Caractérisation d’un variant d’épissage alternatif du
gène FANCE et son impact sur la voie de réparation de
l’ADN FANC-BRCA
Mémoire
Frédérick Bouffard
Maîtrise en médecine moléculaire
Maitre ès sciences (M. Sc.)
Québec, Canada
Résumé
Divers évènements d’épissage alternatif ont été identifiés pour certains gènes de la famille FANC, notamment pour FANCE. La voie de réparation de l’ADN FANC-BRCA nécessite l’intégrité de l’ensemble des protéines Fanconi afin d’assurer l’efficacité de la réparation des pontages inter-brins. Nous avons alors étudié l’impact de l’expression du variant d’épissage alternatif FANCE∆4 au niveau de la voie FANC-BRCA. Son expression exclusive dans des cellules déficientes en FANCE (EUFA130) n’est pas suffisante pour restaurer l’activation de la voie de réparation. À la suite d’un traitement avec un agent pontant (mitomycine C), les cellules EUFA130 complémentées avec FANCE∆4 demeurent bloquées en phase G2/M du cycle cellulaire, la viabilité n’est pas augmentée et la monoubiquitination de FANCD2 et FANCI est absente, contrairement aux cellules EUFA130 complémentées avec FANCE. Ce projet a particulièrement permis de déterminer que FANCE∆4 n’est pas en mesure de se substituer lors de la perte de FANCE.
Abstract
Several alternative splicing events were identified for some genes of the FANC family, such as FANCE. The integrity of the proteins of the FANC-BRCA DNA repair pathway is necessary to maintain efficient ICL repair. We studied the impact of the expression of an alternative splicing isoform, FANCE∆4. Its exclusive expression in FANCE-deficient cells (EUFA130) is not sufficient to restore the activation of the pathway. Following treatment with crosslink agent (mitomycin C), EUFA130 cells complemented with FANCE∆4 are blocked in G2/M phase of the cell cycle, the viability is not increased and the monoubiquitination of FANCD2 and FANCI is absent, in contrast to EUFA130 cells complemented with FANCE. This project highlights FANCE∆4 that cannot replace FANCE in regard to DNA repair.
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vii
Liste des tableaux ... ix
Liste des figures ... xi
Liste des abréviations ... xiii
Remerciements ... xvii Avant-propos ... xix Contributions ... xix Introduction ... 1 1. Anémie de Fanconi ... 1 1.1 Caractéristiques de la maladie ... 1
1.2 Gènes Fanconi et cancers ... 5
1.2.1 Gènes Fanconi et le cancer du sein ... 7
1.3 Pontages inter-brins et agents pontants ... 8
1.3.1 Pontages inter-brins ... 8
1.3.2 Agents pontants et leurs utilisations ... 10
1.4 Voie de réparation FANC-BRCA... 11
1.4.1 Les complexes et sous-complexes ... 13
1.4.2 Le mécanisme de la voie de réparation de l’ADN FANC-BRCA ... 15
1.5 Autres rôles des protéines Fanconi... 21
1.6 FANCE ... 23
1.6.1 Structures et fonctions ... 24
1.6.2 Études d’association et polymorphismes ... 28
2. Épissage alternatif ... 29
2.1 Les principes de l’épissage alternatif ... 29
2.2 Épissage alternatif et cancers ... 33
2.3 Épissage alternatif et gènes Fanconi ... 34
3. Problématique ... 36 3.1 Objectifs de l’étude ... 37 Chapitre 1 ... 39 Graphical abstract ... 40 Résumé ... 41 Abstract ... 42 Highlights ... 43 Abbreviations ... 44
Introduction ... 45
Results ... 47
Discussion ... 53
Materials and methods ... 57
Conflict of interest statement ... 63
Acknowledgements ... 64 References ... 65 Legends to Figures ... 74 Discussion ... 93 Perspectives ... 99 Bibliographie ... 101
Liste des tableaux
IntroductionTableau 1 : Caractéristiques des gènes Fanconi et des protéines produites. ... 12 Tableau 2 : Génotypes de patients atteints de l’anémie de Fanconi pour le gène FANCE ... 29
Chapitre 1
Table 1. FANCE gene sequence variations potentially involved in FANCE∆4 expression and analyzed by minigene assay ... 77 Table 2. In silico analysis of genomic variants potentially involved in the expression of FANCE∆4 alternative isoform ... 78
Supplemental Table 1. Oligonucleotides used for FANCE minigene mRNA quantification ... 91 Supplemental Table 2. Oligonucleotides used for FANCE and FANCE∆4 semi-quantitative PCR ... 91 Supplemental Table 3. Oligonucleotides used for FANCE minigene construct ... 91
Liste des figures
IntroductionFigure 1 : Distribution de la fréquence des patients atteints de l’anémie de Fanconi en
fonction du gène responsable. ... 2
Figure 2 : Implication des protéines Fanconi dans la prévention d’aberrations chromosomiques ... 5
Figure 3 : Schéma simplifié de la voie de réparation de l’ADN FANC-BRCA. ... 13
Figure 4 : Activation de la voie Fanconi par la phosphorylation ATR-dépendante et l’activité de monoubiquitination du complexe I ... 17
Figure 5 : Succession des étapes de la réparation des pontages inter-brins induits à l’ADN. ... 20
Figure 6 : Localisation chromosomique et structure des exons du gène FANCE. ... 24
Figure 7 : Conservation de la séquence de localisation nucléaire du gène FANCE à travers l’évolution. ... 26
Figure 8 : Structure des domaines HEAT, ARM et FANC. ... 27
Figure 9 : Différentes formes d’évènements d’épissage alternatif. ... 30
Figure 10 : Reconnaissance de la jonction intron-exon de la machinerie d’épissage. ... 31
Chapitre 1 Figure 1 : Gene splicing and domains of FANCE and FANCE∆4 proteins ... 80
Figure 2 : Amino acids conservation of FANCE exon 4 in other species ... 81
Figure 3 : FANCE∆4 expression in breast cell lines ... 82
Figure 4 : Polysome analysis of FANCE∆4 mRNA in T-47D cells ... 83
Figure 5 : Tertiary structures of the FANCE∆4 and FANCE proteins ... 84
Figure 7 : FANCD2 and FANCI monoubiquitination allowed by FANCE and FANCE∆4
proteins ... 86
Figure 8 : Cellular localization of FANCE and FANCE∆4 proteins following MMC treatment ... 87
Figure 9 : Co-immunoprecipitation experiments ... 88
Figure 10 : FANCD2 protein regulation ... 89
Liste des abréviations
ADN : acide désoxyribonucléique
ADNc : acide désoxyribonucléique complémentaire ADNg : acide désoxyribonucléique génomique
AML : Acute myeloid leukemia (leucémie myéloïde aiguë) ARN : acide ribonucléique
ARNm : acide ribonucléique messager
ATR : Ataxia telangiectasia and Rad3 related BLM : Bloom syndrome protein
BRCA1/2 : Breast cancer, early onset 1/2
BRIP1/BACH1 : BRCA1-interacting protein 1/BRCA1 associated C-terminal helicase
CHK1 : Checkpoint kinase 1 DEB : Diepoxybutane
EDTA : Éthylène diamine tétraacétique
ER : Récepteur de l’estrogène («Estrogen receptor»)
ERCC4/XPF : Excision repair cross-complementation group 4/Xeroderma pigmentosum group F
FAAP24/100 : Fanconi anemia associated protein 24/100 FACS : Fluorescence activated cell sorting
FAN1 : Fanconi-associated nuclease 1
FANC* : Fanconi anemia complementation group * FANCE : Fanconi anemia complementation group E FANCE∆4 : FANCE présentant une délétion de l’exon 4 HER2 : Human epidermal growth factor receptor 2 hnRNP : Heterogeneous nuclear ribonucleoproteins MHF1/2 : FANCM-associated histone-fold protein 1/2 MMC : Mitomycine C
NER : Réparation par excision de nucléotides («Nucleotide excision repair») NLS : Séquence de localisation nucléaire («Nuclear localisation signal») NMD : Nonsense mediated decay
ORL : Oto-rhino-laryngologie
PALB2 : Partner and localizer of BRCA2 PCNA : proliferating cell nuclear antigen
PCR : Réaction de polymérisation en chaîne («Polymerase chain reaction») PR : Récepteur de la progestérone
RAD51C : Radiation sensitivity abnormal 51 C siARN : Petit acide ribonucléique interférant SIM : SUMO-like domain-interacting motif SLD1/2 : SUMO-like domain
SLX4 : Structure-specific endonuclease subunit homolog SR : serine-rich
SUMO : Small ubiquitin-like modifier TLS : Translesion synthesis
UbeT2 : Ubiquitin-conjugating enzyme E2 T USP1 : Ubiquitin-specific protease 1
VDR : Récepteur vitamine D («Vitamin D receptor»)
Remerciements
D’abord, j’aimerais remercier Dre Francine Durocher de m’avoir accueilli au sein de son équipe de recherche à l’été 2012 afin d’effectuer un stage alors que j’étais au baccalauréat mais également de m’avoir par la suite offert l’opportunité de poursuivre mon cheminement académique à la maîtrise. Merci de m’avoir donné la chance de participer à différents congrès d’envergure. Ton support et ta confiance m’ont permis de me surpasser et je considère que j’en sors grandi en tant que personne.
J’ai eu la chance de côtoyer des personnes extrêmement importantes pour la réussite de mon projet de maîtrise. J’aimerais remercier Dr Yvan Labrie, professionnel de recherche au sein de l’équipe de Dre Durocher. J’ai grandement apprécié tes bons conseils et ton expertise dont j’ai pu bénéficier tout au long de mon cheminement. J’aimerais également prendre le temps de remercier Dre Karine Plourde, sans qui le travail en laboratoire n’aurait pas été le même. J’ai eu le privilège de pouvoir compter sur toi à tout moment et j’en serai toujours reconnaissant.
