© Suryasree Subramania Gangadhara, 2019
L'interaction de SAM68 avec U1 snRNP régule
l'épissage alternatif
Thèse
Suryasree Subramania Gangadhara
Doctorat en biologie cellulaire et moléculaire
Philosophiæ doctor (Ph. D.)
'L'interaction de SAM68 avec U1 snRNP régule
l'épissage alternatif'
Thèse
Doctorat en biologie cellulaire et moleculaire
Philosophiæ Doctor (Ph.D.)
Suryasree Subramania
Sous la direction de :
Résumé
Le profilage transcriptomique global des gènes humains a permis d'estimer que 95% des gènes subissent un épissage alternatif. L'épissage alternatif élargit la diversité de notre génome et le module par des mécanismes de régulation croisée. Les principales petites ribonucléoprotéines nucléaires (snRNP), à savoir U1, U2, U4, U6 et U5, catalysent l'excision des introns d'une manière concertée. Certains des modèles d'épissage prédominants par lesquels l'AS étend la diversité du génome incluent le saut d'exon, les exons mutuellement exclusifs, le site d'épissage alternatif 5' et la sélection du site d'épissage alternatif 3'. Les protéines de liaison à l'ARN (RBP) jouent un rôle majeur dans la régulation de l´épissage alternatif en modulant le recrutement des snRNP aux niveaux des séquences cis-régulatrices; (« enhancer » et « silencer ») situées dans les exons ou les introns. L´objectif actuel dans le domaine de l´épissage est d´établir un « code d´épissage » pour chaque RBP afin de permettre la prédiction de son type d´activité en fonction de la région liée.
Des applications récentes à l'échelle du génome, telles que les microréseaux et l'ARN-Seq, ont mis en lumière des modèles d'épissage souvent négligés, tels que la polyadénylation intronique et la rétention intron. La protéine de liaison à l'ARN, SAM68, module l'épissage alternatif de mTor - qui code mTOR, le régulateur principal de la croissance cellulaire et de l'homéostasie. SAM68 favorise l'épissage intron 5 normal de mTor. Des pré-adipocytes chez des souris déficientes en Sam68 ont montré des défauts de différenciation et une diminution de l'engagement dans la lignée adipocytaire. Ces souris étaient maigres et insensibles à l'obésité d'origine alimentaire. L'analyse à l'échelle d'une micropuce du tissu adipeux blanc de souris Sam68-null a permis d'identifier une régulation à la hausse d'une isoforme tronquée de mTor ; mTori5, qui se termine par transcription dans
l'intron 5, en raison d'un manque d'épissage au site 5´ d´épissage. Cependant, le mécanisme par lequel SAM68 régule les événements d'épissage, en particulier dans le contexte de la reconnaissance du site d'épissage, n'a pas encore été caractérisé à ce jour.
Dans cette thèse de doctorat, je décris une étude approfondie sur le rôle de SAM68 et des régions d'amplificateur intronique dans mTor intron 5 dans la reconnaissance de son site d'épissage 5' amont. Mes résultats mettent en évidence un nouveau rôle du SAM68
dans la modulation du recrutement du snRNP U1 snRNP sur des sites d'épissage de 5'. Je décris la caractérisation biochimique de l'interaction de SAM68 avec U1A, le composant central du snRNP U1 et le rôle de la phosphorylation de la tyrosine SAM68 dans la modulation de cette interaction. Je décris également comment SAM68 par son interaction avec U1 snRNP joue un rôle crucial dans le masquage des signaux de polyadénylation intronique cryptique dans un sous-ensemble de gènes. Collectivement, cette étude contribuera à une meilleure compréhension des éléments introniques et du rôle de SAM68 dans les décisions cruciales en matière d'épissage.
Abstract
Global transcriptome profiling of human genes have led to the estimation that 95% of genes undergo alternative splicing. Alternative splicing expands the diversity of our genome and modulates it by cross-regulatory mechanisms. Major small nuclear ribonucleoproteins (snRNPs) namely U1, U2, U4, U6 and U5 catalyzes intron excision in a concerted manner. Some of the predominant splicing patterns by which alternative splicing expands genome diversity includes include exon skipping, mutually exclusive exons, alternative 5´splice site and alternative 3´splice site selection. RNA binding proteins play a major role in the regulation of alternative splicing by modulating snRNP recruitment and they do so by binding directly to pre-mRNA sequences called splicing enhancers or silencers that are located in exons and/or introns. A current goal in the splicing field is to establish a ‘splicing code’ for each RBP, whereby its activity, as in splicing activation or repression can be predicted based on its binding region relative to splice sites.
Recent genome wide applications such as microarray and RNA-Seq have shed light on the often overlooked splicing patterns such as intronic polyadenylation and intron retention. The RNA binding protein, SAM68, modulates the alternative splicing of mTor – that encodes mTOR, the master regulator of cell growth and homeostasis. SAM68 promotes normal intron 5 splicing of mTor. Pre-adipocytes of Sam68 deficient mice showed differentiation defects and decreased commitment to adipocyte lineage. These mice were lean and unresponsive to dietary induced obesity. Exon-wide microarray analysis of white adipose tissue from Sam68-null mice identified upregulation of a truncated isoform of mTor; mTori5 , that transcriptionally terminates within intron 5 due to lack of splicing at
the upstream 5´splice sites. However, the mechanism by which SAM68 regulates splicing events, particularly in the context of splice site recognition, has not been characterized till date.
In this doctoral thesis, I describe an in-depth study on the role of SAM68 and the intronic enhancer regions in mTor intron 5 in the recognition of its upstream 5´splice site. My results uncover a novel role of SAM68 in modulating U1 snRNP recruitment at 5´splice sites. I describe the biochemical characterization of SAM68 interaction with U1A, the core component of U1 snRNP and the role of SAM68 tyrosine phosphorylation in
modulating this interaction. I also describe how SAM68 by its interaction with U1 snRNP plays a crucial role in masking cryptic intronic polyadenylation signals in a subset of genes. Collectively, this study will contribute to advanced understanding of intronic elements and the role of SAM68 in affecting crucial splicing decisions.
Table of materials
Résumé ... ii Abstract ... iv Table of materials ... vi List of Figures ... viii List of Tables ... x List of Supplementary Figures ... xi List of Abbreviations ... xii Acknowledgements ... xvii Avant-propos ... xix Preface ... xxi INTRODUCTION ... 1 Eukaryotic pre-mRNA processing ... 2 5´ Capping ... 2 Splicing ... 3 3´end processing ... 11 Alternative splicing ... 11 Alternative promoters ... 13 Alternative polyadenylation ... 14 Genome wide technologies to identify alternative splicing events ... 15 Microarrays ... 15 RNA-Sequencing ... 17 Methods to identify RBP binding sites ... 18 Native and cross-linked RNA immunoprecipitation ... 18 In vitro methods ... 22 Non-canonical splicing ... 23 Microexons ... 23 Intronic polyadenylation ... 24 Intron retention ... 25 Regulation of alternative splicing ... 26 Exon and intron sizes in splicing ... 26 Cis determinants of Splicing ... 27 Trans determinants in alternative splicing ... 29 Alternative splicing and disease ... 43 Cis-element mutation and disease ... 43 Trans-factor mutation and disease ... 45Overview of thesis research ... 47 Chapter 1 : SAM68 interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing ... 49 1.1 Avant Propos ... 50 1.2 Preface ... 51 1.3 Résumé ... 52 1.4 Abstract ... 53 1.5 Introduction ... 53 1.6 Results ... 56 1.7 Discussion ... 77 1.8 Material And Methods ... 81 1.9 Supplementary Figures ... 89 Chapter 2: SAM68 and U1 snRNP cooperate to prevent intronic polyadenylation events ... 101 2.1 Avant propos ... 101 2.2 Preface ... 102 2.3 Résumé ... 103 2.4 Abstract ... 104 2.5 Introduction ... 105 2.6 Results ... 106 2.7 Discussion ... 109 2.8 Materials and Methods ... 112 2.9 Supplementary Figures ... 114 Chapter 3 DISCUSSION ... 120 3.1 Role of SAM68 in mTor pre-mRNA splicing ... 120 3.2 Biochemical characterization of SAM68 and U1 snRNP interaction ... 123 3.3 Role of SAM68 and U1A interaction in mTor pre-mRNA splicing. ... 125 3.4 SAM68 orchestrates a splicing program in cooperation with U1 snRNP ... 127 CONCLUSIONS AND FUTURE DIRECTIONS ... 131 Annex A : Tyrosine phosphorylation of SAM68 at (Y386) is critical for U1A interaction 133 Annex B : List of human and mouse genes whose introns contain SAM68 binding motifs close to 5´splice sites ... 135 BIBILIOGRAPHY ... 220
List of Figures
INTRODUCTIONFigure 1 Simplified depiction of constitutive splicing.
