© Jean-Paul Iyombe, 2019
Correction du gène de la dystrophine avec la méthode
CRISPR induced deletion (CinDel)
Thèse
Jean-Paul Iyombe
Doctorat en biologie cellulaire et moléculaire
Philosophiæ doctor (Ph. D.)
Correction du gène de la dystrophine avec la méthode
CRISPR induced deletion (CinDel)
Thèse
Jean-Paul IYOMBE-ENGEMBE
Sous la direction de :
Jacques P TREMBLAY
ii
Résumé
La dystrophie musculaire est une maladie génétique monogénique récessive liée au chromosome X. Elle atteint 1 garçon sur 3500 naissances mâles. Le garçon atteint de la maladie présente des troubles de la locomotion à l’âge de 3-4 ans et la perd vers l’âge de 11 ans. La mort survient entre 18-30 ans suite à des complications cardio-pulmonaires. Il n’existe pas à ce jour un traitement curatif efficace contre cette grave maladie.
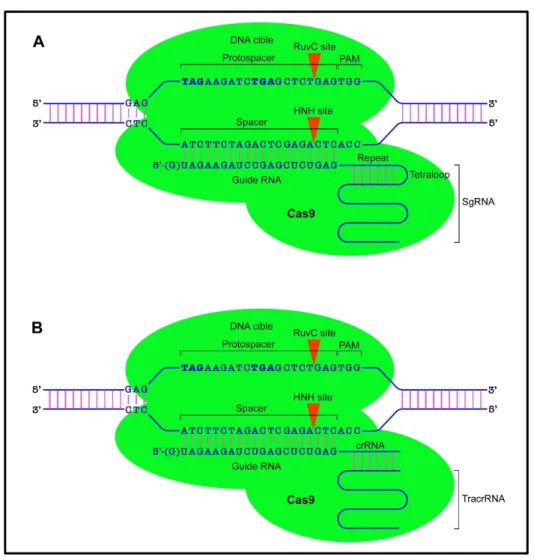
Nous avons développé une approche de thérapie génique appelée CRISPR-induced deletion (CinDel) pour corriger le gène DMD muté. Elle utilise deux ARNg qui ciblent les exons précédant et suivant la délétion responsable du décalage du cadre de lecture. La reconnaissance des sites ciblés par les deux ARNg permet le recrutement de la nucléase Cas9 qui génère des coupures double-brin. Les séquences exoniques et introniques situées entre les deux coupures sont ensuite délétées. Les restes des exons sont joints par la recombinaison non homologue (NHEJ) pour produire un exon hybride, rétablir le cadre de lecture et permettre la synthèse d’une dystrophine tronquée ayant une structure correcte des répetitions de type spectrine (Spectrin-Like Repeat : SLR) et des heptades.
Cette approche CinDel a été utilisée dans le cadre de ce projet d’abord pour corriger le gène DMD muté dans les myoblastes d’un patient avec une délétion des exons 51-53. Les exons 50 et 54 ont été ciblés avec deux ARNg et la Spcas9 pour produire des coupures double-brin et déléter les séquences situées entre ces deux sites et produire par NHEJ un exon hybride 50-54. L’approche a également permis de corriger in vivo le gène DMD muté dans le modèle animal, la souris transgénique avec un gène DMD humain ayant une délétion de l’exon 52 (del52hDMD) en utilisant un vecteur viral AAV9 contenant le gène SpCas9 et deux ARNgs.
Pour vérifier la localisation par rapport au sarcolemme de la dystrophine tronquée avec ou sans une structure correcte des SLR et des heptades, nous avons électroporé les muscles Tibialis anterior de souris mdx/mdx avec des plasmides codant pour les gènes normal et tronqué de la dystrophine fusionnée avec le gène de l’EGFP. Les résultats de cette expérience montrent que les dystrophines tronquées et normale se localisent correctement sous le sarcolemme.
En vue de réprimer efficacement le gène de la SpCas9 et éviter son expression prolongée qui peut être à la base de coupures aléatoires et inattendues (off-target effects) dans le génome, nous avons mis au point une méthode de répression appelée Hara-Kiri moléculaire. Elle utilise la méthode CinDel et consiste à cibler deux régions du gène de SpCas9 avec deux ARNg. Le recrutement de la nucléase permet à celle-ci de couper son propre gène (Hara-Kiri). La séquence située entre les deux sites de coupures est délétée. Par NHEJ, les restes du gène de SpCas9 sont joints en générant un codon stop TAA au point de jonction. Cette approche a permis de réprimer efficacement le gène de SpCas9 in vitro et in vivo.
iii
Abstract
Duchenne Muscular Dystrophy (DMD) is an X-linked genetically recessive genetic disorder. It affects 1 boy out of 3500 male births. The boy with the disorder presents walking disorders at the age of 3-4 years and loses it around the age of 11. Death occurs around 18-30 years of age from cardiopulmonary complications. To date, there is no effective cure for this serious disease.
We have developed a gene therapy approach called CRISPR-induced deletion (CinDel) to correct the mutated DMD gene. It uses two gRNAs that target the exons preceding and following the deletion responsible for the frame shift. The recognition of the target sites by the two gRNAs allows the recruitment of the Cas9 nuclease, which generates double-strand breaks. The exonic and intronic sequences located between the two cuts are then deleted and the remains of the exons are fused by Non-Homologous End Joining (NHEJ) to produce a hybrid exon and restore the reading frame and to allow the synthesis of the truncated dystrophin with correct SLR structure and heptads.
The CinDel approach was used in this project to correct the mutated DMD gene in the myoblasts of a patient with a 51-53 deletion. Exons 50 and 54 were targeted by SpCas9 and two gRNAs and to produce double strand breaks, delete the sequences between the two cleavage sites and produce a hybrid exon 50-54 by NHEJ. This restored the normal reading frame and allowed the expression of truncated dystrophin in the patient's myotubes. The approach also made it possible to correct in vivo the mutated DMD gene in the animal model, the transgenic mouse with a human DMD gene having a deletion of exon 52 (del52hDMD) using an AAV9 viral vector containing the SpCas9 gene and two ARNgs.
To verify the location with respect to the sarcolemma of truncated dystrophin with or without a correct SLR structure and heptads, we electroporated the Tibialis anterior muscles of mdx/mdx mice with the plasmids encoding the normal or the truncated dystrophin gene fused with the eGFP gene. The results of this experiment show that truncated and normal dystrophins were well localized under sarcolemma.
In order to effectively repress the SpCas9 gene and avoid its prolonged expression that may be the basis of random and unexpected (off-target effects) cuts in the genome, we have developed a method of repression called molecular Hara-Kiri. It uses the CinDel method and consists of targeting two regions of the SpCas9 gene with two gRNAs. Recruiting nuclease allows it to cut its own gene (Hara-Kiri). The sequence between the two cleavage sites is deleted. The residues of the SpCas9 gene are then joined by NHEJ generating a TAA stop codon at the junction point. This approach effectively repressed the SpCas9 gene in vitro and in vivo.
