Étude des mécanismes de transfert de chiralité en
catalyse hétérogène
Thèse
Jean-Christian Lemay
Doctorat en chimie
Philosophiæ doctor (Ph. D.)
Québec, Canada
Étude des mécanismes de transfert de chiralité en
catalyse hétérogène
Thèse
Jean-Christian Lemay
Sous la direction de :
Résumé
L’omniprésence de produits de synthèse dans des domaines allant de la pharmaceu-tique à la pétrochimie dépend d’une industrie chimique forte s’appuyant en partie sur la maîtrise de procédés catalytiques. La catalyse hétérogène est particulièrement prisée puisqu’il est facile de récupérer et de réutiliser le catalyseur. Une approche éprouvée pour améliorer l’efficacité en catalyse hétérogène consiste à mieux com-prendre les interactions entre les substrats et les sites catalytiques individuels. De nombreuses questions demeurent quant à l’activité et à la sélectivité des sites actifs en catalyse hétérogène asymétrique. Mes travaux de doctorat concernent spé-cifiquement la caractérisation des sites catalytiques pour l’hydrogénation énantiosé-lective de cétones activées sur une surface de Pt(111) chiralement modifiée par des molécules apparentées à la cinchonidine, la réaction d’Orito, et ce, par microscopie à effet tunnel (STM). La résolution du microscope est suffisante pour cataloguer des dizaines de milliers de complexes selon leur géométrie d’assemblage. Les assem-blages les plus abondants sont comparés aux structures les plus stables calculées selon les calculs DFT du groupe du Pr Hammer, de l’Université d’Aarhus, au Da-nemark. L’adéquation entre les motifs les plus fréquemment observés par STM et les images STM simulées à partir des assemblages les plus stables selon la DFT permet d’assigner une pro-chiralité aux structures cataloguées. La pro-chiralité identifie un complexe selon la chiralité de l’alcool qu’il formerait si le substrat était hydrogéné dans cette géométrie. On peut comparer le ratio pro-chiral (pr) des complexes for-més sur la surface au ratio énantiomérique (er) formé lors de la réaction. L’approche combinée STM-DFT permet de comprendre comment les forces intermoléculaires di-vergent d’un complexe à l’autre, expliquant la stéréosélection.
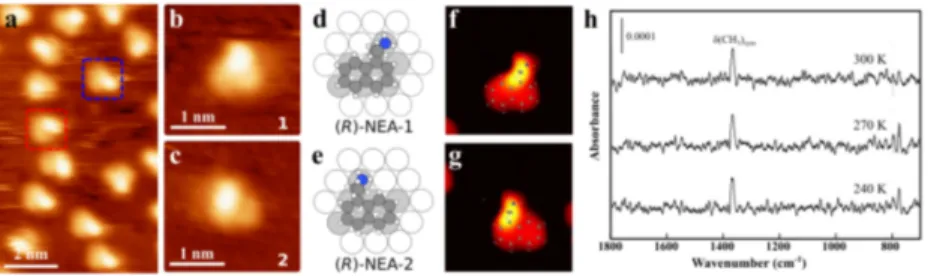
Le chapitre 3 se base sur une série d’expériences comparant les assemblages formés par le substrat cétopantolactone (KPL) avec trois modificateurs chiraux partageant le même groupement ancrant : le 1-(1-naphthyl)éthylamine (NEA), le
(R)-N-Méthyl-1-(1-naphthyl)éthylamine ((R)-MNEA) et le (R)-1-naphthyl-1,2-éthanediol ((R)-NED). Les divergences observées pour les abondances relatives des géométries d’assemblages proviennent de subtiles différences entre les groupements donneurs de liaisons hydrogène des trois modificateurs chiraux. On note qu’autant pour le système (R)-NEA/KPL que pour le système (R)-NED/KPL, le pr et le er concordent, tandis qu’ils divergent pour le système (R)-MNEA/KPL.
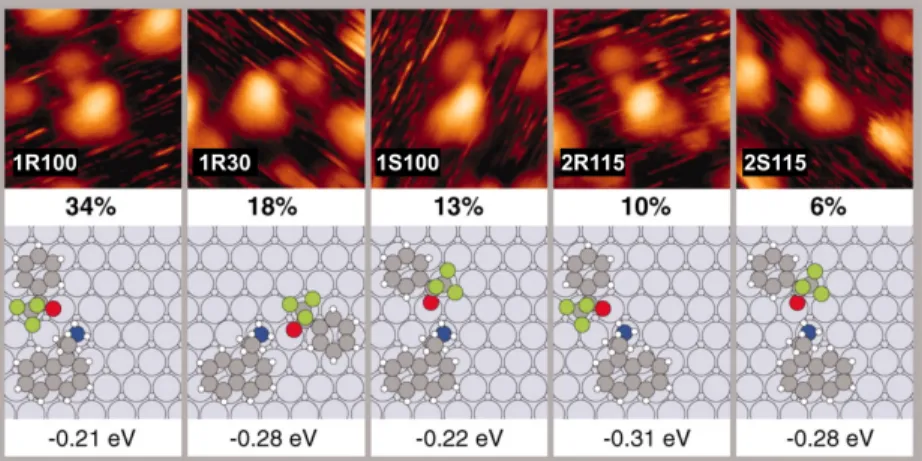
Le chapitre 4 cherche à comprendre l’effet d’une modification simple sur les assem-blages formés par le (R)-NEA et le substrat pro-chiral 2,2,2-trifluoroacétophénone (TFAP). Pour ce faire, un nouveau modificateur chiral a été synthétisé : le (R)-1-(8-methyl-1-naphthyl)éthylamine ((R)-8MeNEA). Celui-ci ne se distingue du (R)-NEA que par l’ajout d’un méthyle sur le groupement aromatique non-substitué. La pré-sence d’un groupement méthyle supplémentaire vient affecter la préorganisation des substrats dans certains sites catalytiques du (R)-NEA, ainsi que la diffusion vers et entre ceux-ci. Évidemment, les assemblages où le TFAP se retrouverait à l’endroit de la substitution sont fortement perturbés par encombrement stérique et sont donc peu observés. De plus, les populations relatives d’assemblages éloignés du site de l’altération changent elles aussi par rapport à celles observées pour le (R)-NEA. De tels effets secondaires doivent être considérés lors de la conception rationnelle de modificateurs chiraux.
Le chapitre 5 compare les complexes formés par deux substrats pro-chiraux, le pyru-vate de méthyle (MP) et le méthyle 3,3,3-trifluoropyrupyru-vate (MTFP), avec le (R)-NEA. Le MP, lorsqu’il y a peu d’hydrogène sur la surface et que la température est suffi-samment élevée, peut former un énol sur la surface de platine. Cet intermédiaire énol n’est pas formé en milieu catalytique, riche en hydrogène. Un moyen d’éviter la for-mation de l’énol est d’utiliser le substrat prochiral MTFP, qui ne diffère du MP que par la substitution d’un groupement méthyle par un trifluorométhyle. Les assem-blages impliquant MP et MTFP diffèrent, y compris aux températures auxquelles les deux substrats sont sous forme cétonique. Ces divergences s’expliquent par la plus grande électronégativité du CF3, qui modifie l’adsorption du carbonyle cétonique, et donc du substrat. Des mesures des populations relatives des diverses géométries d’adsorption des complexes (R)-NEA/MTFP à des températures croissantes révèlent que l’équilibre thermodynamique n’est bien approximé que pour des températures supérieures à 250 K. La mobilité accrue du MTFP à cette température lui permet de sonder plus efficacement les divers sites auxquels il peut se lier sur la surface.
Abstract
The widespread use of chemical synthesis – in domains as varied as the pharmaceu-tical and petrochemical industries – requires a strong chemical industry, relying on the use of many catalytic processes. Heterogeneous catalysis is often favoured as it is easier to recycle and reuse the catalysts. A reliable way to improve efficiency in het-erogeneous catalysis is to better understand how substrates interact with individual catalytic sites.
Many questions remain relating to the activity and selectivity of active sites in asym-metric heterogeneous catalysis. My thesis deals with the characterization of catalytic sites for the enantioselective hydrogenation of activated ketones on chirally modified Pt(111), the so-called Orito reaction, using scanning tunnelling microscopy (STM). The chiral modifiers are structural analogues of cinchonidine. The resolution of STM is sufficient to catalogue tens of thousands of bimolecular complexes according to their interaction geometry. The most abundant motifs are compared to the most sta-ble structures as computed from DFT calculations performed by Pr Hammer’s group in Aarhus University, in Denmark. The agreement between the STM motifs and im-ages simulated from the DFT calculations allow us to assign a pro-chirality to each complexation geometry. The pro-chirality labels complexes according to the chirality of the resulting alcohol if the substrate were to be hydrogenated in this configuration. We can compare the pro-chiral ratio (pr) for complexes observed by STM to the enan-tiomeric ratio (er) measured in a catalytic setting. Combining STM imaging with DFT calculations allow us to better understand why some complexation geometries are favoured, thus explaining stereoselction.
Chapter 3 presents a series of experiments comparing the assemblies formed by the substrate ketopantolactone (KPL) with three chiral modifiers sharing a sim-ilar anchoring moiety: (R)-1-(1-naphthyl)ethylamine ((R)-NEA), (R)-N-Methyl-1-(1-naphthyl) ethylamine ((R)-MNEA) and (R)-1-naphthyl-1,2-ethanediol ((R)-NED).