Figure 2 The core splicing signals of an intron.
Figure 3 Schematic representation of the step-wise assembly of the spliceosome.
Figure 4 Protein composition of the major human snRNPs.
Figure 5 Illustration of the major modes of AS patterns.
Figure 6 Methods to quantify alternative splicing events.
Figure 7 Schematic representation of UV-CLIP and its variants.
Figure 8 Schematic illustration of polyadenylation events in introns.
Figure 9 Exon and intron definition.
Figure 10 Schematic representation of cis and trans determinants of splicing.
Figure 11 Alteration of gene function by splicing mutations.
Chapter 1: SAM68 interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing.
Figure 1-1 In vivo association of SAM68 with U1 snRNP
Figure 1-2 SAM68 associates with U1A in an RNA independent manner
Figure 1-3 SAM68 interaction with U1A is mediated through its C-terminal domain
Figure 1-4 Tyrosine-rich (YY) domain of SAM68 mediates the interaction with U1
snRNP via YXXY repeated motif
Figure 1-5 Both SAM68 and intronic enhancer sequences in mTor are required for U1
snRNP recruitment to 5´splice sites in vitro
Figure 1-6 U1 snRNP is recruited in a SAM68 dependent manner at the exon/intron 5
junction of mTor pre-mRNA
Figure 1-7 SAM68 deleted of ‘ARM binding region’ shows decreased U1A binding
Chapter 2: SAM68 and U1 snRNP cooperate to prevent intronic polyadenylation events.
Figure 2-1 Presence of SAM68 binding bipartite motifs close to 5´splice sites of
mouse and human introns. mouse and human introns
Figure 2-2 SAM68 motifs are enriched in a subset of genes implicated in Sam68
related biology.
Figure 2-3 SAM68 and U1 snRNP co-regulate intronic polyadenylation of a subset of
Chapter 3: Discussion
Figure 3-1 Model depicting splicing of target transcript co-regulated by SAM68 and
U1 snRNP interaction.
Figure 3-2 Model depicting intronic polyadenylation of target transcript upon loss of
SAM68 mediated U1 snRNP recruitment.
Annex A
Figure A-1 Tyrosine phosphorylation of SAM68 at Y386 is critical for its interaction
List of Tables
Chapter 1: SAM68 interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing.
Table 1-1 List of plasmids generated and primers used for this study Table 1-2 List of DNA and RNA oligonucleotides used in the study Table 1-3 List of primers used for RT-PCR and RT-qPCR
Chapter 2: SAM68 and U1 snRNP cooperate to prevent intronic polyadenylation events.
Table 2-1 List of primers used for RT-PCR validation of target transcripts
Annex B: List of human and mouse genes whose introns contain SAM68 binding motifs close to 5´splice sites.
Table B-1 List of genes common to both mouse and humans whose introns contain the
bipartite SAM68 binding motifs close to the respective 5´splice sites
Table B-2 List of genes in humans whose introns contain the bipartite SAM68 binding
motifs close to the respective 5´splice sites
Table B-3 List of genes in mouse whose introns contain the bipartite SAM68 binding
List of Supplementary Figures
Chapter 1: SAM68 interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing.
Supplementary Figure 1-1 Purification steps of hSAM68-Flag Supplementary Figure 1-2 SAM68 interacts with U1A
Supplementary Figure 1-3 SAM68 C-terminal fragment interacts with U1A
Supplementary Figure 1-4 Subcellular localization of the GFP-tagged SAM68
fragments used for the mapping of U1A interaction.
Supplementary Figure 1-5 NMR titrations.
Supplementary Figure 1-6 CLIP assay on endogenous mTor pre-mRNA
Supplementary Figure 1-7 hSAM68(ΔARM) is deficient in promoting splicing of
intron 5 in mTor minigene in shSAM68 HEK 293T cells.
Chapter 2: SAM68 and U1 snRNP cooperate to prevent intronic polyadenylation events.
Supplementary Figure 2-1 Sequences and oligonucleotide positions used to validate
intronic polyadenylation in Sam68-/- MEFs for Ripk1.
Supplementary Figure 2-2 Sequences and oligonucleotide positions used to validate
intronic polyadenylation in Sam68-/- MEFs for HerC1.
Supplementary Figure 2-3 Sequences and oligonucleotide positions used to validate
intronic polyadenylation in Sam68-/- MEFs for Rock2.
Supplementary Figure 2-4 Sequences and oligonucleotide positions used to validate
intronic polyadenylation in Sam68-/- MEFs for Haus6.
Supplementary Figure 2-5 Sequences and oligonucleotide positions used to validate
List of Abbreviations
APC Adenomatous Polyposis Coli
AR Androgen Receptor
AS Alternative Splicing
BPS Branch Point Sequence
BRK Breast Tumor Kinase
bZLM Basic Leucine Zipper-Like RNA Binding Motif
CaMKIV Calcium/Calmodulin Dependent Kinase IV)
CE Cryptic Exon
ChRIP Chromatin RNA Immunoprecipitation
CLIP Cross-Linked Immunoprecipitation
CNS Central Nervous System
CPSF Cleavage/Polyadenylation Specificity Factor
CstF Cleavage Stimulation Factor
CTD Carboxyl Terminal Domain
CTE Constitutive Response Element
cTNT Cardiac Troponin T
DSCAM Down Syndrome Cell Adhesion Molecule
EGF Epidermal Growth Factor
ESE Exonic Splicing Enhancer
ESS Exonic Splicing Silencer
GMP Guanosine monophosphate
GT Guanylyl Transferase
HITS High-Throughput Sequencing
HTS-EQ High-Throughput Sequencing Analysis Of Equilibrium Binding
iCLIP Individual Nucleotide Resolution CLIP
IGHM Immunoglobulin M Heavy Chain
ILS Intron Lariat Spliceosome
IpA Intronic Polyadenylation
IR Intron Retention
IRS Insulin Receptor Substrate
ISE Intronic Splicing Enhancer
ISS Intronic Splicing Silencers
KH K-Homology Domain
LBD Ligand Binding Domain
m3G Trimethylguanosine
MBNL Muscleblind-like protein
MDS Myelodysplastic Syndromes
mRNA Messenger RNA
mTOR Mechanistic Target Of Rapamycin
NLS Nuclear Localization Signal
NMD Nonsense-Mediated Decay
NPC Neural Progenitor Cells
PAP Poly (A) Polymerase
PAR-CLIP Photoactivable Ribonucleoside-Enhanced Crosslinking
PCPA Premature Cleavage And Polyadenylation
PCR Polymerase Chain Reaction
PIE Polyadenylation Inhibition Element
PLP Proteolipid Protein
POMA Paraneoplastic Opscoclonus Myoclonus Ataxia
PPI Protein-Protein Interactions
PPT Polypyrimidine Tract
pre-RNA Precursor RNA
PSA Prostate Specific Antigen Gene
Psi Percent Spliced In´
PTBP1 Polypyrimidine Tract Binding Protein
PTC Premature Termination Codon
PTM Post Translational Modification
QKI Quaking 1
RBD RNA binding Domains
RBPs RNA Binding Proteins
RGG Arginine-Glycine Rich
RNA-IP RNA Immunoprecipitation
RNA-Seq RNA-Sequencing
RNP RNA Protein Complexes
RRE Rev Response Element
RRM RNA Recognition Motif
RRM RNA-Recognition Motifs
rRNA Ribosomal RNA
RS Arginine And Serine
scRNA-Seq Single Cell RNA-Sequencing
SELEX Systematic Evolution Of Ligands By Exponential Amplification
SMN Survival Of Motor Neuron
SREs Splicing Regulatory Elements
SRRM4 Serine/ Arginine Repetitive Matrix 4
tRNA Transfer RNA
TSS Transcription Start Site
WGA Whole Genome tiling Array
ZF Zinc Finger
µ-exons Microexons
3´SS 3´splice site
4-SU 4-thiouridine
“Sometimes, a good idea comes to you when you are least looking for it; through an improbable combination of coincidence, naïveté and lucky mistakes”. – Kary Mullis, on his discovery of Polymerase Chain Reaction.