Merci à Simon Bélanger de m’avoir pris sous son aile lors de mon arrivée au laboratoire. J’ai grandement appris à tes cotés et je t’en remercie. Merci également à Marie-Christine Pouliot de m’avoir supporté dans ces moments d’écriture. J’ai grandement apprécié te côtoyer ces derniers mois. Je tiens aussi à remercier Geneviève Ouellette, Christopher St-Laurent Pedneault, Amélie Quoibion et Charles Étienne Bénard pour votre apport à mon projet de recherche. Un merci spécial à l’équipe de la plateforme de séquençage (Sylvie, Patrick, Annie-Claude, Anne et Nicolas) pour tous les bons moments passés.
Enfin, un énorme merci à ma famille dont j’ai toujours senti le support malgré la distance qui nous sépare.
Avant-propos
Ce mémoire recueille l’ensemble des résultats portant sur la caractérisation d’un variant d’épissage alternatif nouvellement identifié. Ce mémoire de maîtrise est d’ailleurs présenté avec l’insertion d’un article scientifique.
L’introduction de ce mémoire constitue une revue de la littérature des différentes notions nécessaires à la compréhension du projet de recherche. La problématique de recherche y est également présentée en plus des objectifs. Le chapitre 1 est constitué de l’insertion de l’article scientifique qui a notamment été soumis à la revue Journal of Molecular Biology. Ce mémoire se termine avec une discussion des résultats obtenus ainsi que des perspectives quant au projet de recherche.
Contributions
Ce projet de recherche a été réalisé sous la direction de Dre Francine Durocher au Centre du Recherche du CHU de Québec (CHUL). J’ai rédigé l’article scientifique avec l’aide de Dr Yvan Labrie. À l’exception de la culture cellulaire qui a été assurée par Dre Karine Plourde, stagiaire postdoctorale au sein de l’équipe de Dre Durocher, j’ai réalisé l’ensemble des expériences en laboratoire présentées dans l’article inséré. Le séquençage a été réalisé par l’équipe de la plateforme de séquençage et génotypage du CRCHU de Québec. Les observations par immunofluorescence ont pu être réalisées grâce à l’utilisation du microscope à fluorescence de l’équipe de Dre Jasna Kriz. Certains PCRs en temps réel ont été réalisés par Natalie Paquet de la plateforme d’expression génique du CRCHU de Québec. Les photographies des immunobuvardages ont été rendues possible grâce à l’utilisation de l’appareil de l’équipe de Dre Thérèse Di Paolo sous la supervision de Dr Marc Morissette. Enfin, j’aimerais souligner le support technique que m’a offert Dre Karine Plourde tout au long de la réalisation de mon projet de maîtrise.
Introduction
1. Anémie de Fanconi
1.1 Caractéristiques de la maladie
En 1927, Guido Fanconi a été le premier clinicien à décrire l’anémie de Fanconi. À l’époque, ce pédiatre suisse avait décelé la maladie chez trois frères présentant des anormalités physiques ainsi qu’une pancytopénie à la naissance («G. Fanconi, Familiäre infantile perniziosartige anänie (pernizioses blutbild und konstitution), Jahrb, Kinderh 117 (1927) 257- 280»).
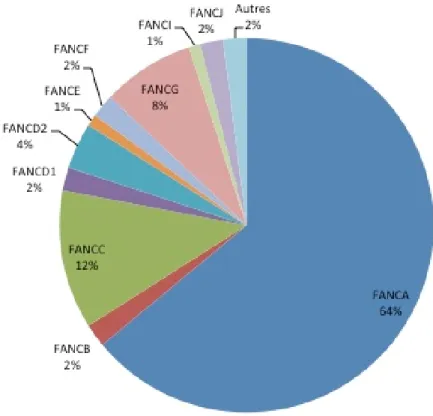
L’anémie de Fanconi est une maladie autosomique récessive, à l’exception du gène FANCB lié au chromosome sexuel X. Une mutation homozygote inactivatrice présente dans l’un des 17 gènes FANC (A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P, Q et S) entraine le développement de la maladie. En 2010, aux États-Unis, on estimait entre 550 à 975 individus atteints de la maladie. La fréquence de porteurs de mutations hétérozygotes pour les gènes Fanconi est évaluée à 1 allèle sur 300 [1,2]. Le gène FANCA est le plus souvent responsable du développement de la maladie, alors que plus de 64% des patients atteints de cette maladie présentent une mutation homozygote pour ce gène [3]. La figure 1 illustre d’ailleurs la proportion de patients appartenant à chacun des groupes de complémentation de l’anémie de Fanconi. Un dix-huitième groupe de complémentation pourrait également être créé. En 2012, des troubles s’apparentant à ceux développés par les patients atteints de l’anémie de Fanconi ont été observés chez un jeune patient, en raison d’une mutation pour le gène XRCC2, un gène encodant pour une protéine impliquée dans la réparation des dommages à l’ADN par recombinaison homologue [4]. Des analyses plus approfondies devront être réalisées afin de désigner XRCC2 comme étant un gène Fanconi et d’y associer un groupe de complémentation.
Figure 1 : Distribution de la fréquence des patients atteints de l’anémie de Fanconi en fonction du gène responsable.
La portion «Autres» rassemble les gènes FANCL (0,4%), FANCM (0,1%), FANCN (0,7%), FANCO (0,1%), FANCP (0,5%), FANCQ (0,1%) et FANCS (0,1%). Valeurs recueillies d’après Wang, A.T., and Smogorzewska, A. (2015). SnapShot: Fanconi Anemia and Associated Proteins. Cell 160, 354–354.e1.
Les manifestations cliniques associées à cette maladie orpheline sont principalement des anomalies congénitales, une insuffisance médullaire, des malformations squelettiques, une augmentation de la prédisposition à développer un cancer ainsi que des troubles hématologiques (pancytopénies, anémies, leucémies et thrombopénies). La leucémie myéloïde aiguë (AML) est particulièrement fréquente chez ces patients alors que les cancers ORL («head and neck squamous cell carcinomas») sont observés chez 3% des patients Fanconi [5,6]. Chez certains patients, la maladie peut également se manifester par une petite stature, une hypo/hyperpigmentation de la peau (taches café au lait), des
d’un patient à l’autre, complexifiant ainsi le diagnostic de la maladie à première vue. Ces mutations créent un désordre important affectant grandement la stabilité génétique. Deux cas de jumeaux portant la même mutation homozygote disposaient de phénotypes complètement différents. Dans un cas, un enfant ne présentait aucune malformation physique visible alors que le deuxième enfant possédait un pouce dupliqué. D’ailleurs, l’un des deux jumeaux souffrait d’atrésie duodénale, un trouble affectant le tube digestif. Dans un second cas de jumeaux, un enfant présentait une absence d’un radius, de la clavicule droite et des deux pouces, alors que le second présentait un pouce droit dupliqué, une hypoplasie du pouce gauche et une absence de la clavicule gauche [7]. Ces phénomènes suggèrent qu’outre le génotype, plusieurs facteurs externes qui surviennent au cours du développement de l’enfant peuvent influencer le phénotype. Le phénomène de mosaïcisme est fréquemment observé chez les patients Fanconi, ce qui peut faire varier le phénotype d’un patient. Par contre, il n’est pas exclu que l’implication de gènes modificateurs, de facteurs environnementaux ou bien des mécanismes épigénétiques pourraient avoir un impact en ce qui a trait à l’hétérogénéité phénotypique observée parmi ces patients [7,8]. Par exemple, chez certains patients appartenant au groupe de complémentation C (FANCC), une progression accélérée de l’insuffisance médullaire a été observée. Ce trait pourrait potentiellement être expliqué par l’implication de gènes modificateurs. En fait, ces patients présentaient également des mutations pour le gène GSTM1 [9]. Par ailleurs, les patients appartenant au groupe de complémentation D2 présentent des troubles hématologiques plus tôt et une progression plus rapide de la maladie comparativement à tous les autres patients non-D2. Ces patients souffrent d’une forme plus sévère d’anémie de Fanconi [10].
L’espérance de vie des patients a grandement augmenté dans les années 90, établie à 19 ans de 1980 à 1989 et augmentant à plus de 30 ans en 2000 [11]. L’avancement des connaissances concernant les différents traitements pouvant être offerts aux patients a fortement contribué à l’augmentation de l’espérance de vie. Par contre, il n’existe aucun traitement miracle afin de guérir ces patients. Généralement, il s’agit plutôt des problèmes qui découlent de l’anémie de Fanconi qui sont traités, comme les problèmes hématopoïétiques ou ceux au niveau de la moelle osseuse par exemple. Pour certains patients, la prise d’androgène ou de facteurs de croissance hématopoïétiques peut être
envisageable, mais seulement une faible quantité de patients reçoivent ce type de traitement [11]. Puisque la plupart des patients présentent des troubles hématopoïétiques importants, la transplantation de cellules souches est employée afin d’améliorer leurs conditions de vie.
Les cellules des patients atteints de l’anémie de Fanconi sont hypersensibles aux agents pontants. À forte concentration, la survie de ces cellules est grandement affectée. Pour poser un diagnostic d’anémie de Fanconi, l’exposition de cellules du patient à un de ces agents, comme le diepoxybutane (DEB) par exemple, demeure une technique encore utilisée aujourd’hui [12,13]. Différentes approches peuvent par la suite être utilisées pour déterminer le groupe de complémentation. La plus courante consiste à infecter des cellules dérivées du patient avec un rétrovirus contenant l’ADN complémentaire (ADNc) d’un groupe de complémentation en particulier. Ensuite, un test de sensibilité, au DEB par exemple, est effectué afin de vérifier si la résistance (phénotype) est rétablie [14]. Par ailleurs, le séquençage des régions codantes peut s’avérer une méthode efficace permettant d’identifier la mutation responsable du développement de la maladie [15].