iv
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures ... vii
Liste des abréviations, sigles, acronymes ... viii
Remerciements ... xii
Avant-propos ... xiii
Introduction générale... 1
Chapitre 1 : Le tissu musculaire, la dystrophie musculaire de Duchenne et le système CRISPR/Cas9 ... 4
1.1 Le tissu musculaire ... 4
1.1.1 Origine embryonnaire du tissu musculaire ... 4
1.1.2 Le tissu musculaire strié squelettique ... 7
1.1.3 Le tissu musculaire strié cardiaque. ... 9
1.2 La dystrophie musculaire de Duchenne ... 10
1.2.1 La dystrophine ... 10
1.2.2 : La dystrophie musculaire de Duchenne et les approches thérapeutiques ... 12
1.2.3 Les vecteurs adéno-associés ... 22
1.3 : Le système CRISPR/Cas9 ... 24
1.3.1 Introduction ... 24
1.3.2 Historique ... 25
1.3.3 La technologie CRISPR/Cas9 : une révolution biologique ... 26
1.3.4 CRISPR/Cas : du mécanisme de défense des procaryotes à l’édition du génome eucaryote . 26 1.3.5 CRISPR/Cas : l’édition des bases, la régulation génique et la régulation épigénétique. ... 34
1.3.6 CRISPR/Cas : Toxicité et les approches de correction ... 35
Chapitre 2 : Le rétablissement du cadre de lecture du gène DMD et de la structure de la dystrophine tronquée dans les myoblastes d’un patient atteint de la dystrophie de Duchenne avec la méthode CinDel ... 38
Résumé ... 38
Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the CinDel method ... 39
Abstract ... 39
Introduction ... 40
Results ... 41
v
Tests of gRNA pairs ... 42
Characterization of the hybrid exon 50–54 in 293T cells ... 43
Characterization of the hybrid exon 50–54 in myoblasts ... 43
DYS expression in myotubes formed by genetically corrected myoblasts ... 44
3-D spectrin-like models resulting from skipping of exon 50 and formation of hybrid exons 2–50/2–54 and 1–50/4–54... 44
Discussion ... 45
Materials and methods... 47
Identification of targets and gRNA cloning. ... 47
Cell culture... 48
Supplementary material ... 51
Acknowledgments ... 51
References ... 51
Tables ... 58
Supplementary table S1 (continous)... 60
Figure legends ... 60
Supplementary material and methods ... 69
In vivo mouse assay ... 69
In vivo correction in the hDMD/mdx mouse ... 69
Chapitre 3 : Expression in vivo de la dystrophine tronquée-EGFP dans la souris modèle mdx/mdx et traitement de la souris transgénique Δ52hDMD/mdx avec la méthode Cindel ... 73
3. 1 Introduction ... 73
3.2 Matériels et méthodes ... 74
3.2.1 Expression in vivo de la dystrophine tronquée-EGFP dans la souris mdx/mdx ... 74
3.2.2 Traitement de la souris transgénique Δ52hDMD/mdx avec la méthode CinDel ... 78
3.3 Résultats ... 80
3.3.1 Expression in vivo des EGFPcDNADMD50-54-1 et EGFPcDNADMD50-54-2 dans la souris mdx/mdx ... 80
3.3.2 Traitement de la souris transgénique Δ52hDMD/mdx avec la méthode CinDel ... 82
3.4 Discussion ... 84
3.5 Conclusion ... 86
Chapitre 4 : Répression du gène SpCas9 avec Hara-Kiri moléculaire ... 87
Résumé ... 87
Abstract ... 88
Introduction ... 88
vi
Discussion ... 94
Materials and methods... 97
Discussion ... 119
Conclusion générale... 122
vii
Liste des figures
Figure 1: Origine embryonnaire du tissu musculaire ... 4
Figure 2 : Organisation générale d’un muscle squelettique ... 8
Figure 3: La dystrophine et le complexe des protéines associées à la dystrophine. ... 11
Figure 4: Correction du gène DMD muté et les types des dystrophines tronquées ... 13
Figure 5: Les α-hélices des répétitions de types spectrine (SLR : spectrin-like repeat) (image modifiée) . 18 Figure 6: Les hélices hybrides des SLRs ... 19
Figure 7: Le système CRISPR/Cas9 : édition du génome et l’interférence ... 27
Figure 8: Le gène SpCas9, Cas9 protéine et le single-guide RNA ... 31
Figure 9: Le plasmide d’expression pCR3.1EGFPcDNADMD. ... 74
Figure 10 : Expression de la dystrophine-EGFP. ... 81
viii
Liste des abréviations, sigles, acronymes
AAV : Virus adéno-associé
ABD : Actin Bindin domain
ADN : Acide désoxyribonucléique
ADNc : Acide désoxyribonucléique complémentaire
ARN : Acide ribonucléique
ARNm : Acide ribonucléique messager
ASO : Anti-sens oligonucleotide
AsCpf1 : Acidoaminococcus sp Cpf1 ATP : adénosine triphosphate
BLAST : Basic local alignment search tool
BH : Bridge helix domain BMD : Becker musculair dystrophy BSA : Bovin serum albumin
BspE I : Bacillus species (Enzyme de restriction)
CASCADE : CRISPR- associated complex for antiviral defence
Cas9 : Clustered regularly interspaced short palindromic repeat-associated 9
CC : cystein rich domain CH1, 2 : Calponin homology 1, 2
CGH : Comparative genome hybridization
Chip-Seq : Chromatin immunoprecipitation- sequencing CinDel : CRISPR-Induced deletion
CjCas9 : Campylobacter jejunii Cas9 CK : Creatine kinase
CKCS-MD : Cavalier King Charles spaniel- muscular dystrophy CMV : Cytomégalovirus
CNV : Copy number variant
CPAD : Complexe des protéines associées à la dystrophine Cpf1 : CRISPR from Provotella and Francisella 1
CRISPR : Clustered regularly interspaced short palindromic repeat
CRISPRa : CRISPR activator CRISPRi : CRISPR inhibitor
CROP-IT : CRISPR/Cas9 off-target prediction and identification tool
crRNA : Clustered regularly interspaced short palindromic repeat ribonucleic acid
ix
DMEM : Dulbecco modified Eagle medium
DSB : Double-strand break
dsODN : Double-strand oligonucleotide dSpCsa9 : deactivated SpCas9
EDTA : Acide éthylènediaminetétraacétique
eGFP : Enhanced green fluorescent protein
ENCODE : Encyclopedia of DNA element eSpCas9 : engineered SpCas9
FACS : Fluorescence actived cell sorting
FBS : Fetal bovin serum
FGF : Fibroblast growth factor GFP : Green fluorescent protein
gRNA : Guide RNA
GRMD : Golden retriever muscular dystrophy
Guide-Seq : genome-wide unbiased idenfication of DSBs enabled by sequencing HBSS : Hanks balanced salt solution
HEK : Human embryonic kidney
hDMD/mdx : Souris mdx avec le gène DMD humain HDR : Homology directed repair
HNH : Histidine-Aspargin motif HRP : Horse-radish peroxidase
HypaSpCas9: hyper accuracy SpCas9
INDELs : Insertions deletions
LB : Luria et Bertani (milieu de)
LbCpf1 : Lachnospiraceae bacterium Cpf 1 ITR : Interverted terminal repeat
MGNs : Méganucléases
mdx : Muscular dystrophy linked X
MHC : Myosin heavy chain
MHCK7 : Myosin heavy chain creatin kinase 7 miRNA : microRNA
MLC : Myosin light chain
MLPA : Multiplex ligation-dependent probe amplification MRF : Myogenic regulator factor
6MWD : 6 minutes walking distance
NCBI : National Center for Biotechnology Information NHEJ : Non-homologous end joining
x
NHGRI : National Human Genome Research Institute NLS : Nuclear localization signal
nNOS : neuronal nitric oxide synthetase NO : Nitric monoxide
NUC : Lobe avec une activité nucléasique OMIM : Online Mendelian inheritance in man 2’OMePS : 2’-O-méthyl phosphorothioate OTEs : off-target effects
PAM : Protospacer adjacent motif
PCR : Polymerase chain reaction
PGH : Projet génome humain PI : PAM interacting domain
PMO : Morpholino-phosphodiamidate oligomère PTC : Premature terminaison codon
RAMP : Repeat associated mysterious protein REC : Lobe de reconnaissance
RNAg : Ribonucleic acid guided
RRM : RNA recognition motif
RT- PCR : Reverse transcriptase polymerase chain reaction SaCas9 : Staphylococcus aureus Cas9
ScCas9 : Streptococcus canis Cas9
SDS PAGE : Sodium dodecyl sulfate polyacrylamide gel electrophoresis sgRNA : Single guide RNA
SLR : Spectrin-like repeat
SpCas9 : Streptococcus pyogenes Cas9
SpCas9-HF : Streptococcus pyogenes Cas9 high fidelity StCas9 : Streptococcus thermophilus Cas9
Swa I : Staphylococcus warneri I (Enzyme de restriction) TALENs : Transcription activator like-effector nucleases
TBE : Tris boric acid EDTA
tcDNA : Tricyclo DNA
TGF-β : Transforming growth factor-beta T4PNK : Phage T4 polynucleotide kinase
tracrRNA : Trans-activating Clustered regularly interspaced short palindromic repeat ribonucleic acid WB : Western blotting
YAC : Yeast artificial chromosome
xi
Citation
« De plus en plus la recherche moderne montre que la maladie n’est que l’aspect superficiel et récent d’un dérèglement biologique beaucoup plus profond et plus ancien »
xii
Remerciements
Au terme de mes études de doctorat en biologie cellulaire et moléculaire, je voudrais remercier mon directeur de recherche, Dr Jacques-P. Tremblay, pour m’avoir accueilli dans son laboratoire, pour sa disponibilité en dépit de ses nombreuses occupations. Le climat serein qui règne dans son laboratoire ainsi que l’encadrement scientifique qu’il m’a fait bénéficier ont été déterminants pour mon épanouissement.
Je voudrais également remercier mon organisme boursier, le Programme canadien des bourses de la francophonie et son Agence d’exécution, le Bureau canadien pour l’éducation internationale (BCEI) (PCBF/BCEI) pour le soutien financier que j’ai pu bénéficier pour ma formation. Mes remerciements s’adressent également à l’Entraide universitaire mondiale Canada (EUMC) aux Affaires mondiales Canada.
Je voudrais enfin remercier toute l’équipe de recherche du Dr Tremblay et plus particulièrement le professionnel de recherche Joël Rousseau, mon encadreur direct pour sons sens aigu de la précision.
Mes remerciements s’adressent également à ma chère épouse, Liliane TOMI-VAIFUA, et à mes enfants pour leur soutien affectif, sans lequel la réalisation d’un travail aussi exigeant n’aurait pas pu s’accomplir.