Differences between the observed populations for competing interaction geometries are ascribed to subtle variations in the hydrogen-bond donors moieties. We note that
pr and er are roughly in agreement for (R)-NEA/KPL and (R)-NED/KPL assemblies,
but not for (R)- MNEA / KPL.
Chapter 4 tries to understand how a small structural alteration can change the com-plexes formed by (R)-NEA and the pro-chiral substrate 2,2,2-trifluoroacetophenone (TFAP). A new chiral modifier (R)-1-(8-methyl-1-naphthyl) ethylamine ((R)-8MeNEA), has been synthesized and differs from (R)-NEA only by an added methyl moiety on the non-substituted aromatic ring. This methyl changes the preorganisa-tion states of chirality transfer complexes, and diffusion among the competing ge-ometries. The binding configurations at the methyl substituent obviously disappear because of steric hindrance. We also record changes in the relative populations of complexation geometries away from the substitution. Such second-order changes must be taken into account for the rational design of chiral modifiers for heteroge-neous catalysis.
Chapter 5 compares chirality transfer complexes formed by two pro-chiral substrates, methyl pyruvate (MP) and methyl 3,3,3-trifluoropyruvate (MTFP), with (R)-NEA. MP can form an enol on Pt(111) in hydrogen poor environment, such as an ultra-high vac-uum system, if the temperature is high enough. An alternative to MP which cannot form the enol is its trifluorinated analogue: MTFP. Populations of competing chirality transfer complexes involving MP and MTFP differ, including at temperatures below which the enol is formed. These divergences arise from the higher electronegativity of the CF3moiety, which modifies the adsorption of the ketonic carbonyl, and hence
of the whole pro-chiral substrate. The populations of competing (R)-NEA/MTFP geometries are found to better approximate the thermodynamic equilibrium at tem-peratures above 250 K. This can be explained by increased MTFP mobility, which allows the pro-chiral substrate to sample more efficiently all competing assemblies on the surface.
Table des matières
Résumé iii
Abstract v
Table des matières vii
Liste des tableaux ix
Liste des figures x
Remerciements xv
Avant-propos xvii
Introduction 1
0.1 Enjeux de société et chimie. . . 1
0.2 Défis de la chimie . . . 4
0.3 Chiralité . . . 6
0.4 Catalyse hétérogène asymétrique . . . 11
0.5 Études catalytiques motivant l’étude du système par les sciences des surfaces . . . 13
0.6 Éclairage apporté par les sciences des surfaces sur la réaction d’Orito 22 0.7 Objectifs de la thèse . . . 30
0.8 Structure de la thèse . . . 30
1 Méthode expérimentale 32 1.1 Sciences des surfaces . . . 32
1.2 Appareillage utilisé et son opération . . . 33
1.3 Caractérisation individuelle des molécules . . . 35
1.4 Caractérisations théoriques : Théorie de la fonctionnelle de la den-sité (DFT). . . 41
2 A comparative study of diastereomeric complexes formed by a pro-chiral substrate and three structurally analogous pro-chiral molecules on
Pt(111) 47
2.1 Résumé . . . 47
2.2 Abstract . . . 48
2.3 Introduction . . . 48
2.4 Material and methods . . . 50
2.5 Theory/calculation . . . 51
2.6 Results and Discussion . . . 51
2.7 Conclusions . . . 60
2.8 Acknowledgments . . . 61
3 Secondary Effects of a Minor Structural Modification in a Chiral Mo-difier on the Formation of Chemisorbed Chirality Transfer Complexes on Pt(111) 62 3.1 Résumé . . . 62
3.2 Abstract . . . 63
3.3 Introduction . . . 64
3.4 Material and methods . . . 65
3.5 Results . . . 66
3.6 Discussion . . . 71
3.7 Conclusions . . . 75
4 Equilibration of Chirality Transfer Complexes formed by MTFP and the Chiral Modifier (R)-NEA. Evaluation of the Accuracy of DFT Cal-culated Complexation Energies 77 4.1 Résumé . . . 77
4.2 Abstract . . . 78
4.3 Introduction . . . 78
4.4 Material and methods . . . 81
4.5 Results and Discussion . . . 82
4.6 Conclusion . . . 100
Conclusion 102
Liste des tableaux
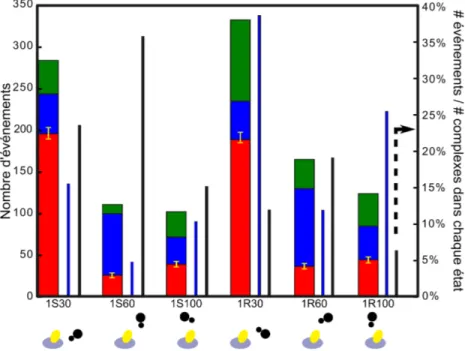
4.1 Comparaison du er et du pr pour différentes paires de modificateurs
Liste des figures
Chapitre 0
0.1 Chiralité . . . 7
0.2 Bioactivité des énantiomères du praziquantel . . . 9
0.3 Hydrogénation d’une cétone activée . . . 12
0.4 Réaction d’Orito . . . 14
0.5 Modificateurs chiraux pour la réaction d’Orito . . . 15
0.6 Ajustement induit de la cinchonidine. . . 16
0.7 Substrats pour la réaction d’Orito . . . 18
0.8 Signal RAIRS pour le (S)-NEA et d’autres molécules apparentées. . . . 20
0.9 Spectres infrarouges pour l’adsorption du (R)-NEA et du PNEA . . . . 21
0.10 Motifs observés par STM pour la cinchonidine . . . 22
0.11 Effet de la pression d’hydrogène sur la mobilité de la cinchonidine . . 23
0.12 Géométrie d’adsorption du (R)-NEA . . . 24
0.13 Substrats pro-chiraux étudiés . . . 25
0.14 Simulations Tersoff-Hamann des dimères de TFAP . . . 26
0.15 Géométries d’interaction pour les complexes (R)-NEA/TFAP . . . 28
0.16 Interconvertion entre les différentes géométries pour un adduit cata-lytique (R)-NEA/TFAP . . . 29
Chapitre 1 1.1 Surface utilisée : Pt(111) . . . 33
1.2 Système du système UHV utilisé . . . 34
1.3 Schéma du fonctionnement de la boucle de contrôle du STM . . . 36
1.4 Images STM et AFM de pentacène sur du Cu(111) . . . 36
1.5 Effet tunnel. . . 38
1.6 Schéma de l’imagerie STM . . . 38
1.7 Résolution des orbitales du pentacène par STM . . . 40
1.8 Modélisation de liaisons chimiques en surface par DFT . . . 44
1.9 Caractérisation de complexes de transfert de chiralité par STM et DFT 45 1.10 Juxtaposition des données STM et DFT pour les assemblages TFAP– (R)-NEA . . . 46
2.1 Complexes formed by three structurally analogous chiral modifiers
and a prochiral substrate on Pt(111). . . 49
2.2 Enantioselective hydrogenation of KPL induced by NEA,
(R)-MNEA and (R)-NED on Pt . . . 50
2.3 Chiral modifiers (R)-NEA, (R)-MNEA and (R)-NED on Pt(111) . . . 52
2.4 STM complexation motifs for (R)-NEA/KPL, (R)-MNEA/KPL and
(R)-NED/KPL 1 :1 complexes on Pt(111) at room temperature . . . 53
2.5 Summary of DFT-calculated most stable (R)-NEA/KPL structures . . . 54
2.6 Schematic categorization of STM motifs for ensembles of (R)-NEA/KPL, (R)-MNEA/KPL and (R)-NED/KPL 1 :1 complexes in
terms of the location and prochirality of complexed KPL . . . 56
2.7 DFT calculated (R)-MNEA-1/KPL structures . . . 58
2.8 DFT calculated (R)-MNEA-1/hy-KPL structures . . . 59
Chapitre 3
3.1 STM images and DFT structures for (R)-NEA and(R)-8Me-NEA on
Pt(111) . . . 67
3.2 STM motifs for the six most abundant complexation geometries
bet-ween (R)-8Me-NEA-1 and TFAP. . . 68
3.3 Schematic depiction of the labeling system used to describe
TFAP/(R)-8Me-NEA and TFAP/(R)-NEA complexes . . . 69
3.4 Method used to correlate STM data for NEA and
TFAP/(R)-8Me-NEAcomplexes . . . 70
3.5 Relative abundances of complexes formed between TFAP and (R)-NEA
and (R)-8Me-NEA on Pt(111) . . . 71
3.6 DFT calculated structures of TFAP/(R)-8Me-NEA complexes . . . 72
3.7 Structural changes in the ethylamine group resulting from adding a
methyl substituent at the 8-naphthyl position . . . 74
Chapitre 4
4.1 STM motifs for the six most abundant complexation geometries
bet-ween (R)-NEA-1 and MTFP . . . 83
4.2 Example of (θ, ϕ) density plots for complexes involving MTFP with
(R)-NEA . . . 84
4.3 DFT calculated structures of MTFP/(R)-NEA . . . 85
4.4 Relative abundances of complexes formed between MTFP and
(R)-NEA on Pt(111) at four temperatures . . . 86
4.5 Hydratation of MTFP to form an hemiketal . . . 86
4.6 Ratios of complexes involving the majority conformer and the minority
conformer . . . 88
4.7 Termolecular assemblies . . . 90
4.8 Time evolution of some (R)-NEA-2+MTFP complexes . . . 92
4.10 Corresponding chemisorbed MTFP and MP structures on Pt(111) . . . 94
4.11 TM motifs for the abundant complexation geometries between
(R)-NEA and MP . . . 95
4.12 Methyl pyruvate transforms to an enolate on Pt(111) . . . 96
4.13 Relative abundances of complexes formed between MP and (R)-NEA
Ce sont souvent les petites choses qui font toute la différence.