En plus de leur sensibilité aux agents pontants à fortes concentrations, les cellules des patients Fanconi présentent un blocage en G2/M du cycle cellulaire à concentrations moyennes [16]. En fait, en raison de la réparation des pontages inter-brins inefficace, les cellules ne sont pas en mesure d’outrepasser ce point de contrôle et il y a blocage du cycle cellulaire. Par la technique de FACS («fluorescence-activated cell sorting»), il est possible de trier les cellules selon la phase du cycle cellulaire et ainsi vérifier la présence d’un blocage en G2/M à la suite d’un traitement avec un agent pontant.
Les cellules des patients atteints de l’anémie de Fanconi présentent davantage d’aberrations chromosomiques, tel qu’illustré à la figure 2A. En effet, il est connu que les protéines Fanconi et BLM collaborent durant la mitose afin de prévenir la formation de micro-noyaux et d’assurer la stabilité chromosomique [17]. De plus, les différentes protéines Fanconi ont été observées par microscopie aux centrosomes (en colocalisation avec BRCA1) et aux microtubules. La répression de l’expression des gènes Fanconi par des petits ARN interférents (siARN) a démontré une augmentation significative du phénomène
mène également à des aberrations chromosomiques [18]. Les protéines Fanconi auraient alors une importance dans le processus de division cellulaire selon cette étude.
Figure 2 : Implication des protéines Fanconi dans la prévention d’aberrations chromosomiques
A) Mécanismes potentiels pouvant provoquer le développement de cellules possédant plus d’un noyau pour les cellules Fanconi. B) Localisation des protéines Fanconi aux centrosomes et aux microtubules (mitotic spindle). Images traduites de Nalepa, G., Enzor, R., Sun, Z., Marchal, C., Park, S.-J., Yang, Y., Tedeschi, L., Kelich, S., Hanenberg, H., and Clapp, D.W. (2013). Fanconi anemia signaling network regulates the spindle assembly checkpoint. J. Clin. Invest. 123, 3839–3847.
1.2 Gènes Fanconi et cancers
Des anomalies pour les gènes Fanconi ont été observées dans le contexte de différentes pathologies et cancers. Parmi ces anormalités, on peut penser à la méthylation, au niveau d’expression des gènes ainsi qu’à la présence de variations de séquences (mutations, délétions, insertions). Dans le cadre de certains cancers, certaines régions promotrices de gènes Fanconi sont hyperméthylées, ce qui mène à une réduction importante de l’expression de ces gènes. À l’opposé, l’hypométhylation mènerait à une augmentation
de l’expression de ces gènes. Ainsi, la fonction des gènes Fanconi n’est pas pleinement assurée, ce qui fait en sorte que les cellules de ces patients sont potentiellement plus sensibles aux traitements avec des agents pontants, sachant que les cellules FANC-déficientes sont hypersensibles à ce type d’agents. Tel que mentionné précédemment, la leucémie myéloïde aiguë (AML) est fréquemment développée chez les patients Fanconi. Par contre, certaines aberrations pour quelques gènes Fanconi ont été rapportées pour des patients non-Fanconi atteints de cette leucémie. Entre autre, des réductions de l’expression de FANCA et FANCF ont été observées [19-21]. Quant à FANCF, la répression serait influencée par une hyperméthylation. En effet, la méthylation du gène FANCF semble être un processus rattaché à diverses pathologies, ce qui fait en sorte que sa régulation est importante. En fait, une hyperméthylation du promoteur serait associée à une augmentation de la sensibilité des cellules face à un traitement à la cisplatine chez les patientes atteintes d’un cancer ovarien [22,23]. De plus, la méthylation de FANCF serait potentiellement associée au cancer du sein, au cancer du col de l’utérus, au cancer du poumon, ainsi qu’aux cancers développés au niveau de la tête et du cou (fréquemment développés par les patients Fanconi) [22,24-26]. Quant à FANCC et FANCG, des mutations et des délétions ont été observées dans certains cas de cancers pancréatiques [27,28].
Il a également été démontré que différentes cytokines/interleukines, dont IL-6, IL-8, MMP-2 et MMP-9, s’avèrent surexprimées dans les cellules de patients atteints de l’anémie de Fanconi. D’ailleurs, cette surexpression est sous le contrôle de la voie de signalisation NF-kB/TNF-alpha. TNF-alpha est aussi surexprimé dans les cellules de patients atteints de l’anémie de Fanconi [29,30]. La surexpression de ces cytokines/interleukines aurait un impact potentiel concernant le développement de certains cancers, ce qui est en accord avec ce que l’on peut observer chez les patients Fanconi. Ces cellules présentent des caractéristiques particulières aux cellules cancéreuses, soit une augmentation de la prolifération, de la migration et de l’invasion [31].
1.2.1 Gènes Fanconi et le cancer du sein
Tel que mentionné précédemment, des mutations homozygotes dans un gène Fanconi peuvent entraîner le développement de l’anémie de Fanconi. Par ailleurs, des mutations hétérozygotes peuvent aussi conférer une prédisposition accrue de développer un cancer du sein. Parmi les 17 gènes Fanconi, cinq sont bien connus pour leur association à un risque accru de développer un cancer du sein, soit FANCD1 (connu sous le nom de BRCA2), FANCJ (BACH1/BRIP1), FANCN (PALB2), FANCO (RAD51C, un paralogue de RAD51) et FANCS (BRCA1) [32-36]. À la base, ces gènes étaient déjà connus pour leur implication dans la réparation des dommages à l’ADN par recombinaison homologue et leur lien avec le cancer du sein, alors que leur association à un groupe de complémentation de l’anémie de Fanconi s’est réalisée ultérieurement. Cette découverte a entraîné une vague d’études d’association pour les autres gènes de cette famille afin d’identifier de nouvelles allèles potentiellement associées au risque de développer un cancer du sein.
Plusieurs gènes Fanconi ont ainsi fait l’objet de séquençage chez des patientes atteintes du cancer du sein sans mutation BRCA1/2. En 2003, Seal et al ont procédé au séquençage des gènes FANCA, FANCC, FANCD2, FANCE, FANCF et FANCG dans une cohorte de 88 familles du Royaume-Uni dont aux moins 3 femmes étaient atteintes du cancer du sein sans mutations dans les gènes BRCA1 et BRCA2. Deux mutations (1 pour FANCA et 1 pour FANCE) ont été identifiées à une reprise parmi les 88 cas alors qu’elles étaient absentes chez les contrôles. Aucune corrélation statistiquement significative en regard à une association au cancer du sein n’a pu être établie [37]. En 2005, Lewis et al. ont procédé au séquençage de FANCD2 dans une cohorte de 30 patientes à risque familial, sans mutation BRCA1/2, et aucune mutation n’a été identifiée [38]. Par contre, d’après Barraso et al., une mutation localisée dans la région du promoteur de FANCD2 pourrait avoir un impact et une association avec le développement d’un cancer du sein sporadique dans la population espagnole [39]. Dans la même étude, aucune mutation pour FANCA et FANCL n’a été décelée et dans laquelle 897 patientes atteintes du cancer du sein et 1033 contrôles ont été analysés. Plus récemment, une étude d’association menée au sein d’une cohorte finlandaise a permis d’identifier une mutation non-sens pour le gène FANCM (c.5101C>T) significativement associée au cancer du sein. Il s’agit d’une mutation rare très peu retrouvée
dans d’autres populations. De plus, l’association la plus forte était observée dans le regroupement des patientes triples négatives, soit ER-, PR- et HER2- [40]. Quant à FANCP (SLX4), une étude d’association menée au sein d’une cohorte de patientes juives a démontré qu’une mutation de type «perte de fonction» pouvait contribuer au développement d’un cancer du sein, mais très rarement [41].
Dans l’optique d’identifier de nouveaux gènes conférant une prédisposition accrue de développer un cancer du sein chez les familles canadiennes-françaises sans mutation BRCA1/2, l’équipe du Dre Durocher s’est intéressée au séquençage de plusieurs gènes Fanconi. Parmi les gènes analysés, on retrouve FANCA, FANCC, FANCJ, FANCL et FANCN [42-46]. À l’issue des analyses, plusieurs mutations non répertoriées ont été identifiées, sans toutefois être corrélées avec une prédisposition accrue de développer un cancer du sein chez ces familles à risque élevé.
Outre la présence de variations de séquence, des variations épigénétiques parmi les gènes Fanconi peuvent avoir un impact sur le développement du cancer du sein. Il a notamment été démontré que la région promotrice de RAD51C (FANCO) pouvait être hyperméthylée dans certains cas de cancers du sein sporadiques [47].
1.3 Pontages inter-brins et agents pontants 1.3.1 Pontages inter-brins
Les pontages inter-brins induits à l’ADN représentent un type de dommages parmi les plus toxiques pour la cellule. La création d’un pontage inter-brin n’entraîne aucune modification de la séquence nucléotidique [48]. En fait, ces dommages peuvent provoquer une distorsion de la double hélice d’ADN par la création du lien covalent établi entre deux bases azotées de brins complémentaires. Lorsqu’un lien covalent est établi entre deux bases d’un même brin de l’ADN, comme les dimères de thymine, il s’agit plutôt d’un pontage intra-brin. L’intensité du lien créé par un pontage inter-brin nuit à la séparation des deux brins de l’ADN, ce qui empêche la machinerie de transcription ou de réplication de compléter leurs fonctions [49]. La réparation de ces dommages est primordiale afin
Quelques méthodes existent pour détecter des pontages inter-brins. De façon plus générale, les essais de type COMET permettent de détecter et de mesurer l’efficacité de la réparation de toutes lésions induites à l’ADN [50]. Les dommages causés à l’ADN perturbent l’homéostasie cellulaire et peuvent donc entraîner le développement de différentes maladies et cancers lorsqu’ils ne sont pas réparés adéquatement. On peut notamment penser au cancer du sein dont plusieurs gènes de susceptibilité sont impliqués dans la réparation de l’ADN par recombinaison homologue (BRCA1, BRCA2, etc.) ou bien aux gènes impliqués dans la réparation de l’ADN par excision de nucléotides qui sont associés au cancer de la peau (XPF, XPG, etc.) [51,52].
