012). Les méthodes non spécifiques, quant à elles, correspondent à la détection des substances qui occasionnent une certaine réponse, par exemple, les
substances contenant du carbone (Kumar et al., 2012). Les méthodes analytiques peuvent également se diviser en deux autres catégories qui correspondent aux méthodes avec ou sans prise d’échantillon.
1.4.2 Méthodes non spécifiques
Une des méthodes non spécifiques les plus utilisées est le carbone organique total (COT). Cette méthode consiste à déterminer la quantité de dioxyde de carbone suite à l’oxydation des carbones contenus dans les résidus sur la surface et permet d’obtenir un résultat plus rapidement qu’avec une analyse par HPLC (Kaiser & Minowitz, 2014). Cette méthode permet d’obtenir un résultat qui correspond au pire scénario puisque la méthode est non spécifique, donc toutes les substances contenant du carbone sont détectées (Clark, 2014). Le COT a été utilisé pour quantifier un résidu suite à sa collecte par écouvillon (Sajid, Arayne, & Sultana, 2010) ainsi que pour effectuer le contrôle des systèmes de nettoyage automatique en analysant le solvant après chaque nettoyage avec un détecteur à infrarouge moyen (Siegmann-Hegerfeld et al., 2013). Cette méthode a pour avantage d’être sensible, d’offrir un bon taux de récupération et est affectée par les interférences de façon minime (Sajid et al., 2010). Par contre, cette méthode nécessite des composés qui sont solubles dans l’eau et ne peut être utilisée avec des solvants organiques puisque ceux-ci contiennent des carbones (Kaiser & Minowitz, 2014). Comme l’ibuprofène est peu soluble dans l’eau (Science Lab, 2013), cette technique ne pourra pas être utilisée pour effectuer la vérification de nettoyage de cette substance. D’autres exemples de méthodes non spécifiques incluent l’utilisation du pH, de la conductivité et du titrage (Kaiser & Minowitz, 2014).
1.4.3 Méthodes spécifiques
1.4.3.1 Méthode avec prise d’échantillon par écouvillon
Il existe des méthodes plus rapides que la méthode traditionnelle, mais elles demandent tout de même une prise d’échantillonnage par écouvillon. Ces méthodes s’apparentent à des méthodes de type « at-line » où une prise d’échantillon est réalisée et celui-ci est analysé à côté de l’équipement de production ou à l’aide d’une méthode de laboratoire plus rapide qu’une HPLC.
1.4.3.1.1
Spectrométrie de mobilité ionique
La spectrométrie de mobilité ionique (SMI) consiste à la séparation en phase gazeuse de la substance ionisée sous un champ électrique en fonction de sa mobilité (Armenta, Alcala, Blanco, & Gonzalez, 2013). L’utilisation de la SMI demande que l’échantillon soit récolté avec un écouvillon avant d’être analysé (Excellims Corporation, 2010). Les avantages de cette technique sont qu’elle est 50 fois plus rapide que la chromatographie liquide (IVT, 2013), qu’elle ne demande pas l’utilisation additionnelle de solvant et que différentes techniques d’ionisation sont disponibles pour pouvoir l’utiliser avec des composés non volatiles,
thermiquement instable, etc. (Excellims Corporation, 2010). L’inconvénient de cette technique est qu’une prise d’échantillon est nécessaire avant d’effectuer l’analyse, donc le problème relié à la collecte d’échantillon n’est pas réglé.
1.4.3.1.2
Spectroscopie Raman
Une méthode utilisant la spectroscopie Raman de type SERS (« surface enhanced Raman spectroscopy ») a été développée pour la quantification de la nélarabine (Corrigan et al., 2010). Cette technique consiste à prendre un échantillon avec un écouvillon, à extraire le résidu et à le faire sécher sur la surface active du spectromètre avant de prendre son spectre Raman. Les résultats ont démontré que la méthode peut être utilisée de façon qualitative en combinaison avec la méthode traditionnelle puisque l’erreur entre les réplicas d’une même concentration est grande. Cette erreur est occasionnée par la façon dont le résidu sèche sur la surface du spectromètre. Cette technique permet seulement d’obtenir une première estimation de la quantité de résidu sur la surface. Elle ne peut donc pas être utilisée de façon autonome ce qui limite son application pour la vérification de nettoyage.
1.4.3.1.3
Méthode capillaire
La méthode capillaire la plus utilisée correspond à l’électrophorèse par zone capillaire (Kaiser & Minowitz, 2014). L’instrumentation comprend une source de haute tension, un capillaire et un détecteur. La méthode consiste à l’application d’un potentiel électrique entre deux solutions pour que la substance à analyser passe d’une solution à l’autre en passant par le détecteur. L’inconvénient de cette technique est que la limite de détection est généralement plus grande que pour la méthode la plus utilisée, soit une HPLC. Par contre, cette technique facilite la détection des substances ayant une faible absorption UV. Comme la limite de détection est plus grande que la méthode traditionnelle et que cette technique demande une prise d’échantillon, la méthode capillaire ne permet pas de résoudre les problèmes reliés à la méthode conventionnelle.