Enfin, que tous ceux qui ont contribué d’une manière ou d’une autre à ma formation veuillent trouver ici l’expression de ma gratitude.
xiii
Avant-propos
Le présent projet de doctorat a porté sur la correction du gène de la dystrophine avec la méthode CRISPR-induced deletion. Cette approche vise à corriger le gène de la dystrophine tout en maintenant la structure de la dystrophine tronquée semblable à celle de la dystrophine normale.
La thèse comporte deux parties. La première partie est constituée d’un chapitre divisé en trois sections. La première section rappelle brièvement l’origine embryonnaire du tissu musculaire ainsi la structure du muscle squelettique strié et cardiaque. La deuxième section présente la structure de la dystrophine et ses interactions avec les autres protéines du cytosol et de la matrice extracellulaire, la dystrophie musculaire de Duchenne ainsi que les différentes approches thérapeutiques ont été passées en revue. La troisième section traite du système CRISPR/Cas9 de ses origines à l’édition du génome en passant par la structure du gène ainsi que ses diverses applications dans l’édition du génome.
La deuxième partie comprend les chapitres 2, 3, et 4. Au chapitre 2, j’ai présenté les résultats des essais
in vitro réalisés sur les myoblastes du patient dystrophique avec une délétion des exons 51-53. Ces
résultats ont été publiés dans le journal Molecular Therapy and Nucleic Acids. Ils montrent, après le traitement informatique en 3D avec le logiciel I-Tasser, que la dystrophine tronquée obtenue après formation de l’exon hybride 50-54 avec l’approche CinDel respecte les répétitions de type spectrine (Spectrin-like repeat, SLR) et les heptades.
Le troisième chapitre présente les résultats obtenus in vivo dans les modèles animaux, les souris mdx/mdx et hDMDΔ52/mdx. Dans la souris mdx/mdx, nous avons montré la localisation de la dystrophine-eGFP tronquée avec ou sans structure correcte des SLR et des heptades par rapport au sarcolemme. Dans la souris hDMDΔ52/mdx, les résultats obtenus montrent la correction du gène DMD avec la méthode CinDel dans les muscles squelettiques et cardiaque et la synthèse de la dystrophine tronquée dans les différents tissus.
Dans le quatrième chapitre, nous avons présenté les résultats in vitro et in vivo obtenus avec l’approche
Molecular Hara-Kiri visant à réprimer efficacement le gène SpCas9 après l’édition du génome en utilisant
la méthode CinDel. La réduction de coupures aléatoires (off-target effects, OTEs) a été évaluée avec la technique Guide-Seq [1] et les résultats ont été soumis au journal Molecular Therapy.
La thèse se termine avec une discussion sur l’apport de l’approche CinDel dans le traitement de la DMD par rapport à des approches similaires et une conclusion générale.
1
Introduction générale
Il y a un peu plus de trente ans, Sir David J Weatherall affirmait que le monde vivait une période extraordinaire en matière de développement des sciences biologiques. Il prédisait que l’application des techniques de biologie cellulaire et moléculaire permettra la résolution de plusieurs énigmes de la pathologie humaine dans un avenir proche [2]. Depuis lors les sciences biologiques connaissent une véritable révolution avec des avancées sans précédent. L’une de ces avancées est sans conteste l’aboutissement du Projet Génome Humain (PGH) en 2004 [3] [4]. Après avoir mobilisé plusieurs chercheurs et des fonds tant publics que privés, le PGH a permis le séquençage de 3 milliards de nucléotides constituant le génome humain. Mais cette étape bien qu’elle soit essentielle dans notre compréhension de l’organisation de l’information au niveau moléculaire, n’est, selon certains auteurs, que le début du déchiffrage du « code » [5]. Pour comprendre comment la cellule utilise l’information contenue dans le génome, le National Humain Genome Research Institute (NHGRI) a mis sur pied en 2003 un ambitieux projet appelé le Projet ENCODE (ENCyclopedia Of DNA Elements) [6]. Parmi les résultats obtenus lors de l’analyse du génome, il a été établi que le génome humain ne renferme que 20 000 gènes. Cette partie codante ne représente qu’un peu plus de 1% de l’ensemble du génome. On estime à plus de 10 000 maladies génétiques monogéniques [7] qui peuvent affecter le génome humain. La dystrophie musculaire de Duchenne est l’une de ces maladies génétiques monogéniques.
La dystrophie musculaire de Duchenne (DMD, OMIM #310200) est une maladie monogénique récessive liée au chromosome X. Elle est l’une des plus fréquentes avec une incidence d’un cas pour environ 3500 naissances mâles [8]. La maladie est le résultat d’une série de mutations qui affectent le gène de la dystrophine, gène DMD, situé sur le bras court du chromosome X (locus Xp21) [9, 10]. La conséquence de toutes ces mutations est l’absence de la dystrophine dans les fibres musculaires du patient. Il a été démontré que la dystrophine joue le rôle d’amortisseur de choc lors de contractions musculaires. Elle stabilise le sarcolemme et préserve ainsi l’intégrité de la fibre musculaire. En l’absence de la dystrophine, les contractions des muscles squelettiques et cardiaques peuvent provoquer la rupture du sarcolemme et entrainer la mort de la fibre musculaire.
La recherche à ce jour n’a pu mettre sur pied une thérapie curative de cette grave maladie. Plusieurs approches thérapeutiques sont cependant en cours d’étude pour le traitement de la maladie. Ces approches peuvent être classées en trois catégories : thérapie pharmacologique, thérapie cellulaire et thérapie génique.
La thérapie pharmacologique vise à améliorer l’état du patient en retardant la survenue des complications telles que la perte de la marche, les atteintes cardiaques et respiratoires [11].
2
La thérapie cellulaire consiste dans la majorité des cas à transplanter les myoblastes allogéniques chez le patient. Cette approche est confrontée à un défi majeur : la forte mortalité des myoblastes transplantés dans les heures qui suivent la transplantation [12].
Depuis la découverte du gène DMD et d’une forme rare et modérée de la maladie, la dystrophie musculaire de Becker (BMD, OMIM #300376) chez un patient dystrophique [13], plusieurs approches de thérapies géniques ont été développées. Ces approches visent soit à corriger le gène DMD muté, soit à corriger l’ARNm transcrit du gène DMD muté, soit encore à livrer une copie du gène tronqué avec des vecteurs viraux. Le but ultime de ces approches, dont l’une fait l’objet de la présente thèse, est de transformer un patient dystrophique Duchenne (forme sévère) en un patient dystrophique de type Becker avec une forme légère de la maladie. La première difficulté associée à cette transformation est celle liée aux différents phénotypes de BMD. En effet si la forme sévère (DMD) présente un seul phénotype, la forme légère (BMD) est caractérisée par l’existence de plusieurs phénotypes dont certains ont la même sévérité que la DMD [14]. La distinction des patients dystrophiques en deux groupes, patients avec le gène DMD muté sans et avec décalage du cadre de lecture (in-frame et out-of-frame) selon la règle de Monaco [15], ne peut pas expliquer la sévérité de la maladie observée chez certains patients Becker. Chez certains patients dystrophiques Becker, la synthèse de la dystrophine tronquée dans les fibres musculaires n’améliore pas leur phénotype. En d’autres termes, la dystrophine tronquée ne serait pas fonctionnelle pour ces patients. En effet, certains auteurs ont démontré que la fonctionnalité ou non de la dystrophine tronquée était liée à la structure secondaire de sa partie centrale (rod domain) [16, 17]. La partie centrale de la dystrophine est constituée de 24 répétitions de type spectrine dont chacune des répétions est formée de trois α-hélices (hélices A, B et C). Le repliement des hélices se fait autour d’un noyau central (core) formé de 7 acides aminés a, b, c, d, e, f et g, appelé heptade. Les acides aminés a et d sont essentiellement hydrophobes et sont situés à l’intérieur de la structure où ils sont liés entre eux par les interactions hydrophobes. Au vu de la complexité de la structure de la dystrophine, nous avons formulé l’hypothèse selon laquelle, la dystrophine tronquée devrait, pour être fonctionnelle, respectée l’organisation structurale des répétitions de type spectrine et des heptades. À cet effet, j’ai utilisé l’approche développée par Zheng et coll.[18]. Elle utilise le système CRISPR/Cas9 (Cluster regularly interspaced short palindromic repeat / CRISPR associated nuclease 9) avec deux ARN guides (gRNA) pour cibler d’une manière précise les exons précédant (exon 50) et suivant (exon 54) la délétion responsable du décalage du cadre de lecture chez un patient dystrophique Duchenne avec une délétion des exons 51-53, générer des coupures double-brin, déléter les parties des exons et des introns situées entre les deux coupures. Enfin, par la réparation non homologue (Non- homologous end joining, NHEJ), faire la jonction des restes des exons 50 et 54 pour produire un exon hybride 50-54 et rétablir le cadre de lecture. Cette approche appelée
3
structurale des répétitions de type spectrine et les heptades dans la dystrophine tronquée, éléments essentiels pour sa fonctionnalité.