La réparation des pontages inter-brins est effectuée par différents mécanismes dépendamment de la phase du cycle cellulaire. En phase G1, le mécanisme d’excision de nucléotides (NER) est employé alors qu’en phase de réplication (phase S), la réparation s’effectue via la voie FANC-BRCA, tel que vu précédemment. Le processus du NER vise à créer une coupure de part et d’autre des nucléotides à changer afin de remplacer cette courte séquence. Par la suite, la synthèse du brin s’effectue à partir du brin complémentaire [53]. Certaines endonucléases impliquées dans ce processus procèdent également à la création de l’incision dans le mécanisme de la voie FANC-BRCA, qui a été abordé à la section 1.1. La voie FANC-BRCA et le mécanisme de réparation par excision de nucléotides (NER) sont particulièrement reliés [54]. FANCQ (ERCC4/XPF) est d’ailleurs bien connue pour son implication dans la réparation de l’ADN par le mécanisme NER [55]. Enfin, lorsque les pontages inter-brins doivent être réparés en phase G2, un processus est orchestré par quelques protéines Fanconi, dont FANCM. Par contre, le mécanisme d’activation est différent et la réfection de la lésion est assurée par les polymérases de translésion TLS («translesion synthesis polymerase») ainsi qu’un complexe formé de BLM («Bloom Syndrome Protein») [56,57]. La réparation des pontages inter-brins dans cette phase du cycle cellulaire demeure encore peu caractérisée.
Un pontage inter-brin peut être provoqué par différents facteurs, notamment des agents chimiques endogènes ou exogènes, ou bien par des irradiations. L’exposition au rayonnement ultraviolet peut entraîner des dommages à l’ADN, dont les pontages
inter-brins [58-60]. Les divers agents pontants connus ainsi que les utilisations que peuvent avoir ceux-ci seront notamment abordés dans la section 2.2.
1.3.2 Agents pontants et leurs utilisations
Divers composés endogènes peuvent créer des pontages inter-brins. Les plus connus sont les dérivés d’aldéhydes (acétaldéhydes, formaldéhydes, etc.). En effet, ceux-ci sont formés par différentes réactions chimiques du métabolisme, dont la peroxydation des lipides, l’auto-oxydation des ascorbates et carbohydrates, l’activité catalytique du cytochrome P450, etc. [61]. Il a été démontré à plusieurs reprises que les acétaldéhydes ou bien le formaldéhyde par exemple entraînaient une hypersensibilité et une accumulation de dommages à l’ADN en absence d’une protéine Fanconi [62-64].
Par ailleurs, divers agents exogènes peuvent également créer ce type de dommages, tels la mitomycine C (MMC), les moutardes azotées, les composés nitrosourés, le psoralène, les composés platinium (cisplatine, carboplatine, etc.) ou bien le diepoxybutane (DEB) [12,13,57,65]. Les bases liées à la suite de l’exposition à l’agent peuvent varier dépendamment de la nature de cet agent. Par exemple, la mitomycine C, utilisée dans le cadre de cette étude, crée un lien covalent entre deux guanines (G) de brins complémentaires [66].
Étant donné que les pontages inter-brins sont très toxiques et dommageables pour la cellule, celle-ci entre en apoptose lorsqu’il y a une accumulation de ces dommages. L’induction de ce type de dommages par l’utilisation d’agents pontants de type exogène est alors devenue une technique efficace et couramment utilisée pour traiter plusieurs cancers. La création d’un pontage inter-brin bloque ainsi la réplication de l’ADN et induit donc l’apoptose via p53. Les agents pontants sont couramment utilisés en chimiothérapie en combinaison avec d’autres agents, et ce pour différents types de cancers. Parmi les plus connus, le cisplatine et le carboplatine sont utilisés pour traiter les cancers ovariens alors que la mitomycine C est utilisée pour les cancers de l’œsophage [56].
1.4 Voie de réparation FANC-BRCA
Un disfonctionnement de la voie de réparation de l’ADN FANC-BRCA est observé chez les patients atteints de l’anémie de Fanconi, ce qui entraine une accumulation d’aberrations chromosomiques [67]. Les 17 protéines Fanconi mentionnées précédemment collaborent dans cette voie de réparation de l’ADN. Le mécanisme s’enclenche en réponse aux pontages inter-brins induits à l’ADN en phase S du cycle cellulaire. La formation de divers complexes protéiques est déterminante pour le bon déroulement de la cascade mécanistique permettant une réparation efficace de ce type de dommages. Cette voie de réparation est d’ailleurs conservée du poisson zèbre jusqu’à l’humain, autant pour la structure des protéines que du fonctionnement général [68]. La réparation des pontages inter-brins par la voie FANC-BRCA implique des étapes de la réparation par excision de nucléotides afin de créer l’incision de part et d’autre du pontage, du «translesion synthesis» (TLS) pour combler la lésion sur l’un des brins ainsi que de la recombinaison homologue afin de combler la cassure double brin [65]. Il s’agit de trois mécanismes indépendants qui s’enclenchent de manière successive pour restaurer un pontage inter-brins.
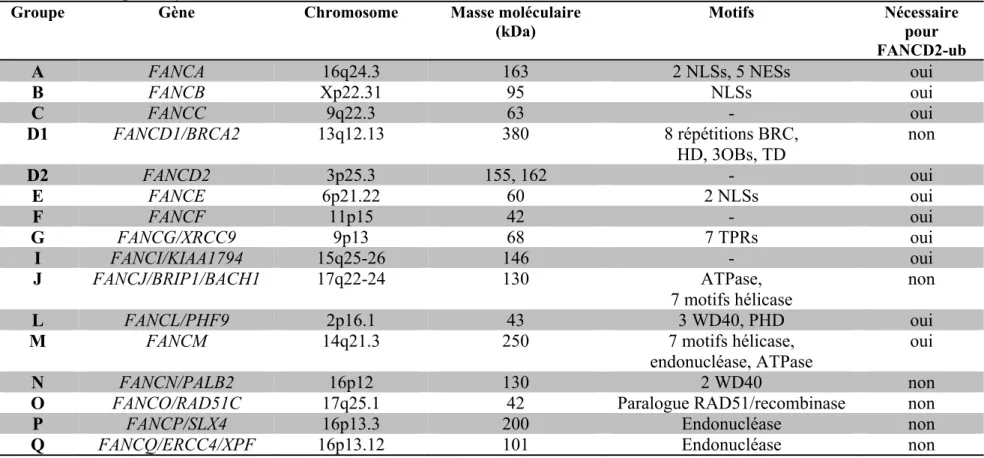
Les protéines Fanconi, toutes impliquées dans la voie de réparation de l’ADN FANC-BRCA, sont présentées dans le Tableau 1, à l’exception de FANCS qui a récemment été identifiée [36]. La section suivante portera sur la composition des complexes formés au cours de l’activation du processus de réparation de l’ADN de la voie FANC-BRCA alors que le mécanisme sera abordé dans la section 1.1.2.
Tableau 1 : Caractéristiques des gènes Fanconi et des protéines produites.
(Tableau traduit et adapté de Longerich, S., Li, J., Xiong, Y., Sung, P., and Kupfer, G.M. (2014). Stress and DNA repair biology of the Fanconi anemia pathway. Blood 124, 2812–2819.)
Groupe Gène Chromosome Masse moléculaire (kDa)
Motifs Nécessaire pour
FANCD2-ub
A FANCA 16q24.3 163 2 NLSs, 5 NESs oui
B FANCB Xp22.31 95 NLSs oui C FANCC 9q22.3 63 - oui D1 FANCD1/BRCA2 13q12.13 380 8 répétitions BRC, HD, 3OBs, TD non D2 FANCD2 3p25.3 155, 162 - oui E FANCE 6p21.22 60 2 NLSs oui F FANCF 11p15 42 - oui G FANCG/XRCC9 9p13 68 7 TPRs oui I FANCI/KIAA1794 15q25-26 146 - oui J FANCJ/BRIP1/BACH1 17q22-24 130 ATPase, 7 motifs hélicase non L FANCL/PHF9 2p16.1 43 3 WD40, PHD oui
M FANCM 14q21.3 250 7 motifs hélicase,
endonucléase, ATPase
oui
N FANCN/PALB2 16p12 130 2 WD40 non
O FANCO/RAD51C 17q25.1 42 Paralogue RAD51/recombinase non
P FANCP/SLX4 16p13.3 200 Endonucléase non
Q FANCQ/ERCC4/XPF 16p13.12 101 Endonucléase non
HD : domaine hélical, NES : signal d’exportation nucléaire, NLS : séquence de localisation nucléaire, TPR : répétition tétratricopeptide, PHD : «plant homeodomain», OB : «oligonucleotide binding», TD : «tower domain»
Figure 3 : Schéma simplifié de la voie de réparation de l’ADN FANC-BRCA.