1.4.3.1.4
Fluorescence
Peles et al (Peles et al., 2013) ont développés une méthode « at-line » pour effectuer la vérification des procédures de nettoyage d’un IPA en utilisant un détecteur qui emploie la fluorescence induite par laser (LIF). Le détecteur possède 4 diodes électroluminescentes (DEL) et l’excitation des DEL est contrôlable. La méthode consiste à prélever un échantillon par écouvillon, à extraire le résidu et le placer dans une cuvette pour l’analyser. L’analyse se fait dans des solutions qui émettent un faible signal en fluorescence (« background » en anglais ou bruit de fond en français). Le bruit de fond correspond au signal émis par la solution lorsqu’aucun résidu n’est présent. Le fait que le détecteur possède une source d’excitation contrôlable permet la détection de faibles concentrations de résidu puisque l’intensité d’émission est proportionnelle à la
source d’excitation. L’inconvénient de cette technique est qu’elle demande la collecte du résidu à l’aide d’un écouvillon, donc ne permet pas d’effectuer l’analyse du résidu directement sur la surface. Malgré cet inconvénient, cette méthode permet d’accélérer l’analyse puisque la détection avec le LIF prend moins d’une minute. Le filtre d’émission choisi permet la détection dans la région d’intérêt de la molécule et permet le rejet du rayonnement parasitaire, ce qui fait en sorte que la méthode est spécifique. L’étude conclut que la méthode est spécifique puisque ni l’agent de nettoyage, ni les excipients utilisés dans la formulation n’avait d’impact sur le signal. Cela s’explique par le fait que la plupart des excipients ne fluorescent pas contrairement aux IPA. De plus, si les excipients fluorescent, il y a peu de chances que cela se produise aux mêmes conditions que l’IPA à quantifier (mêmes combinaisons de longueurs d’onde d’excitation et d’émission). En conclusion, cette méthode est plus rapide qu’une HPLC, mais ne permet pas d’éviter l’utilisation de l’écouvillon pour la collecte de l’échantillon. La méthode montre tout de même le potentiel de la fluorescence pour la quantification des ingrédients actifs.
1.4.3.1.5
Conclusion sur les méthodes demandant une prise d’échantillon
Les méthodes avec prise d’échantillons présentées ont l’avantage d’être plus rapide que la méthode traditionnelle, malgré le fait qu’elles demandent tout de même une prise d’échantillon par écouvillon. Comme le but est de remplacer la méthode traditionnelle et d’éviter l’utilisation de solvant ainsi que l’utilisation d’écouvillon, ces méthodes ne permettent pas d’atteindre cet objectif. Il faut donc se tourner vers des méthodes qui ne demandent pas de prise d’échantillon.
1.4.3.2 Méthode analysant le solvant du rinçage
La vérification de nettoyage peut s’effectuer de manière « on-line » ou « in-line » en analysant le solvant du dernier rinçage. Ce type de technique ne demande pas de collecte d’échantillon sur la surface. L’inconvénient de ce type de méthode est que le résidu doit se retrouver dans le solvant et ne doit pas rester sur la surface de l’équipement (Jones, 2013). La spectrophotométrie UV-visible a été utilisée pour réaliser ce type de vérification de nettoyage et est généralement utilisée pour déterminer le moment où le nettoyage peut être arrêté (Jones, 2013). Le COT est également utilisé pour effectuer ce type de contrôle.
Comme la plupart des vérifications de nettoyage utilisent l’échantillonnage par écouvillon suivi d’une HPLC, ce type de méthode ne semble pas être la bonne alternative à la méthode traditionnelle pour l’application à l’étude. Il reste seulement à examiner ce qui a été fait jusqu’à présent pour réaliser la vérification de nettoyage sans prise d’échantillon.
1.4.3.3 Méthodes sans prise d’échantillon
Les méthodes directes correspondent à celles qui ne demandent pas de prise d’échantillon, c’est-à-dire que la collecte des données s’effectue directement sur la surface de l’équipement de production. L’utilisation d’une méthode directe permet d’éviter l’utilisation d’un écouvillon. Ces méthodes comprennent notamment l’utilisation de technologies infrarouge, de la spectroscopie de masse et de la fluorescence.
1.4.3.3.1
Technologie infrarouge
Les méthodes infrarouges (IR) peuvent se diviser en deux grandes catégories, soit les méthodes qui utilisent le proche infrarouge (PIR) ou les méthodes qui utilisent l’infrarouge moyen. L’intervalle de longueurs d’onde employé pour le PIR est de 800 à 2500 nm (Tasumi & Sakamoto, 2015) et celui de l’infrarouge moyen va de 2500 à 25 000 nm (Šašić & Ozaki, 2010).