La méthode CinDel exploite la précision des coupures double-brin générées avec la technologie CRISPR/Cas9 d’une part et la quasi absence de micro-délétions et de micro-insertions aux sites de jonction des restes exoniques pour permettre non seulement le rétablissement du cadre de lecture mais aussi une structure adéquate des hélices des SLRs hybrides résultant des exons hybrides. La méthode CinDel permet également d’induire une délétion d’une séquence de petite taille (délétion des parties des exons au lieu des exons entiers) pour maintenir intacte la partie de la dystrophine qui interagit avec les protéines du cytosol. Pour obtenir une si grande précision dans les sites de coupure des exons 50 et 54, nous avons utilisé les séquences des exons 50 et 54 [19] afin de construire les différents ARNg ciblant ces deux exons. Les ARN guides ont été ensuite utilisés par paire pour identifier les sites de coupure double-brin à la fois sur les exons 50 et 54 et prédire la séquence délétée ainsi que la jonction des restes des exons permettant la formation de l’exon hybride 50-54. Les SLRs résultant des exons hybrides 50-54 issus des différentes paires des ARNg ont été ensuite soumises à la modélisation in silico en utilisant le logiciel I-tasser. Celle-ci a confirmé la bonne conformation de certaines SLRs hybrides ainsi que des heptades obtenues avec certaines paires d’ARNg. Ces paires d’ARNg et le gène de la SpCas9 ont été insérés dans les vecteurs adéno-associés et ont été utilisés pour la correction in vivo du gène DMD muté dans le modèle animal.
4
Chapitre 1 : Le tissu musculaire, la dystrophie
musculaire de Duchenne et le système
CRISPR/Cas9
1.1 Le tissu musculaire
1.1.1 Origine embryonnaire du tissu musculaire
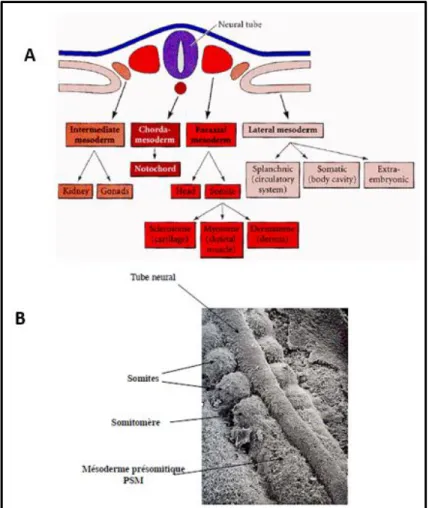
Le tissu musculaire dérive de mésoderme paraxial ou somitique. Situé de part et d’autre du tube neural, le mésoderme paraxial est à l’origine de l’ensemble des muscles du corps et de nombreux tissus conjonctifs du dos et de la face du crâne (Figure 1A) [20].
Figure 1: Origine embryonnaire du tissu musculaire
A) Le mésoderme et les grandes lignées
5
Pendant la somitogènese qui se déroule en plusieurs étapes (fissuration, périodicité, épithélialisation, spécification et différentiation), le mésoderme présomitique se segmente, à intervalles réguliers, en somitomères précurseurs de somites. Après maturation, les somitomères se transforment en somites matures multipotents (Figure 1B) [21]
Les somites matures vont ensuite se différencier en sclérotome et en dermamyotome. Le sclérotome est le mésenchyme ventral à partir duquel se forment le cartilage des côtes, les tendons, les vertèbres, les disques intervertébraux, les articulations. Le dermamyotome se subdivise en trois parties : le myotome formé des deux régions latérales du dermamyotome et le dermatome formé à partir de la région centrale. Les cellules précurseures de muscles dérivant du myotome migrent ensuite entre le dermamyotome et le sclérotome pour constituer un mince feuillet formé par les cellules myogéniques primaires. Ces dernières se différencient finalement en myoblastes capables de produire les facteurs de régulation myogéniques tels que Myf5, MyoD et MRF4 (en anglais, myogenic regulatory factors (MRFs) [22].
Après une phase de multiplication médiée essentiellement par les Fibroblasts growth factors (FGFs), les myoblastes post-mitotiques mononucléés fusionnent pour former les myotubes multinucléés. La fusion des myoblastes post-mitotiques se déroule en quatre étapes. La première étape permet la sortie des myoblastes du cycle cellulaire [23]. Dans la deuxième étape, les myoblastes secrètent la fibronectine et d’autres protéines dans la matrice extracellulaire. L’interaction entre les myoblastes et le complexe des protéines de la matrice extracellulaire se fait avec l’intégrine α5β1 [24] [25]. L’interaction fibronectine-intégrine est une étape critique pour induire la différenciation des myoblastes en cellules musculaires. La troisième étape se caractérise par l’alignement en chaine des myoblastes. Cette étape est médiée par les glycoprotéines membranaires dont plusieurs cadhérines [26]. La dernière étape permet la fusion membranaire des myoblastes. La fusion de myoblastes est activée par les protéines du transport de calcium à travers la membrane plasmique [27]. D’autres protéines telles que les métalloprotéinases meltrines, interviennent également dans la fusion membranaire [28]. La fusion des myoblastes permet l’activation de myogénine qui, à son tour, active l’expression de plusieurs gènes codant les facteurs intervenant dans la différenciation des cellules musculaires [29]. La fin de la fusion des myoblastes intervient lorsque les nouvelles membranes cytoplasmiques sont formées sous l’action des protéines telles que myoferline et dysferline qui jouent les rôles de stabilisateurs des phospholipides membranaires [30]. Les myotubes ainsi formés vont se différencier et accumuler les protéines contractiles, l’actine et la myosine. Les deux protéines sont arrangées en faisceaux formant ainsi les myofibrilles. La disposition des myofibrilles en filaments montre une alternance entre les filaments minces d’actine et les filaments épais de myosine le long des myofibrilles. L’organisation des myofibrilles en filaments minces et épais est à la base des unités contractiles répétées appelées sarcomères. Au début de leur formation, les myofibrilles sont localisées à la périphérie des myotubes et les noyaux au centre. Mais au fur et
6
à mesure de la maturation des myotubes, les myofibrilles repoussent les noyaux à la périphérie et finissent par occuper le centre où elles sont disposées longitudinalement. Les myotubes synthétisent alors les protéines régulatrices (troponine et tropomyosine) et les enzymes cytosoliques intervenant dans le métabolisme énergétique de la cellule musculaire.
Les fibres musculaires nouvellement formées secrètent IL4 qui permet le recrutement de nouveaux myoblastes et la maturation des fibres musculaires [31].
Il est à noter que la myostatine, un membre de la famille des TGF-β, régule négativement le nombre et la taille des fibres musculaires. Une mutation par perte de fonction de ce gène est à la base du phénotype herculéen avec hyperplasie et hypertrophie de muscles [32].
1.1.1.1 Les cellules satellites
Toutes les cellules dérivant du myotome ne se différencient pas en myoblastes pour fusionner et former les fibres musculaires. Il existe une lignée de cellules souches et progénitrices qui se localisent entre la membrane de fibre musculaire et lamina basal extracellulaire. Ce sont les cellules satellites [33]. Les cellules satellites permettent l’accroissement de la masse musculaire lors d’exercices physiques et la régénération musculaire à la suite des dommages [34].
Pendant que les cellules du myotome qui se différencient en myoblastes et expriment Myf5 et MyoD, celles destinées à devenir les cellules satellites expriment Pax3, Pax7 et surtout les miRNA-489 et miRNA-31. L’expression combinée de Pax3 et Pax7 inhibe MyoD et empêche la différenciation de ces cellules en myoblastes. Pax7 protège également ces cellules contre l’apoptose [35]. Les deux microRNAs (miRNA-489 et mRNA-31) préviennent la traduction des mRNAs de MRFs tels que Myf5 qui interviennent également dans la différentiation des cellules musculaires [36, 37]. Les études récentes ont montré que les cellules satellites ne forment pas une population homogène en ce qui concerne leur stade de maturation. On distingue à la fois les cellules souches et les cellules progénitrices. Les cellules souches représentent environ 10 % de cellules satellites. Elles expriment Pax7 et non Myf5 et sont appelées Pax7+/Myf5-. Elles se divisent d’une manière asymétrique donnant ainsi naissance à deux types de cellules satellites : Pax7+/Myf5- et Pax7+/Myf5+. Le premier type, comme nous l’avons indiqué ci-dessus, est la cellule satellite souche tandis que le deuxième type est la cellule satellite progénitrice qui peut se différencier en cellule musculaire. Le facteur qui régit la division asymétrique est miRNA-489. Il est présent dans les cellules souches filles et absent dans celles qui deviennent progénitrices et qui peuvent se différencier en cellules musculaires. Il maintient ainsi les cellules satellites souches à l’état quiescent chez l’adulte.