Les trois complexes majeurs sont illustrés en vert (complexe 1), en orange (complexe ID2) et en bleu (complexe III). Mise à jour d’après une figure issue du Mémoire de maîtrise de Charles Joly Beauparlant (2011) Université Laval
1.4.1 Les complexes et sous-complexes
Tel que mentionné précédemment, on retrouve 17 protéines Fanconi toutes impliquées dans la voie de réparation de l’ADN FANC-BRCA, en plus des protéines associées, dont les FAAPs («Fanconi Anemia Associated Proteins»). On retrouve trois complexes protéiques majeurs, soit le complexe I (connu en anglais sous le nom de «core complex»), le complexe ID2 et le complexe III, tel qu’illustré à la figure 3.
Le complexe I est constitué de quatre principaux complexes. Un premier sous-complexe est localisé à la chromatine, et est formé de FANCM et FAAP24. Le domaine C-terminal de FANCM est important pour l’interaction avec FAAP24 [69]. Il s’agit donc du premier sous-complexe à intervenir dans la voie de réparation, permettant la reconnaissance du dommage et le recrutement des autres protéines du complexe I [70]. Ensuite, il a été
démontré dans un système double-hybride en levure qu’une forte interaction était établie entre FANCA et FANCG, créant ainsi un second sous-complexe [71]. De plus, une interaction FANCG-FANCC plus faible a été observée, suggérant ainsi une liaison à un troisième sous-complexe [72]. Ce troisième sous-complexe est d’ailleurs formé de FANCC-FANCE-FANCF. L’interaction entre FANCE et FANCC est bien documentée comme le démontre les diverses expériences de co-immunoprécipitation et les essais double hybride en levure [73]. Quant à FANCF, il jouerait un rôle d’adaptateur flexible important pour l’assemblage des sous-complexes, notamment en stabilisant FANCC-FANCE avec le complexe FANCA-FANCG [74]. Enfin, FAAP100-FANCB-FANCL forme le quatrième sous-complexe [24,25]. L’attachement de ce sous-complexe au complexe I n’est pas encore parfaitement défini, mais l’interaction pourrait possiblement avoir lieu entre FANCA et FANCL, alors que FANCB jouerait un rôle de stabilisateur [75]. Bref, il est bien connu que la présence de tous les membres de ces sous-complexes est nécessaire pour assurer la stabilité du complexe I et permettre l’activation et le bon fonctionnement de la voie de réparation de l’ADN FANC-BRCA.
Le complexe ID2 est formé de deux protéines, soit FANCD2 et FANCI. L’interaction entre ces deux partenaires a été démontrée par co-immunoprécipitation [26]. De plus, il est bien connu que ces deux protéines sont modifiées de façon post-traductionnelle par monoubiquitination [76,77]. En effet, la monoubiquitination de ces deux protéines survient après l’établissement de leur interaction. Pour FANCI, l’ubiquitine est ajoutée sur la lysine 523, quant à FANCD2, il s’agit de la lysine 561 [77,78]. La cristallographie de ce complexe a particulièrement permis de visualiser le domaine d’interaction entre ces deux protéines ainsi que le positionnement des acides aminés phosphorylés ou monoubiquitinés [79].
Différents essais de double hybride en levure et des co-immunoprécipitations ont démontré une interaction entre FANCE (du complexe I) et FANCD2 (du complexe ID2). Cette interaction impliquerait la portion C-terminale de FANCE et favoriserait le recrutement du complexe ID2 en vue d’être monoubiquitiné par le complexe I grâce à l’activité ubiquitine ligase de FANCL [80].
Enfin, au sein du complexe III, on retrouve toutes les protéines impliquées dans le processus de création de l’incision ainsi que de la réparation de l’ADN. Chacune des protéines de ce complexe possède une fonction indépendante, comparativement aux protéines du complexe I. Ce groupe notamment comprend les 7 protéines Fanconi suivantes : FANCD1, FANCJ, FANCN, FANCO, FANCP, FANCQ et FANCS. Le recrutement à la chromatine des protéines de réparation sera abordé dans la prochaine section, en présentant leur fonction dans le processus de réparation des dommages à l’ADN par la voie FANC-BRCA.
1.4.2 Le mécanisme de la voie de réparation de l’ADN FANC-BRCA
La reconnaissance d’un pontage inter-brin s’effectue via le complexe FANCM-FAAP24. Cet hétérodimère joue de multiples rôles pour assurer le maintien de la stabilité de la fourche de réplication. FANCM possède des domaines hélicase/ATPase et endonucléases hautement conservés à travers les espèces et constitue également l’une des deux protéines du protéome humain à présenter ces deux domaines sur un même polypeptide [81,82]. FAAP24 possède un domaine d’interaction avec l’ADN simple brin, facilitant ainsi la reconnaissance d’un blocage de la fourche de réplication dû à un pontage inter-brin [83]. Le domaine tandem «helix-hairpin-helix» (HhH)2 de FAAP24 est essentiel pour la localisation du dimère à la chromatine [70]. Une fois lié à la chromatine, le complexe FANCM-FAAP24 est stabilisé par MHF1 et MHF2, des «histone-fold proteins» [84]. FAAP24 stimule l’action de la voie de signalisation ATR («Ataxia telangiectasia and Rad3 related»), un point de contrôle qui est activé en réponse à un stress réplicatif [85]. L’activation de la voie de signalisation ATR engendre la phosphorylation de quelques protéines Fanconi, grâce à son activité kinase ou celle de son partenaire CHK1 [86]. Parmi celles-ci, FANCA, FANCD2, FANCE, FANCG, FANCI et FANCM sont phosphorylées [87-92]. Les protéines FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL et FAAP100 seront alors recrutées aux sites de dommages à l’ADN où se trouvent FANCM-FAAP24, et constitueront le complexe I. La formation des sous-complexes constituant le complexe I a été abordée dans la section précédente.
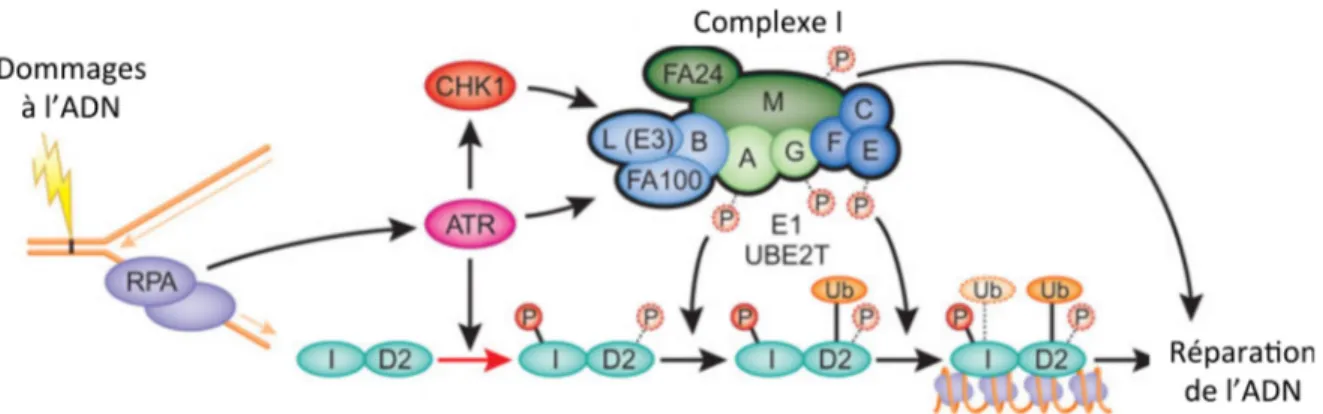
Le complexe I a pour rôle principal de procéder à la monoubiquitination de FANCD2 et de FANCI, soit les deux constituants du complexe ID2. Il s’agit d’une étape clé dans la cascade réactionnelle de cette voie de réparation de l’ADN afin de permettre son activation. Une interaction entre FANCE et FANCD2 favoriserait le rapprochement du complexe ID2 au complexe I, ce qui faciliterait la réaction de monoubiquitination [80,93]. La réaction surviendrait alors que FANCD2 et FANCI formeraient un hétérodimère. La monoubiquitination de FANCD2 est grandement augmentée lorsque FANCI est impliquée. Un niveau de monoubiquitination de FANCD2 faible est observé lorsque seul celui-ci est présent [94,95]. La réaction de monoubiquitination nécessite l’implication d’une enzyme de type E1 («ubiquitin activating enzyme»), E2 («ubiquitin conjugating enzyme») et E3 («ubiquitin ligase»). Dans le cadre de la voie FANC-BRCA, Ube2T possède l’activité E2 et est associé à FANCL qui possède l’activité E3 grâce à son domaine «RING finger» [96,97]. Ube2T transporte alors l’ubiquitine utilisée lors de la réaction de monoubiquitination. La figure 4 illustre l’activation de la voie de réparation FANC-BRCA par phosphorylation et par monoubiquitination de FANCD2 et FANCI via le complexe I («core complex»).
Figure 4 : Activation de la voie Fanconi par la phosphorylation ATR-dépendante et l’activité de monoubiquitination du complexe I
Schématisation de l’activation de la voie de réparation FANC-BRCA via la phosphorylation par ATR et CHK1 de FANCA, FANCE, FANCG, FANCI et FANCD2. La formation des sous-complexes du complexe I («FA core complex») est mise en évidence par les différentes couleurs. La monoubiquitination de FANCD2 et FANCI par le complexe I permet ensuite de procéder à la réparation des dommages à l’ADN grâce au recrutement des protéines du complexe III (non-illustrées). Image traduite de Wang, W. (2008). A major switch for the Fanconi anemia DNA damage-response pathway. Nat Struct Mol Biol 15, 1128–1130.
Il est de plus en plus évident que le complexe FANCD2-FANCI est aussi modifié par sumoylation. Cette modification post-traductionnelle surviendrait après la phosphorylation et la monoubiquitination. En fait, il s’agit d’un ajout d’une chaine peptidique SUMO («small ubiquitin-like modifier») pouvant être de longueur variable. La sumoylation jouerait également un rôle important dans la régulation de l’activation et de la répression de la voie FANC-BRCA [98].
