© Reza Rahimian, 2019
Mécanismes régulant l'activation microgliale et la
réponse immunitaire après l'ischémie cérébrale
Thèse
Reza Rahimian
Doctorat en neurobiologie
Philosophiæ doctor (Ph. D.)
Mécanismes régulant l'activation microgliale
et la réponse immunitaire après l'ischémie
cérébrale
Thèse
Reza Rahimian
Sous la direction de :
Résumé
Le développement d’une stratégie thérapeutique efficace contre les accidents cérébrovasculaires (ACV) est une priorité de la recherche en neurosciences depuis des décennies. L’indentification de nouvelles voies de signalisation moléculaires et cellulaires influençant la pathogénèse des ACV pourrait mener au développement de nouvelles approches thérapeutiques. Une étape cruciale et limitante dans la conception de thérapies efficace contre les ACV vient du fait que les acteurs majeurs, comme l’immunité innée et l’inflammation, peuvent agit comme une épée à double tranchant dans ces circonstances pathologiques complexes. Bien que les rôles précis de la microglie dans la neuroinflammation ne sont pas complètement déchiffrés, de récentes études suggèrent que ses différentes fonctions sont essentielles dans la pathogénèse des ACV. Galectin-3 (Gal-3) est une lectine liant le -galactosidase. Des recherches récentes montrent que les galectines peuvent agir en tant que modulateurs endogènes des réponses inflammatoires suivant une ischémie cérébrale. Gal-3 est impliquée dans l’activation cellulaire, la prolifération, la migration, l’apoptose et a été associée avec la réponse immunitaire et l’inflammation. En effet, la surexpression de Gal-3 est une caractéristique de l’activation alternative des macrophages et est associée avec le phénotype des macrophages médié par l’interleukin-4. Dans le chapitre 2 de cette thèse, nous avons étudié le rôle de Gal-3 en tant que modulateur du phénotype microglial dans des conditions physiologiques et pathologiques à la fois in
vitro et in vivo. In vitro, l’utilisation de cellules microgliales primaires démontrent que
Gal-3 augmente le nombre et la longueur des filopodes par un mécanisme dépendant de l’interleukin-4. Chez la souris, suite à un ACV et à une injection intracérébroventriculaire de Gal-3, les niveaux de cytokines pro-inflammatoires et d’iNOS diminuent et le niveau de Ym1 augmente. Ces changements sont accompagnés par une diminution notable du signal de TLR2. Dans le chapitre 3, nous investiguons l’action protectrice de la glucosamine, un ligand potentiel de Gal-3, sur un modèle expérimental de l’ACV ischémique. Dans ce contexte, le dimorphisme sexuel de la réponse microgliale à la glucosamine à la suite de l’occlusion de l’artère cérébrale moyenne a aussi été étudié. Nos résultats démontrent que la glucosamine mène à des actions protectrices seulement chez les sujets mâles à la suite de l’ischémie cérébrale. Dans le chapitre 4, nous avons étudié comment des mécanismes cellulaires associés aux ribosomes contrôlent la réponse immunitaire innée et l’activation
microgliale suivant une stimulation inflammatoire. Après cette stimulation (l’injection de LPS), nous avons observé une dissociation marquée du transcriptome et du protéome microglial. Finalement, nous avons remarqué que la protéine liant l’ARN SRSF3 est responsable des modifications post-trancriptionnelles de l’ARNm par un mécanisme impliquant le 3’UTR de ce dernier.
Abstract
Development of an effective therapeutic strategy for stroke has been a priority of neuroscience research for decades. Elucidation of novel cellular and molecular pathways that influence the pathogenesis of stroke could lead to development of new therapeutic approaches. A crucial rate limiting step in designing effective therapeutics for stroke may stem from the fact that major players, such as innate immunity and inflammation, can act as a double edged sword in the complex pathological circumstances. Although the precise roles of microglia in neuroinflammation have not been yet fully deciphered, recent studies suggest their various essential functions in stroke pathology. Galectin-3(Gal-3) is a -galactosidase binding lectin. Mounting evidence suggests that galectins may act as endogenous modulators of the innate inflammatory responses following brain ischemia. Gal-3 is involved in cell activation, proliferation, migration and apoptosis and has been associated with immune response and inflammation. Indeed, evidence suggests that Gal-3 overexpression is a feature of alternative macrophage polarization and is associated with the IL-4 mediated macrophage phenotype. In chapter 2 of this thesis, we studied the role Gal-3 as a modulator of microglia phenotype in physiological and pathological conditions in both in vitro and in vivo settings. In vitro measurements in primary microglia culture showed that Gal-3 increased the number and length of filopodia via an IL4-dependent mechanism. In stroke condition, after i.c.v injection of Gal-3 levels of pro-inflammatory cytokines and iNOS diminished and the content of alternative macrophage marker Ym1 increased. These changes were accompanied by the notable decrease in TLR2 signals. In chapter 3, we investigated the protective actions of Glucosamine, as a potential Gal-3 ligand, in experimental model of ischemic stroke. In this context sexual dimorphism in the microglia responses to glucosamine following MCAO was also studied. Our findings indicated that Glucosamine elicits protective actions only in male subjects following cerebral ischemia. In chapter 4, we studied how ribosome-based mechanisms control the innate immune response and microglia activation following inflammatory challenge. Following innate immune challenge (LPS injection) we observed a marked dissociation of microglia mRNA and protein networks. Finally we saw that RNA-binding protein, SRSF3 is responsible for mRNA post-transcriptional modifications through 3' UTR-mediated mechanisms.
Table of contents
Résumé ... iii Abstract ... v Table of contents ... vi List of figures... ix List of abbreviations ... x Acknowledgement ... xii Foreword ... xiii 1 GENERAL INTRODUCTION ... 1 1.1 CEREBRAL ISCHEMIA ... 21.1.1 Pathophysiology of ischemic stroke ... 3
1.1.2 Excitotoxicity... 3
1.1.3 Oxidative stress... 4
1.1.4 Animal models of ischemic stroke ... 5
1.1.5 Inflammation following brain ischemia ... 7
1.2 TLRS AND NEUROINFLAMMATORY RESPONSES ... 10
1.2.1 TLRs endogenous ligand and signaling... 10
1.2.2 Crucial roles of TLR2 in stroke pathogenesis ... 10
1.3 ROLE OF MICROGLIA IN PHYSIOLOGICAL CONDITION AND FOLLOWING BRAIN ISCHEMIA ... 11
1.3.1 Role of Microglia in healthy brain ... 12
1.3.2 Role of microglia in stroke pathogenesis ... 13
1.3.3 Classification of activated microglia ... 16
1.3.4 Different characteristics of resident microglia versus monocyte-derived macrophages following brain ischemia ... 18
1.4 TARGETING INFLAMMATION AS THE THERAPEUTIC APPROACH FOR BRAIN ISCHEMIA 19 1.4.1 Role of cytokines in stroke pathology ... 20
1.4.2 Role of PPAR-γ in stroke pathology ... 24
1.4.3 Gal-3; the modulator of inflammation and microglia activity after ischemic insult 26 1.4.4 Importance of Gal-3 in modulation of neuroinflammation after brain ischemia 28 1.5 IMPORTANCE OF SEXUAL DIMORPHISM IN STROKE PATHOLOGY AND THERAPY ... 31
1.5.1 Sexual dimorphism in innate immunity and microglia responses ... 33
1.6 TECHNIQUES TO INVESTIGATE DIFFERENT ASPECTS OF MICROGLIA IN COMPLEX NEURODEGENERATIVE CONDITIONS ... 35
1.6.1 In vitro methods to study microglia ... 35
1.6.2 In vivo methods to study microglia ... 36
1.7 POTENTIAL APPLICATIONS OF BIOMARKERS IN ACUTE CEREBROVASCULAR EVENTS 41 1.8 HYPOTHESIS AND AIMS ... 44
2 CHAPTER: DELAYED GALECTIN-3 MEDIATED REPROGRAMMING OF