7 1.1.2 Le tissu musculaire strié squelettique
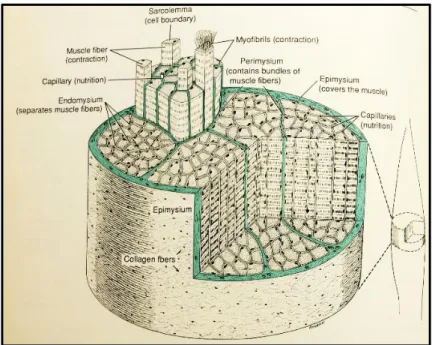
L’ensemble des muscles squelettiques représente environ 40-45% de la masse totale du corps. Le muscle est formé par le regroupement des fibres musculaires réunies par un tissu conjonctif. Ces dernières ont la forme d’un cylindre dont le diamètre varie entre 10 et 100 µm et la longueur peut atteindre 30 cm [38]. Le regroupement des fibres musculaires qui forment les différents muscles ne se produit pas d’une manière aléatoire. De la périphérie vers le centre, on distingue trois enveloppes de tissu conjonctif qui entourent les différentes parties du muscle. Il s’agit de l’épimysium (enveloppe tout le muscle), du périmysium (délimite les faisceaux musculaires) et de l’endomysium (entoure chaque fibre musculaire) [39] (Figure 2).
La cellule musculaire ou fibre musculaire est l’unité fonctionnelle et morphologique du muscle. C’est une cellule cylindrique allongée, plurinucléée avec les noyaux situés à la périphérie de la cellule contre le sarcolemme (membrane cytoplasmique de la cellule musculaire). Ce dernier est entouré d’une membrane basale. Le sarcoplasme se caractérise essentiellement par la présence des protéines fibrillaires contractiles regroupées en myofibrilles. Dans la section qui suit, nous allons examiner les myofibrilles ainsi que les différentes protéines contractiles qui les composent.
1.1.2.1 Les myofibrilles
Les myofibrilles sont des structures protéiques tubulaires, parallèles et allongées dans le sens de la cellule musculaire. Le sarcomère, unité contractile des myofibrilles, est constitué des myofilaments de deux types : les filaments minces d’actine associée à la tropomyosine et à la troponine et les filaments épais de myosine. Ces différents filaments sont réunis selon une organisation géométrique en trois dimensions très particulière. Dans le sarcomère, les filaments minces sont attachés de part et d’autre d’une structure protéique appelée disque Z. Ce disque comprend l’α-actinine et des protéines d’ancrage des filaments d’actine. Ces filaments d’actine sont disposés parallèlement sans qu’ils se touchent entre eux. Entre deux disques Z et dans les espaces entre les filaments minces, on trouve des filaments épais de myosine [39].
8
Figure 2 : Organisation générale d’un muscle squelettique
De la périphérie vers le centre, on distingue les différentes enveloppes formées de tissu conjonctif entourant les différentes parties du muscle : épimysium, périmysium et l’endomysium (Avec l’aimable autorisation de l’éditeur) [39].
1.1.2.1.1 Les filaments minces d’actine
L’actine est un polypeptide de forme globulaire (forme G). La polymérisation des monomères d’actine se fait sous une forme filamenteuse (forme F). Les polymères d’actine s’associent par deux pour former une longue double hélice. Les myofilaments minces sont formés par l’association de cette double hélice d’actine de deux protéines régulatrices : la tropomyosine, un dimère filamenteux rigide de renforcement, et la troponine, un complexe de trois sous-unités polypeptidiques disposées à intervalles réguliers. Le long des filaments d’actine, en regard de chaque tête de myosine, la tropomyosine et la troponine sont impliquées dans la régulation la contraction musculaire par le calcium. La nébuline associée à chaque filament mince du muscle strié squelettique, détermine la longueur de filaments en servant de guide pour la polymérisation de l’actine.
1.1.2.1.2 Les filaments épais de myosine
La myosine est un composant important de l’appareil contractile des fibres musculaires squelettiques. La forme fonctionnelle de la myosine est un hexamère composé de deux chaines lourdes (Myosin heavy Chain, MHC) de 200 kDa chacune et de deux chaines légères (Myosin Light Chain, MLC). On distingue MLC 20 de 20 kDa et MLC 17 de 17 kDa. Il existe cependant de nombreuses isoformes de MHC qui sont exprimées à différentes étapes de la myogenèse et qui rendent compte également du caractère plus ou moins rapide de la contraction de différents types des fibres musculaires. Les deux MCH de la même protéine sont identiques et
9
accolées l’une à l’autre. Chaque chaine est constituée d’une tête globulaire et d’une longue queue en hélice α [38]. La tête appelée également domaine moteur a une activité ATPasique et peut transformer l’énergie chimique due à l’hydrolyse de l’ATP en une énergie mécanique capable de générer une contraction musculaire. Les queues en hélice α forment des tiges enroulées qui permettent l’assemblage d’une molécule de myosine individuelle en un filament fonctionnel.
La connectine ou titine est une protéine qui, dans chaque demi-sarcomère, relie chaque filament épais à la strie Z. Composante élastique, la connectine maintient l’alignement des filaments épais et oppose une résistance à l’étirement excessif du sarcomère.
1.1.3 Le tissu musculaire strié cardiaque.
Le tissu musculaire strié cardiaque compose la paroi du cœur. Cependant bien qu’il s’agit d’un muscle strié, ses contractions sont involontaires. De plus, certains muscles qui le composent sont dotés d’autorythmicité qui leur permet d’établir une cadence inhérente et alternative de contractions et de relâchements [38]. Les fibres du tissu musculaire cardiaque sont de forme parallélépipédique et font près de 14 µm de large [39]. Elles sont composées de cellules mononucléées appelées cardiomyocytes. Les noyaux de ces cellules sont au centre du sarcolemme. Bien que ressemblant à celui de muscles striés squelettiques, le sarcolemme des myotubes cardiaques contient plus de mitochondries. Ces mitochondries sont par ailleurs plus volumineuses. Enfin, l’unité sarcomérique est également semblable à celle observée dans le tissu musculaire strié squelettique. L’ensemble des fibres musculaires cardiaques se ramifient et s’anastomosent formant ainsi deux réseaux distincts. Les oreillettes, définissant la partie supérieure des cloisons cardiaques, constituent le premier réseau. Les ventricules qui constituent la partie inférieure du cœur, composent quant à elles le deuxième réseau. Pour un réseau donné, chaque fibre s’interconnecte au niveau d’épaississements traverses irréguliers du sarcolemme appelés disques intercalaires. Ces disques comprennent des desmosomes et jonctions lacunaires. Ces structures assurent réciproquement entre les fibres, un maintien structural et un passage des potentiels d’action musculaires qui sont à l’origine des contractions cardiaques. Ainsi lorsqu’une fibre est stimulée, toutes les autres fibres du réseau le sont également. De ce fait, chaque réseau se compose comme une unité fonctionnelle.
Les fibres cardiaques sont des fibres à contractions perpétuelles et extrêmement rapides. Cette distinction majeure avec les fibres qui composent le tissu musculaire strié squelettique implique que le tissu musculaire cardiaque bénéficie d’une bonne vascularisation et de la présence d’un nombre conséquent de mitochondrie de grande taille. Conséquemment, le muscle cardiaque utilise la voie de synthèse aérobie comme mode principal de production de l’ATP. Enfin, à la différence du muscle strié squelettique, le muscle cardiaque peut
10
se contracter sans stimulation nerveuse extrinsèque ou hormonale. En effet, des fibres cardiaques spécialisées lui confèrent une capacité contractile autonome.
1.2 La dystrophie musculaire de Duchenne
Avant que nous puissions examiner la dystrophie musculaire de Duchenne, il parait utile de revenir brièvement sur la dystrophine qui est la protéine affectée par la maladie.
1.2.1 La dystrophine
1.2.1.1 Le gène de la dystrophine et la structure de la protéine
Le gène DMD codant la dystrophine est situé sur le bras court du chromosome X [9, 10]. Il s’agit du plus long gène de l’organisme avec 2,4 Mb. Il représente environ 1% du chromosome X. Le gène DMD est formé de 79 exons séparés par de larges séquences introniques (Leiden muscular dystrophy)[19]. La partie codante ne représente qu’environ 0,6% du gène avec un cDNA de 14 kb [40]. La transcription du gène DMD se fait sur 7 promoteurs tissu-spécifiques et génère des transcrits longs et courts qui, une fois traduits, correspondent aux différentes isoformes longues et courtes de la dystrophine.