Remerciements
Je tiens à exprimer ici ma reconnaissance à tous ceux et celles qui m’ont soutenu et encouragé au cours des dernières années. Votre support et vos conseils m’ont permis de beaucoup apprendre et de me développer. J’aimerais spécialement témoigner ma gratitude à certaines personnes qui m’ont permis de grandement cheminer durant mon doctorat, autant du point de vue scientifique qu’humain.
Tout d’abord, je tiens à remercier mon directeur de recherche, Peter, pour son sou-tien et ses conseils tout au long de mes études aux cycles supérieurs. Je lui suis re-connaissant de m’avoir laissé beaucoup de latitude et laissé découvrir des trucs par moi-même, tout en m’appelant à corriger le tir lorsque nécessaire. Merci aussi pour toutes les opportunités que tu m’as offertes et qui m’ont beaucoup appris !
Je tiens à remercier en particulier ceux qui m’ont initié aux méthodes expérimentales de sciences des surfaces et formé sur le microscope : Vincent et Guillaume. De plus, j’aimerais exprimer ma gratitude à Yi, avec qui j’ai aussi passé de longues soirées devant le microscope.
J’aimerais aussi exprimer ma sollicitude envers mes collègues que j’ai eu le plaisir de côtoyer au laboratoire : Raphaël, Yang, Tianchi, Carole-Anne et Keramat. Un merci tout spécial aussi à nos pauvres stagiaires qui ont passé des étés à faire du comptage, soit l’assignation manuelle des masques sur tous les motifs moléculaires : Carole-Anne, Mireille, Mathis, Jean-Simon et Ginette.
J’ai une grande dette scientifique envers nos collaborateurs, qui ont grandement bo-nifié mes travaux. Michael Groves deserves high praise for his DFT calculations, as well as his insightful comments for the articles. Je tiens aussi à remercier Vincent Albert du groupe Boukouvalas pour l’élégante synthèse du (R)-8MeNEA.
Un merci tout spécial aussi au personnel de soutien du Département et de la Faculté : Magali, Denyse, Mélanie, Jean, Sébastien, André et Normand. Merci de m’avoir
dé-panné et aidé à régler tous les petits problèmes administratifs et techniques auxquels j’ai pu faire face. I would also like to thank George Lengel, from SPECS, for his recur-ring help in fixing the STM.
Je tiens aussi à remercier mon comité de thèse, qui ont su formuler bonifier ma thèse par leurs commentaires. Je tiens à leur faire part de ma gratitude pour leur disponi-bilité au moment de la défense.
Je tiens aussi à remercier le fonds de soutien départemental ainsi que le FQRNT pour le support financier.
Merci aussi à mes compagnes de rédaction au cours de la dernière année. La rédac-tion en groupe est toujours plus motivante que seul ! Je conclus mes remerciements par mes parents, mon frère et mon ex-copine, qui ont toujours été là pour moi. Un merci spécial à Marie-Ève, mon ex-copine, pour ses conseils qui m’ont permis de rendre plus attrayantes beaucoup des figures de cette thèse. Mes pensées vont aussi à mon oncle Jeannot, qui nous a quittés alors que je m’apprêtais à défendre cette thèse.
Avant-propos
Les chapitres 3 à 5 présentent des résultats sous forme d’articles, dont je suis le seul auteur principal. Pour ces trois chapitres, les observations par microscope à effet tun-nel, leur analyse et leur interprétation ont été réalisées au sein de laboratoire des sciences des surfaces de l’Université Laval sous l’égide du Pr McBreen. L’analyse au-tomatisée des complexes utilisée pour les chapitres 4 et 5 emploie l’algorithme déve-loppé par Guillaume Goubert et Yi Dong, auquel j’ai apporté quelques ajustements. J’ai réalisé les figures et les schémas présentant ces données. Les calculs théoriques sont l’œuvre du Pr Michael Groves, alors qu’il était stagiaire postdoctoral au sein du groupe du Pr Hammer à l’Université d’Aarhus. Le chapitre 3 a été publié en avril 2016, tandis que les chapitres 4 et 5 sont en cours de préparation et devraient être soumis sous peu.
J’ai écrit le manuscrit du chapitre 3 avec le Pr McBreen, Vincent Demers-Carpentier et le Pr Michael Groves. Les expériences STM et leur analyse ont été menées par Vincent Demers-Carpentier, Guillaume Goubert, Yi Dong et moi-même.
Le manuscrit du chapitre 4 est ma réalisation, avec plusieurs commentaires, sugges-tions et révisions des Pr McBreen et Groves. Vincent Albert, sous la supervision du Pr Boukouvalas, s’est chargé de la synthèse, de la caractérisation et de l’identification du (R)-8MeNEA. J’ai planifié et réalisé les expériences STM avec Yi Dong, et supervisé le comptage accompli par les stagiaires d’été, Carole-Anne Fortin, Ginette N’guessan et Jean-Simon Frenière, en plus d’en exécuter une large part.
J’ai écrit le manuscrit du chapitre 5 en collaboration avec le Pr McBreen en utilisant des résultats DFT du Pr Michael Groves. J’ai réalisé et planifié toutes les expériences STM, dont certaines avec Yi Dong. Je me suis chargé de l’analyse et de l’interprétation des données ainsi que de la majorité du comptage, avec l’appui de Carole-Anne Fortin et de Mireille Ouellet.
Introduction
L’expansion de la population humaine au cours du dernier siècle s’est faite largement en parallèle avec l’amélioration des conditions de vie matérielles pour la grande ma-jorité de gens. Ce fait – inusité dans l’histoire humaine – repose sur plusieurs facteurs, auxquels les progrès en chimie ne sont pas étrangers. Une meilleure compréhension et un meilleur contrôle des processus chimiques permirent la synthèse des matériaux à la base de notre technologie électronique, des fibres de nos vêtements, des engrais sans lesquels nous ne pourrions pas nourrir près de huit milliards d’êtres humains ou encore des traitements médicaux qui ont allongé notre espérance de vie au-delà de 80 ans dans les pays développés.
La chimie verte est une nouvelle façon d’aborder la chimie qui se propose de déve-lopper de nouvelles approches moins polluantes que les méthodes conventionnelles de synthèse. La catalyse hétérogène, qui est moins énergivore et requiert moins de solvants, s’inscrit dans cette tendance [1–5]. Des progrès en catalyse pourraient donc contribuer à la résolution de certains enjeux environnementaux et de santé auxquels nous faisons toujours face. Le champ d’application de la catalyse hétérogène demeure assez réduit et ses mécanismes fondamentaux, souvent peu compris. Les travaux pré-sentés dans cette thèse tentent d’apporter un éclairage sur une étape clé d’une réac-tion de catalyse hétérogène asymétrique.
0.1
Enjeux de société et chimie
0.1.1 Environnement
Les progrès technologiques fulgurants du XXesiècle ne furent pas sans coûts,
par-ticulièrement élevés au niveau environnemental. Le taux de disparition des espèces – indicateur de la biodiversité – est comparable à celui observé lors de l’extinction Crétacé-Tertiaire qui a mené à la disparition des dinosaures [6]. La cause principale
de cette extinction de masse semble être l’activité humaine. Par exemple, le monarque d’Amérique est menacé par la disparition de l’asclépiade – une source essentielle de nourriture pour les chenilles [7] – due à l’emploi massif de désherbants ; tandis que le béluga du Saint-Laurent l’est par la présence d’hydrocarbures aromatiques cycliques, de composés organochlorés ou encore de métaux lourds dans l’estuaire du Saint-Laurent [8]. Cela est sans mentionner l’inquiétante diminution de la quan-tité d’insectes récemment rapportée, insectes qui sont à la base de larges pans de la pyramide alimentaire [9].
Le réchauffement de la planète reste la crise environnementale la plus discutée de notre époque [10]. Une large part de l’énergie consommée par nos sociétés provient de la combustion de carburants fossiles. Les produits de cette combustion sont l’eau et le dioxyde de carbone ; ce dernier étant le principal gaz à effet de serre. Il est donc primordial de trouver des moyens de limiter notre consommation d’énergie. Plu-sieurs biens courants améliorant notre qualité de vie utilisent des matériaux dont la synthèse est énergivore. L’approche catalytique – par définition moins énergivore – peut donc aider à réduire la production de gaz à effet de serre.
L’extraction de ressources naturelles – renouvelables et non renouvelables – peut en-dommager l’écosystème dans lequel nous évoluons. Cet écosystème nous rend de grands services qui seraient autrement très dispendieux. Ne pensons qu’aux Marais du Nord à la tête du lac Saint-Charles. Ils filtrent et oxygénisent – gratuitement et efficacement – les eaux de surface qui ruissèlent vers ce lac, qui est la source d’eau potable pour la ville de Québec. Cet exemple illustre pourquoi il est primordial de bien mesurer, et limiter, l’impact des activités humaines sur le milieu, afin d’assurer la qualité de vie de tous.