Acknowledgements
I am indebted to several people who have supported me during my graduate studies since 2014. I firstly thank Dr. Marc-Étienne Huot for giving me this opportunity, for believing in me and for shaping my scientific thinking and training. He has been an exceptional mentor and has always been supportive of his students. The care and concern he has towards us would fondly be remembered. I extend my thanks to my pre-doctoral committee members, Dr. Martin Simard, Dr. Darren Richard, and Dr. Josée Lavoie, whose feedback on my project had been extremely helpful. I thank Dr. Rachid Mazroui, Dr. Jean Yves Masson, and Dr. Samer Hussein for their inputs and fruitful collaborations. I also extend my thanks to Victoire Fort, Julia, and Yan Coulombe for helping me with several aspects of my Ph.D. project. I also thank my jury members, Dr. Marc-Etienne Huot, Dr. Martin Simard, Dr. Rachid Mazroui, Dr. François-Michelle Boisvert and the president of the committee, Dr. Amine Nourani.
I am indebted to late Dr. Govindan and Dr. Nalini for helping me settle down and acclimatize with the Québec culture and winter. I have been fortunate to have motivated and encouraging team members; Jonathan Bergeman, Laurence and Alexia. Thanks for introducing me to the beautiful Québec! Our trips to Val Cartier, Arbraska, les vallées bras du nord and sugar shacks will fondly be remembered! We also had good scientific discussions that helped to shape my perspectives on various topics. I am also thankful for the opportunity to mentor Maude Valliancourte Audet, an undergraduate student of Dr. Samer Hussein. The opportunity enabled us to learn more about long non-coding RNA biology. I was fortunate to mentor Karel Mocaer from University of Rennes, France. I thank her for the encouragement, friendship and will cherish our time together. I also thank Pauline Adjibade for all the help and support. You are a very helpful person and I admire your multi-tasking skills! I am lucky to have met Deepthi who has been very kind and supportive. I thank my friends in Québec; Ramesh, Sai, Razan Sheta, Anisha, Nupur, and Nikunj – for their incredible parties, great food and discussions. I thank Anahita Lashgari, Jonathan Humbert, Victoria, Ananditha, Hemantha and Niraj for the wonderful times together.
Finally, I am thankful to my family for encouraging me to cross continents for my scientific endeavors. I thank my beloved grandmother, Indira Devi Amma, and my aunt, S. Jalaja, who have always encouraged me to follow my heart. I am falling short of words to describe the unconditional support and love of my husband, Sastha, without whom, it would have been difficult to complete my graduate studies. Thank you for your unwavering support! I thank my brother, Swaroop, for his encouragement. I also thank my uncles Sadasivan and Unni for their support. I extend my thanks to my family in Canada including Vimal, Manjari, Madhavi and Shylaja for their support and encouragement.
Avant-propos
Le laboratoire du Dr. Marc-Étienne Huot tente a) de comprendre les facteurs régulant la protéine mTOR (Mechanistic target of rapamycin), au niveau transcriptionnel et traductionnel et b) de caractériser le rôle des complexes protéiques de l´ARN (RNP) dans la dissémination des cellules cancéreuses et les métastases. Mon projet de doctorat est centré sur la compréhension du rôle de la protéine de liaison à l´ARN, SAM68, dans la modulation de l´épissage alternatif du mTor. Dans ce contexte, les découvertes scientifiques décrites dans cette thèse sont le résultat de mes études supérieures avec le Dr. Marc-Étienne Huot. Ma thèse comprend les sections suivantes :
Introduction sur l'épissage alternatif, les types d' l'épissage alternatif, le
spliceosome, l'assemblage des spliceosomes, les petites ribonucléoprotéines nucléaires (snRNP), les RBP et leur rôle dans l'AS. Je mentionne aussi brièvement le rôle de SAM68 dans l´épissage alternatif de mTor.
Le Chapitr, 1 présente le manuscrit de mes travaux publiés en tant que premier auteur dans Nucleic Acids Research et intitulé «L´interaction de SAM68 avec U1A module le recrutement de U1 snRNP et régule l´épissage des pré-ARNm de mTor». J'ai conceptualisé l'étude, générée et interprétée méthodiquement les données avec l'aide du Dr. Marc-Étienne Huot. J'ai contribué à la rédaction du manuscrit qui a ensuite été corrigé par les Drs. Marc-Étienne Huot et Dr. Samer Hussein. Néanmoins, ces travaux ont largement bénéficié des collaborations fructueuses avec les équipes du Dr. Fréderic Allain (ETH Zurich), du Dr. Jean-Yves Masson et du Dr. Samer Hussein.
Le Chapitre, 2 intitulé ‘SAM68 et U1 snRNP coopèrent pour prévenir les événements de polyadénylation intronique’, fera l'objet d'une publication dans le futur. J'ai conceptualisé l'étude avec les commentaires du Dr. Marc-Étienne Huot. Dr. Samer Hussein a fait l'analyse bio-informatique. Victoire Fort, étudiante au doctorat avec le Dr. Samer Hussein, et moi-même avons validé les cibles identifiées par le pipeline bio-informatique. Nous décrivons ici un vaste programme d'épissage orchestré par SAM68 en coopération avec U1 snRNP.
Le Chapitre, 3 est une discussion de tous les travaux scientifiques présentés dans les chapitres 1 et 2, suivie de la conclusion et des orientations futures du projet. Enfin, à
l'annexe A, j'ai présenté des données préliminaires qui constituent des domaines de
recherche pour l'avenir. Dans l'annexe B, j'ai présenté la liste des gènes humains et de souris identifiés par l'analyse in silico dont les introns contiennent les motifs de liaison bipartites SAM68 à proximité des sites de jonction 5'.
Preface
Dr. Marc-Étienne Huot’s laboratory attempts to a) understand factors that regulate mechanistic target of rapamycin (mTOR) at the transcriptional and translational level and b) to characterize the role of RNA-protein complexes (RNP) in cancer cell dissemination and metastases. My Ph.D. project is centered to understand the role of the RNA binding protein, SAM68, in modulating the alternative splicing of mTor pre-mRNA. In this context, the scientific findings described in this thesis are the outcome of my graduate studies with Dr. Marc-Étienne Huot. My thesis comprises the following sections:
Introduction on alternative splicing, types of alternative splicing, spliceosome,
spliceosome assembly, small nuclear ribonucleoproteins (snRNPs), RNA binding proteins and their role in alternative splicing. I also briefly mention the role of SAM68 in the alternative splicing of mTor.
Chapter 1 is my first author publication in Nucleic Acids Research titled ‘SAM68
interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing’. I conceptualized the study, methodically generated, and interpreted the data with inputs from Dr. Marc-Étienne Huot. I have contributed towards the writing of the manuscript that was later corrected by Dr. Marc-Étienne Huot and Dr. Samer Hussein. In addition, this work significantly benefited from the fruitful collaborations with the teams of Dr. Fréderic Allain (ETH Zurich), Dr. Jean-Yves Masson, and Dr. Samer Hussein.
Chapter 2, titled ‘SAM68 and U1 snRNP cooperate to prevent intronic
polyadenylation events’ will be part of a publication in the future. I conceptualized the study with inputs from Dr. Marc-Étienne Huot. Dr. Samer Hussein did the bioinformatics analysis. Victoire Fort, Ph.D. student of Dr. Samer Hussein, and I validated the targets identified by the bioinformatics pipeline. Here, I describe an extensive splicing program orchestrated by SAM68 in cooperation with U1 snRNP.
Chapter 3 is a discussion of all the scientific works, presented in chapters 1 and 2,
followed by conclusion and future directions of the project. Lastly, in Annex A, I have presented data that are preliminary and are areas of future research. In Annex B, I have
presented the list of human and mouse genes identified by the in silico analysis whose introns contain the bipartite SAM68 binding motifs in proximity to 5´splice sites.