Il existe 3 isoformes longues de 427 kDa chacune: Dp 427 B, Dp 427 M et Dp 427 P respectivement pour le cerveau, les muscles et les cellules de Purkinje [41]. L’isoforme 427 kDa également appelée dystrophine normale (full length, en anglais), est constituée de 3685 acides aminés. Outre les isoformes longues dont les promoteurs sont situés dans la région promotrice du gène DMD et à l’intron1 respectivement pour Dp427 B et Dp427 M d’une part et, Dp427 P, d’autre part, il existe quatre isoformes courtes. Il s’agit de l’isoforme de la rétine ou Dp 260, R; de l’isoforme spécifique du cerveau ou Dp 140, B; de l’isoforme de la cellule de Schwann ou Dp 116, S et l’isoforme générale ou Dp 71, G dont les promoteurs sont situés respectivement sur l’intron 29, l’intron 44, l’intron 55 et l’intron 62 [41].
La dystrophine est formée de quatre principaux domaines : domaine N-terminal, domaine central, domaine riche en cystéine et le domaine C-terminal [42].
Le domaine N-terminal est codé par les exons 1 à 7 et se lie à l’actine (ABD1, actin-binding domain1). Cette région interagit également avec la kératine 19 et les filaments intermédiaires. Elle est constituée des deux séquences CH1 et CH2 (calponin-homology 1 and 2) ayant une homologie avec la calponine. Les acides aminés 9 à 246 appartiennent à ce domaine.
Le domaine central (rod domain) est codé par les exons 8 à 64. Il est formé par 24 répétitions de type spectrine (R1-24) (SLR : spectrin-like repeat, en anglais) chacune contenant trois hélices-α [43, 44]. On note
11
également la présence de quatre séquences riche en proline appelées hinge (H1, H2, H3 et H4) localisées respectivement entre CH2 et R1, R3 et R4, R19 et R20 et, R24 et le domaine riche en cystéine. Cette région interagit avec plusieurs partenaires. Les répétitions R1 à R4 ainsi que R4 à R19 interagissent avec les phospholipides membranaires. La kinase PAR-1b interagit avec les répétitions R8 et R9. Le deuxième site de liaison à l’actine (ABD2) fait intervenir les R11 à R18. Cette partie se lie également aux filaments intermédiaires. Les répétitions R16 et R17 se lient à nNOS (neuronal nitric oxide synthetase) et les microtubules aux répétitions R20 à R24.
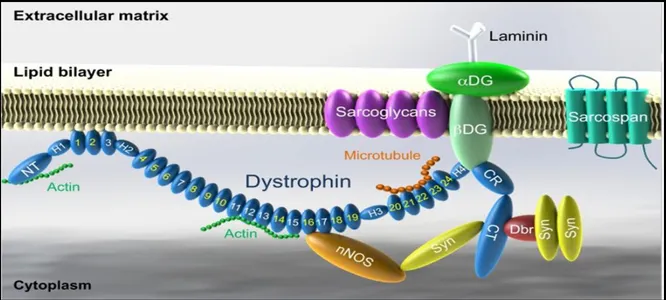
Le domaine riche en cystéine ou CC domaine et la région C-terminale sont formés de 280 acides aminés dont 15 résidus cystéines pour CC domaine et de 325 acides aminés pour la région C-terminale. Cette partie interagit avec les différentes protéines du complexe des protéines associées à la dystrophine telles que les α et β-dystroglycane qui permettent la jonction avec la laminine et la matrice extracellulaire.[45] (Figure 3).
Figure 3: La dystrophine et le complexe des protéines associées à la dystrophine.
La dystrophine intéragit avec les différentes protéines du cytosol et, grâce à la bêta-dystroglycane (βDG), avec la matrice extracellulaire. NT: N-terminus, H1, H2, H3 et H4 : hinges, nNOS : neuronal nitric oxyde synthetase, Syn : syntrophin, CR : cystein rich domain, CT : C-terminus, Dbr : Dibrobrevin, αDG : alpha-dystroglycan, 1-24 : spectrin like-repeats [46].
12
1.2.2 : La dystrophie musculaire de Duchenne et les approches thérapeutiques
1.2.2.1 Introduction
À ce jour, il n’existe pas un traitement curatif efficace contre la dystrophie musculaire de Duchenne. Il existe cependant plusieurs approches thérapeutiques qu’on classe habituellement en trois groupes : la thérapie symptomatique, la thérapie cellulaire et la thérapie génique. Si la première approche vise seulement à améliorer les aspects cliniques les plus sévères de la maladie, les deux dernières visent soit à apporter le gène DMD normal présent dans les myoblastes allogéniques, soit à corriger le gène DMD muté ou le produit d’expression du gène DMD muté (mRNA) présent dans le tissu musculaire du patient. Dans ce chapitre, nous examinerons les aspects cliniques de la dystrophie musculaire de Duchenne, les différentes approches thérapies en indiquant les avantages et les inconvénients des uns et autres. Nous terminerons la section sur les modèles animaux utilisés pour valider les expérimentations in vivo avant les essais cliniques les vecteurs adéno-associés.
1.2.2.2 La dystrophie musculaire de Duchenne
1.2.2.2.1 Les aspects cliniques
La dystrophie musculaire de Duchenne ou myopathie de Duchenne est une maladie génétique récessive liée au chromosome X. Elle a été décrite pour la première fois en 1858 par le médecin français Duchenne de Boulogne [47]. La maladie se manifeste vers l’âge de 2 à 4 ans par les troubles de la marche. Le garçon atteint de la maladie marche tardivement, tombe souvent et utilise la « manœuvre de Gowers » pour se relever. En effet le patient assis ou couché se sert de ses mains et de ses bras pour se relever. La manœuvre qui se déroule en quatre temps s’explique par la faiblesse des muscles des cuisses et des hanches [48]. Il éprouve beaucoup de difficultés à monter les escaliers et on peut noter une pseudo-hypertrophie des mollets. Vers l’âge de 11 à 12 ans, il perd la marche et ne peut plus se déplacer que sur une chaise roulante. La mort survient vers l’âge de 20 à 30 ans suite à des complications cardio-pulmonaires [49].
Le mérite de Duchenne de Boulogne est celui d’être le premier à avoir pratiqué le prélèvement des biopsies chez les patients et le suivi en temps réel de l’évolution de la maladie. L’examen de la biopsie montre que le tissu musculaire chez le patient est progressivement détruit et remplacé avec un tissu conjonctif et/ou adipeux non fonctionnel avec infiltration des cellules inflammatoires [47]. Si à l’époque, le caractère héréditaire de la maladie et son mode de transmission lié à au chromosome X (la très grande majorité des patients sont des garçons) sont déjà établis, il faudra attendre plus d’un siècle pour connaitre la cause de cette grave maladie [9, 10]. Il s’agit d’une mutation du gène de la dystrophine, gène DMD, situé sur le locus Xp21 et dont la conséquence est l’absence de la dystrophine dans les fibres musculaires du patient. De par sa localisation dans une structure formée par plusieurs protéines appelée le complexe des protéines associées à la
13
dystrophine (CPAD) [45], la dystrophine permet la jonction entre les protéines de la matrice extracellulaire avec l’actine située sous le sarcolemme (Figure 3). Ainsi lors de la contraction musculaire, la dystrophine joue le rôle d’amortisseur de chocs en préservant l’intégrité du sarcolemme [50]. L’absence de la dystrophine chez les patients DMD va se traduire par la rupture du sarcolemme lors des contractions musculaires, le déversement du contenu des fibres musculaires dans le courant circulatoire (augmentation de la créatine kinase dans le sérum) et par la mort de la cellule musculaire [49] (Figure 4). L’absence de la dystrophine comme le montrent les études récentes [11] va également affecter l’organisation structurale des complexes protéiques cytosoliques auxquels elle est rattachée et également plusieurs voies de signalisation dans lesquelles elle intervient [51].
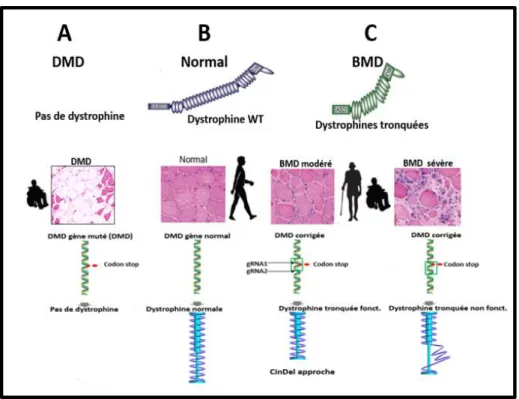
Figure 4: Correction du gène DMD muté et les types des dystrophines tronquées
A) DMD avec une destruction des fibres musculaires (zone claire dans une coupe de muscle squelettique
colorée à l’hématoxyline-éosine). Le patient perd la locommotion et est cloué sur un fauteuil. La mutation (point rouge sur le gène DMD) n’a pas été corrigée et on observe l’absence de la dystrophine représentée ici sous forme d’amortisseur de choc. B) Sujet non dystrophique. C) Patient BMD. À gauche la forme modérée avec le tissu musculaire moins atteint et une dystrophine tronquée avec une structure correcte après correction du gène muté avec CinDel. À droite la forme sévère avec un tissu musculaire détruit et une dystrophine tronquée avec une structure incorrecte (image modifiée) [46].