Les questions environnementales deviennent de plus en plus importantes dans les discours publics, et avec raison. Les risques de déversements pétroliers pouvant dé-truire des pêcheries, des aquifères ou des terres agricoles expliquent une large part de l’opposition aux projets d’oléoducs ou d’exploitation pétrolière [11]. Conjuguer progrès technologique et protection de l’écosystème est un défi majeur pour nos ci-vilisations. La chimie se doit de faire partie de la solution, autant pour réduire le stress posé sur notre environnement, ou mieux employer et réutiliser les ressources matérielles et énergétiques limitées dont nous disposons.
0.1.2
Santé
L’augmentation remarquable de l’espérance de vie au cours du siècle dernier n’aurait pas été possible sans la mise au point de nouvelles classes de médicaments et l’amé-lioration des conditions hygiéniques. Un environnement sain demeure évidemment un déterminant important d’une bonne santé. Les défis de santé auxquels nos socié-tés post-industrielles font face sont le manque d’activité physique et une alimentation de mauvaise qualité ou la consommation, généralement involontaire, de substances nocives. Un effort de prévention et de changement de mauvaises habitudes est mani-festement de mise. Développer de nouvelles méthodes de production, de transport et de préservation des fruits et légumes frais pourrait diminuer leur coût et augmen-ter leur disponibilité, particulièrement au sein de communautés défavorisées. Quant au développement de nouveaux médicaments plus sélectifs, moins coûteux et dont la production est moins polluante, beaucoup d’espoir est engendré par les progrès importants en synthèse.
0.1.3 Rôle de la chimie dans la recherche de solutions
Plusieurs problèmes sociaux contemporains peuvent être liés aux enjeux environ-nementaux ou de santé. Pauvreté, guerres et épidémies sont amplifiées, voire cau-sées par l’épuisement de ressources fondamentales [12] . La chimie peut contribuer à l’amélioration de l’extraction ou de la réutilisation de ces ressources non renou-velables, mais elle peut surtout développer des façons d’utiliser des ressources re-nouvelables de façon durable pour répondre à nos besoins. Par exemple, l’épuise-ment éventuel des ressources pétrolières fait craindre la disparition des précurseurs pour la synthèse de plastiques. Des méthodes de synthèse alternatives – utilisant de monomères provenant de ressources renouvelables – permettront de pérenniser la production de plastiques.
Les efforts en recherche et en développement de l’industrie pharmaceutique, tout en permettant le développement de nouveaux médicaments, sont un facteur important de l’explosion des coûts de santé. Cette augmentation des coûts est une préoccupa-tion majeure pour bien des gens et des sociétés, en particulier dans un contexte de vieillissement de la population. Cet exemple permet de cerner la question plus géné-rale du partage des risques, des coûts et des bénéfices associés à l’industrie chimique. L’acceptabilité sociale de la chimie découle des réponses que la communauté scien-tifique sera en mesure d’apporter à cette question [13].
À mon avis, la pertinence et l’acceptabilité sociale de la science dépendent des solu-tions que les chercheurs peuvent développer relativement aux défis contemporains en environnement et en santé. L’approche catalytique – moins énergivore – sera ap-pelée à jouer un grand rôle dans la recherche de solution, et à plus forte raison la ca-talyse hétérogène, pour laquelle la récupération et la réutilisation du caca-talyseur sont plus faciles [14]. Les travaux présentés dans cette thèse essaient justement d’élucider le mécanisme d’une réaction de catalyse hétérogène.
0.2 Défis de la chimie
Les défis environnementaux et dans le domaine de la santé se traduisent par des dé-fis plus spécifiques en chimie [14]. Permettez-moi de me concentrer exclusivement sur les défis auxquels les travaux présentés dans cette thèse touchent, parfois direc-tement, parfois tangentiellement :
— une meilleure compréhension de la catalyse hétérogène ; — le design rationnel de médicaments ;
— la simulation de systèmes complexes ;
— le développement de meilleures méthodes en chimie théorique.
L’utilisation de catalyseurs, qui abaissent l’énergie d’activation d’une réaction sans être eux-mêmes consommés, permet d’améliorer l’efficacité énergétique des réac-tions chimiques. Une approche catalytique est donc à privilégier autant que pos-sible dans une perspective environnementale. La catalyse hétérogène, où le cataly-seur n’est pas dans la même phase que les réactifs et produits, offre souvent une voie de synthèse plus économique et écologique, et est donc favorisée en industrie. Ce-pendant, la conception de nouveaux catalyseurs hétérogènes s’avère difficile et les progrès dans ce domaine sont lents, ce qui s’explique, du moins en partie, par une connaissance insuffisante des mécanismes réactionnels. Une meilleure compréhen-sion de la catalyse hétérogène par une meilleure compréhencompréhen-sion de ses mécanismes est donc le premier des défis spécifiques de la chimie ici exposé. Un meilleur contrôle d’une réaction catalytique hétérogène, et de la rétroaction relativement aux condi-tions dans le réacteur catalytique qui évoluent, sont difficiles à mettre en œuvre en catalyse hétérogène. Ces concepts de contrôle et rétroaction permettent robustesse et sélectivité, et font la force de réactions catalytiques enzymatiques. Les découvertes
dans ce domaine ouvrent des portes pour le développement de nouvelles avenues catalytiques hétérogènes.
La vague d’optimisme qui a suivi le séquençage de l’ADN laissait espérer le dévelop-pement rapide et efficient de médicaments pour traiter la plupart des maux qui nous affligent. Malgré le tour de force que représente le séquençage du génome humain, son application directe à la synthèse de médicaments reposait sur une relation di-recte et univoque entre l’ADN, la structure des protéines synthétisées à partir de brins d’ADN spécifiques, et la définition d’un site actif bien défini sur ces protéines. Cepen-dant, ces relations sont très complexes et non évidentes. De la même façon qu’avoir un dictionnaire ne permet pas de comprendre le cursus littéraire d’une langue, le séquençage de l’ADN ne permet pas de comprendre la biologie moléculaire. Bien que le défi de définir les mécanismes moléculaires sur lesquels nous pouvons inter-venir pour soigner les patients appartienne au domaine biomédical, la chimie a un défi connexe à relever : le design rationnel de médicaments. Pour avoir un contrôle adéquat de la forme et de la fonction des molécules thérapeutique, il est nécessaire d’avoir des synthèses qui soient sélectives, et plus spécifiquement pour les travaux ici présentés, énantiosélectives (voir section0.3).
Plusieurs réactions chimiques sont désormais mieux comprises grâce aux progrès en chimie quantique, et particulièrement par l’application de la théorie de la fonc-tionnelle de la densité (DFT : Density Functional Theory). Kohn s’est d’ailleurs vu remettre le Prix Nobel de chimie en 1998 pour le développement de la DFT [15]. Une part des interactions, les interactions d’échange-corrélation, n’est pas connue. On doit donc tester la validité des différentes approximations utilisées pour modéliser ces in-teractions. C’est pourquoi il est primordial de recourir à des systèmes tests permet-tant de comparer les prédictions de la chimie computationnelle aux résultats expé-rimentaux. Un exemple illustrant la pertinence de tels systèmes tests est notre col-laboration fructueuse avec l’équipe du Pr Hammer, dont les prédictions théoriques par DFT ont été mises à l’épreuve et validées par l’analyse statistique de nos images STM [16].
Ces défis de la chimie peuvent être, en partie, relevés par un dur labeur impliquant les méthodes et les instruments à notre disposition. Le développement de nouvelles techniques analytiques pourrait permettre d’obtenir plus d’informations pertinentes à la résolution des défis énoncés ci-haut, et même d’ouvrir de nouveaux domaines de recherche [17]. La croissance remarquable des connaissances en chimie des surfaces
à la suite du développement et de l’adoption de la microscopie à sonde locale illustre l’impact qu’une nouvelle technologie peut avoir sur un domaine de recherche. Mes travaux de doctorat portent sur l’étude des mécanismes de transfert de chiralité en surface, une étape clé lors de réactions de catalyse hétérogène asymétrique. Plus spécifiquement, j’étudie l’hydrogénation énantiosélective de cétones activées sur une surface chiralement modifiée par la cinchonidine ou des molécules structurellement apparentées. La recherche fondamentale ne se fait pas en vase clos : elle se doit de répondre à des préoccupations sociétaires et du domaine d’étude. Mes travaux, bien que fondamentaux, contribuent à l’avancement des connaissances en catalyse hétéro-gène. À terme, ils peuvent aider à développer des méthodes de synthèse plus vertes pour des précurseurs de médicaments. Par exemple, le (R)-pantolactone généré par la synthèse asymétrique de cétopantolactone peut être un précursuer pour la syn-thèse de vitamine B12 ou de la coenzyme B [18, 19]. Grâce à un microscope à effet tunnel, j’étudie une étape clé d’une réaction de catalyse hétérogène en cataloguant les géométries d’assemblages de différents complexes bimoléculaires. Ces résultats sont ensuite comparés avec des structures calculées par DFT pour mieux comprendre les interactions chimiques expliquant ces géométries particulières. Ces calculs sont ef-fectués par nos collaborateurs du groupe Hammer, à l’Université d’Aarhus. J’espère avoir réussi à établir par cette section la pertinence de mes travaux de doctorat. Il convient toutefois de rappeler ici que mes contributions ne répondent que très par-tiellement aux défis énoncés, et ce n’est qu’avec les contributions d’une large commu-nauté de chercheurs que des solutions globales aux défis énoncés plus haut pourront être développées et mises en place.