INTRODUCTION
The 20th century brought to the world key discoveries that revolutionized our understanding of how information flows between the three key biological polymers DNA, RNA, and proteins (1-3). The information stored in the DNA is read in two steps: transcription and translation. During transcription, the double-stranded DNA template gives rise to single-stranded RNA molecules which can regulate important cellular functions or accomplish this by producing proteins (4). These findings inspired several research groups to focus on key areas of gene regulation, one of which was the relationship between the DNA, its transcribed product, heterogenous RNA (hnRNA), and the mature messenger RNA (mRNA).
Prevalent in the 1970s was Crick’s ‘sequence hypothesis’ where the mRNA sequence was assumed to be completely complementary to its corresponding gene sequence (5). Later in 1977, laboratories of Dr. Philip Sharp and Dr. Richard Roberts independently proved that genes are split and did not display a co-linear relationship with its mRNA (6, 7). They observed using electron microscopy that when adenovirus mRNAs were hybridized to its corresponding gene, the latter produced three distinct loops which were absent in the mRNA (6, 7). This was found to be the case with other cellular genes in higher eukaryotes (8, 9).
Shortly after the discovery that genes were split in nature, Walter Gilbert proposed that the notion of the ‘cistron’, the genetic unit of function that one thought corresponded to a polypeptide chain, now must be replaced by that of a transcription unit containing regions called introns that will be lost from the mature mRNA alternating with regions which will be expressed – exons (10). He also intuitively suggested that single base changes at the exon-intron boundaries can alter splicing patterns making splicing less efficient so that a single transcription unit can produce new gene products (10). The discovery of RNA splicing thus improved our understanding of gene organization and eukaryotic pre-mRNA processing.
In eukaryotes, the three major types of RNA – ribosomal RNA (rRNA), messenger RNA (mRNA) and transfer RNA (tRNA) are derived from precursor RNA molecules
(pre-RNA) by RNA polymerase I, II and III respectively (11). In RNA Pol II derived transcripts, the corresponding segment from which the pre-RNA or heterogenous RNA is produced is termed a transcription unit. Upstream of the transcription start site (TSS) lies the promoter and further upstream, dispersed over 100 base pairs, are several 10bp sequences called enhancers to which transcription factors bind (12). Before being transported to the cytoplasm for translation, the initial heteregenous RNA produced by RNA Pol II transcription has to undergo several maturation steps that comprises the addition of a 5´cap, splicing and processing of 3´ends (13).
Eukaryotic pre-mRNA processing
5´ Capping
RNA Pol II comprises an enlarged central active site wherein the double stranded DNA is forced apart as a single stranded bubble while channels to this active site permit nucleotide access and RNA exit (13, 14). Located below the RNA exit channel is the unstructured largest subunit of Pol II, the carboxyl terminal domain (CTD), that has 52 tandem repeats in mammals and 26 in yeast, of the consensus heptad ‘YSPTSPS’ (15). Phosphorylation of Serine 5 and Serine 2 is linked to transcriptional initiation and elongation respectively (15).
Increasing evidence suggests that 5´capping of Pol II nascent transcripts (20-25nt long) is the first co-transcriptional pre-mRNA processing step and that Pol II pauses at few nucleotides downstream the transcription start site to recruit capping enzymes to its phosphorylated CTD (16).
The cap usually a mono-methyl guanosine (m7GpppN, except snRNAs) identifies transcription start sites and play critical roles in mRNA maturation, translation and stability (17). In mammals, the 5´capping is catalyzed by two enzymes: guanylyltransferase (GT) and guanine-N7 methyltransferase (MT) (17). Guanylyltransferase has dual activities; one is RNA 5´- tri-phosphatase that removes the terminal phosphate of the initial ribonulceotide from the nascent chain and the second is guanylyltransferase activity that transfers 5-phosphoguanosine (GMP) to the 5´- diphosphate RNA of the nascent chain. Guanine-N7
methyltransferase catalyzes the addition of a single methyl group at position 7 of the terminal guanine (17, 18). The reaction is summed up as follows:
5´Gppp + 5´pppApNpNp…. Gppp5´-5´ApNpNP…+ pp +p
The 5´cap protects the nascent RNA chain from 5´- 3´ exonuclease attack. It also makes sure that Pol II engages in a productive transcription and the initial pausing is a checkpoint for transcription reinitiation (19). The 5´m7G cap also functions as unique identifier to recruit proteins for splicing, polyadenylation, nuclear export of mRNA and cap-dependent translation (17).
Pol II CTD and its regulatory phosphorylation play a critical role in the efficiency of splicing. Promoter swapping experiments with Pol II transcribed genes put under the control of Pol III promoters resulted in poor splicing and polyadenylation (20). Truncation of Pol II CTD caused defects in subsequent RNA processing steps including capping, splicing and cleavage/polyadenylation (21).
Splicing
Eukaryotic genes can be constitutively spliced which refers to the process of intron excision and ligation of exons in the order they appear in a gene as shown in Figure 1 (22). Alternative splicing is a deviation from this preferred pattern where the splicing machinery has to differentiate between multiple splice site choices. They are of several types which are discussed later on. Gilbert’s 1978 prediction of alternative splicing is currently the mechanism that explains the discrepancy between the number of protein coding genes (~ 20,000) in humans and its generation of > 90,000 protein products (23).
Mechanism of pre-mRNA splicing
Spliceosomal introns occur only in eukaryotic nuclear genomes and are excised by the macromolecular machinery called the ‘Spliceosome’ (24). The spliceosome is a repertoire of small nuclear RNAs (snRNAs) in complex with their core proteins called snRNPs (24). It excises introns precisely by employing a series of RNA-RNA, protein-protein and RNA-protein-protein interactions (25).
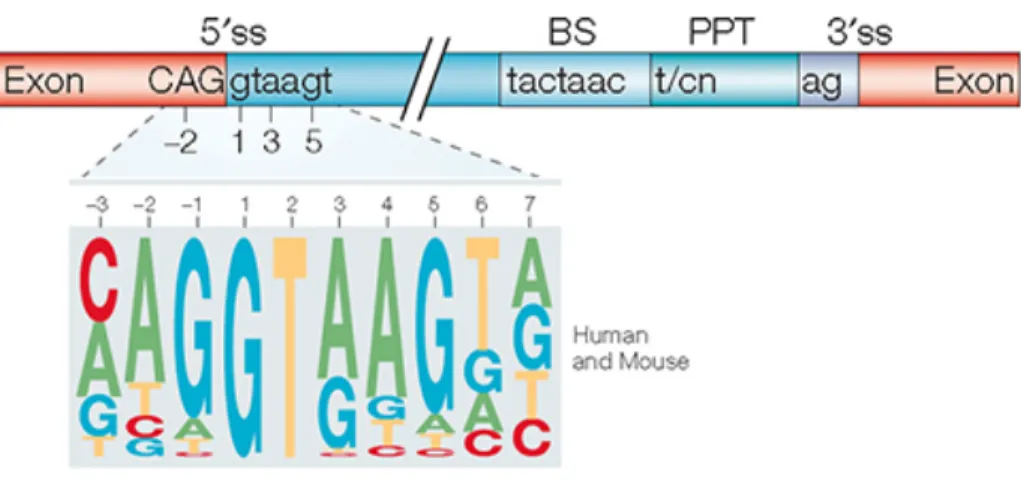
Each intron has four conserved sequences i) the 5´splice site (5´SS) at the 5´end of exon-intron junction ii) the branch point sequence (BPS) within the intron that includes a functionally important adenosine iii) a polypyrimidine tract (PPT) and iv) the 3´splice site (3´SS) at the 3´ end of intron-exon junction (25), as shown in Figure 2.
Figure 1: Simplified depiction of constitutive splicing. In constitutive splicing, all the
exons of pre-mRNA are included in the transcript or mRNA in the same order which codes for a full-length protein.
Figure 2: The core splicing signals of an intron. (Top); The consensus sequences of the
5´splice site with relative positions upstream and downstream, branch point sequence, polypyrimidine tract, and the 3´splice site is shown. (Below); Logo showing the relative frequency of each nucleotide across 49,778 human and mouse 5´splice sites. Figure is taken with permission from Springer Nature, Nature Reviews Genetics, Gil Ast, 2004.