14
1.2.2.2.2 Le diagnostic moléculaire
Le diagnostic clinique de la dystrophie musculaire de Duchenne se fait dans la majorité des cas vers l’âge de 2-4 ans lorsque l’enfant commence à marcher. Les difficultés de marche ou encore la marche tardive, la difficulté à monter les escaliers sont les premiers signes cliniques qui conduisent les parents à consulter le pédiatre. A cet âge, un examen clinique montre une faiblesse musculaire, parfois une hypertrophie des mollets. La détermination de l’activité de créatine-kinase (CK) dans le sérum est fortement augmentée. L’augmentation de l’activité de CK est un diagnostic présomptif qui doit être confirmé par un diagnostic moléculaire. Ces dernières années, plusieurs méthodes ont été développées pour préciser le type de mutation de la dystrophie musculaire de Duchenne. La recherche d’une délétion d’un ou de plusieurs exons peut être effectuée par amplification PCR des exons (Multiplex ligation-dependent probe amplification, MLPA) [52]. Cette méthode permet de poser le diagnostic moléculaire de la dystrophie musculaire de Duchenne chez 65 % de patients et permet de déterminer la variation du nombre de copie d’exons (copy number variant, CNV, en anglais). La diminution du nombre de copies indique une délétion tandis qu’une augmentation du nombre de copie montre une duplication.
La méthode MLPA, comme on peut le remarquer, ne permet pas de poser le diagnostic des mutations ponctuelles. Pour une plus grande précision, il faut utiliser la technique du Comparative genomic hybridization (CGH) qui utilise une micropuce et qui permet de faire une analyse complète du gène DMD. On peut de cette manière mettre en évidence les variations du nombre de copies (CNV) des introns, des exons et des régions de jonction intron-exon.
1.2.2.3 Les approches thérapeutiques
1.2.2.3.1 La thérapie symptomatique
La thérapie symptomatique vise à ralentir la progression de la maladie et à améliorer le bien-être du patient dystrophique Duchenne. Elle fait essentiellement appel aux corticostéroïdes et dans certains cas à la physiothérapie.
Les corticostéroïdes ont été utilisés depuis de nombreuses années comme traitement symptomatique contre la DMD [53]. Ils agissent comme des anti-inflammatoires stéroïdiens. Les corticostéroïdes les plus utilisés sont prednisone/prednisolone et Déflazacort, un dérivé de prednisolone. Ces deux médicaments ont permis d’améliorer d’une manière significative les symptômes des patients dystrophiques comme le montrent plusieurs études [54, 55]. L’utilisation sur une longue période de ces corticostéroïdes a cependant montré des effets secondaires tels que les troubles nerveux et l’ostéoporose [56] qui peuvent limiter le temps de leur utilisation.
15
Pour faire face aux effets secondaires des corticostéroïdes, d’autres molécules anti-inflammatoires ont été développées. C’est le cas de Givinostat, un inhibiteur de l’histone désacétylase de classe 1 (HDAC2) [57].
La dystrophie musculaire de Duchenne régule négativement la synthèse de l’oxyde nitrique (NO) par la synthétase neuronale de l’oxyde nitrique (nNOS), certaines molécules sont utilisées comme source de NO et améliorent ainsi le phénotype des patients. L’isosorbide dinitrate est l’une de ces molécules et est utilisé comme une source de NO [58].
12.2.3.2 La thérapie cellulaire
La transplantation des myoblastes allogéniques ou autologue corrigés génétiquement ex vivo, ou de cellules souches dans les muscles du patient dystrophique vise à apporter le gène DMD normal ou corrigé dans les fibres musculaires [59, 60] [61]. La transplantation locale des myoblastes allogéniques est confrontée à un problème majeur : la forte mortalité des myoblastes transplantés [12]. Malheureusement jusqu’à ce jour, les causes de cette forte mortalité n’ont pas encore été élucidées [62].
Les myoblastes allogéniques ne sont pas les seules cellules qui peuvent être utilisées dans la thérapie cellulaire pour la dystrophie musculaire de Duchenne. Différentes cellules souches qui peuvent se différencier en myoblastes ont été identifiées. Il s’agit notamment des mésoangioblastes [63]. Ces cellules souches et progénitrices myogéniques qui dérivent des vaisseaux sanguins auraient la capacité de franchir la paroi des vaisseaux et de migrer vers les fibres musculaires. Outre les mésoangioblastes, d’autres cellules souches ont été identifiées. C’est le cas des péricytes (cellules souches progénitrices isolées sur les parois des vaisseaux sanguins et capables de se différencier en cellules musculaires) [64] et des CD33+ (population hétérogène des cellules myéloides immatures pouvant se différencier en cellules musculaires) [65].
1.2.2.3.3 La thérapie génique
La grande difficulté à surmonter dans la thérapie génique de la dystrophie musculaire de Duchenne est la très grande taille du gène. Avec 2,4 Mb et un cDNA de 13 kb [45], seuls les adénovirus peuvent inclure ce cDNA pour l’introduire dans l’organisme du patient. Cependant ce vecteur ne peut pas infecter les fibres musculaires d’une souris adulte. La découverte d’un patient avec une forme légère de la maladie [13], la dystrophie musculaire de Becker, dans laquelle une forme tronquée de dystrophine était synthétisée dans les fibres musculaires, a ouvert la voie à plusieurs approches de thérapie géniques. Elles visent toutes à transformer le patient Duchenne en un patient de type Becker. En tenant compte de la partie ciblée à corriger, on peut distinguer 3 types de thérapie génique : thérapie génique par correction des transcrits du gène DMD muté, la thérapie génique par l’apport d’un gène tronqué avec des vecteurs associés à l’adénovirus (AAV) et la thérapie génique par la correction in situ du gène DMD muté.
16
1.2.2.3.3.1 La correction des transcrits du gène DMD muté
La présence de fibres révertantes dans les muscles des patients dystrophiques a amené certains chercheurs à développer un mécanisme d’épissage alternatif pour déléter une partie du pré-mRNA et rétablir le cadre de lecture [52]. La découverte d’un patient dystrophique avec une délétion de 46% du gène DMD [13], a également conforté les chercheurs à l’idée que la délétion d’une partie du gène DMD ou de son transcrit pouvait être compatible avec une forme légère de la maladie. Étant donnée que cette approche porte sur le transcrit (pré-mRNA) et non sur le gène DMD muté, elle ne peut être que transitoire et nécessite donc une administration répétée de l’agent thérapeutique (ASOs de synthèse). Mais l’administration répétée et sur une longue période du produit chimique peut comporter des effets secondaires qui peuvent affecter gravement l’état de santé du patient.
1.2.2.3.3.2 Le saut d’exon
Le saut d’exon (exon skipping en anglais) est l’une des approches qui exploitent la correction du transcrit du gène DMD muté. Il consiste à masquer les régions d’épissage (splicers donor et acceptor en anglais) avec les oligonucléotides antisens (ASOs) d’un ou des plusieurs exons. Les séquences à épisser qui forment les hétéroduplex ASO-transcrit ne sont pas reconnues (sont « sautées ») par la machinerie d’épissage. L’épissage va alors se réaliser en amont et en aval des hétéroduplex permettant ainsi la délétion de certains exons du pré-mRNA. La délétion de l’exon ou des exons responsables du décalage du cadre de lecture permet la traduction du mRNA tronqué en une protéine amputée de plusieurs acides aminés qui pourrait être fonctionnelle [66, 67].
La première difficulté liée à l’utilisation des ASOs est leur dégradation par les RNAse H intracellulaire. Pour surmonter cette difficulté, des modifications chimiques ont été apportées lors de la production des ASOs pour qu’ils résistent à l’action de la RNAse H. C’est le cas de 2’-O-méthyl phosphorothioate oligonucléotide antisens (2’OMePS) et de morpholino-phosphorodiamidate oligomères (PMO), les deux ASOs les plus utilisés. Ce dernier (PMO) sous le nom d’Exondys51R (Eteplirsen) de la firme Serepta Therapeutics, a obtenu en 2016 une approbation accélérée controversée pour la commercialisation de FDA (Food and Drug administration) suite à un essai clinique de phase I sur un petit nombre de patients DMD [68]. Pour obtenir l’approbation définitive de la FDA, Serepta Therapeutics doit démontrer l’efficacité et l’innocuité de l’Exondys51R dans l’essai clinique NCT02255552 [69].
Pour améliorer les propriétés pharmacologiques des ASOs, de nouvelles molécules ont été produites telles que Tricyclo-DNA (tcDNA). Cette molécule présente une structure analogue à celle de l’ADN avec comme particularité la présence de trois atomes de carbone supplémentaires entre C5’ et C3’ [70]. Elle peut pénétrer
17
le muscle cardiaque et franchir la barrière hémato-encéphalique dans le modèle mdx en comparaison aux deux ASOs PMO et 2-O-Me-PS [71].