0.3
Chiralité
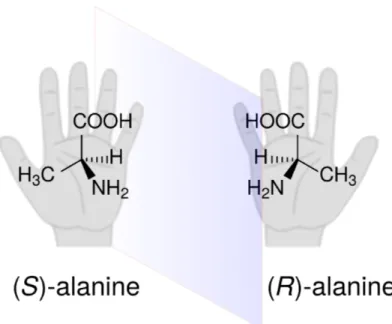
Un objet sera dit chiral s’il n’est pas superposable à son image miroir. Nos mains sont chirales ; la main droite est l’image miroir de la main gauche et elles ne peuvent être superposées. Cette propriété de nos mains est d’ailleurs reliée à l’origine étymolo-gique du mot chiralité, qui provient du terme grec 𝜒𝜖𝜄𝜌, signifiant main. La Figure0.1
illustre d’ailleurs cette analogie entre une molécule chirale, l’acide aminé alanine, et nos mains. Chaque forme chirale d’une molécule sera nommée énantiomère. Donc pour l’alanine nous avons l’énantiomère (R) et l’énantiomère (S).
Figure 0.1 – L’acide aminé alanine est présent en deux énantiomères, soit le (R) et le (S). Ces deux énantiomères sont constitués des mêmes groupements, et sont l’image miroir non superposable l’un de l’autre, tout comme nos mains.
de rotation impropre (𝑆𝑁)1, où 𝑁 est un entier positif [20]. Bien que les molécules
chi-rales les plus communes sont celles impliquant des carbones tétravalents, plusieurs classes de molécules peuvent être chirales. Par exemple, certains complexes métal-liques et métallocènes, ou des molécules possédant des axes de chiralité, de l’hélicité ou des plans de chiralité peuvent être chiraux [21]. Pour un carbone asymétrique, deux arrangements équivalents de quatre substituants distincts existent, et ne sont pas superposables. Dans un tel cas, ce carbone sera alors nommé centre chiral. Les molécules chirales présentées dans cette thèse comportent toutes un carbone agissant comme centre chiral.
Au chapitre précédent, nous avons brièvement mentionné qu’une réaction pouvait être énantiosélective, c’est-à-dire qu’elle ne forme qu’un seul énantiomère. La no-menclature (R,S) utilisée jusqu’ici pour nommer les deux énantiomères repose sur les règles de Cahn, Ingold et Prelog qui décrivent la configuration absolue d’une mo-lécule [22]. Une priorité – principalement basée sur le numéro atomique – est assignée à chaque substituant autour du centre chiral. Lorsque la molécule est placée de ma-nière à ce que le substituant de plus faible priorité soit derrière le centre chiral. Si le sens de la rotation joignant les substituants en ordre de priorité décroissante (1 → 3)
1. 𝑆𝑁consiste en une rotation de 2𝜋/𝑁 autour d’un axe suivie d’une réflexion par rapport à un
est horaire, la molécule sera (R), du latin Rectus pour droite ; si elle est anti-horaire, la molécule sera (S) du latin Sinister pour gauche. Une molécule peut avoir plusieurs centres chiraux, chacun pouvant avoir une configuration distincte. Une molécule avec deux centres chiraux sera d’ailleurs nommée diastéréoisomère.
Il existe deux autres nomenclatures pour la chiralité. Pasteur a découvert que les sels d’acide tartrique forment deux types de cristaux, qui sont l’image miroir l’un de l’autre [23]. Le plan de polarisation de la lumière polarisée qui passe au travers de ces deux types de cristaux est dévié dans des directions opposées ; le sens et la magni-tude de cette déviation est nommée pouvoir rotatoire. Si le plan de polarisation est dévié vers la droite pour un observateur vers lequel le faisceau lumineux se dirige, la substance étudiée sera dextrogyre et notée (+). Dans le cas d’un pouvoir rotatoire opposé, la substance sera dite lévogyre et notée (-). Il est intéressant de noter qu’avec des méthodes prédatant la preuve de l’existence des atomes ou la cristallographie, il était possible de distinguer deux énantiomères. Par contre, on ne peut conclure de la configuration absolue d’une molécule par son pouvoir rotatoire ; une molécule (R) peut être dextrogyre ou lévogyre, et une substance dextrogyre peut être composée de molécules (R) ou (S). Une dernière nomenclature, utilisée encore en biologie, iden-tifie chacun des énantiomères en utilisant les lettres D ou L selon qu’ils partagent la même configuration relative que le (+)-glycéraldéhyde ou le (-)-glycéraldéhyde, respectivement [24]. Dans cette thèse, la nomenclature (R, S) sera utilisée presque exclusivement.
Ayant la même composition, deux énantiomères d’une même molécule auront les mêmes propriétés chimiques et physiques, à l’exception de leur effet sur le plan de polarisation de la lumière et du spin [25]. À l’opposé, leurs propriétés biologiques peuvent différer de façon importante, et parfois même dramatiquement. Il appert que la biochimie du vivant repose sur l’homochiralité de nombreuses classes de mo-lécules. Par exemple, tous les glucides naturels sont D tandis que les acides aminés à la base des protéines sont L [24]. Cette homochiralité du vivant se traduit par une sensibilité de certains récepteurs biochimiques variant selon l’énantiomère lié. Par exemple, les récepteurs olfactifs vont identifier un arôme de térébenthine lorsque que le (S)-limonène est détecté, et d’agrumes en présence du (R)-limonène. Dans le sec-teur agroalimentaire, seul l’énantiomère (R) de l’herbicide dichlorpop est actif [26]. De manière plus générale, il est fréquent que deux énantiomères soient métabolisés selon deux chemins réactionnels différents résultant en des résultats pharmacolo-giques autres [27]. La Figure 0.2 illustre l’activité biologique divergente des deux
énantiomères du praziquantel. Seul l’énantiomère (R) permet d’éliminer les schisto-somes, un parasite, tandis que l’énantiomère (S) a un goût amer [28–30]. Peu de pa-tients terminent leur traitement de praziquantel – dont les comprimés comprennent les deux énantiomères – étant donné la grande taille des comprimés et leur goût amer. Isoler le (R)-praziquantel permettrait de réduire la taille des comprimés et d’éviter le goût amer tout en maximisant l’efficacité anti-parasitaire. Un énantiomère peut être actif pour une réaction biochimique particulière tandis que l’autre peut trouver un autre récepteur ailleurs dans le corps et avoir un effet secondaire possiblement toxique. Par exemple, l’énantiomère (R) de la thalidomide est utile pour contrer les nausées matinales de femmes enceintes, l’énantiomère (S) est quant à lui tératogène et cause des malformations chez l’enfant qu’elle porte [31]. C’est pour ces raisons que près de la moitié des médicaments les plus vendus – incluant le Lipitor, le Plavix et le Nexium – sont désormais énantiopurs [32].
Figure 0.2 – Structure des deux énantiomères du praziquantel. Seul l’énantiomère (R) est actif contre les schistosomes, tandis que l’énantiomère (S) laisse un goût amer en bouche.
Certains énantiomères de molécules sont facilement extraits de supports biologiques. Pour des applications à l’activité humaine, les quantités requises requièrent souvent l’utilisation de plus grandes quantités que ce qui peut être extrait du milieu naturel ; c’est d’ailleurs pourquoi l’industrie chimique est si développée et répandue. Consi-dérons le praziquantel, pour lequel il n’existe pas encore, à notre connaissance, de méthode économique et verte pour isoler l’énantiomère (R) [28]. Deux stratégies se-raient plausibles, soit la séparation des énantiomères ou la synthèse sélective d’un seul énantiomère.
0.3.1
Séparation
La méthode de séparation d’énantiomères la plus élémentaire est la chromatogra-phie chirale [33]. La phase stationnaire est choisie de façon à ce qu’elle interagisse différemment avec chacun des deux énantiomères que l’on veut séparer ; elle se doit donc d’être chirale. L’énantiomère interagissant le plus fortement avec la phase sta-tionnaire passera plus de temps dans la colonne et sera extrait plus tard.
On peut aussi former des complexes — de façon ionique ou covalente — entre un seul énantiomère et une autre molécule. Les nouvelles propriétés physico-chimiques, comme la solubilité, de ces complexes permettent la séparation. On peut aussi former ces complexes de façon à ce qu’ils soient plus faciles à cristalliser, et que les cristaux ainsi formés soient énantiopurs.
La résolution cinétique de deux énantiomères est aussi possible. Dans ce cas, on consomme spécifiquement un énantiomère, souvent par l’utilisation d’un enzyme sélective. La solution finale sera donc plus riche en l’énantiomère le moins réactif pour la réaction ciblée.
On peut aussi combiner plusieurs approches pour séparer les énantiomères. On peut ainsi former des diastéréoisomères conçus afin qu’ils puissent interagir plus forte-ment avec une phase stationnaire non chirale que l’énantiomère non ciblé. Il est à noter que la plupart de ces méthodes requièrent l’utilisation d’importants volumes de solvants, et impliquent donc un coût environnemental élevé. D’importants efforts sont d’ailleurs en cours afin d’optimiser la consommation de solvants [34].