Spliceosomal introns are of two types - U12 and U2 introns. The latter accounts for 99% of introns and are also called GT-AG introns; with the highly conserved di-nucleotide GT and AG at 5´ and 3´splice sites respectively (26). They are processed by the major U2-dependent or canonical spliceosome that comprises five small nuclear ribonucleoproteins (snRNPs) namely U1, U2, U4, U5 and U6 snRNPs along with other accessory non-snRNP proteins (27). The other, less abundant, U12-dependent or minor spliceosome comprises the U11, U12, U4atac, U5 and U6atac (28). They splice both GT-AG and AT-AC introns that are longer, possess tightly constrained consensus sequences at the 5´SS and branch point sites and lacks the polypyrimidine tract upstream of the 3´SS (29).
U2-spliceosome catalyzes splicing through two consecutive trans-esterification reactions namely branching and exon ligation (30, 31). In branching reaction, the branch point adenosine mounts a nucleophilic attack on the 5´ SS producing an intermediate intron lariat-3´ exon and a 5´exon. In the exon ligation reaction, the 5´ exon mounts a nucleophilic attack on the 3´ exon generating a ligated exon-exon junction and releasing the intron lariat (31). This mechanism is strikingly similar to self-splicing ribozymes - Group II introns are thought to have evolved to spliceosomal introns during the course of evolution (32, 33). However, in the spliceosome, there is a division of labor to numerous protein components
to organize and activate the catalytic site RNAs to pair the splice sites together (32). Unlike most stable ribonucleoprotein (RNP) enzymes that employ preformed active sites, spliceosome employs structural and compositional dynamics for catalytic activation and active site remodeling (34).
It is currently not clear if spliceosome assembly occurs in a step-wise or in a pre-assembled manner, although there are numerous studies (based on in vitro native gel-electrophoresis, affinity selection, and glycerol gradient centrifugation) supporting the former (32, 34, 35). In the step-wise assembly, as illustrated in Figure 3, the catalytic spliceosome matures through different complexes; commitment E, spliceosome A, pre-catalytic spliceosome B, activated Bact, catalytically activated B* that catalyzes the step I or
branching reaction, catalytic step 1 spliceosome C, the C* complex that catalyzes the step II or exon ligation, the post-catalytic spliceosome P that sheds the ligated exons and the intron lariat spliceosome (ILS) which disassembles the intron lariat from the snRNPs and proteins (31, 35, 36). During each step, there is sequential recruitment of snRNPs and accessory RNA binding proteins (RBPs) (31).
The E or commitment complex is formed by the recognition of the 5´splice site, branch point sequence, polypyrimidine tract and 3´splice site as shown in Figure 3. It involves a) ATP independent recruitment of U1 snRNP, stabilization of base-pairing interactions of 5´end of U1 snRNA with the 5´splice site of the intron b) branch point binding protein; BBP or SF1 to the branch point sequence, c) U2 auxiliary factors; U2AF65 to the polypyrimidine tract and U2AF35 to the 3´SS of intron (34). Numerous studies have demonstrated that U1 snRNP interaction with pre-mRNA is stabilized by its interaction with other RBPs (37-39).
In complex A, SF1 is replaced by U2 snRNP in an ATP dependent manner where the U2 snRNA base pairs with the BPS, the interaction of which is strengthened by its associated heteromeric protein complexes; SF3a and SF3b (40). This promotes the recruitment of the pre-assembled tri-snRNP, U4/U6.U5 to form the B complex, where the U1 snRNP is still bound to the 5´SS (40). In the tri-snRNP nomenclature, the slash indicates base pairing between U4 and U6 snRNAs while the period conveys that U5 snRNP binds to U4/U6 di-snRNP through protein-protein and RNA-protein interactions
(41). The B complex is not yet catalytically active although it comprises all the snRNPs; U1, U2, and U4/U6.U5 (25).
Figure 3: Schematic representation of the step-wise assembly of the spliceosome.
Spliceosome assembly proceeds through E, A, B, Bact, B*, C, C* and the post-lariat intron complex to yield the final mature mRNA. Also highlighted are the four core splicing signals; the 5´SS - GU, the branch point adenosine, the polypyrimidine tract – YRYYRY where Y is pyrimidine and R is purine and the 3´SS – AG. Figure is modified with permission from Cold Spring Harbor Press, Cold Spring Harbor Perspectives in Biology, Will and Lührmann, 2011.
Complex B undergoes conformational rearrangements initiated by the U5 snRNP proteins; Prp8 and RNA helicase, Brr2, leading to the ATP-dependent unwinding of U4/U6 helix to form the Bact complex (42, 43). Along with the displacement of U1 and U4 snRNPs, U6 snRNA base pairs with both the 5´SS and U2 snRNA (43). The dissociation of
U1 snRNP lets U6 snRNA juxtapose with the 5´SS while the release of U4 snRNP serves as the rate-limiting step to proceed to the activated state or the B* complex (44). The catalytic B* complex comprises the U2, U6 and U5 snRNPs that catalyzes the first trans-esterification reaction where the branch site adenosine attacks and cleaves at the 5´ SS of the intron (35).
The resulting C complex contains the 5´ exon and the intron lariat - 3´ exon intermediates (45). The 5´exon is base paired with U5 snRNA to retain it in the active site, U6 snRNA interacts with the branched 5´ SS while the U2 snRNA immobilizes the intron lariat with branch point adenosine covalently linked to the 5´ end of the intron (46, 47).
The transition of C to the catalytically active step II complex, C*, is triggered by the ATPase, Prp16, in yeast (30). This catalyzes the exon ligation reaction, where the 3´-hydroxyl group of 5´ exon attacks the first residue of the 3´ exon producing the P complex (45). Upon ATP hydrolysis, the step II factors and ligated mRNA dissociates leaving the ILS and associated factors that are later recycled for the next round of splicing (35).
Proteomic analysis of the human spliceosome indicates that more than 170 proteins associates with the spliceosome in the course of splicing with individual assembly intermediates, B and C complex having fewer up to 110 proteins (48).There is a massive and dynamic exchange of snRNPs and accessory proteins during this process and often involves homologous proteins indicating an evolutionarily conserved design of these compositional changes (49).
The design of spliceosome renders it to be regulated by mainly two factors; 1) the functional sites of pre-mRNA are recognized multiple times by different factors to ensure splicing fidelity and 2) multiple weak interactions can function together to activate or suppress splice-site choices (34). These structural and compositional dynamics of spliceosome assembly allows for remarkable plasticity to regulation (34). This fact might have helped in the evolution of alternative splicing (AS) which is discussed later on.
Small nuclear ribonucleoproteins (snRNPs)
Each snRNP comprises one (U1, U2, U5) or two (U4/U6) small nuclear RNA, a common set of seven Sm or Smith antigen (B/B´, D3, D2, D1, E, F and G) proteins and its associated core proteins (41). They undergo substantial remodeling during splicing (50). Except for U6, all snRNAs possess a 5´-trimethylguanosine (m3G)cap and single-stranded uridine-rich sequence flanked by two hairpin loops called the Sm site around which the Sm ring assembles (50). The U6 snRNA has a 5´- mono-methylguanosine cap and does not bind to Sm proteins as it lacks the Sm site(51).
In eukaryotes, the survival of motor neuron (SMN) complex mediates the biogenesis of snRNPs and reduction of SMN protein levels results in decreased snRNP assembly and the formation of the heptameric Sm cores on snRNAs (52). Reduction in SMN protein levels causes a severe motor neuron degenerative disease termed spinal muscular atrophy in humans (53). Figure 4 depicts the composition and structure of each of these snRNPs.
The U1 snRNP is a 12S particle and comprises U1 snRNA, its associated core proteins; U1-70K, U1A, and U1C, in addition to the common Sm proteins (54). Two distinct forms of U2 snRNP exists in HeLa cells, the 12S U2 snRNP comprises B´´ and A´ while the 17S U2 snRNP comprises 9 additional proteins and is thought to be the active U2 snRNP. The U4/U6 and U5 have been isolated as 12S or as 25S U4/U6.U5 tri-snRNP and the latter enters the spliceosome (54). In the next section, I describe the U1 snRNP.