1.4.3.3.1.1 Proche infrarouge
L’utilisation l’imagerie chimique par proche infrarouge (IC-PIR) pour effectuer les vérifications de nettoyage de façon directe a été proposée par Cullen et al (Cullen, Jones, Alvarez-Jubte, Mishra, & Sullivan, 2013). L’utilisation de cette technique permet d’avoir de l’information à la fois spectrale et spatiale ce qui permet de déterminer la concentration de la substance et sa distribution sur la surface. Avec cette technique, le nombre de spectres obtenus correspond au nombre de pixels du système et la limite de détection (LD) dépend de la résolution spatiale, de la dimension des particules et de la distribution de l’échantillon sur la surface. Cette méthode demande une classification pour distinguer les pixels du résidu par rapport à ceux de la surface sur laquelle le résidu est déposé (Cullen, Jones, Alvarez-Jubte, Mishra, & Sullivan, 2013). L’absorption pour l’acier inoxydable est plus grande que celui des IPA, donc le nombre de pixel est plus grand pour la surface que pour les IPA (CORDIS, 2015). Cette technique demande donc l’application de la normalisation SNV («standard normal variates») ou le prétraitement par MSC («multiplicative scatter correction») et d’un lissage Savitzky-Golay comme prétraitement des données (CORDIS, 2015) ainsi que l’utilisation d’une fonction de classification. Une relation linéaire a été observée entre le nombre de pixels et la concentration du résidu (Cullen, Jones, Alvarez-Jubte, Mishra, & Sullivan, 2013).
Un prototype de laboratoire a été utilisé en premier lieu pour tester la méthode. Le prototype de laboratoire, qui n’est pas portable, possède des LD allant de 36,4 à 96,84 µg/cm2 pour la caféine sur différentes surfaces. Cette limite élevée est dû au fait qu’il y a une grande variabilité des réplicas d’une même concentration causée par la façon dont la substance à l’étude sèche sur la surface. Selon le rapport, il serait possible de réduire cette limite à 2 µg/cm2 en réduisant la variabilité entre les réplicas. Afin de passer au test en production, un nouveau prototype a été fabriqué, celui-ci est maintenant portable et permet d’analyser une section de 44 mm2. Des problèmes ont été rencontrés lors de l’évaluation de la technique in situ dû au fait que la qualité
des images obtenues n’était pas satisfaisante et que des anomalies dans les spectres étaient présentes. La méthode permet, en ce moment, de détecter des concentrations allant jusqu’à 25 µg/cm2 (CORDIS, 2015). En résumé, cette méthode ne permet pas d’atteindre les limites acceptables des résidus qui sont de l’ordre de quelques microgrammes par centimètre carré avec sa configuration actuelle. De plus, l’application de cette méthode demande un prétraitement des spectres avant de pouvoir obtenir le signal du résidu.
1.4.3.3.1.2 Infrarouge moyen
Block engineering (Block Engineering, 2012) propose un spectromètre portable qui utilise l’infrarouge moyen pour effectuer des vérifications de nettoyage sur des surfaces métalliques à l’aide d’un laser à cascade quantique. L’appareil repose sur le principe que la lumière est reflétée ou absorbée par la substance, ce qui est une caractéristique à la substance étudiée. Grâce à des librairies internes, il est possible de détecter ou d’analyser la substance à une distance supérieure à 15 cm. Avec cette technique, il est également possible de voir comment le résidu est distribué sur la surface. Par contre, cette méthode est utilisée pour la détection et l’indentification des contaminants et non pour la quantification.
1.4.3.3.1.3 Spectroscopie d’absorption infrarouge en réflexion
Une technique de vérification de nettoyage in situ par infrarouge moyen utilisant la transformation de Fourrier (TF-IR) a été étudiée sur des surfaces métalliques (Mehta et al., 2003). La technique utilisée est la spectroscopie d’absorption infrarouge en réflexion (IRRAS). L’appareil à l’étude consiste à une sonde IR moyen avec un angle d’incidence connecté par fibre optique (2 m de long) à un spectromètre non portable («benchtop») de type TF-IR qui détecte sur une section de 4,5 cm2. Il a été démontré qu’il existe une relation linéaire entre l’aire des pics des spectres et la concentration de la substance sur la surface, mais l’étalonnage obtenu n’est pas assez robuste. Les résultats montrent le potentiel que cette méthode pourrait avoir comme remplacement de la méthode traditionnelle. Par contre, cette méthode ne permet pas d’avoir un détecteur portable pour effectuer les vérifications de nettoyage.
L’IRRAS a aussi été étudié pour la quantification de l’acétaminophène et l’ibuprofène sur le verre et sur des surfaces métalliques en utilisant la régression par moindres carrés partiels (PLS) pour construire la courbe d’étalonnage (Hamilton et al., 2005). Cette méthode utilise le même instrument décrit plus haut pour la mesure in situ. La chimiométrie permet de caractériser les comportements non linéaires qui sont observés lorsque l’IRRAS est utilisé, de tenir compte des interférences des molécules atmosphériques ainsi que la possibilité de quantifier plusieurs composés simultanément. Les interférences des molécules atmosphériques, principalement l’eau, sont occasionnées par le fait que le rayon IR passe à travers l’air avant de rejoindre la surface et lorsqu’ils sont réfléchis par la surface. L’utilisation de corrections ne permet pas d’éliminer complètement l’effet des interférences atmosphériques. L’étude sur l’impact des autres substances en