1.2.2.3.3.3 La traduction de l’ARNm contenant un codon non-sens (stop).
Il a été démontré que certaines molécules telles que les aminoglycosides ou non-aminoglycosides tel que PTC124 (ancien Ataluren ou Tranlsarna) pouvaient permettre la traduction par les ribosomes des ARNm contenant un codon non-sens. Il y a incorporation dans la protéine dystrophine d’un acide aminé qui peut ne pas être celui rencontré habituellement à cette position dans la protéine. Il y aurait une sorte de « mutation faux sens » au niveau de l’ARNm. L’Atularen a déjà été utilisé dans les essais pré-cliniques et cliniques [56]. L’essai clinique en phase 3 (NCT01826487) n’a pas montré une amélioration significative chez les patients traités par rapport à ceux ayant reçu le placébo en ce qui concerne le test de 6 minutes de marche (6MWD en anglais)[72].
L’Atularen est également utilisé dans le traitement de la fibrose kystique. L’essai clinique en phase 3 (ACT CF NCT02139306) dont les résultats ont été publiés en 2017 n’ont malheureusement pas montré une amélioration de la capacité pulmonaire chez les patients traités par rapport à ceux ayant reçu le placébo [73]. 1.2.2.3.3.4 La thérapie génique par l’apport d’un gène DMD tronqué
Cette approche utilise un gène DMD délété d’une large partie de la partie centrale (rod domain) et ayant conservé les extrémités N et C terminales. Ce gène DMD tronqué est ensuite inséré dans un vecteur adéno-associé et injecté par voie systémique dans l’organisme du patient dystrophique. Suivant la taille du gène
DMD tronqué, on peut synthétiser soit une micro-dystrophine (taille de l’insert environ 4 kb), soit une
mini-dystrophine (taille de l’insert 6-8 kb) [46]. En 2006, un essai clinique de phase I a été conduit avec l’utilisation d’un mini gène DMD inséré dans un AAV2.5 [74]. Le patient a malheureusement développé une réaction immunitaire contre le vecteur viral et même contre la mini-dystrophine. En vue d’améliorer l’efficacité et la sécurité de l’utilisation de microgène, trois essais cliniques ont commencé en décembre 2017 dont celui dirigé par l’équipe du Dr Mendell (NCT03375164). Le micro-gène inséré dans le vecteur viral AAV-rh74-micro-dystrophine (similaire à AA8) sera administré en dose unique de 2 x 1014 vg/kg par voie systémique. Le transgène sera sous un promoteur tissu-spécifique MHCK7. Les résultats de ces essais seront connus en janvier 2020. Cette approche exige également une administration répétée de mini gène [75].
1.2.2.3.3.5 La thérapie génique par la correction in situ du gène DMD muté
Les délétions d’un ou de plusieurs exons localisés dans la « partie chaude » du gène DMD située entre les exons 44 et 56 sont les causes les plus fréquentes de la DMD dans plus de 60 % des cas [76]. Les délétions responsables de la DMD sont celles dont le cadre de lecture a été interompue par la présence d’un codon stop prématuré en aval du site de délétion. Le codon stop prématuré entraine l’arrêt dans la continuité des codons
18
codant les acides aminés lors de la jonction des exons adjacents pendant la maturation de l’ARNm. Ce manque de continuité est le résultat d’une délétion d’un ou de plusieurs exons dont le nombre total de nucléotides délétés n’est pas un multiple de trois [15]. Le rétablissement du cadre de lecture consiste à assurer la continuité des codons codant les acides aminés lors de la jonction des exons qui se suivent dans le gène DMD tronqué après avoir délété un ou plusieurs exons dont le nombre total de nucléotides est un multiple de trois. Comme nous l’avons souligné plus haut, la partie centrale est formée de 24 SLRs comprenant les exons 8 à 64 [46]. Chaque SLR est formée des trois α-hélices A, B et C. Ces hélices sont en continuité avec les hélices de la SLR suivante dans l’ordre suivant : A B C A’ B’ C’ (Figure 5). Ainsi chaque SLR est consituée d’environ 100 acides aminés provenant de plus ou moins trois exons. Les 30 acides aminés consitutifs de chaque hélice (formant 4 heptades de 7 acide aminés chacun) peuvent provenir de deux exons différents qui se suivent.
Figure 5: Les α-hélices des répétitions de types spectrine (SLR : spectrin-like repeat) (image modifiée)
[77]
La thérapie génique in situ vise à corriger d’une manière permanente le gène DMD muté dans les myoblastes, les fibres musculaires du patient et/ ou dans les cellules souches et progénitrices du muscle. Elle fait essentiellement appelle aux endonucléases recombinantes telles que les méganucléases (MGNs), les Zinc
finger nucleases (ZFNs), les Transcription activator like-effector nucleases (TALENs) et surtout le système
CRISPR/Cas9. Les différents gènes d’endonucléases recombinantes sont insérés dans les vecteurs plasmidiques ou viraux plus particulièrement dans les AAV (virus adéno-associés). Ils peuvent alors être transfectés, électroporés ou administrés par voie systémique.
19 La correction de la mutation se fait
- soit en délétant (exon knock-out) le ou les exons entiers responsables du décalage du cadre ou porteur d’une mutation non-sens. On peut alors cibler les séquences des introns qui sont situés de part et d’autre de l’exon à déléter avec deux gRNAs [78] [79] [80]. Mais on peut également cibler le site d’épissage (splicer donor et splicer acceptor en anglais) de l’exon à déléter avec un seul gRNA, une stratégie similaire à celle du saut d’exon [81].
Les délétions des exons entiers en vue de corriger le gène DMD muté peuvent parfois rétablir le cadre de lecture. Certaines de ces délétions ne permettent pas cependant une continuité entre les acides aminés
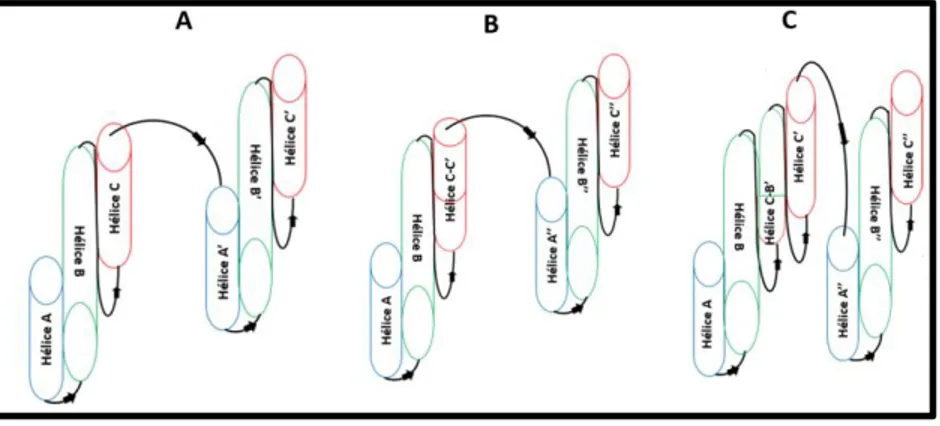
constituant leshélices des SLRs. On observe ainsi les liaisons A B CB’ C’ A’’ B’’ C’’ à la base des SLRs hybrides fractionnées non fonctionnelles (Figure 6C).
- soit en insérant un exon (exon knock-in) pour non seulement rétablir le cadre de lecture mais aussi introduire la partie manquante du gène. Dans ce cas il doit y avoir un ADN donneur avec la séquence du ou des exons à insérer ainsi que les séquences d’homologie de part et d’autre du site d’insertion. L’insertion se fait dans ce cas par la recombinaison homologue (HR) [82]. L’insertion peut se faire également sans séquences d’homologie [83].
Figure 6: Les hélices hybrides des SLRs
A) Les hélices des répétitions de types spectrine normales. On note la jonction des A, B et C. L’hélice C de la
première répétition est en continuité avec l’hélice A’ de la répétition adjacente. B) La présence de l’hélice C-C’ hybride issu de l’exon hybride après correction du gène DMD muté avec la méthode CinDel. Cette SLR a une structure semblable à celle de SLRs de la figure A. Elle est également liée à la répétition suivante par la jonction CC’-A’’. C) Formation d’une SLR fractionnée avec la formation d’une hélice hybride A-CB’-C’.
- soit enfin en générant des délétions des parties d’exons situés de part et d’autre de la délétion avec deux gRNAs, en faisant la jonction des restes d’exons pour générer un exon hybride afin de rétablir le cadre de
![Figure 5: Les α-hélices des répétitions de types spectrine (SLR : spectrin-like repeat) (image modifiée) [77]](https://thumb-eu.123doks.com/thumbv2/123doknet/3451242.100808/32.918.142.788.481.736/figure-hélices-répétitions-types-spectrine-spectrin-repeat-modifiée.webp)