U1 snRNP
The human U1 snRNP (245kDa, 11 subunits) consists of a 164 nucleotide U1 snRNA, the seven Sm proteins that form a ring around the single-stranded Sm site and core factors, U1-70K that binds to stem-loop I, U1A that binds to stem-loop II and U1C that interacts with the particle via U1-70K (55). The 5´ end of U1 snRNA base pairs with the 5´splice site promoting the ordered assembly of the remaining four snRNPs to form the catalytically active spliceosome that triggers pre-mRNA splicing (50).
Figure 4: Protein composition of the major human spliceosomal snRNPs. All seven Sm
(Smith Antigen) proteins (B/B´, D3, D2, D1, E, F, and G) or LSm (Like Smith antigen) proteins (Lsm2-8) are indicated by “Sm” or “LSm” at the top of the boxes showing the proteins associated with each snRNP. The U4/U6.U5 tri-snRNP contains two sets of Sm proteins and one set of LSm proteins. Figure is modified with permission from Cold Spring Harbor Press, Cold Spring Harbor perspectives in biology, Will and Lührmann, 2011.
U1-70K has an unstructured N-terminus, an RNA binding domain that mediates its interaction with stem-loop I of U1 snRNA and a C-terminus rich in arginine and serine residues (RS domain) as well as R-(D/E) residues (56). Phosphorylation of U1-70K RS domain is crucial for its interaction with the non-snRNP protein, SF2, for subsequent splicing activation (57).
U1C has an N-terminus zinc finger domain and a C-terminus rich in arginine/glycine (RG) residues and it binds to the U1 snRNP via U1-70K (58). U1A has RNA recognition motifs at N and C-terminus, a central nuclear localization signal (NLS)
and it binds to stem-loop II of U1 snRNA via its N-terminus RRM1 with a great affinity (59). In fact, the 7nt AUUGCAC was the winner sequence of U1A as identified by in vitro RNA binding experiments (60).
3´end processing
Most eukaryotic mRNAs except mRNAs encoding core histone proteins have strings of poly (A) tails at their 3´ends. Differential inhibition of poly (A) addition of hnRNA and mRNA synthesis confirmed that poly (A) addition occurs post-transcriptionally by the template-independent poly (A) polymerase (PAP) (61). Other conserved factors include cleavage stimulation factor (CstF) and cleavage/polyadenylation specificity factor (CPSF). The sequence surrounding the poly (A) region has various cis- elements; a) upstream hexamers called PAS, such as AAUAAA, AUUUAA and other variants (62) and b) downstream U-rich and GU-rich elements (63). Experiments with viral mRNA templates showed that transcription occurs past the consensus poly (A) signal (PAS): AAUAAA (64). The poly (A) signal triggers an endonucleolytic cleavage further downstream at a GU-rich sequence followed by the addition of around 200 adenosine residues (61). CstF comprises 3 subunits CstF-50 CstF-64 and CstF-77 and functions in recognizing the GU-rich downstream sequence. CPSF comprises four subunits - CPSF-160, CPSF-100, CPSF-73, and CPSF-30. It binds the AAUAAA sequence and interacts with CstF to recruit PAP (65).
Alternative splicing
Alternative splicing is the phenomenon where different combinations of splice sites from pre-mRNA are selected to produce multiple transcripts significantly expands the coding power of genomes (66), generating in some instances, several thousands of transcript isoforms from single genes (67, 68). An incredible example is the Down syndrome cell adhesion molecule or Dscam gene in Drosophila melanogaster, that generates 38,016 alternative splice variants which exceeds the total number of genes (~15,500) in the organism (66). AS, in addition, also regulates the spatiotemporal regulation of mRNAs by modulating its stability, translational efficiency, and localization of transcripts (69, 70).
Initial experimental proof for Gilbert’s prediction of alternative splicing (10) came from the work of Berk and Sharp with DNA viruses (71). Later in the 1980s, the first cellular examples of alternative splicing came from the laboratories of Baltimore and Hood by their discovery of isotype switching in immunoglobulin genes (72, 73). Recent advances in transcriptome deep sequencing and splice-sensitive microarray platforms allowed quantitative profiling of alternative splicing events that estimate 90-95% of human genes are spliced (74). Although it is still not known if all the splice variants identified by RNA-Seq have protein-coding potential, both quantitative proteomics and ribosome-association studies have confirmed the production of protein isoforms in mammalian organisms (23). Alternative splicing, thus, explains the morphological and behavioral complexity of humans when compared to other model organisms like mice, fruit flies and, worms despite having around 19000-25000 genes (75).
Alternative splicing occurs very rarely in the Saccharomyces cerevisiae genome as only a subset of genes has introns (76). On the other hand, most alternative splicing events in mammals are evolutionarily conserved and thus are implicated in the functional specialization of cell types and tissues (77, 78). With the advent of high-throughput sequencing, alternative splicing profiling have catapulted from the study of single splicing events to the unraveling of global splicing regulatory networks and their coordination (78). The role of AS in cell differentiation, lineage determination, tissue identity, and organ development is now well established (23, 79).
Figure 5 shows the major modes by which alternative splicing exert combinatorial diversity. These are a) cassette-exons that are either included or skipped in transcripts, b) mutually exclusive exons, where two exons do not occur together in a transcript, c) alternative 5´splice-site d) alternative 3´splice-site that can change part of an exon or intron sequence and d) intron retention where an intron is not excised and is thus retained in the transcript (80) .
Figure 5: Schematic illustration of the major modes of AS patterns. a) Exon
skipping/inclusion/cassette exons, b) alternative 5´splice site selection, c) alternative 3´splice site selection, d) mutually exclusive exons and, e) intron retention. Figure is modified with permission from Springer Nature, Nature Reviews Genetics, Cartegni et al., 2002.
In addition to AS, alternative promoter usage and alternative polyadenylation also contribute to transcriptome diversity in higher eukaryotes (81) which are briefly discussed below.
Alternative promoters
Most eukaryotic genes contain multiple promoters and each promoter determines a different start site and first exon (82). Tan et al., found that 35% of erythroid genes had alternative first exons generating proteins with distinct N-terminus (83). Alternative
promoter usage permits differential transcriptional regulation of genes especially involved in the development and immune regulation while single promoter usage is found in genes involved in RNA processing, DNA repair and protein biosynthesis (84). Alternative promoters can generate mRNA transcripts with identical protein coding sequence with different 5´-UTRs or transcripts with different protein coding sequences (85). The former affects mRNA stability and translation efficiency although it encodes the same protein while the latter could generate proteins with different and even antagonistic functions (85). Another way by which alternative first exons affect translation is by the use of upstream open reading frames (µORF) that precedes the real ‘start’ codon as they can compete with the downstream ORF for the translation machinery (86).
Alternative polyadenylation
Polyadenylation of mRNA is a crucial step in its maturation and is tightly coupled with transcription to define the 3´end of genes. Over half of the human genes have multiple pA signals (87). While mutually exclusive alternative splicing of terminal exons produces isoforms with different sequences, alternative pA selection leads to the production of isoforms with identical protein-coding sequences but different 3´-UTRs (88, 89). Thus, alternative polyadenylation adds another layer of regulation of mRNA expression by regulating their translation, stability, localization, and function (90). In fact, recent studies have shown that proliferating cells such as T-lymphocytes (91) and tumor cells (88) express isoforms with short 3´-UTRs to evade micro-RNA mediated regulation while terminally differentiated tissues like brain express isoforms with longer 3´-UTRs (79, 92). An additional layer of AS regulation includes tissue-specific control and regulation by developmental or differentiation cues and external stimuli (23).
The advent of new genome-wide technologies has led to a more profound understanding of alternative splicing events and their regulation. In the following section, I describe some of the key technologies that have helped in uncovering unconventional splicing events.
Genome wide technologies to identify alternative
splicing events
The two powerful methods currently used to identify and quantify alternative splicing events by high-throughput sequencing are microarrays and RNA-Seq.