Le recrutement des protéines de réparation est orchestré par le complexe ID2 monoubiquitiné, plus particulièrement FANCD2. Peu d’études ont été réalisées concernant la fonction de FANCI monoubiquitinée, comparativement à FANCD2. Parmi les protéines recrutées, on retrouve 7 protéines Fanconi, soit FANCD1 (BRCA2), FANCJ (BACH1/BRIP1), FANCN (PALB2), FANCO (RAD51C), FANCP (SLX4), FANCQ (ERCC4) et FANCS (BRCA1) en plus de certaines nucléases dont FAN1 et MUS81-EME1. Ces protéines agissent de concert afin de réparer le pontage inter-brin induit à l’ADN. Dans un premier temps, les nucléases interviennent à la lésion. D’abord, grâce à
son domaine UBZ («ubiquitin binding domain»), FAN1 «Fanconi-associated Nuclease 1» est recruté au pontage inter-brin par FANCD2 monoubiquitinée [99]. Elle agit avec l’hétérodimère MUS81–EME1, possédant une activité endonucléase, permettant de convertir le pontage inter-brin en cassure double brin. FAN1 possède une activité endonucléase pour créer l’incision en 5’, alors que MUS81-EME1 agit en 3’ [100,101]. Pour ce qui est de FANCP, celle-ci interagit directement avec FANCD2 monoubiquitinée grâce à son domaine UBZ4 et coopèrerait avec MUS81-EME1 pour créer l’incision [102,103]. FANCP est aussi reconnue pour être une protéine d’échafaudage. De plus, une étude suggère que SLX4, soit FANCP, agisse à titre de SUMO E3 ligase pour elle-même ainsi que pour XPF [104].
Dans un second temps, à la suite de la création de la cassure double brin créée par le décrochage du pontage inter-brin, les polymérases de translésion (TLS) sont recrutées à la lésion créée sur le brin sens. Cette famille consiste en des polymérases de faible fidélité, généralement utilisées au moment de la réplication lorsqu’il y a dommage à l’ADN pour combler les nucléotides manquant sur un brin. Ainsi la cellule est en mesure de compléter son cycle cellulaire [105]. Dans le contexte de la réparation d’un pontage inter-brin, les polymérases REV1 et POLζ sont recrutées par PCNA monoubiquitinée («proliferating cell nuclear antigen»). La monoubiquitination de PCNA n’est pas effectuée par le complexe I de la voie FANC-BRCA mais plutôt par RAD6 alors que RAD18 favoriserait son recrutement [106]. Par contre, FAAP20, membre du complexe I de la voie FANC-BRCA qui stabilise FANCA, serait aussi important pour le recrutement de REV1 à la lésion [107]. SNM1A, grâce à son activité exonucléase 5’-3’, permet de dégrader les nucléotides impliqués dans le pontage inter-brin, qui sont devenus excédants [108]. Cette étape facilite l’action des polymérases de translésion.
La réfection du brin sens par les polymérases TLS servira ensuite de modèle («template») pour la synthèse du brin complémentaire par la recombinaison homologue. D’abord FANCJ, possédant une activité hélicase, permet de défaire certaines structures induites à l’ADN afin de favoriser la recombinaison. Recrutée par FANCD2 monoubiquitinée, FANCJ participe également au recrutement de FANCS (BRCA1) grâce à
FANCD1 (BRCA2), une protéine qui se lie à l’ADN simple brin [110,111]. FANCD1 permet le recrutement de FANCO (RAD51C), la recombinase impliquée dans la voie FANC-BRCA [34,111]. Enfin, FANCN (PALB2) est connue pour interagir directement avec BRCA2 et RAD51, ce qui mènerait à son recrutement à la chromatine [112]. Bref, FANCD2 agit à titre de chef d’orchestre pour le recrutement des protéines de réparation de l’ADN et grâce aux diverses interactions établies entre les protéines du complexe III et FANCD2, celles-ci sont recrutées à la chromatine. Pour ce qui est de FANCQ (ERCC4), cette protéine possède aussi un domaine nucléase, mais une investigation plus approfondie pour mieux comprendre son implication dans la voie FANC-BRCA devrait être réalisée. Les différentes interactions entre ces protéines sont très importantes pour garantir leur recrutement à la lésion tout en assurant l’efficacité de leur fonction dans la réparation des dommages à l’ADN. La figure 5 permet d’illustrer de façon simplifiée les différentes étapes menant à la réparation d’un pontage inter-brin induit à l’ADN.
Figure 5 : Succession des étapes de la réparation des pontages inter-brins induits à l’ADN.
La réparation de l’ADN via la voie FANC-BRCA peut se diviser en 4 étapes, soit 1. L’activation, 2. La création de l’incision par les nucléases, 3. La réfection de la cassure double brin, 4. La recombinaison homologue. Traduit de Kottemann, M.C., and Smogorzewska, A. (2013). Fanconi anaemia and the repair of Watson and Crick DNA
Une fois le pontage inter-brin réparé, la voie FANC-BRCA est inactivée par la déubiquitination de FANCD2 et la dissociation du complexe I. Le dimère USP1/UAF1 a été identifié comme étant responsable du retrait de l’ubiquitine sur FANCD2 puisque la répression de USP1 a démontré une hyper-accumulation de FANCD2 monoubiquitinée. De plus, cette répression crée une instabilité génomique importante, ce qui suggère que autant la régulation de la monoubiquitination que la déubiquitination de FANCD2 est importante pour la réparation des pontages inter-brins et la stabilité cellulaire [113]. USP1 procéderait également à la déubiquitination de FANCI, mais l’importance de cette réaction n’a pas été démontrée en ce qui a trait à la désactivation de la voie FANC-BRCA [114]. L’activité enzymatique du dimère est présente grâce à USP1 [115]. Une étude a démontré que la forme sumoylée de FANCI favoriserait le recrutement de USP1/UAF1 via UAF1, en raison de son domaine tandem SLD («SUMO-like domains»), soit SLD1 et SLD2. Le domaine SIM («SUMO-like domain-interacting motif») de FANCI interagit alors avec UAF1, entraînant la déubiquitination du complexe ID2 [116,117]. La délétion du domaine SLD2 de UAF2 ou des mutations dans le domaine SIM de FANCI provoque une diminution de la déubiquitination de FANCD2 et de la réparation de l’ADN [116]. Par la déubiquitination du complexe ID2, le recrutement des protéines de réparation à la chromatine est alors inhibé. En plus, le complexe I est dissocié à la suite de la réparation de la lésion. L’hyperphosphorylation de FANCM jouerait un rôle dans le contrôle négatif de la voie FANC-BRCA. En fait, cela créerait la dissociation du complexe I ce qui entraînerait une perte de monoubiquitination de FANCD2 et de FANCI. Il a été démontré que Plk1 phosphoryle FANCM et provoque sa dégradation à la suite de la réparation des dommages à l’ADN. Ainsi, la protéine agissant à titre d’ancrage à la chromatine pour le complexe I, soit FANCM, est dégradée, ce qui induit la dissociation du complexe I [118].
1.5 Autres rôles des protéines Fanconi
Les potentielles fonctions individuelles des protéines du complexe I sont très peu caractérisées, voire très peu connues. Outre leur fonction dans la voie de réparation de l’ADN FANC-BRCA, les protéines Fanconi ont d’autres implications. Ces protéines semblent de plus en plus importantes pour l’homéostasie de la cellule. L’identification de
partenaires d’interaction protéique est souvent une méthode utilisée pour déterminer de nouvelles fonctions d’une protéine. La spectrométrie de masse ou bien le criblage par essai double hybride sont deux techniques fréquemment utilisées dans ce genre d’expérimentations. Par exemple, un criblage par un essai double hybride en levure avec une banque d’ADN complémentaire a permis d’identifier plusieurs partenaires d’interaction possibles pour FANCA, FANCC et FANCG. Selon cette étude, des protéines impliquées dans la transcription, la signalisation cellulaire, le transport cellulaire et le métabolisme d’oxydation ont été identifiées [119]. De plus, tel que mentionné dans la section 1.1, les protéines Fanconi se localisent aux centrosomes ainsi qu’aux microtubules, suggérant un rôle dans le processus de division. Rappelons que des aberrations surviennent au moment de la division cellulaire dans les cellules Fanconi, notamment par la création de micronoyaux. L’implication des protéines Fanconi à ce processus, qui demeure encore plutôt méconnue, se montre plutôt importante avec les récentes études [18,120,121].
Dans un autre ordre d’idée, il a été démontré que FANCC interagit directement avec STAT1, et aurait donc un effet important en ce qui a trait à l’activation de STAT1 en réponse aux cytokines et autres facteurs de croissance [122,123]. La portion centrale de la protéine FANCC serait importante pour son interaction avec STAT1. L’interaction entre FANCC et STAT1 permettrait de protéger les cellules hématopoïétiques de l’apoptose induite par IFN-gamma et TNF-alpha [123].
Les patients atteints de l’anémie de Fanconi présentent un taux de dommages suite à un stress oxydatif supérieurs à la normale. FANCA, FANCC et FANCG interagissent avec des membres de la famille des cytochromes P450, grandement connus pour leur implication dans les réactions d’oxydoréduction [124]. FANCG localise dans les mitochondries et interagit également avec PRDX3, qui est bien caractérisé pour son implication dans les mitochondries, où plusieurs réactions d’oxydoréduction ont lieu [125]. L’implication directe des protéines Fanconi dans le métabolisme du stress oxydatif demeure plutôt méconnue, alors que des études plus approfondies permettraient d’avoir des meilleures connaissances générales sur les diverses implications des protéines Fanconi. Toujours concernant le stress oxydatif, il a été démontré que FANCD2 interagit avec FOXO3a, un
[127]. La présence de FANCD2 est particulièrement importante pour la rétention de FOXO3a dans le noyau des cellules souches hématopoïétiques. De plus, il est suggéré que la voie FANC-BRCA et celle de FOXO3a interagiraient ensemble dans le maintien de la stabilité des cellules souches hématopoïétiques [128].