0.3.2 Synthèse
Plusieurs méthodes existent permettant de ne synthétiser qu’un énantiomère d’une molécule [33]. Il est aussi possible de mettre en valeur le métabolisme de micro-organismes pour produire à grande échelle certains acides aminés de façon énan-tiosélective. Il s’agit de la méthode de synthèse chirale par fermentation, dont le champ d’action est limité aux molécules pouvant être synthétisées par des micro-organismes, génétiquement modifiés ou pas.
On peut utiliser un précurseur chiral auquel on applique des réactifs achiraux. D’autre part, il est possible de former des complexes impliquant un seul énantio-mère et une autre molécule chirale, appelée auxiliaire chiral pour l’application qui suit. Ces diastéréoisomères peuvent alors être soit favoriser ou inhiber certaines voies
de synthèse. Ces deux approches permettent donc à divers groupements d’être fixés sélectivement à un squelette chiral. Ces méthodes requièrent des précurseurs et des réactifs facilement disponibles et abordables ainsi qu’un bon rendement des réactions appliquées au squelette chiral.
La dernière méthode présentée pour obtenir un seul énantiomère est la catalyse asy-métrique. On peut obtenir de la sélectivité — et de l’énantiosélectivité — par les trois types de catalyseurs présentés au chapitre précédent, soit la catalyse enzymatique, la catalyse homogène et la catalyse hétérogène. La plus faible énergie d’activation des réactions catalytiques permet certaines synthèses autrement difficilement réalisables. Cependant, la voie catalytique pour la synthèse de certains produits requiert parfois des catalyseurs chers ou n’a pas encore été développée. Cette thèse porte spécifique-ment sur une réaction de catalyse hétérogène asymétrique ; la prochaine section por-tera donc sur la chiralité et sa transmission à la surface d’un catalyseur hétérogène.
0.4 Catalyse hétérogène asymétrique
L’objectif de la catalyse hétérogène asymétrique est de transformer une molécule achi-rale en une molécule chiachi-rale par une réaction énantiosélective. L’adsorption d’une molécule planaire dont le plan moléculaire est parallèle à la surface sur une surface cause nécessairement un bris de symétrie. Une seule face est sélectivement exposée à la surface, comme la cétone activée présentée à la Figure0.3[35]. Pour l’hydrogéna-tion de la cétone du TFAP, l’hydrogène atomique proviendra de la surface de platine et viendra attaquer le lien double entre l’oxygène et le carbone. Deux orientations non équivalentes de ce lien C –– O existent sur la surface ; l’hydrogénation de chacune de ces orientations donnera un énantiomère différent de l’alcool. Pour l’alcool (S), on nommera le précurseur en surface pro-S, tandis que la cétone pro-R donne l’alcool (R).
On peut utiliser la surface directement pour induire la chiralité. La surface d’un so-lide chiral cristallisé sera nécessairement chirale, mais notre discussion ici porte sur les métaux, dont la structure est achirale. Il est possible de couper des cristaux métal-liques selon certains axes afin d’exposer certains sites intrinsèquement chiraux [36–
38]. Watson et Attard ont démontré que ce type de surface peut électro-oxyder sélec-tivement un seul énantiomère du glucose [39].
Figure 0.3 – La cétone activée TFAP peut être adsorbée selon deux formes sur la sur-face de platine. L’hydrogénation de la cétone de chacune de ces deux formes aboutit à un énantiomère différent de l’alcool. Chaque forme sur la surface est nommée d’après l’énantiomère de l’alcool résultant de son hydrogénation. Tiré de [35].
à fixer des molécules chirales sur une surface. Par exemple, des catalyseurs asymé-triques homogènes peuvent être fixés sur des surfaces d’oxydes métalliques [40]. Les systèmes catalytiques résultants sont malheureusement souvent compliqués et dis-pendieux [41]. L’induction de chiralité par l’adsorption de molécules chirales sur une surface métallique est une approche qui a eu du succès pour quatre systèmes :
— l’hydrogénation des β-cétoesters sur une surface de nickel de Raney modifiée par de l’acide tartrique (réaction d’Izumi) [42] ;
— l’hydrogénation de cétones activées sur du palladium ou du platine chira-lement modifié par la cinchonidine ou une molécule apparentée (réaction d’Orito) [43] ;
— l’hydrogénation d’oléfines sur du palladium modifié par de la cinchonidine ou la (S)-proline [44] ;
— l’addition d’acides arylboroniques à des composés carbonyles α,β-insaturés sur un catalyseur de rhodium modifié par un ligand bifonctionnel comprenant un groupement diène et un groupement amide [45].
Seuls les deux premiers ont fait l’objet d’études en sciences des surfaces afin d’en établir le mécanisme de transfert de chiralité. Pour la réaction d’Orito. des complexes 1 :1 seront formés par le substrat pro-chiral avec une molécule chirale, le modificateur chiral. L’interaction entre substrat et modificateur chiral viendra davantage activer la cétone à hydrogéner. Cette activation supplémentaire permet une hydrogénation
beaucoup plus rapide que sur les autres sites catalytiques racémiques nécessairement présents sur la surface. L’information chirale transmise au substrat pro-chiral lors de l’hydrogénation des liens C –– C sur le Pd (111) semble aussi ne provenir que d’une molécule [46].
Les travaux présentés dans cette thèse ont été obtenus dans le cadre de l’étude de la réaction d’Orito. Avant de détailler davantage les phénomènes de surface, nous présentons certains résultats catalytiques qui ont apporté un éclairage pertinent sur la réaction et ont nourri et éclairé les études en sciences des surfaces.
On mesure l’énantiosélectivité d’une réaction soit par le ee, l’excès énantiomérique, ou le er, le ratio énantiomérique, calculés comme suit :
𝑒𝑒 = [𝑅] − [𝑆]
[𝑅] + [𝑆] (1)
𝑒𝑟 = [𝑅] ∶ [𝑆] (2)
On note que le [𝑅] et [𝑆] sont normalisés de telle sorte que [𝑅] + [𝑆] = 100.
0.5
Études catalytiques motivant l’étude du système
par les sciences des surfaces
La réaction d’Orito, illustrée à la Figure 0.4, se déroule dans un milieu catalytique complexe comprenant une surface métallique et son support catalytique, un solvant et parfois des additifs, un modificateur chiral, un réactif et son alcool correspondant produit par la réaction, et bien entendu de l’hydrogène [43]. L’approche des sciences des surfaces présentée dans cette thèse se veut réductionniste ; nous ne nous concen-trons que sur les interactions entre modificateur chiral et substrat pro-chiral sur un monocristal de platine dans un environnement ultra-haut vide. Évidemment, plu-sieurs interactions importantes pour améliorer le rendement et la sélectivité de la réaction sont négligées par notre approche. La brève présentation des résultats obte-nus sur le système catalytique ici présentée ne se veut pas exhaustive ; elle permet de mieux cerner les questions auxquelles nos travaux peuvent contribuer à répondre. Tout d’abord, les nanoparticules de platine utilisées en catalyse se retrouvent sur un support d’oxyde d’aluminium ou de silicium, qui peut avoir une influence sur la
ré-Figure 0.4 – Réaction d’Orito. Une cétone activée est hydrogénée de façon énantiosé-lective sur des nanoparticules de platine (ou de palladium) sur lesquelles est adsor-bée une molécule chirale appelée modificateur chiral. La réaction se déroule dans un solvant dans lequel de l’hydrogène est dissous [43].
action. En effet, le groupe de Baiker a pu démontrer que l’acidité du support était corrélée positivement avec l’énantiosélectivité et négativement avec la vitesse de ré-action [47]. Baiker et collab. proposent comme explication un transfert électronique du platine vers le support. Ce changement de l’état électronique du platine vient alors influer sur la chimisorption de l’hydrogène, du substrat pro-chiral et du modificateur chiral. Cependant, la magnitude de ces effets n’a pas été quantifiée, et l’hypothèse de Baiker, bien que fort plausible, ne demeure qu’une supposition.
Les meilleures sélectivités et activités obtenues pour la réaction d’Orito l’ont été sur des nanoparticules de platine d’environ 3 nm de diamètre, soit une taille suffisante pour que la cinchonidine ou une molécule apparentée puisse être adsorbée sur une terrasse avec un faible indice de Miller [48]. Malgré la plus faible réactivité des atomes sur des terrasses comparativement à ceux sur des décrochements, la présence du mo-dificateur chiral permet à la réaction d’avoir lieu avec une bonne activité sur la ter-rasse. Baiker et collab. ont comparé le rendement catalytique de nanoparticules de formes différentes, et donc de différents ratios de terrasses de faible indice de Mil-ler [49]. Ils ont pu établir que c’était à la face (111) du platine que se déroulait la ré-action avec la meilleure énantiosélectivité. Ce résultat — en déterminant la meilleure surface pour la réaction — nous a permis de sélectionner le bon cristal sur lequel effectuer nos études en sciences des surfaces : le Pt (111).