MICROGLIA AFTER STROKE IS PROTECTIVE ... 60
2.1 RÉSUMÉ ... 61
2.2 ABSTRACT ... 61
2.3 INTRODUCTION ... 62
2.4 METHODS ... 63
2.5 RESULTS ... 70
2.5.1 Gal-3 Alters Microglia Secretory Profile, Morphology and Migration Patterns In vitro 70 2.5.2 Gal-3 Changes Microglia Morphology in Vivo ... 73
2.5.3 Gal-3 Induces Alternative Microglia Activation after MCAO ... 74
2.5.4 Single Gal-3 injection induces marked shift in the cytokine expression profiles and significantly decreases ischemic lesion ... 77
2.5.5 Glucosamine increases Gal-3 ligand availability after MCAO and alters morphology and phenotype of microglia... 79
2.5.6 Glucosamine Increases Microglia Proliferation and IGF-1 Levels Following MCAO ... 81
2.6 DISCUSSION ... 82
2.7 CONCLUSION ... 86
2.8 ACKNOWLEDGMENTS ... 87
2.9 REFERENCES ... 87
3 CHAPTER: SEXUAL DIMORPHISM IN THE MICROGLIA RESPONSES TO GLUCOSAMINE FOLLOWING MIDDLE CEREBRAL ARTERY OCCLUSION . 92 3.1 RÉSUMÉ ... 94
3.2 ABSTRACT ... 95
3.3 INTRODUCTION ... 96
3.4 MATERIALS AND METHODS ... 97
3.5 RESULTS ... 102
3.5.1 Glucosamine alters morphology and phenotype of cultured microglia via IL-4 dependent signaling ... 102
3.5.2 Glucosamine induces delayed induction of the TLR2 signals in male mice after stroke 105 3.5.3 Glucosamine decreases pro-inflammatory cytokines level after stroke and decreases ischemic lesion in male mice. ... 107
3.5.4 Sexual dimorphism in response to glucosamine after stroke ... 109
3.6 DISCUSSION ... 113
3.7 FUNDING ... 116
3.8 REFERENCES ... 116
4 CHAPTER: DIVERGING MRNA AND PROTEIN NETWORKS IN ACTIVATED MICROGLIA REVEAL SRSF3 SUPRESSES TRANSLATION OF HIGHLY UP-REGULATED INNATE IMMUNE TRANSCRIPTS ... 120
4.1 RÉSUMÉ ... 121
4.2 ABSTRACT ... 121
4.4 RESULTS ... 123
4.4.1 Generation and Characterization of the CD11brGFP Transgenic Mice ... 123
4.4.2 Translational Profiling of Activated Microglia Reveals a Cluster of the Highly Regulated Innate Immune Genes ... 125
4.4.3 LPS-Activated Microglial Cells Exhibit Distinct Molecular Signatures for mRNAs and Proteins ... 129
4.4.4 Translational Regulation of Gene Expression in Innate Immune Response ... 131
4.4.5 Serine/Arginine-Rich Splicing Factor 3 Serves as a Master Regulator of the Innate Immune Gene Translation ... 134
4.4.6 SRSF3 Controls Innate Immune Cascade in Vivo ... 137
4.5 DISCUSSION ... 140
4.6 MATERIAL AND METHODS ... 142
4.7 ACKNOWLEDGEMENTS ... 144
4.8 REFERENCES ... 144
5 GENERAL DISCUSSION ... 149
5.1 ROLE OF GAL-3 IN STROKE PATHOGENESIS; A DOUBLE-EDGED SWORD ... 151
5.2 GLUCOSAMINE; A TRANSLATIONAL INTERVENTION FOR STROKE PHARMACOTHERAPY ... 154
5.3 SEXUAL DIMORPHISM FOLLOWING CEREBRAL ISCHEMIA ... 157
5.4 RNA-SEQ STUDIES TO DECODE GENDER DIFFERENCE IN GLIAL IMMUNE RESPONSES AFTER STROKE ... 159
5.5 IMPORTANCE OF CNS MICROENVIRONMENT ON MICROGLIA PHENOTYPE ... 160
5.6 DISCREPANCY IN TRANSCRIPTOMIC AND PROTEOMIC PROFILE OF ACTIVATED MICROGLIA ... 161
5.7 CONCLUSION AND FUTURE PERSPECTIVES ... 164
List of figures
Figure 1-1 Pathophysiology of brain damage following ischemia and reperfusion ... 4
Figure 1-2. Classification of different experimental models of ischemic stroke... 7
Figure 1-3. Inflammatory mechanisms after stroke... 9
Figure 1-4. Microglial-neuronal interactions in health and disease ... 15
Figure 1-5. Central nervous system myeloid cells and their defining lineage markers in steady state and following brain inflammation... 17
Figure 1-6. IL-4 signaling pathways... 24
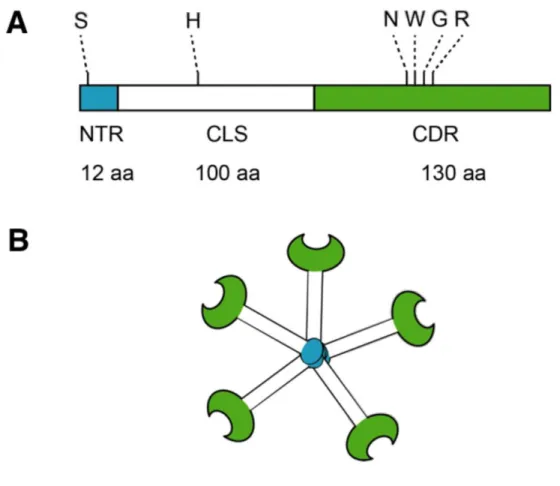
Figure 1-7. Structure of Gal-3 ... 27
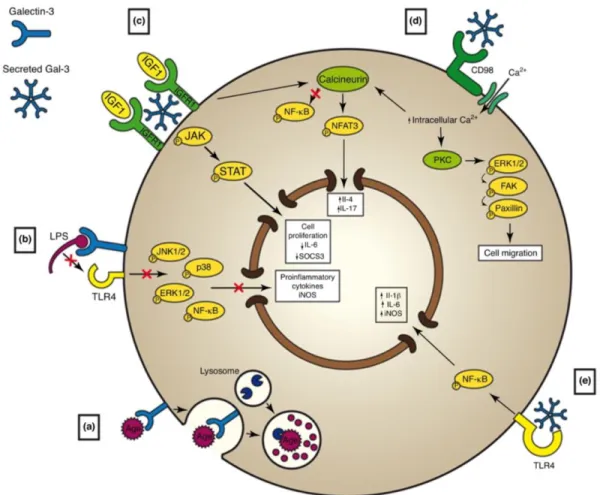
Figure 1-8. Possible mechanisms underpinning the effects of Gal-3 on microglia polarization ... 30
Figure 1-9. Sexually dimorphic pathways of cell death after cerebral ischemia ... 32
Figure 1-10. Potential biomarkers of stroke may be categorized by their role in the ischemic cascade ... 44
Figure 2-1. Gal-3 alters the secretory profile, morphology and migration pattern of cultured microglia ... 73
Figure 2-2. Gal-3 alters the morphology of microglia in vivo ... 74
Figure 2-3. Gal-3 ameliorates microglia activation/TLR2 response and changes their phenotype after ischemia in vivo ... 76
Figure 2-4. Gal-3 decreases the content of pro-inflammatory cytokines and ischemic size after MCAO ... 78
Figure 2-5. Glucosamine treatment increases Gal-3 ligand avidity after stroke and induces neuroprotective markers ... 80
Figure 2-6. Glucosamine treatment induces microglia proliferation and upregulates IGF-1 following MCAO ... 82
Figure 3-1. Glucosamine changes morphology and phenotype of primary cultured microglia ... 104
Figure 3-2. Glucosamine enhances microglia activation/TLR2 responses following MCAO in male mice... 106
Figure 3-3. Glucosamine treatment decreases the content of pro-inflammatory cytokines and ischemic size after MCAO in male mice ... 108
Figure 3-4. Glucosamine do not alter microglia TLR2 responses after MCAO in female mice ... 111
Figure 3-5. Glucosamine treatment induces the production of the pro-inflammatory cytokines following MCAO in female mice... 113
Figure 4-1. Characterization of the CD11brGFP Transgenic Mice ... 124
Figure 4-2. Highly Upregulated Immune Genes are Not Translated ... 127
Figure 4-3. Diverging mRNA and Protein Networks in Activated Microglia ... 131
Figure 4-4. Inhibition of Luciferase Reporter Gene Activity by Saa3-3’UTR ... 133
Figure 4-5. SRSF3 Regulates Inflammatory Genes Expression via 3’UTR ... 136
Figure 4-6. SRSF3 is a Translational Regulator of Inflammatory Genes in Vivo ... 139
List of abbreviations
ATP : Adenosine triphosphateAGE : Advanced glycation end-product ALS : Amyo-trophic lateral sclerosis BBB : Blood–brain barrier
Mgat5 : β1,6 N-acetylglucosaminyltransferase V CRD : Carbohydrate-recognition domain CREB : cAMP response element binding protein CNO : Clozapine-N-oxide
DAMPs : Damage-associated molecular patterns
DREADD : Designer receptors exclusively activated by designer drugs FACS : Fluorescence activated cell sorting
EAE : Experimental autoimmune encephalomyelitis EGFP : Enhanced green fluorescent protein
ET-1 : Endothelin-1
ERK1/2 : Extracellular signal-regulated kinase 1/2 Gal-3 : Galectin-3
GAP-43 : Growth associated protein-43 GF : Growth factor
GPCRs : G-protein coupled receptors GFRs : Growth factor receptors IGF-1 : Insulin-like growth factor-1 IL-1β : Interleukin 1 beta
IL-4 : Interleukin 4 IL4R : IL-4 receptor IFN-𝛾 : Interferon-𝛾
ILK : Interleukin kinase
iPSCs : induced pluripotent stem cells
MCA : Middle cerebral artery
MCAO : Middle cerebral artery occlusion MCP-1 : Monocyte chemotactic protein-1 MS : Multiple sclerosis
NFAT : Nuclear factor of activated T-cells
NF-κB : Nuclear factor kappa-light-chain-enhancer of activated B cells NOS : Nitric oxide synthase
OPC : Oligodendrocyte progenitors POA : Preoptic area
PDCD4 : Programmed cell death 4
PPAR-γ : Peroxisome proliferator activated receptor gamma RAGE : Advanced glycation end-product receptor
ROS : Reactive oxygen species RRM : RNA recognition motif
SRSF3 : Serine and arginine rich splicing factor 3 STAT3 : Transducer and activator of transcription TDG : Thiodigalactoside
TGF-𝛽 : Transforming growth factor-beta TH1 : Type 1 helper
TH2 : Type 2 helper
TNF-𝛼 : Tumor necrosis factor-alpha TLRs : Toll-like receptors
Tmem119 : Transmembrane protein 119 TSPO : Translocator protein
tPAs : Tissue plasminogen activators VEGF : Vascular endothelial growth factor
Acknowledgement
I would like to thank my supervisor Dr. Jasna Kriz for giving me an opportunity to do my ph.D in her lab. Her constant support motivated me to finish my thesis as productive as possible. Every scientific discussion with her enhanced my knowledge in the field of my research and helped me to think and act independently.