Il s’agit d’une minime fraction des autres rôles des protéines Fanconi. Cette famille de protéines est multifonctionnelle et ces diverses fonctions sont majoritairement en lien avec la stabilité cellulaire. Enfin, ces protéines sont exprimées dans une large gamme de tissus et types cellulaires, ce qui renforce l’importance de leur expression adéquate [129,130].
1.6 FANCE
La mise en évidence d’un cinquième groupe de complémentation de l’anémie de Fanconi, soit le groupe E (FANCE), a été réalisée en 1995. La complémentation avec les quatre groupes existant à cette époque (FANCA, FANCB, FANCC et FANCD) avait échouée, ce qui a engendré l’hypothèse de l’existence d’un cinquième groupe de complémentation [131]. Il s’agissait d’un patient né en Turquie présentant un retard de croissance, des anormalités squelettiques (hypoplasie du pouce droit), une hyperpigmentation de la peau, une microcéphalie en plus de souffrir de thrombopénie et de pancytopénie [132]. D’ailleurs, la lignée cellulaire FANCE-déficiente, EUFA130, qui est couramment utilisée afin de réaliser des tests spécifiques à FANCE, est issue de ce patient. Une mutation non-sens est présente dans le deuxième exon, menant à une protéine tronquée et non-fonctionnelle. Ses parents et son frère présentaient cette mutation de façon hétérozygote [133]. Environ 1 % des patients Fanconi appartiennent au groupe de complémentation E [3].
1.6.1 Structures et fonctions
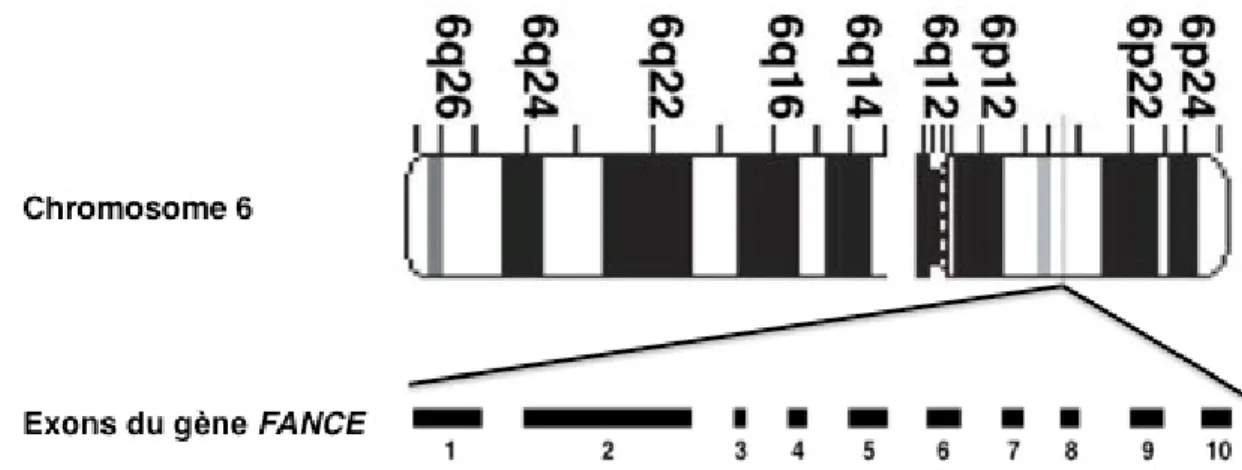
C’est en 1999 que le locus 6p21-22 a été associé au gène FANCE [134]. Il s’agit d’un gène composé de 10 exons dont la région codante contient 1611 paires de bases et produit une protéine de 536 acides aminés (poids moléculaire d’environ 60 kDa).
Figure 6 : Localisation chromosomique et structure des exons du gène FANCE.
Image adaptée de Titus, T.A., Selvig, D.R., Qin, B., Wilson, C., Starks, A.M., Roe, B.A., and Postlethwait, J.H. (2006). The Fanconi anemia gene network is conserved from zebrafish to human. Gene 371, 211–223.
Une étude a démontré que la transcription de FANCE serait potentiellement induite par les facteurs STAT1beta, SP1, XCPE et GATA3 [135]. Également, une étude a démontré que le locus du gène FANCE présentait un motif de liaison du facteur de transcription VDR (Récepteur vitamine D), ce qui fait de FANCE une cible de VDR [136]. 1α,25-dihydroxyvitamin D3 permet d’activer VDR. Ainsi, lorsque des cellules sont traitées avec ce dernier, l’expression de FANCE est augmentée de façon significative. Concernant les modifications post-traductionnelles, CHK1 (Checkpoint kinase 1) phosphoryle FANCE aux positions T346 et S374 dans les 30 minutes suivant l’apparition des dommages à l’ADN. La phosphorylation de ces deux sites est essentielle pour assurer la résistance aux agents pontants (MMC), mais n’est pas primordiale pour la monoubiquitination et la
avoir un effet négatif pour la régulation de la protéine. En fait, à la suite d’un traitement aux ultraviolets, une dégradation de la protéine par le protéasome était observée. Le double mutant pour les deux sites de phosphorylation était quant à lui stable. En bloquant le mécanisme de dégradation des protéines par un traitement au MG132, le niveau d’expression de la protéine FANCE était augmenté. Ainsi, FANCE serait phosphorylée en réponse à un traitement avec un agent pontant dans les 30 minutes suivant la création du pontage inter-brin. Par la suite, il y aurait poly-ubiquitination de FANCE afin de procéder à sa dégradation, ce qui pourrait potentiellement être un mécanisme de régulation de l’activité de la voie FANC-BRCA [89].
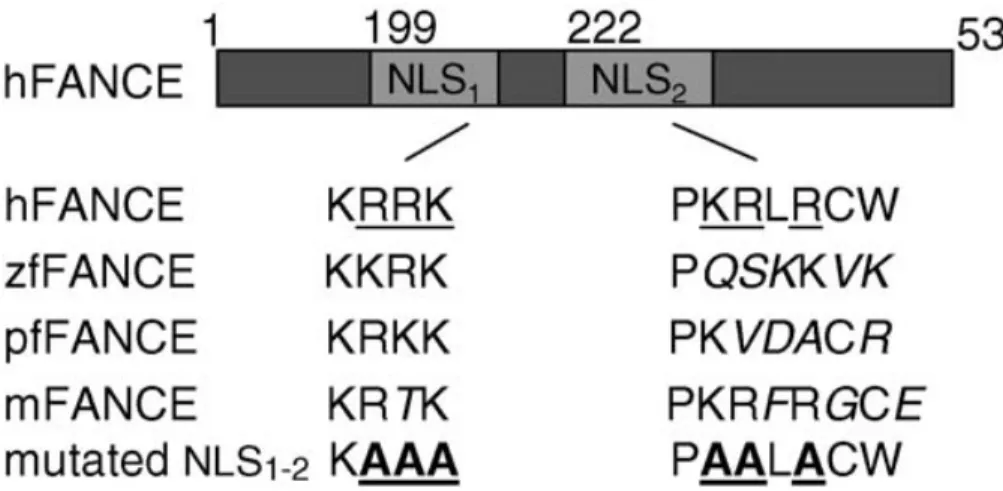
Quant à la localisation cellulaire, FANCE est une protéine retrouvée de façon majoritaire au noyau. Tel qu’illustré à la figure 7, on retrouve un motif de signal de localisation nucléaire (NLS) bipartite correspondant aux acides aminés 199-202 et 222-228, qui est hautement conservé à travers les espèces [68,93,133]. Cette séquence favorise ainsi le transport de FANCE vers le noyau, mais il a été démontré que FANCC y jouait également un rôle important. À noter que FANCE muté pour les deux positions du motif de localisation nucléaire est toujours en mesure de migrer vers le noyau. Par contre, ce double mutant se retrouve au cytoplasme dans une cellule déficiente en FANCC [73,93]. Cette dépendance semble réciproque puisque FANCC demeure dans le cytoplasme dans la lignée déficiente FANCE (EUFA130) [137]. La section centrale (acides aminés 149-371) de FANCE est nécessaire pour l’interaction avec FANCC [73].
Figure 7 : Conservation de la séquence de localisation nucléaire du gène FANCE à travers l’évolution.
(hFANCE : humain, zfFANCE : poisson-zèbre, pfFANCE : poisson-ballon, mFANCE : souris). Image tirée de Léveillé, F., Ferrer, M., Medhurst, A.L., Laghmani, E.H., Rooimans, M.A., Bier, P., Steltenpool, J., Titus, T.A., Postlethwait, J.H., Hoatlin, M.E., et al. (2006). The nuclear accumulation of the Fanconi anemia protein FANCE depends on FANCC. DNA Repair 5, 556–565.
FANCE est un membre du complexe I et son interaction avec FANCD2 permettrait alors de créer le pont entre le complexe I et le complexe ID2 dans le but de procéder à la monoubiquitination de FANCD2 et de FANCI [138]. La cristallographie de la protéine FANCE permet notamment de visualiser ces deux domaines d’interaction (avec FANCC et FANCD2) et d’appuyer ces modèles d’interaction proposés [138]. Contrairement à d’autres protéines du complexe I qui sont connues pour avoir des fonctions dans d’autres mécanismes (voir section 1.4), FANCE n’est pas reconnue pour avoir d’autres implications que la voie FANC-BRCA. Peu d’études ont été réalisées afin de caractériser le gène et la protéine FANCE. Par contre, il n’est pas impossible qu’elle ait d’autres rôles importants au sein de la cellule qui demeurent encore méconnus à ce jour.