Microarrays
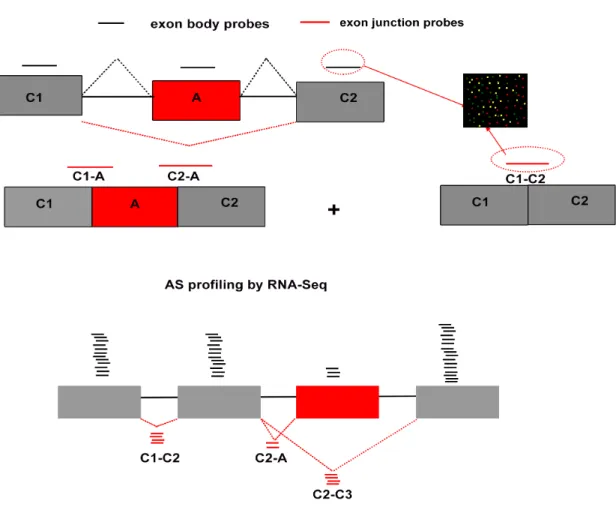
Oligonucleotide microarrays contain millions of probes, typically 25-mers on Affymetrix arrays and 60-mers on Agilent arrays, that are complementary to the target transcripts (93, 94). Multiple probes (gapped or overlapping) spanning an entire transcript, several transcripts implicated in the same pathway, or an entire organism is spotted on a surface with a known pattern. Then, RNA extracted from control and an experimental sample (e.g., cells depleted of an RBP) is sheared and ligated with different fluorophores. The fluorescent labeled RNA molecules are allowed to flow across the array. The fragments hybridize with its complementary probes and the fluorescence of each spot is used as readout for the expression level of the target. There are several variations of microarrays based on the probe design to detect alternative splicing events (95). Figure 6 represents an example of the use of microarrays to profile exon inclusion.
Exon junction microarray, where the probes are complementary to exon-exon junctions are used to quantitatively determine the expression of alternatively spliced isoforms (96). Thus, to find the expression of a cassette exon, six probe sets are designed in total, three of which span each exon and the other three span the three splice-junctions. Exon inclusion or ‘percent spliced in’ or Psi (ψ) is determined by comparing the signals from alternative exon and its upstream and downstream junction against the one obtained from constitutive exon junctions (97). Johnson et al., profiled alternative splicing events among 10,000 human genes across 52 human tissues and cell lines using exon-junction microarrays (98). Thereafter, splicing-sensitive microarrays have been widely used to profile AS events upon knockdown/knockout of RNA binding proteins (99-101) to study stimulus-induced changes in transcript diversity (102), to deduce evolutionary features governing co-regulated exons and for the organization of transcription networks (103).
Figure 6: Methods to quantify alternative splicing events. (Top) Microarray design to
identify cassette exon inclusion. Six oligonucleotide probes are used, three (black) exon body probes spanning constitutive exons, C1, C2, and A spanning alternative exon while the other three (red) are exon junction probes, C1-C2 spanning both C1 and C2 to measure exon skipping event while C1-A and C2-A are used to measure alternative exon inclusion levels. The probes are attached to glass array (on right) and total mRNA isolated from cells are sheared, labeled with fluorophores allowed to hybridize. The level of exon inclusion is calculated from the fluorescence intensity as the ratio of signals obtained from C1-A, C2-A and A to C1-C2. To profile alternative splicing events by RNA Seq (bottom), total mRNA is isolated, sheared to small pieces and converted to cDNA and sequenced. The reads are then mapped to the existing genome or de novo. As shown above, reads from constitutive exons, C1 and C2 (black) are more than from alternative exon, A. Reads aligned across exon junction C1-C2, C2-C3 are also more than reads across C2-A. Collectively, the exon inclusion levels are low in this cell-type. Figure is modified with permission from Elsevier, Molecular Cell, Pan et al., 2004.
Whole genome tiling array (WGA), on the other hand, comprises probes that span an entire genome in an overlapping manner or with spaced with an equal distance apart as in quasi-WGA (104). Tiling arrays have the potential to identify novel transcripts, protein coding, or non-translated, especially arising from intronic regions (105). Reliance on existing genome information and hybridization issues such as cross-hybridization with transcripts with highly similar sequences are some of the disadvantages with microarray technologies. Thus, the predictions obtained by microarray needs to be validated by RT-PCR and sequencing (98).
RNA-Sequencing
High-throughput RNA-Seq is best-suited for discovery-based experiments and gets updated as new sequence information comes in, or in other words, it helps build de novo transcriptome assembly (106). An example of RNA-Seq analysis to study exon inclusion is shown in Figure 6. Other advantages are that it offers very high signal to noise ratio as the reads obtained can be readily mapped to unique regions of the genome and unlike microarrays, it does not have any hybridization issues (106). RNA-Seq is thus widely used to identify transcript types, their splicing patterns, to quantify transcript abundance and their changes in experimental samples and to identify novel splice variants (106).
Mortazavi et al., analyzed mouse tissue transcriptome using the Illumina platform to obtain 52 million reads of 25nt each and found a) approximately 17,000 previously unannotated regions of known genes, many of which were in extended 5´ or 3´ UTRs b) 1,45,000 novel splice site junctions and c) new alternative splicing events in 3500 genes (107). Cloonan et al., profiled transcriptomic changes during mouse embryonic stem cell and embryoid body differentiation using the Applied Biosystems SOLiD technology generating 100 million 25-nucleotide reads (108). One-third of those reads were mapped outside of already annotated exons thus highlighting the potential of mRNA-Seq to identify novel splicing events.
Human transcriptome analyses followed by aligning reads, that do not match genomic sequences, to synthetic or computationally selected splice-site junctions revealed that 92% to 97% of multi-exon genes are alternatively spliced with an average of seven
alternative splicing variants per gene (74, 79, 109). RNA-Seq has also helped redefine transcriptome complexity in metazoans and to probe the evolution of alternative splicing in vertebrates and invertebrates (110).
An exciting feat of RNA-Seq is its potential to profile gene expression and alternative splicing events in single cells – scRNA-Seq (111). Contrary to bulk mRNA-Seq of a mixed pool of cells that indicates multiple isoforms per gene, scRNA-Seq of the same cells show only one or only a few isoforms per gene (112, 113). This emerging picture of splicing heterogeneity in single cells will undoubtedly play a pivotal role in the functional characterization of cell subpopulations (114).
Collectively, these methods have highlighted the fact that most of the alternative splicing events are tissue-regulated and that canonical splicing regulators and other RBPs can co-regulate RNA processing events such as alternative splicing coupled polyadenylation. These technologies together with methods that are aimed at the identification of RBP binding sites described below will continue to revolutionize transcriptome-wide studies.
Methods to identify RBP binding sites
The human genome encodes around 1500 RBPs that contain well-defined RNA binding domains (RBD) such as RNA recognition motif (RRM, 240 RBPs), the hnRNP K-homology domain (KH, 60 RBPs), C3H1-zinc finger (ZF, 50RBPs), DEAD motifs, and the double-stranded RNA binding motifs (115, 116). They engage with RNA in a sequence and/or structure specific manner (115). Identification of RNA targets is thus a crucial step towards characterizing the molecular and cellular functions of RBPs. Both in vitro methods and methods that preserves the context of living cells are currently being employed to catalog the binding sites of RBPs. These methods along with recent advances in high throughput sequencing are instrumental in deciphering the roles of novel RBPs.
Native and cross-linked RNA immunoprecipitation
RNA immunoprecipitation (RNA-IP) is a powerful method to study the physical association between individual proteins and RNA molecules in vivo (117). It involves
immunoprecipitating the RBP of interest along with its bound RNA after cell lysis. The associated RNA is isolated and analyzed further by PCR, hybridization or sequencing. The two variations are native RIP and cross-linked immunoprecipitation (CLIP). Native RIP allows determining the identity and quantity of RNAs bound by the RBP of interest. Additional steps such as mild sonication and ribonuclease treatment can be included to map the binding site of the RBP. Unlike native RIP, cross-linked immunoprecipitation (CLIP) employs formaldehyde, ultra-violet light or combinations of both to prevent post-lysis re-association or “mixing” of RNA-protein complexes and enables stringent purification conditions (118).
In UV-CLIP, UVC (254nm) irradiation of cultured cells or organisms cross-links the RBP to RNA that are in direct contact followed by denaturing cell lysis and capture of RNPs by immunoprecipitating the target RBP with its specific antibody (119). If no antibody is available, the RBP is fused to epitope tag that is then expressed as a transgene for affinity purification. The co-purified RNA after treatment with proteinase K is attached to 5´ and 3´ adaptors, reverse transcribed to cDNA, which are subjected to Sanger sequencing and aligned to the reference genome to identify the RBP binding sites within transcripts (120). This method was used to study NOVA dependent splicing regulation in the brain (119).