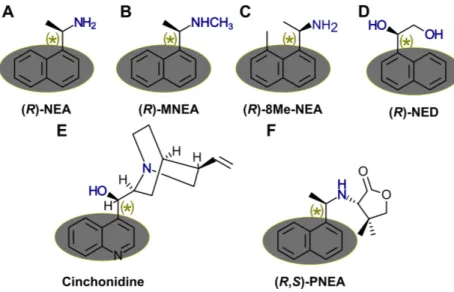
La présence d’une molécule chirale — le modificateur chiral — est nécessaire pour induire préférentiellement la formation d’un énantiomère. Le premier modificateur chiral utilisé, et souvent le plus efficace, est la cinchonidine [43,46]. La Figure0.5 pré-sente certains des modificateurs chiraux testés pour la réaction d’Orito, La cinchoni-dine représente les modificateurs chiraux naturels issus de la famille des alcaloïdes tandis que les autres sont des modificateurs chiraux synthétiques. Tous partagent
certaines caractéristiques structurelles : un centre chiral, un groupement aromatique pour ancrer la molécule, et une amine donneuse de liens hydrogène [46,50]. Les liens C – H aromatiques peuvent aussi agir comme source de liaisons hydrogène pour un éventuel substrat pro-chiral.
Figure 0.5 – Différents modificateurs chiraux pour la réaction d’Orito. Les masques ovales indiquent le groupement aromatique ancrant, l’astérisque verte le centre chiral et les donneurs de liens hydrogène apparaissent en bleu. (A) (R)-1-(1-naphthyl)éthylamine ((R)-NEA). (B) (R)-N-Méthyl-1-(1-naphthyl)éthylamine ((R)-MNEA). (C) (R)-1-(8-méthyl-1-naphthyl)éthylamine ((R)-8MeNEA). (D) (R)-1-naphthyl-1,2-éthanediol ((R)-NED.) (E) Cinchonidine. (F) (R, S)-pantoyl-naphthyléthylamine ((R, S)-PNEA).
La configuration générale des membres de la famille des alcaloïdes a été étudiée en variant la chiralité des centres stéréogéniques et en introduisant différents groupe-ments périphériques. Tout d’abord, il semble que la protonation de l’amine tertiaire soit nécessaire pour que la réaction énantiosélective ait lieu [51]. La protonation fige certains des degrés de liberté internes de la cinchonidine et la rend plus rigide [52]. Le positionnement relatif des différentes sections du modificateur chiral crée différents sites catalytiques où peut venir se lier un substrat pro-chiral. Différentes configura-tions se soldent nécessairement par des sites catalytiques d’efficacités et de sélecti-vités variables pour chaque substrat [46]. La grande flexibilité structurelle des alca-loïdes leur permet d’adopter différentes configurations selon le milieu ; la tempéra-ture, la protonation de l’amine et la nature du solvant influent toutes sur la structure des alcaloïdes [51–53], et donc de présenter différents sites catalytiques. Le substrat pro-chiral sur une surface contrôle lui aussi, en partie, la configuration du modifica-teur chiral adsorbé, et donc sa réactivité. Schmidt et collab. ont en effet pu observer
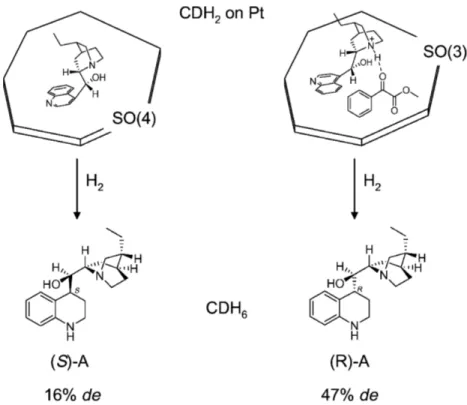
la configuration de la cinchonidine en étudiant les produits de son hydrogénation en présence de divers substrats pro-chiraux [54]. Celle-ci donne des produits distincts selon le substrat auquel elle est liée, comme présenté à la Figure0.6. Il s’agit ici d’un phénomène d’ajustement induit du modificateur.
Figure 0.6 – La présence d’un substrat pro-chiral change le conformère majoritaire de la cinchonidine sur la surface, tel qu’indiqué par le produit de l’hydrogénation de la cinchonidine qui change de chiralité. Issu de [54].
Des modificateurs chiraux synthétiques peuvent aussi être employés pour l’hydro-génation énantiosélective de cétones. Des dérivés de la proline, d’imidazolidinone et de tryptophane ont été étudiés en milieu catalytique et induisent une certaine sé-lectivité, bien que les ee obtenus ne soient pas comparables à ceux atteints avec la cinchonidine [55,56]. Des études catalytiques plus poussées ont été effectuées sur le NEA et ses dérivés [18, 57–60]. Le NEA partage avec la cinchonidine les caractéris-tiques requises pour être un modificateur chiral efficace : un groupement aromatique pouvant servir d’ancre, un centre stéréogénique, et une amine à laquelle un substrat peut se lier par des interactions non covalentes. Le NEA dispose de beaucoup moins de liberté conformationnelle que la cinchonidine en surface. Son adsorption sera dis-cutée à la section suivante. Cette liberté de mouvement moins grande ne signifie pas que le NEA soit incapable de se modifier afin de mieux accommoder un substrat pro-chiral ; des calculs DFT montrent que la géométrie d’adsorption du (R)-NEA
va-rie légèrement en fonction du substrat auquel il est lié [61, 62]. Le NEA peut aussi s’adapter au substrat, et même se lier de façon covalente à ce dernier. Dans le cas du pyruvate d’éthyle (EP : ethyl pyruvate), la réaction de condensation est inévitable dans le toluène [18,59,60].
Le (R)-NED est un modificateur chiral efficace pour l’hydrogénation énantiosélective du KPL, atteignant des ee de 30 % [58]. Sa structure rappelle celle de la cinchonidine et du (R)-NEA. Deux groupements hydroxyles viennent ici remplacer l’amine comme donneur de liaisons hydrogène. Ce changement, combiné à la sélectivité du (R)-NED, soulève la question du mécanisme réactionnel, et donc du mode d’adsorption de ce modificateur chiral. En particulier, si l’hydroxyle le plus près du centre chiral est adsorbé parallèlement à la surface, l’autre en sera nécessairement éloigné.
Orglmeister et collab. ont condensé plusieurs molécules sur l’amine du (R)-NEA afin de trouver un modificateur chiral synthétique efficace pour l’hydrogénation asymé-trique du KPL [18]. Pour un méthyle condensé sur l’amine, le modificateur chiral résultant, le (R)-MNEA, n’a qu’un ee de 32 %, inférieur au ee de 52 % obtenu par le (R)-NEA. Le (R, S)-PNEA, obtenu par la condensation du KPL sur le (R)-NEA, permet d’atteindre un ee de 79 % [18,63].
La Figure 0.7 présente les substrats pro-chiraux, et leurs ee avec la cinchonidine et le (R)-NEA, pertinents pour cette thèse [46]. Ceux-ci partagent certaines similarités structurelles. Tout d’abord, ce sont des molécules planes. La présence d’un groupe-ment électroattracteur en α de la cétone ciblée par la réaction est aussi un point com-mun des substrats efficaces, et semble nécessaire afin d’activer la cétone. Des études en sciences des surfaces et des calculs théoriques ont apporté un éclairage sur la géo-métrie d’adsorption du KPL, du TFAP et du MTFP et seront présentées à la section suivante.
La solubilité — autant du substrat que du modificateur — contrôle la disponibilité de ces molécules sur la surface [46]. Le solvant employé influe directement sur la conformation de la cinchonidine, soit par des interactions directes ou en favorisant la protonation de l’amine tertiaire. Cette amine devient plus à même à faire des liai-sons hydrogène, autant avec d’autres parties de la cinchonidine — permettant une certaine stabilisation intramoléculaire — qu’avec le substrat pro-chiral. Le solvant peut avoir une influence sur la réaction globale en interagissant avec le substrat. Par exemple, l’hydrogénation du TFAP en présence de cinchonidine donne l’alcool (S) pour les solvants plus basiques et l’énantiomère (R) dans les plus acides [66].
L’ad-Figure 0.7 – Différents substrats pro-chiraux pour lesquels la réaction d’Orito est effi-cace. (A) Méthyle 3,3,3-trifluoropyruvate (MTFP). (B) Pyruvate de méthyle. (MP) (C) Cétopantolactone (KPL). (D) 2,2,2-trifluoroacétophénone (TFAP). La cétone activée apparait en bleu et les groupements électroattracteurs en vert. Le meilleur ee obtenu — et l’énantiomère favorisé — sont indiqués sous les schémas de chaque molécule pour la cinchonidine et le (R)-NEA [46,61,64]. Aucun résultat catalytique n’a été pu-blié pour l’hydrogénation énantiosélective du MTFP. L’ ee indiqué pour le MP et le (R)-NEA (*) a plutôt été mesuré pour le pyruvate d’éthyle, tandis que le modificateur chiral effectif est un condensat du (R)-NEA et du pyruvate d’éthyle [65].
duit catalytique implique un hémiacétal pour un solvant basique, qui se liera néces-sairement de façon différente à la cinchonidine. L’étude de l’effet des solvants sur les complexes catalytiques individuels en surface n’en est qu’à ses balbutiements, bien que des études théoriques, comme celle de Steinmann et collab. ont pu démon-trer que l’énergie des différents modes d’adsorption du COOH varie en fonction du solvant [67]. Des études STM en phase liquide sur la solvatation de molécules orga-niques permettent de croire que des études molécule-par-molécule pourront appor-ter un nouvel éclairage sur la contribution du solvant au mécanisme de transfert de chiralité en surface [68].