I would like to thank Dr. Sachiko Sato and her research assistants for their great help to run some experiments and providing recombinant Galectin-3 for our experiments.
I express my heartfelt thanks to my jury members; Prof. Jean Pierre Julien, Dr. Benoit Labonte and Dr. Tiina Kauppinen for accepting to evaluate my thesis.
I would like to extend my special thanks to our research professional, Yuan Cheng Weng for his great and kind help for all stroke surgery and tissue processing.
I would also like to acknowledge Dr. Melanie-Lalancette Hebert and Dr. Hejer Boutej for their end-less support and help in preparing and analysing of results. On their personal characters, both of them have a warm personality and their presence made the lab a pleasant place for me to work
My huge thanks to our wonderful research assistant, Geneviève Soucy for her help in my experiments. I thank Esssam Abdelhamid for his kind help in doing some experiments. Special thanks to my great lab mates Sai Sampath, Louis and Pierre for their kind helps and support. I also express my heartfelt thanks to Kallol and Silvia for their help and support. Big thanks to my wife, Ghazal for her end-less help and support. Her positive mind and emotional supports gave me lots of energy to peruse my study.
At the end, I dedicate this thesis to my parents and lovely sister.
Thank you Reza Rahimian
Foreword
This thesis includes a literature review that outlines the current knowledge and recent findings about roles of microglia in pathogenesis of stroke. The first part of the introduction explains the stroke pathology, animal models of stroke and important role of neuro-inflammation following brain damage. The second part of the introduction reveals the pivotal role of microglia and its modulators in post-stroke recovery. In this context sexual dimorphism in innate immune responses is also discussed. Third part of the introduction deals mostly with the importance of new techniques that help studying microglia involvement in complex in vivo settings. The final part of the introduction explains the potential of using biomarkers as crucial diagnostic tools in clinical settings.
In the chapter four the article entitled “Delayed galectin-3 mediated reprogramming of
microglia after stroke is protective” was accepted and published in Molecular Neurobiology (2019 Feb 23. doi: 10.1007/s12035-019-1527-0). There no difference
between the published version and the version included in this thesis.
Authors: Reza Rahimian, Star Lively, Essam Abdelhamid, Melanie Lalancette Hébert, Lyanne Schlicter, Sachiko Sato and Jasna Kriz.
Authors contributions: Reza Rahimian did more than 90% of the experiments, analyzed the results and wrote the manuscript. Star Lively from Prof. Schlicter lab did microglia migration assay. Essam Abdelhamid performed primary microglia culture and Real time PCR. Melanie Lalancette Hébert plotted all figures and formatted the manuscript. Prof. Kriz supervised the project, read and corrected the manuscript for final submission.
The third chapter of this thesis is presented in the form of a research manuscript where I am the principal author. The manuscript will be submitted to the Stroke journal in near future.
“Sexual dimorphism in the microglia responses to Glucosamine following middle cerebral artery occlusion”
Reza Rahimian, Melanie Lalancette Hébert, Yuan Cheng Weng and Jasna Kriz.
Authors contributions: Reza Rahimian did more than 90% of the experiments, analyzed the results and wrote the manuscript. Yuan Cheng Weng did all stroke surgeries and cutting the perfused brains. Melanie Lalancette Hébert plotted all figures and formatted the
manuscript. Prof. Jasna Kriz supervised the project, read and corrected the manuscript for final submission.
In the chapter four the article entitled “Diverging mRNA and Protein Networks in Activated
Microglia Reveal SRSF3 Supresses Translation of Highly Up-Regulated Innate Immune Transcripts” was accepted and published in Cell Reports (2017 Dec 12;21(11):3220-3233. doi: 10.1016/j.celrep.2017.11.058). There no difference between the published
version and the version included in this thesis.
Authors: Hejer Boutej, Reza Rahimian, Sai Sampath Thammisetty1, Louis-Charles Béland, Mélanie Lalancette-Hébert and Jasna Kriz.
Authors contributions: Hejer Boutej generated transgenic model, designed the study, performed proteomic and transcriptomic experiments, analyzed the data. Reza Rahimian performed in vitro studies, live imaging studies and RNA quality experiments. Sai Sampath Thammisetty performed the proteomic validation/western blot analysis. Louis-Charles Beland and Melanie Lalancette Hébert helped with analysis and validation of transgenic model. Jasna Kriz designed the study and wrote the manuscript.
1.1 Cerebral ischemia
Brain injury following transient or permanent focal cerebral ischemia (stroke) develops from a complex series of pathophysiological events that evolve in time. After heart disease and cancer, stroke is the third leading cause of death and a major cause of disability in developed countries (Dirnagl et al. 1999). Although the mortality rate of stroke has decreased significantly in recent years, the total number of stroke cases is not in decline (Kriz and Lalancette-Hebert 2009). Stroke imposes lots of costs to social health systems. In fact, only a minority of stroke patients will completely recover and in most cases ischemic brain injuries are associated with mild to severe permanent neurological deficits. These deficits can include partial paralysis and difficulties with memory and movements (Kriz and Lalancette-Hebert 2009). Stroke is a heterogeneous and multifactorial disease. Several modifiable and non-modifiable risk factors might have influence on stroke incidence and outcomes. Modifiable factors include a history of diabetes mellitus, hypertension, high blood cholesterol levels and ischemic heart diseases. Non-modifiable factors include age, gender and race. Other less-well documented risk factors include alcoholism and socioeconomic status. Roughly 80% of stroke events could be prevented by making simple lifestyle changes (Allen and Bayraktutan 2008).
