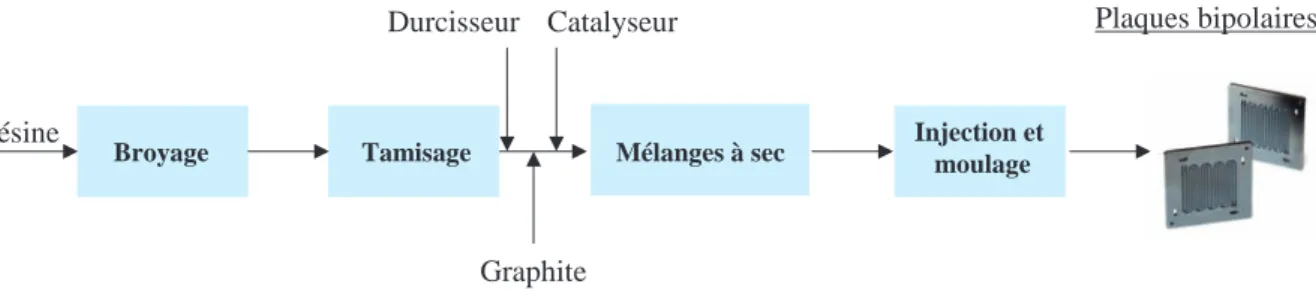
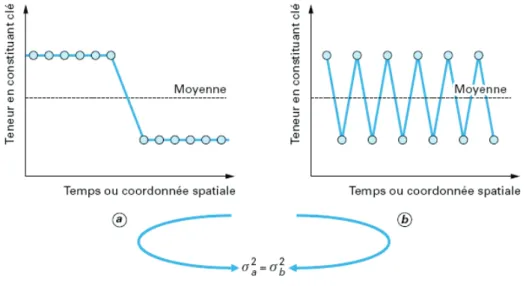
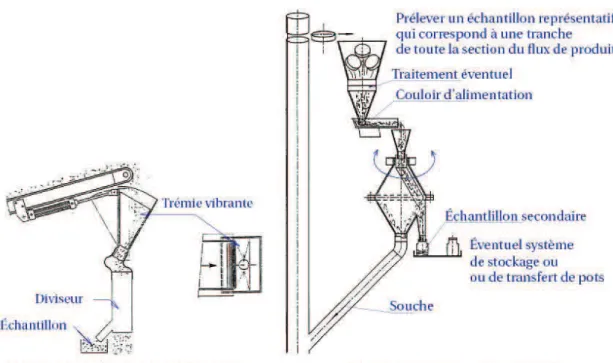
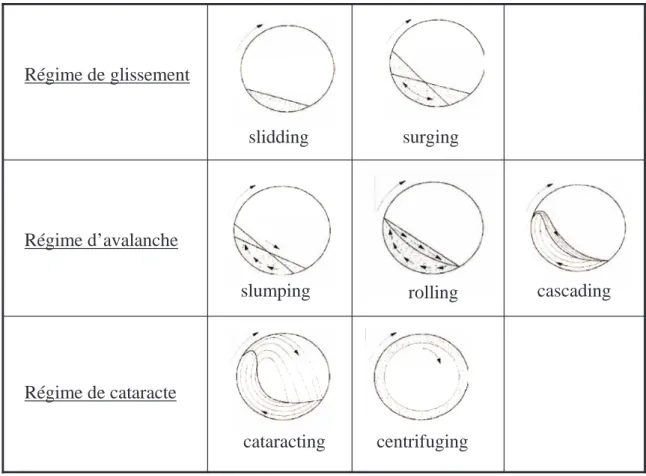
Étude dynamique et effet du changement d'échelle pour plusieurs systèmes particulaires en mélangeur Turbula® : application à un mélange destiné à la fabrication de plaques composites
249
0
0
Texte intégral
Figure

+7


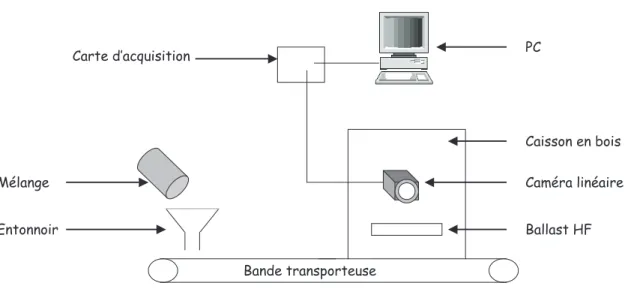

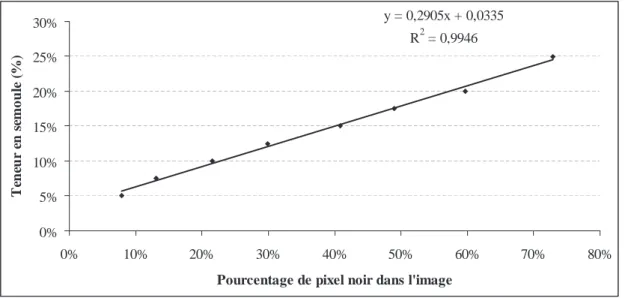
Documents relatifs