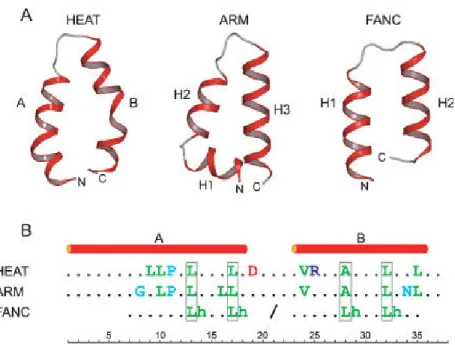
Sur le plan structurel, FANCE est une protéine majoritairement constituée d’hélices alpha, mais d’aucun domaine fonctionnel connu. Il s’agit d’une protéine qui fait partie de la famille des «non-globular protein». On retrouve 5 motifs de répétition FANC («FANC repeats») constitués de deux hélices alpha, des structures de la même famille que les motifs ARM et HEAT [138]. Les motifs ARM et HEAT ont été davantage étudiés et ont une
localisés en C-terminale de FANCE, dont le premier débute en position L357 et le cinquième se termine en position G535. Étant donné qu’il est connu que FANCD2 interagit avec l’extrémité C-terminale de FANCE, ces séquences répétées seraient alors importantes pour cette interaction. La figure 8 illustre d’ailleurs la comparaison entre les trois motifs (HEAT, ARM et FANC) pour ce qui est de la structure primaire protéique ainsi que la structure secondaire (positionnement des hélices alpha). Il a été démontré que les deux régions terminales de la protéine, soit les régions correspondant aux acides aminés 1-170 et 270-536 s’avéraient ordonnées alors que la région 170-270 était plutôt désordonnée [138]. Cette caractéristique suggère que la protéine FANCE est plutôt statique, tel qu’observé chez les protéines ordonnées [140]. Les acides aminés en C-terminale sont grandement conservés à travers l’évolution, ce qui confère une importance à cette région pour l’activité ou le repliement de la protéine [138].
Figure 8 : Structure des domaines HEAT, ARM et FANC.
A) Illustration de la structure secondaire des trois domaines HEAT, ARM et FANC B) Comparaison de la structure primaire des trois domaines HEAT, ARM et FANC. Image tirée de Nookala, R.K., Hussain, S., and Pellegrini, L. (2007). Insights into Fanconi Anaemia from the structure of human FANCE. Nucleic Acids Research 35, 1638–1648.
1.6.2 Études d’association et polymorphismes
Selon une étude d’association menée au sein d’une cohorte Turque atteinte du cancer de l’œsophage, une mutation hétérozygote (p.Val311SerfsX2) du gène FANCE a été identifiée chez un patient sur 190. Il s’agit d’une insertion d’une cytosine dans le quatrième exon qui entraine l’apparition d’un codon stop prématuré. Cette mutation n’a pas été détectée parmi les 811 contrôles [141]. Selon l’étude, p.Val311SerfsX2 serait donc potentiellement associée à une augmentation du risque de développer un cancer de l’œsophage. Dans une étude menée au sein d’une cohorte de patients chinois atteints d’un cancer de l’œsophage («esophageal squamous cell carcinoma»), on suggère une association entre le gène FANCE et ce cancer. 1942 patients ont participé au projet ainsi que 2111 individus contrôles [142]. D’avantages d’études devront toutefois être réalisées afin de valider FANCE en tant que gène de susceptibilité du cancer de l’œsophage. Concernant l’augmentation du risque de développer un cancer du sein, contrairement à FANCD1 (BRCA2), FANCJ (BACH1/BRIP1), FANCN (PALB2), FANCO (RAD51C) et FANCS (BRCA1), aucune association claire n’a été prouvée pour le gène FANCE.
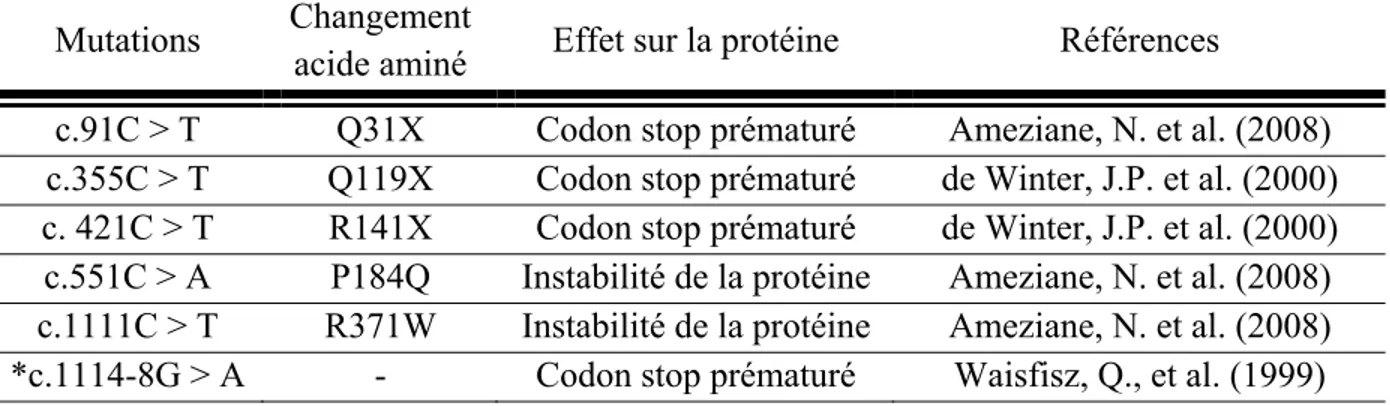
Quelques mutations ont été rapportées pour le gène FANCE chez des patients atteints de l’anémie de Fanconi. Certains présentent un codon stop prématuré à la suite d’une mutation dans une région codante ou de la rétention d’une région intronique présentant un codon stop, alors que dans certains cas, des mutations affectent la conformation de la structure tertiaire de la protéine, produisant une protéine non-fonctionnelle et instable. Par exemple, la mutation c.1111C>T n’empêche pas la production de la protéine et ne transforme pas un codon en un codon stop. Par contre, la protéine produite n’est pas en mesure de remplir pleinement sa fonction. En fait, l’interaction avec FANCD2 est perdue en raison de la modification de la conformation de FANCE due au changement d’acide aminé (R371W) [138]. D’ailleurs, puisqu’il est connu que la région C-terminale de FANCE est cruciale pour l’interaction avec FANCD2, les variants tronqués dus à la présence d’un codon stop prématuré ne seront pas en mesure de conserver leur interaction avec FANCD2, et seront alors considérés inactifs. Il existe une base de données qui regroupe les différentes variations génétiques observées parmi les gènes Fanconi, soit
«The Rockefeller University Fanconi Anemia Mutation Database». Quelques exemples des mutations causales les plus fréquentes pour le gène FANCE sont listés au tableau 2.
Tableau 2 : Génotypes de patients atteints de l’anémie de Fanconi pour le gène FANCE
Mutations Changement
acide aminé Effet sur la protéine Références
c.91C > T Q31X Codon stop prématuré Ameziane, N. et al. (2008)
c.355C > T Q119X Codon stop prématuré de Winter, J.P. et al. (2000)
c. 421C > T R141X Codon stop prématuré de Winter, J.P. et al. (2000)
c.551C > A P184Q Instabilité de la protéine Ameziane, N. et al. (2008)
c.1111C > T R371W Instabilité de la protéine Ameziane, N. et al. (2008)
*c.1114-8G > A - Codon stop prématuré Waisfisz, Q., et al. (1999)
*Rétention d’une région intronique
2. Épissage alternatif
2.1 Les principes de l’épissage alternatif
La mise en évidence d’un processus complexe visant à produire plusieurs ARN messagers (ARNm) différents à partir d’une même séquence d’ADN a été réalisée en 1977 [143]. Il a été démontré qu’il était possible de produire 5 ARNm d’adénovirus différents à partir d’une même séquence d’ADN dans des cellules humaines. Plusieurs études ont ensuite découlé de cette découverte, permettant ainsi de caractériser ce processus de maturation de l’ARN pré-messager qui vise à éliminer les régions introniques afin d’obtenir un ARNm prêt à être traduit en protéine par les ribosomes. Le déséquilibre entre le nombre de gènes et le nombre de protéines existantes a donc pu être expliqué par ce processus d’épissage alternatif. Le processus d’épissage alternatif est d’ailleurs reconnu pour générer une diversité protéique chez l’humain [144]. La vaste majorité des gènes possédant plus d’un exon (plus de 90% de ces gènes) sont sujet à l’épissage alternatif [145]. Selon la figure 9, différentes formes de variants d’épissage alternatif peuvent être produites ; le saut d’un ou plusieurs exons (a et e), la rétention d’un intron, (d) un nouveau site 3’ ou 5’ alternatif (b et c), un promoteur alternatif (f) et un nouveau site de polyadénylation (g) [146].
Figure 9 : Différentes formes d’évènements d’épissage alternatif.
Illustrations de 7 différentes formes d’épissage alternatif pouvant survenir au sein de la cellule. Les régions alternatives sont illustrées en violet. Traduit de Keren, H., Lev-Maor, G., and Ast, G. (2010). Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet. 11, 345–355.
La machinerie d’épissage reconnaît des séquences consensus qui délimitent la jonction intron/exon (sites donneurs et sites accepteurs). L’intégrité de ces sites est importante pour assurer un épissage adéquat. La figure 10 illustre notamment la reconnaissance de cette machinerie d’épissage pour un ARN, via les séquences consensus