Development of an effective therapeutic strategy for stroke has been a priority of neuroscience research for decades. Unfortunately, to date, the results of hundreds of clinical trials for pharmacotherapy of stroke have been frustrating. Recombinant tissue plasminogen activators (tPAs) are the only agents approved by the FDA as thrombolytic in ischemic stroke. Elucidation of novel cellular and molecular pathways that influence the pathogenesis of stroke could lead to development of new therapeutic approaches. A crucial rate limiting step in designing effective therapeutics for stroke may stem from the fact that major players, such as inflammation, can act as a double edged sword in the complex pathological circumstances (Kriz and Lalancette-Hebert 2009; Lee et al. 2014),
1.1.1 Pathophysiology of ischemic stroke
Depending on the underlying pathology, stroke can be classified as ischemic or hemorrhagic stroke. More than 80% of strokes are ischemic.Cerebral ischemia results from blood flow deficiency to a portion of the brain because of cerebral artery occlusion (Khoshnam et al. 2017). Following ischemia, neurons are not capable of keeping their transmembrane ionic gradient and homoeostasis. This malfunction triggers several mechanisms including excitotoxicity, oxidative stress, inflammation, and apoptosis that lead to cell death (Besancon et al. 2008; Ouyang et al. 2007). Due to these pathological events, a part of the brain tissue affects by irreversible neuronal damage due to necrotic cell death, while the adjacent tissues contain salvageable and metabolically active cells (penumbra), in which cell death happens less quickly (Bandera et al. 2006; Jung et al. 2013; Moskowitz et al. 2010). The pathological cascades that occur following ischemia-reperfusion is explained in figure 1.1.
1.1.2 Excitotoxicity
Brain ischemia causes the impairment of adenosine triphosphate (ATP) synthesis. This adversely affects Na+/K+ ATPase pump, resulting in plasma membrane depolarization,
release of potassium into the extracellular space and entry of sodium into cells. In addition, the Ca2+ pump also fails that causing a huge rise in intracellular calcium concentration
during ischemia. The rise in the intracellular calcium levels activates different cell death associated molecules such as lipases and calcium-dependent proteases (Khoshnam et al. 2017). Excitotoxicity, the specific type of neurotoxicity mediated by glutamate, may be the missing link between ischemia and neuronal death. Interest in excitotoxicity began when glutamate was found to be neurotoxic. Evidence soon demonstrated that glutamate is not only the primary excitatory neurotransmitter in the adult brain, but also a critical transmitter for signaling neurons to degenerate following stroke. The finding led to a number of clinical trials that tested inhibitors of excitotoxicity in stroke patients. Glutamate exerts its function in large by activating the calcium-permeable ionotropic NMDA receptor (NMDAR), and different subpopulations of the NMDAR may generate different functional
outputs, depending on the signaling proteins directly bound or indirectly coupled to its large cytoplasmic tail (Lai et al. 2014).
1.1.3 Oxidative stress
It has been shown that reactive oxygen species (ROS) are crucial mediators of tissue damage following acute stroke. Several experimental studies indicated that free radical formation is increased after middle cerebral artery occlusion (MCAO). The important sources of free radical are glutamate-induced excitotoxicity, mitochondrial dysfunction, Ca2+ influx and neuronal nitric oxide synthase (NOS) activation. ROS have extensive
cellular effects, inducing cell death by activating the processes including lipid peroxidation, DNA damage, protein destruction, release of Ca2+ from intracellular stores and chemotaxis.
In addition, ROS have notable effects on the cerebral vasculature which finally influence cerebral blood flow. Superoxide molecules increase endothelial permeability, vasodilation and aggregation of the platelets in affected area by ischemia (Allen and Bayraktutan 2009; Khoshnam et al. 2017; Wei et al. 1996).
Figure 1-1 Pathophysiology of brain damage following ischemia and reperfusion
platelet aggregation and cytokine (IL-1β) release. Translocation of P-selectin on the surface of platelets and endothelial cells leads to platelet-leukocyte aggregation. Complement is activated, and arachidonic acid metabolites (AA) are released. In the vascular wall, upregulation of E- and P-selectin on endothelial cells provides a platform for low affinity leukocyte binding through interaction with glycoproteins expressed on leukocytes, for example, P-selectin glycoprotein ligand-1. Firm adhesion is obtained after endothelial expression of ICAM-1 interacting with leukocyte b2 integrins (LFA-1 and Mac-1). Loss of NO promotes vasoconstriction and enhances leukocyte and platelet aggregation. MMP activation could lead to BBB breakdown and matrix proteolysis, facilitating leukocyte extravasation. In the perivascular space, chemotactic complement subunits (C5a) acting on mast cell complement receptors (CD88) leads to degranulation and release of histamine and proteases, contributing to BBB leakiness. Cytokines (TNF, IL-1β) are produced by mast cells and perivascular macrophages, providing further signals to guide leukocyte migration across the vessel wall. In the brain parenchyma, injured cells release purines (ATP), which act as early proinflammatory signals leading to production of cytokines and chemokines. Disruption of neuronal-microglial interaction (CX3CL1, CD200) and increases in extracellular glutamate (Glu) acting on microglial GluR1 metabotropic receptor27 also contribute to the proinflammatory milieu (Iadecola and Anrather 2011a).
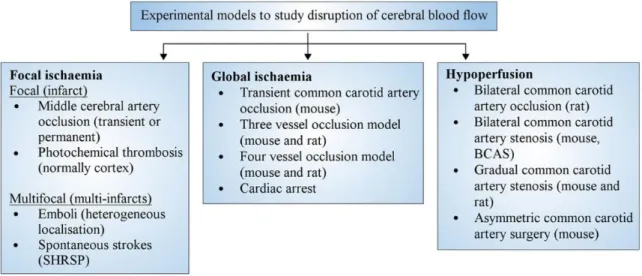
1.1.4 Animal models of ischemic stroke
Several models of focal cerebral ischemia have been developed. Each model has its own strengths and limitations in comparison to what happens in human stroke. The majority of experimental stroke researches have been done in small rodents such as mice & rats. Ischaemic stroke in humans most commonly occurs through an occlusion of the middle cerebral artery (MCA) and therefore MCAO is the favorite model in experimental studies. One important concern is that he translational neuroprotective strategies of stroke therapy from bench to bed have been failed so far. This problem has questioned the validity of animal models of stroke in recent years. Here, some important experimental models of ischemic stroke are explained in the following sections (McCabe et al. 2018).
The intraluminal filament model is the most commonly used model of ischemic stroke. In order to induce MCAO an intraluminal filament or suture is introduced into the internal carotid artery and advanced until it occludes the origin of the MCA. The filament model of MCAO is commonly used to investigate the efficacy of different therapeutic interventions. Depending on the length of occlusion time the striatum and cortical damage have been reported with the filament model of MCAO. This model mimics human ischemic stroke in the origination and exhibits a penumbra that is similar to that of human stroke. Although this model is very well-established, but it has few downsides. One of the disadvantages of the filament model of MCAO is the risk of incomplete occlusions of the MCA. This problem happens when filament size is not proper for the diameter of blood vessel at the point of occlusion. Sometimes the filament has not reached the MCA following insertion. Another limitation of intraluminal filament MCAO model is that it does not completely mimic the human stroke because in the animal model following filament removal a drastic increasing of reperfusion happens while in human reperfusion occurs gradually. Indeed thrombolysis with t-PA results in a gradual lysis of the clot which can take up to several hours (Hossmann 2012; McCabe et al. 2018).
Another experimental model which has been in the center of attention is Endothelin-1 (ET-1)-induced MCAO. ET-1 is a potent vasoconstrictor and is a suitable tool for inducing focal cerebral ischemia. ET-1 is injected stereotaxically into tissue adjacent to the MCA. One advantage of this model is that the surgery is fast. Also it does not damage facial muscles. Another advantage is that ET-1 can be injected in conscious animals thereby removing limitation of anaesthesia. ET-1 can be stereotaxically injected into any region of brain to induce a site-specific ischaemic lesion and consequently a specific behavioural deficit. Photothrombosis model of MCAO is another important technique has been used is in experimental stroke studies. Rose Bengal as a photosensitive dye should be injected intravenously then the skull should be illuminated of by a laser in a specific cortical location. This illumination induces the production of highly reactive oxygen species by the dye. These free radicals induce platelet activation and endothelial damage which leads to formation of thrombi. Interestingly this method is fast and the variability is low. It mimics
the thrombi production in human stroke and a sizeable volume of penumbra has been reported with this method (Qian et al. 2016).
In order to mimic the human stroke pathology, a number of embolic models of ischemia have been developed. The most widely used thromboembolic model is based on the generation of an embolus for occlusion of the MCA. Emboli are created outside the body using autologous blood (Zhang et al. 2015). The most Advantage of this model is that it can be used to investigate the efficacy of thrombolytics. However, the embolic model is associated with higher mortality and variability in lesion size. Another problem of this model is that the clot can break up and result in multifocal ischaemic lesions. Alternatively, the clot may block the vessel other than the MCA such as the posterior cerebral artery (Macrae 2011; McCabe et al. 2018). Alongside focal models of ischemia different models of global ischemia and hypoperfusion have been also developed (figure 1.2). In the present thesis we employed the transient middle cerebral artery occlusion model which is explained in detailed in the material and methods section of the chapter 2 and chapter 3.