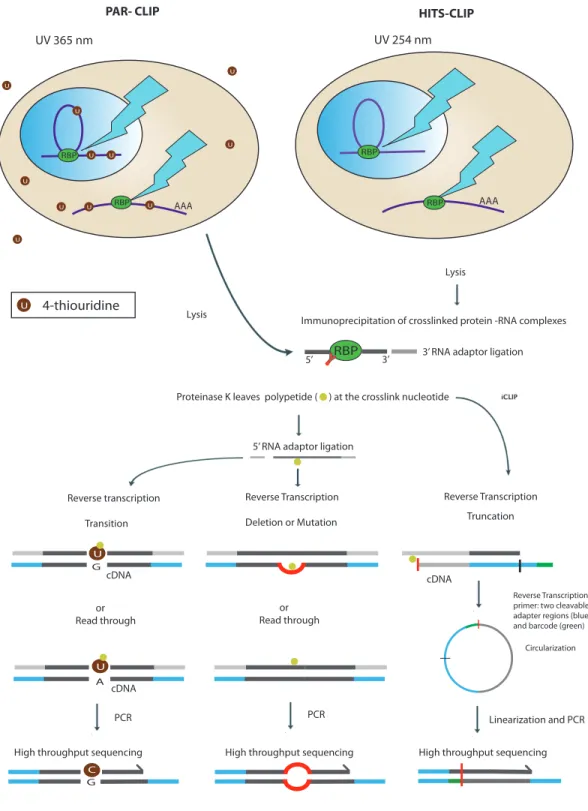
Licatalosi and colleagues combined CLIP with high-throughput sequencing in HITS-CLIP or CLIP-Seq that provided more comprehensive binding information on NOVA’s role in alternative polyadenylation (92). Granneman and colleagues demonstrated that UV crosslink induced point mutations and deletions can be used to identify cross-link sites of RBPs within transcripts (121). Recently, two strategies were developed based on this fact to gear up CLIP-Seq to nucleotide resolution namely, photoactivable ribonucleoside-enhanced crosslinking (PAR-CLIP) and individual nucleotide resolution CLIP (iCLIP). UV-CLIP and its variants are outlined in Figure 7.
PAR-CLIP uses the nucleoside analogs, 4-thiouridine (4SU) or 6-thioguanosine, that are incorporated into nascent transcripts that upon UVA irradiation (365nm) cross-links the transcripts to the RBP (122). Some of the advantages of PAR-CLIP are that a)
purification conditions b) UVA irradiation causes less photodamage to the cells and c) nucleoside analogs create base transitions at cross-link sites during reverse transcription and analysis of cDNA sequences for this mutation pinpoints the cross-link site at nucleotide resolution (123). However, a major pitfall of PAR-CLIP is the reported cytotoxicity of cells and tissues to nucleoside analogs (124).
Availability of RBP specific antibodies and the chance of disruption of binding site by extensive enzymatic steps for the library preparation are some of the difficulties that still need to be addressed for all CLIP variants. Nonetheless, it has the potential to compile a comprehensive list of RNA interactions of multiple RBPs. Methods to identify RBP binding sites combined with analyses to address how RNA-protein interactions are involved in differential regulation of the targets will help us better understand the molecular basis of phenotypes observed during loss or gain of function of RBP.
Figure 7: Schematic representation of UV-CLIP and its variants. In PAR-CLIP, cells
are grown in media with 4-thiouridine (4SU) that is incorporated in the elongating transcript. UV irradiation at 365nm causes cross-links at 4-SU sites, followed by cell-lysis, digestion of RNA to 30-50 nucleotides long, and then immunoprecpitating the
protein-RBP U U RBP U U U U U U U U AAA AAA UV 365 nm UV 254 nm
PAR- CLIP HITS-CLIP
U 4-thiouridine
RBP
U U RBP
Lysis
Immunoprecipitation of crosslinked protein -RNA complexes 3’ RNA adaptor ligation
Proteinase K leaves polypetide ( ) at the crosslink nucleotide
5’ RNA adaptor ligation
Reverse transcription Transition Reverse Transcription Deletion or Mutation Reverse Transcription Truncation or
Read through orRead through
Reverse Transcription primer: two cleavable adapter regions (blue) and barcode (green)
Circularization
Lysis
RBP
5’ 3’
High throughput sequencing High throughput sequencing High throughput sequencing PCR PCR Linearization and PCR U U C G A G cDNA cDNA cDNA
the co-purified RNA is then attached to 5´ and 3´ adaptors and upon reverse transcription, 4SU causes base transition that can later be used to identify cross-link sites. In HITS-CLIP, growing cells are UV irradiated at 254nm and the same steps are followed. However, addition of 5´-adaptor captures only cDNAs that bypasses the link sites while cross-link sites are identified by deletions or mutations in the reads. In iCLIP, in order to capture the cDNAs that truncate at the peptide that remains at the cross-linked nucleotide after proteinase K digestion, the RT-primer is designed to comprise two cleavable adaptors with barcode and used for reverse transcription. After reverse transcription, the cDNA is circularized, linearized, and subjected to PCR followed by sequencing. The cross-link sites are identified at single nucleotide resolution. Figure is modified with permission from Springer Nature, Nature Reviews Genetics, Konig et al., 2012.
In vitro methods
Systematic Evolution of Ligands by Exponential Amplification (SELEX) is an in vitro method to identify RBP binding sites and was developed in the 1990s by two independent laboratories of Ellington (125) and Turek (126).
The first step consists of single-stranded DNA/RNA aptamer synthesis to create an oligonucleotide library. A typical library consists of random sequences up to 60 nucleotides in length flanked by constant sequences to align primers for polymerase chain reaction. In order to identify high-affinity aptamers for an RBP, in vitro purified RBP is incubated with a library of chemically or in vitro synthesized RNA oligos, following which the unbound molecules are washed and the bound molecules are reverse transcribed to DNA for PCR amplification. This process is repeated at least twenty times to enrich aptamers with a high target affinity (125, 126).
In 2012, Ohuchi (127) introduced ‘cell-SELEX’ that involves incubating the oligonucleotide library with whole cells. Unbound molecules are washed away, the aptamers are separated from the target and amplified by PCR, and the process repeated at least 35 times. This method enabled the identification of RNA aptamers that are capable of binding breast cancer cells. Another variation termed ‘in vivo SELEX’ involves generating the aptamers in live cells or organisms (128). For example, mRNAs extracted from human brain samples led to the discovery of the RNA recognition element for the RBP, ELAVL2 (also known as HuB) by repetitious binding and purification strategy (129). SELEX
identifies a few high affinity “winner” sequences but is unable to identify a full spectrum of RNA targets or their associated affinities (130).
Recently, methods have been developed that are coupled to high-throughput sequencing and microarray analysis to measure affinities and reaction kinetics of thousands of RNA simultaneously. Some of them are high-throughput sequencing analysis of equilibrium binding (HTS-EQ), RNA Bind-n-Seq, and RNA MaP (131). These methods provide global information on the surrounding context of a given sequence motif, bypass the selection and amplification cycles of the SELEX procedure and measure the amount of RNA species that are both weakly and tightly bound (132, 133).
I introduce below some variants of alternative splicing produced by non-canonical splicing mechanisms which are currently being identified with modified RNA-Seq pipelines.
Non-canonical splicing
Non-canonical splicing refers to unconventional splicing mechanisms and occurs by cryptic splice site usage that deviates from the standard rules of splicing. The advent of RNA-Seq technologies and modified analysis pipelines have led to the discovery of some of the products of non-canonical splicing like micro-exons, intron retention, and intronic polyadenylation.
Microexons
Microexons (µ-exons) are shorter than 30 nucleotides (134) encoding protein domains implicated in protein-protein interactions (PPI) related to cell-signaling and are flanked by intronic motifs bound by RBPs such as polypyrimidine tract protein (PTBP1) and serine/ arginine repetitive matrix 4 (SRRM4 or nSR100) that binds specifically to PPT upstream of microexons (135, 136). Microexons are also more conserved and frame preserving than longer neural exons (136).