Il est aussi à noter que la réaction d’hydrogénation énantiosélective du pyruvate de méthyle sur du platine chiralement modifié fonctionne dans un environnement ga-zeux, et que le solvant n’est pas nécessaire à la réaction [69]. Il existe donc certaines interactions fondamentales entre substrat pro-chiral et modificateur chiral qui ne dé-pendent pas nécessairement du solvant, et qui peuvent donc être sondées par des études en environnement ultra-haut vide.
L’ajout d’additifs à la réaction peut aider à en améliorer le rendement et la sélecti-vité. Dans le cadre de la réaction d’Orito, la présence d’acide trifluoroacétique (TFA) peut jouer ce rôle très efficacement, comme pour l’hydrogénation du TFAP par la cinchonidine, dont le ee passe de 50% à 92% en présence de TFA [70–74]. Le groupe
de Baiker a observé que le TFA vient perturber les assemblages de TFAP pro-S avec la cinchonidine, tandis que Brunelle et al. ont relevé que le TFA vient perturber les dimères homochiraux — autant pro-R que pro-S — de TFAP et ainsi réduire leur activité [75,76].
L’adsorption du groupement aromatique des alcaloïdes de la famille des cinchonas ou du (R)-NEA et de ses dérivés a longtemps été assumée comme étant parallèle à la surface. C’est effectivement le cas pour les résultats ultra-haut vide [61, 77, 78]. Les calculs théoriques ne tenant pas compte du solvant supportent d’ailleurs cette hypothèse [61,77–79]. Un point important soulevé par le groupe de Sautet pour l’ad-sorption du naphtalène sur une surface de platine est l’activation des liens C – H aro-matiques comme conséquence de la chimisorption ; liens qui peuvent ensuite agir comme donneurs de liaisons hydrogène [80].
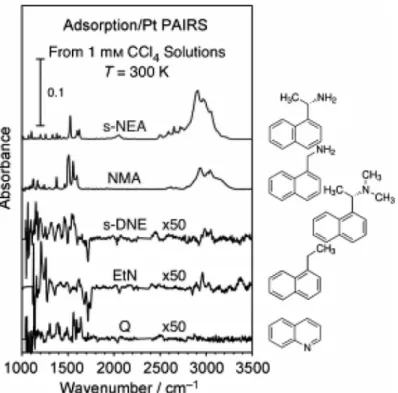
Le solvant semble cependant avoir un effet sur le mode d’adsorption du modificateur chiral. Des travaux menés sous l’égide du Pr Zaera suggèrent que le (S)-NEA adsorbe avec une composante perpendiculaire à la surface dans le tétrachlorométhane [81]. La Figure0.8 relève un signal RAIRS fort pour les vibrations de l’aromatique que pour les molécules possédant une amine pouvant se lier à la surface. La conclusion tirée est que le groupement aromatique du NEA est fortement incliné pour ce substrat et que la liaison à la surface se fait au niveau de l’amine.
Des études in situ et operando par le groupe du Pr Baiker ont été menés pour obtenir plus d’informations sur la géométrie d’adsorption des modificateurs chiraux dans un environnement catalytique. Les modificateurs chiraux ciblés par leurs méthodes spectroscopiques se trouvent à l’interface liquide-solide et subissent à la fois l’in-fluence de la surface catalytique et du solvant. Autant pour la cinchonidine que pour le NEA, les géométries d’adsorption favorisées sont celles pour lesquelles le groupe-ment aromatique est parallèle à la surface, lorsque l’influence du solvant est négligée. Comme pour toutes les mesures spectroscopiques IR en surface, seules les vibrations perpendiculaires à la surface peuvent être observées. Pour le (R)-NEA et le PNEA en phase liquide, des modes de vibration spécifiques du naphtyle apparaissent à 1510 cm-1et à 1597 cm-1, pour des vibrations selon l’axe long et l’axe court, respectivement. Le spectre d’adsorption du (R)-NEA, à la Figure 0.9, relève la présence de ces deux bandes vibratoires, indiquant que chacun des deux axes principaux du naphtyle est incliné par rapport à la surface. Les bandes sont assez larges, suggérant une variété de modes d’adsorption distincts en surface.
Figure 0.8 – Signal RAIRS pour le (S)-NEA et d’autres molécules apparentées. Un signal n’est mesuré que lorsque l’amine est disponible pour se lier à la surface. Tiré de [81].
Pour le PNEA, la bande vibratoire associée à une vibration selon l’axe long du naph-tyle — la bande à 1510 cm-1— est fortement atténuée, indiquant une inclinaison ma-joritairement selon l’axe long. L’absence de la vibration du carbonyle à 1765 cm-1 suggère que le groupement pantoyle est majoritairement parallèle à la surface, si on considère que celui-ci demeure rigide [82].
La cinchonidine dont le groupement quinoléine est incliné par rapport à la surface semble jouer un rôle primordial pour l’hydrogénation énantiosélective de cétones activées. Dans le cas de l’hydrogénation du 4-méthoxy-6-méthyl-2-pyrone sur palla-dium, Rodriguez-Garcia et collab. ont pu démontrer que la cinchonidine « inclinée » permettait une plus grande sélectivité, bien qu’elle soit adsorbée moins fortement que la cinchonidine à plat sur la surface [83]. Un phénomène similaire a été observé par la même équipe [84] pour l’hydrogénation du KPL sur du platine ; ils ont relevé une forte corrélation entre l’intensité du signal associé à la cinchonidine inclinée et une énantiosélectivité accrue.
La cinchonidine inclinée est moins stable que la cinchonidine à plat sur la surface, mais demeure responsable d’une grande part de la réaction sélective. Par contre, si la
Figure 0.9 – Spectres infrarouges pour l’adsorption du (R)-NEA et du PNEA. Le spectre supérieur est obtenu pour les molécules en solution dans le toluène. Ceux du milieu sont obtenus lors d’une exposition croissante de la surface au modifica-teur chiral, tandis que les spectres inférieurs sont obtenus après que la surface ait été rincée pendant 30 minutes. Issu de [81].
différence d’énergie d’adsorption entre deux modificateurs chiraux est trop grande, le modificateur le plus stable occupera majoritairement la surface et sera responsable de la majorité de la réaction. Des mélanges binaires de modificateurs dont l’énergie d’adsorption diffère beaucoup et qui produisent des énantiomères opposés lors de l’hydrogénation d’une cétone particulière permettent d’étudier le lien entre l’énergie d’adsorption du modificateur et son rendement catalytique. L’hydrogénation du py-ruvate d’éthyle en présence de la quinidine donne majoritairement du lactate d’éthyle (S) ; l’addition d’un peu de cinchonidine – plus fortement liée à la surface – entraine la synthèse presque exclusive de l’énantiomère (R) [85].
De nombreux facteurs influencent le rendement et la sélectivité pour la réaction d’ Orito, et une grande variété d’approches — systématiques et réductrices — semblent nécessaires afin d’élucider son mécanisme réactionnel. Les méthodes des sciences des surfaces peuvent sonder avec acuité les interactions entre le substrat et le modifica-teur sur une surface catalytique idéalisée. C’est donc sur ces interactions que porte la prochaine section. L’accent sera mis sur les résultats obtenus par STM, en accord avec leur pertinence relative au contenu de cette thèse.
0.6
Éclairage apporté par les sciences des surfaces sur
la réaction d’Orito
Le modificateur chiral le plus étudié en système catalytique est, comme mentionné à la section précédente, la cinchonidine. La Figure0.10, issue du mémoire de J. Bru-nelle, présente quelques exemples de motifs obtenus pour la cinchonidine [86]. Ce qui frappe l’œil est leur grande variété ; plus de six motifs distincts sont obtenus. Il est fort plausible que seulement un de ces conformères observés soit actif catalyti-quement, si même la forme active de la cinchonidine a été observée lors de ces ex-périences. Aucune étude complète de la cinchonidine sur le platine n’a été présentée dans la littérature jusqu’à maintenant.
Figure 0.10 – Une grande variété de motifs est observée par STM pour la cinchoni-dine [86].
Les travaux de von Arx et collab. indiquent que l’introduction de l’hydrogène dans le système UHV cause une transformation qualitative de certains motifs obtenus pour la cinchonidine [87]. La Figure 0.11 illustre que les motifs apparaissant seulement après l’ajout d’hydrogène sont beaucoup plus mobiles. Si l’on suppose que cet hy-drogène permet la protonation de l’amine, on peut conclure que la cinchonidine dont l’amine est protonée diffuse davantage sur la surface [87]. Sachant que cette proto-nation de l’amine est nécessaire pour que la cinchonidine soit un modificateur chiral efficace, il apparait nécessaire que toute expérience étudiant les complexes qu’elle peut former avec des substrats pro-chiraux doive être effectuée soit sous pression
![Figure 0.10 – Une grande variété de motifs est observée par STM pour la cinchoni- cinchoni-dine [86].](https://thumb-eu.123doks.com/thumbv2/123doknet/3400824.98496/39.918.230.687.427.738/figure-grande-variété-motifs-observée-stm-cinchoni-cinchoni.webp)