Figure 1-2. Classification of different experimental models of ischemic stroke
1.1.5 Inflammation following brain ischemia
The inflammatory response after ischemic stroke is a well-documented and broadly studied phenomenon. Pathological characteristics of brain injury such as cell death and ROS can
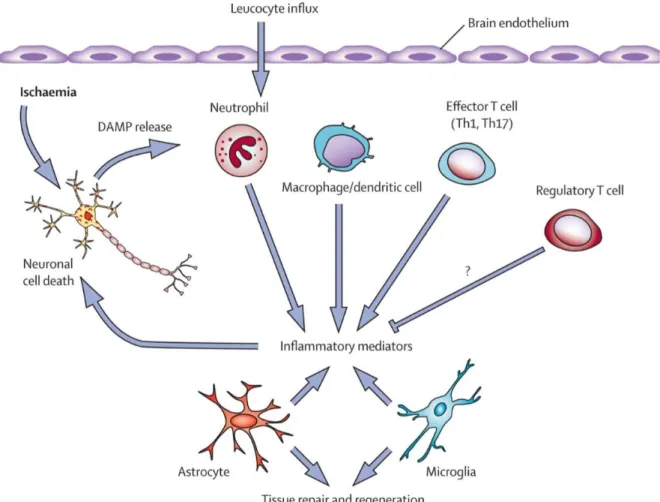
trigger inflammatory cascades by activating resident microglia and astrocytes as well as attracting infiltrating leukocytes from circulating blood. Microglia and astrocytes are the first cells get activated after ischemic insult. Activated microglia phagocyte toxic substances thereby help to maintain normal cellular functions in the brain. Moreover, activated microglia secrete several pro-inflammatory cytokines, anti-inflammatory cytokines and growth factors within 24 hours of stroke (Kim et al. 2016b). Astrocytes are the most abundant cells in the brain. In healthy brain, these cells provide structural and nutritive support for neurons. After ischemic stroke, astrocytes contribute in wound healing and repair by mediating reactive gliosis and glial scar formation (Kim et al. 2016b; Pekny and Nilsson 2005). Leucocytes are another group of cells that have important roles in post-stroke inflammation and damage. Different mechanisms are involved in infiltrated leukocytes-induced brain injury after ischemic insults. 1). Phospholipase activation in leukocytes leads to production of biologically active substances, such as leukotrienes and eicosanoids and platelet-activating factor which result in vasoconstriction and platelet aggregation. 2). Activated leukocytes at the surface of the endothelium liberate ROS and proteases that impair potentially brain tissues that can be saved. 3). Adhesion of leukocytes to the endothelium can reduce the flow of red blood cells through the microvasculature leading to the cerebral no-reflow phenomenon (Hartl et al. 1996; Kim et al. 2016b; Yilmaz and Granger 2008). Figure 1.3 has summarized how different types of immune cells initiate post-stroke inflammatory cascades in brain. Different investigations indicated that post ischemic inflammation has different important components. One pivotal component is glia cells which contribute to post stroke inflammatory and repair mechanisms. In this context Toll-like receptors (TLRs) have well documented roles in initiation of glial inflammatory response after ischemic insults (Kriz 2006). In the next following sections, the importance of these components is discussed.
Figure 1-3. Inflammatory mechanisms after stroke
Ischemia causes cell death in the brain parenchyma and subsequent release of endogenous molecules termed damage-associated molecular patterns (DAMPs) from dying cells. DAMPs trigger a cascade of inflammatory events that contribute to activation of resident microglia and astrocytes and recruitment of circulating leucocytes (neutrophils, macrophages, dendritic cells, and T lymphocytes). Production of inflammatory mediators exacerbates neuronal injury. Conversely activated resident cells might produce trophic factors that promote tissue repair and recovery. A potential role of regulatory T cells in restricting brain ischaemic injury has been proposed (Macrez et al. 2011).
1.2 TLRs and neuroinflammatory responses
1.2.1 TLRs endogenous ligand and signaling
TLRs are components of the innate immune system that are involved in ischemic damage following stroke. TLRs, so-called because of their similarity to the Drosophila Toll receptors, were first characterized in mammals by their ability to recognize pathogen-associated molecular patterns such as those found in the bacterial cell wall components peptidoglycan and lipopolysaccharide (LPS). Different types of endogenous ligands have been identified for TLRs. Namely HSP60 and fibrinogen activate TLR4, while HMGB1 activates TLR2. The TLRs belong to tyrosine kinase family of receptors. Most of TLRs initiates their signaling via recruitment the adaptor protein, MyD88. This adaptor protein creates a complex with members of the IRAK family, including IRAK1, IRAK2, and IRAK4, to begin a process of auto- and cross-phosphorylation among the IRAK molecules. IRAKs dissociate from MyD88 when they got phosphorylated and bind to TRAF6. TRAF6 activates TAK1 which itself activates the IKK complex. The IKK complex, composed of IKKα, IKKβ and the regulatory subunit IKKγ/NEMO, phosphorylates IκB proteins. This phosphorylation is instrumental for the ubiquitination and proteosomal degradation of IκBs and the subsequent nuclear translocation of the transcription factor NFκB (Marsh et al. 2009).
1.2.2 Crucial roles of TLR2 in stroke pathogenesis
Several evidences indicate that post-stroke inflammation plays an important role in the evolution of brain injury after ischemia. However, it is still unclear that to what extent inflammatory processes are harmful and/or beneficial for brain recovery (Bohacek et al. 2012; Iadecola and Anrather 2011b; Lo et al. 2003). TLRs, especially TLR2, might contribute to the evolution of brain damage following cerebral ischemia (Ziegler et al. 2011; Ziegler et al. 2007). While previous reports were focused on the role of TLR2 only in the acute phase of stroke, the latter phases of the tissue response to ischemia remained unexplored (Bohacek et al. 2012; Hua et al. 2009). Using a live imaging approach, it has
Hebert et al. 2009). For the first time Bohacek et al. investigated the involvement of TLR2 receptors in acute and chronic phase of ischemic stroke. The temporal analysis of the microglia activation profiles in TLR2 knockout mice showed the reduced capacity of resident microglia to proliferate, the decreased microglia activation after stroke, as well as less amounts of monocyte chemotactic protein-1 (MCP-1). Interestingly, although TLR2 knock out showed smaller stroke volume in acute ischemic phase of stroke (24 to 72 h) but the observed alterations in innate immune response were more noticeable in chronic phase of stroke (at day 7), which finally resulted in delayed aggravation of ischemic lesion leading to larger chronic infarctions as compared with wild-type mice (Bohacek et al. 2012). Furthermore TLR2 deficiency was associated with significant decrease in the levels of neurotrophic factor insulin-like growth factor 1 (IGF-1), expressed by microglia in the stroked areas (Bohacek et al. 2012). Importance of TLR2 in neuro-glia responses after brain ischemia is still the matter of debate and controversy. Tang et al. showed that neurons express several TLRs, and that the levels of TLR2 and TLR4 are up-regulated in neurons in response to IFN-𝛾 stimulation and energy deprivation. Intriguingly, their study revealed that TLR2 and TLR4 expression was increased in cerebral cortical neurons in response to ischemia/reperfusion damage, and the levels of neurological deficits caused by a stroke were significantly less in TLR2 and TLR4 knockout mice compared with WT control mice (Tang et al. 2007).
1.3 Role of Microglia in physiological condition and following
brain ischemia
Beyond the crucial role of microglia in pathological condition like brain ischemia, recent investigations revealed that microglia are emerging as an important element normal of brain physiology. Indeed microglia play pivotal role in shaping the brain. Experimental and clinical findings show that microglia interact with neurons and glia within the developing CNS and they have influences on synaptic maturation and brain wiring (Hammond et al. 2018; Tremblay et al. 2011). Different types of interactions have been reported with microglia in healthy brain. Namely phagocytosis of newborn neurons during adult neurogenesis and phagocytosis of synaptic structures during development (Tremblay et al.
2011). In this section the importance of microglia in healthy brain responses and following ischemic insult is discussed.
1.3.1 Role of Microglia in healthy brain
An important aspect of microglia is the rapid identification of dying cells, followed by migration and clearance of apoptotic material by phagocytosis (Peri and Nusslein-Volhard 2008). This process is pivotal to inhibit the deleterious effects of apoptotic debris and degradation products (Lauber et al. 2004). It is still unclear how microglia identify apoptotic cells in physiological conditions. It has been shown that in pathophysiological condition like neural injury, P2Y6 purinergic receptors induce microglial phagocytosis. Microglia express the metabotropic P2Y6 receptor. Activation of these receptors by their endogenous ligand UDP triggers microglial phagocytosis. Systemic administration of kainic acid in experimental animals resulted in neuronal cell death in the hippocampal regions. This cell death is accompanied with P2Y6 receptor upregulation. Thus, P2Y6 receptor could act as a sensor for phagocytosis by identifying UDP signals (Koizumi et al. 2007).
Recent investigations indicate that microglia can eliminate axons and synaptic terminals as part of the pruning process. Using electron microscopy and high-resolution in vivo engulfment assays presynaptic and postsynaptic structures have been identified inside microglia in the mouse visual system (Schafer et al. 2012; Tremblay et al. 2010). Schafer et al. showed that deregulation of microglial pruning leads to impairment of synaptic development and neuroplasticity (Schafer et al. 2012). It has been shown that classical complement pathway, which mediates the removal of cellular debris and pathogens, has the pivotal role in synaptic pruning. C1q and C3 are the most important elements of complement system which are involved in synaptic pruning. These molecules localize to subsets of immature synapses. Microglia are the only CNS cell type that expresses the C3 receptor. These receptors target immature synapsis for phagocytosis (Stevens et al. 2007). Interestingly recently in Alzheimer mouse model, it has been shown that complement-dependent pathway and microglia that prune excess synapses in development are
inhibition of C1q, C3, or the microglial complement receptor CR3 decreases the number of phagocytic microglia and the early synapse loss (Hammond et al. 2018; Hong et al. 2016). Microglia also play crucial roles in the development of oligodendrocyte. Several researches have indicated that the microglia derived proteins influence oligodendrocyte progenitors (OPC) cell development. Namely, through vascular endothelial growth factor (VEGF) and IGF-1 microglia increase OPCs survival and maturation (Hammond et al. 2018; Nicholas et al. 2001; Pang et al. 2013). Recent studies have also confirmed that a subpopulation of microglia could be important for the generation of OPCs and myelination. These Cd11c-expressing microglia is located only in the white matter tracts of the early postnatal mouse brain and their transcriptomic profile is completely different from cortical microglia transcriptome. The subpopulation of Cd11c-expressing microglia in white matter express proteins that are involved in differentiation of OPCs and oligodendrocytes (Hagemeyer et al. 2017; Wlodarczyk et al. 2017). Intriguingly, iron is essential for the appropriate proliferation and maturation OPCs and microglia are the primary source of iron to OPCs during myelination (Todorich et al. 2009)
1.3.2 Role of microglia in stroke pathogenesis
Microglia are activated within few minutes in the acute phase of brain ischemic. Ischemia initiates excitotoxic signals that activates microglia in brain lesions. Furthermore, ATP liberated by dying neurons can activate microglia via interaction with purinergic receptors on microglia cell membrane. Once activated, microglia adopt diverse morphologies. For instance the round shape amoeboid morphology is found in the peri-infarct regions in the post-acute phases. In addition to the morphological change, gene expression pattern and secretary profile of microglia are affected by ischemic insult (Guruswamy and ElAli 2017). Interestingly it has been shown that proliferating microglia have neuroprotective properties in acute phase of stroke. In this context selective ablation of proliferating microglia is associated with exacerbation of brain damage following ischemica. In a pioneer investigation done by Lalancette-Hébert et al, they showed that a post ischemic proliferation of the resident microglia might function as a crucial modulator of a
neuroinflammatory responses. These proliferating microglia can be supplier of neurotrophic molecules such as IGF-1 (Lalancette-Hebert et al. 2007).
Another important issue after MCAO is microglia-neuron cross talk. After ischemic insult, many neurons die because of reduced blood flow and energy depletion. Neuro-glia interaction is complicated and different types of ligands and receptors are involved in this interaction (Correa et al. 2013). Dying cells liberate glutamate and damage-associated molecules that induce neuronal damage (Neher et al. 2013). Interestingly, new investigations demonstrated that damaged neurons could induce microglia polarization. Namely, damaged neurons induced by ischemia could release interleukin 4 (IL-4). The interaction of this cytokine with IL-4 receptor expressing microglia enhances microglia polarization to anti-inflammatory phenotype (Ma et al. 2017; Zhao et al. 2015).
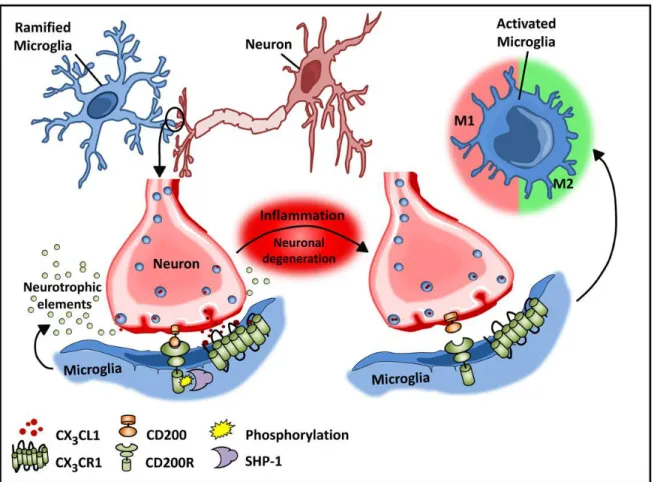
CD200/CD200R and CX3CL1/CX3CR1 are among the most investigated pair of ligand- involving in controlling microglia polarization. CD200 is constitutively expressed on neurons and CD200R is expressed on microglia. CD200 deficiency in mice induces microglia activation and exacerbates neuroinflammation in different experimental models such as experimental autoimmune encephalomyelitis (EAE) (Ma et al. 2017). CX3CL1/CX3CR1 is another ligand-receptor pair that inhibits microglia activation. CX3CL1 is expressed on neurons and CX3CR1 is only expressed on microglia in the brain. Lack of CX3CR1 in mice increases microglia mediated neurotoxicity in animal models of ALS and Parkinson disease (Cardona et al. 2006b). Figure 1.4 shows how neuro-glia interaction contributes to post stroke pathology.
Figure 1-4. Microglial-neuronal interactions in health and disease
Healthy neurons expressing chemokine fractalkine (CX3CL1) and CD200 membrane proteins intimately interact with their respective transmembrane protein receptors on microglia, CX3CR1, and CD200R to sustain a down-regulated microglial phenotype. Microglial receptors have immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which upon ligand–receptor activation suppresses downstream immune signaling through the recruitment of phosphatases including SHP-1. Inflammation disrupts this intimate neuronal–glial interaction, thus releasing the microglial cells from a down-regulated inhibited state to an activated phenotype (Dey et al. 2014)
1.3.3 Classification of activated microglia
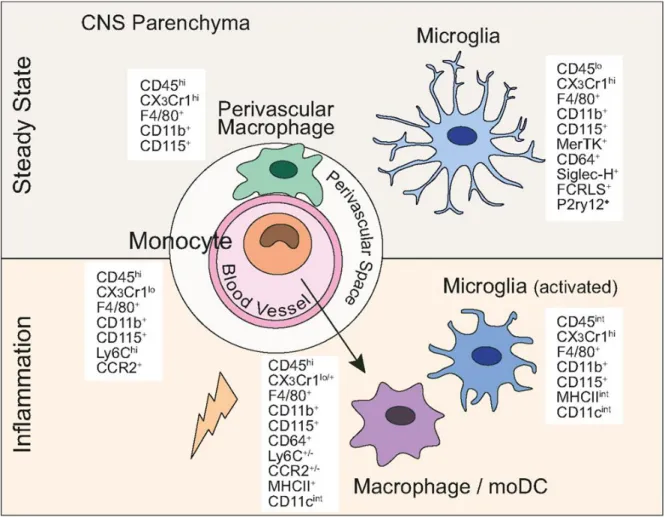
During microglial activation after an inflammatory insult, cell morphology is altered either to M1, the typically activated phenotype, or to M2, an alternatively activated phenotype; this phenotypic switch depends on the type of stimulation. M1 microglia are considered as proinflammatory, producing mediators such as tumor necrosis factor-alpha (TNF-𝛼), interleukin 1 beta (IL-1β), and interferon-𝛾 (IFN-𝛾) (Patel et al. 2013). M2 microglia are regarded as “healing cells” that contribute to recovery after damage and secrete anti-inflammatory mediators such as interleukin- IL-10, transforming growth factor-beta (TGF-𝛽), IL-4, IL-13, and IGF-1. Contents of TGF-β, IL-10 and CD206 increased in early phase of stroke and their peak of increase is 4-6 days after stroke. Furthermore, TGF-β liberated by microglia induces anti-inflammatory signaling which is accompanied with increased proliferation in the ischemic regions (Hanisch and Kettenmann 2007). Several proteins have been identified as markers for M1 or M2. MHC II, involves in antigen presentation, and CD86 are considered as a marker for M1 (Ponomarev et al. 2005). Ym-1 and CD206 have been associated with the protective phenotype of microglia following stroke (Perego et al. 2011). CNS myeloid cells express different markers in healthy steady state condition and after an inflammatory insult (figure 1.5).
It has been known for a long time that microglia transform to reactive states in response to inflammatory insult. In both transcriptional features and functional consequences, microglia seem to be more complicated and dynamic than ever thought. This aspects might elucidate why engagement of microglia can be either neuroprotective or neurotoxic, leading to attenuation or exacerbation of disease progression (Hanisch and Kettenmann 2007). Several experimental and clinical studies showed that different factors including age, gender and disease context affect microglia phenotype and behavior. In complex settings like stroke we need to use sensitive, specific and well-advanced technology to address heterogenic and context dependent properties of microglia. Now it is well-established that activated microglia express the canonical gene products of both M1 and M2 states at the same time. It has been demonstrated that after brain injury microglia do not simply switch to a
cell RNA-seq researches on microglia uncover the existence of the mixed phenotype at the single-cell level (Kim et al. 2016a). We can identify microglia phenotype in responses to ischemia by using state of the art technologies such as single-cell RNA-seq, translating ribosome purification analyses and single cell live imaging. These methods might give us the chance to depict a more realistic picture on important roles of microglia in complex and very heterogenic pathologies like brain injury (Kim et al. 2016a; Ransohoff 2016).
Figure 1-5. Central nervous system myeloid cells and their defining lineage markers in steady state and following brain inflammation
1.3.4 Different characteristics of resident microglia versus
monocyte-derived macrophages following brain ischemia
The immune cells infiltration to the brain following ischemia is a time dependent phenomenon. Peripheral immune cells such as monocytes/macrophages, myeloid and neutrophils are detectable in brain a day after stroke. Their peak is 3 days post stroke and they remain elevated until 7 days after stroke. Disruption of the blood–brain barrier (BBB) following ischemia facilitates the migration of immune cells into the brain. Peripheral monocytes reach the brain parenchyma via endothelial migration and they differentiate into tissue macrophages. Monocyte/macrophage and resident microglia share several common functions including expression of common antigens and the morphological similarity. Additionally, in general both cell types can polarize toward pro-inflammatory and/or anti-inflammatory phenotypes (Kim and Cho 2016). In the stroke are, there are several different macrophage phenotypes and these macrophages can transform to other phenotypes depending on the ischemic environment (Perego et al. 2011). In early phase of stroke infiltrated peripheral monocytes/macrophages are mostly Ly-6Chi (CCR2+) cells. In the
ipsilateral stroked area these cells become macrophages expressing pro-inflammatory markers. Once settled, these cells lose Ly-6C and CCR2 expression and release VEGF and TGF-𝛽 which are important mediators of brain repair. It has been shown that monocyte-derived macrophages recruited to the injured brain early after ischemic stroke contribute to long-term functional recovery through inflammation-resolving activity (Wattananit et al. 2016). Biochemical analyses demonstrated that at day 7 after MCAO, monocyte-derived macrophages exhibited a mixed pro-inflammatory/anti-inflammatory phenotype but 2 weeks after stroke, monocyte-derived macrophages with an anti-inflammatory phenotype are dominated. Using the anti-CCR2 antibody that prevents monocytes migration to brain during the first week after stroke, long-term brain recovery was diminished (Wattananit et al. 2016). These findings show the crucial and complex roles of monocyte-derived macrophages in stroke pathogenesis.
transgenic fate mapping, the responses of these cell types to mild brain ischemia has been investigated. Single cell transcriptomic analysis of gene expression at 7 days after MCAO revealed 472 transcripts that exclusively expressed in blood-derived macrophages as well as 970 transcripts for resident microglia. This investigation confirms differences in activation state between resident microglia and blood-derived macrophages after stroke and identifies unique molecular signatures for either cell type. Both cell types showed a mixture of phenotypes in vivo but the blood-derived macrophages generally displayed more neuroprotective phenotype. This finding might open a new avenue to understand the role of macrophages in pathophysiology of ischemic stroke (Kronenberg et al. 2018).
1.4 Targeting inflammation as the therapeutic approach for
brain ischemia
The role of inflammation in stroke pathology has been in the centre of attention in recent neuroscience researches. Inflammation plays different and diverse roles in acute and chronic phase of brain ischemia. In the early phases, different studies showed that inflammation is mostly detrimental. However, inflammatory processes are also important for repair mechanisms, the processes that occur in subacute and chronic phase of stroke. In this context pharmacotherapeutic interventions should target the specific phase of stroke (Kim et al. 2014). This double-edged sword concept of inflammation might stem from the fact that important mediators of inflammation like resident microglia have very complicated and diverse role in stroke pathology. General immunosuppressants and anti-inflammatory agents such as corticosteroids, nonsteroidal anti-anti-inflammatory drugs and minocycline have failed in stroke therapy. Failure of such therapies might be due to different possibilities. Namely these anti-inflammatory drugs might be too broad and/or they might not restore CNS cell function. In this paradigm immunosuppressants can completely suppress the immune response and anti-inflammatory interventions could inhibit initial phases of the innate-immune system response (Masgrau et al. 2017).Recent gene profiling of microglia isolated from experimental models of stroke, multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS) and aging indicated molecular heterogeneity among diseases (Chiu et al. 2013; Yamasaki et al. 2014). The effects of these differences in
disease pathogenesis are not fully understood but this complexity suggests that general anti-inflammatory agents might not show any clinical protection after ischemic insult (Masgrau et al. 2017).
Another important point is that restoring cellular function in brain ischemia is mostly recognized for neurons and not for glia cells although signs of functional impairment in glia cells have been reported. Deregulation of microglia function has been reported in the aged ovariectomized mice following stroke (Cordeau et al. 2016). In other neurodegenerative conditions also the malfunctions of microglia have been studied. In the mouse Alzheimer model microglia showed diminished capacity to clear amyloid-β (Iaccarino et al. 2016). Transcriptomic analysis elucidated a signature of suppressed metabolism in microglia isolated from a mouse model of MS (Masgrau et al. 2017; Yamasaki et al. 2014). According to this complexity specific drugs rather than general immunosuppressive or anti-inflammatory drugs might be necessary to restore cell function in microglia after brain injury (Masgrau et al. 2017).
The complex roles of microglia suggest that aim of stroke therapy should be shifted from simple inhibition of microglia toward fine-tuning the balance between protective and deleterious microglia responses (Hu et al. 2012). To gain this aim, understanding the roles of modulators that determine microglia phenotype is pivotal. Galectin-3 (Gal-3) is one of such assumed modulators that. It has been considered as a canonical tuner of macrophages/microglia activity. In the following sections the important roles pro-inflammatory cytokines, anti-pro-inflammatory cytokines and Gal-3 in stroke pathology are discussed in detail.
1.4.1 Role of cytokines in stroke pathology
Cytokines are glycoproteins that are produced by cells in the brain in response to damaged tissue after ischemia and are responsible for regulating the innate and adaptive immune response (Doll et al. 2014). Different types of cytokines including pro-inflammatory and anti-inflammatory ones are secreted by astrocytes, microglia and neurons. It has been
Therefore, mononuclear phagocytes, T lymphocytes and polymorphonuclear leukocytes produce and secrete cytokines and might take part in ischemia associated brain inflammation (Ferrarese et al. 1999).
The shift in the balance of pro- and anti-inflammatory cytokines production is associated with a larger stroke size in experimental models of brain ischemia (Vila et al. 2003). Different cytokines such as TNF-α, IL-1β, IL-4, and IL-6 have important roles in the pathogenesis of stroke and have been targeted as therapeutic options for brain recovery. How these cytokines contribute in stroke pathogenesis is important because different studies showed the time dependent pattern of cytokine secretion and the complex roles of these molecules (Doll et al. 2014). While TNF-α and IL-1β appear to aggravate brain damage, IL-4 and IL-10 might be protective.
Increasing the levels of TNF-α has been repeated in different experimental models of brain injury. Up-regulation of TNF-α mRNA and protein occurs 1-3 hours following middle MCAO in experimental models. Interestingly, the early expression of TNF-α mRNA preceding leukocyte infiltration (del Zoppo et al. 2000). TNF-α has been shown to be either neurotoxic or neuroprotective in stroke studies. Namely, TNF-α knock-out mice show larger infarct size compared to control mice suggesting that TNF-α is neuroprotective using the permanent model of MCAO (Doll et al. 2014; Lambertsen et al. 2009). In this context it has been shown that microglia derived TNF-α protects neuron against ischemia (Lambertsen et al. 2009). Other studies proved that TNF-α is neurotoxic in stroke condition. Antibodies that target TNF-α have exerted neuroprotective action following MCAO and caused a dose-dependent decrease in infarct volume (Nawashiro et al. 1997). IL-1β is another potent pro-inflammatory cytokine and involves in activation of different inflammatory processes. IL-1β has shown neurotoxicity in different experimental stroke models by using different interventions to potentiate or inhibit IL-1β effects (Doll et al. 2014). mRNA and protein level of IL-1β increase following ischemic insult. In addition, it has been shown that intraventricular injection of recombinant IL-1β after MCAO induces brain edema and the influx of neutrophils (Yamasaki et al. 1995).
IL-6 is an inflammatory cytokine that binds to class I cytokine receptors. IL-6 signaling activates JAK which activates STAT family of transcription factors and subsequently activates RAS-RAF-MAPK pathways (Doll et al. 2014; Erta et al. 2012). These signalling pathways can influence microglia activation which are important for tissue remodeling and recovery after stroke (Erta et al. 2012). Neuroprotective and neurotoxicity of IL-6 is a subject of controversy and debate. IL-6 can be detrimental by increasing body temperature which has been shown to increase brain damage after stroke (Azzimondi et al. 1995). IL-10 and IL-4 are among the most important anti-inflammatory cytokines that have been studied in stroke pathology. When the IL-10 receptor is activated by IL-10, tyrosine residues on the intracellular domain of IL-10 receptor are phosphorylated and consequently JAK1 and Tyk2 become activated. Stat3 interacts with these phosphorylated tyrosines to phosphorylate other Stat proteins that translocate to the nucleus to activate transcription of Stat3-responsive genes (Doll et al. 2014; Finbloom and Winestock 1995). Indeed JAK-STAT pathway is activated following activation of cytokine receptors and this activation might polarize microglia to anti- or inflammatory phenotype. IL-10 can inhibit pro-inflammatory cytokine production, chemokine secretion, and can inhibit antigen presentation by macrophages and microglia (Ooboshi et al. 2005). Protective aspects of IL-10 have been also reported in neurodegenerative conditions like ALS. In SOD1 mutation model of ALS our group previously showed that blocking IL-10 increased inflammation and aggravates clinical disease onset, whereas overexpression of IL-10 in microglia using a gene therapy approach significantly delayed disease onset and increased survival of ALS mice. Based on this finding, targeted overexpression of IL-10 in microglia may have therapeutic potential in neurodegenerative contexts (Gravel et al. 2016). Regarding the pivotal role of IL-4 in microglia polarization, IL-4 receptor signaling pathway is discussed separately in the following section.
1.4.1.a IL-4; a pivotal cytokine for microglia/macrophage polarization following cerebral ischemia
macrophages polarization and it plays pivotal roles in brain function following ischemic insult (Liu et al. 2016). Experimental and clinical studies have explained the importance of IL-4 in the acute stages of stroke. Namely, in the patient’s serum levels of IL-4 increase significantly several hours after stroke onset (Kim et al. 2000). Animal studies indicated that IL-4 has as an early endogenous neuroprotective effect after MCAO and IL-4 deficiency aggravates brain injury (Xiong et al. 2011). Interestingly it has been shown that IL-4 also improves long-term neurological outcomes after stroke, perhaps through induction of alternative activated microglia/macrophages. These findings highlight the importance of IL-4 in promoting long-term functional recovery after stroke (Liu et al. 2016).
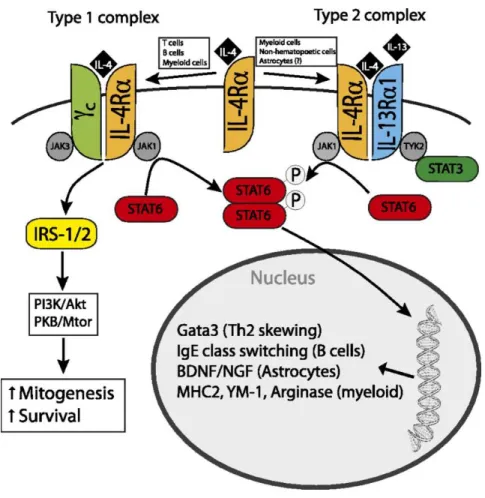
The effect of IL-4 signaling is mediated through the IL-4 receptor alpha chain (IL-4Rα). Following binding to its ligand, IL-4Rα dimerizes either with the common gamma chain (γc) to produce the type-1 signaling complex located mainly on hematopoietic cells, or with the IL-13 receptor alpha 1 (IL-13Rα1) to produce the type-2 complex, which is expressed also on non-hematopoietic cells. The type-1 signaling complex is pivotal for alternatively activated macrophages (Gadani et al. 2012). Upon activation, the type-1 complex signals through Janus family kinases (JAK1 and JAK3), which phosphorylate and create docking sites for the transcription factor STAT6, which then dimerizes and translocate to the cell nucleus (Gadani et al. 2012). STAT6 has important role in transcription of anti-inflammatory genes downstream of IL-4 receptor, the effect that causes in induction of neuroprotective phonotype in glia. On the other hand, STAT3 induces the pathway that might increase the level of inflammatory mediators in microglia. Figure 1.6 summarized the type-1 and type-2 signalling following IL-4 receptor activation.
Peroxisome proliferator-activated receptors (PPAR)-gamma (PPAR-γ) is an important and canonical transcriptional factor in the induction of anti-inflammatory signaling pathways. It has been reported that IL-4 receptor activation might activate PPAR-γ in microglia and induce microglia alternative activation (Rahimian et al. 2018b). Regarding the pivotal role of this transcriptional factor in post stroke anti-inflammatory signaling, in the next section, properties of PPAR-γ is discussed in detail.
Figure 1-6. IL-4 signaling pathways
After IL-4 binding IL-4Rα, the IL-4 receptor is created by dimerization with the common γ chain (γc) to create the type 1 signaling complex or with IL-13Rα1 to create the type 2 complex. Both receptors signal through STAT6, which is phosphorylated, dimerizes, and traffics to the nucleus to function as a transcription factor for Th2, IgE, and alternatively activated macrophage associated genes. The type 1 receptor also activates insulin receptor substrate-1 and/or 2, leading to increased mitogenesis and inhibition of apoptosis through multiple signaling pathways. The type 2 complex also responds to IL-13, and it can signal other STAT molecules (STAT3) through the JAK family kinase TYK2 (Gadani et al. 2012).
and a DNA binding domain. When their endogenous or exogenous ligands bind to PPAR, they create a heterodimeric complex which recruits other coactivators including PPAR coactivator-1, PPAR-interacting protein, PPAR-binding protein, steroid receptor coactivator-1 and CREB binding protein. This complex binds to the promoter regions of specific genes that contain a regulatory element known as the peroxisome proliferator response element which either activates or transrepresses the target genes (Vemuganti 2008). In the mammalian body, PPARs control glucose metabolism, cell proliferation and differentiation (Berger and Moller 2002). Several studies showed that PPARγ ligands elicit anti-inflammatory and neuroprotective actions in various experimental models of neurodegenerative diseases (Landreth and Heneka 2001). In this paradigm beneficial properties of PPARγ agonists such as rosiglitazone and pioglitazone have been demonstrated in brain ischemia models. These interventions are currently approved by FDA for the treatment of type-2 diabetes. Following MCAO in mice, PPARγ expression significantly up-regulates in brain damaged area and treatment with PPARγ agonists inhibits the expression of many pro-inflammatory proteins (Vemuganti 2008). In mice treatment with rosiglitazone or pioglitazone, the attenuation of microglial activation and down regulation of pro-inflammatory proteins like cyclooxygenase-2, iNOS and IL-1β in the ipsilateral brain have been reported (Sundararajan et al. 2005).
Indeed, PPARγ activation inhibits the activity of transcription factors including NF-κB, AP1 and STAT. Some studies indicate that IL-4 receptor signaling increases the endogenous level of PPAR-γ ligands such as prostaglandin J2 and subsequently amplifies transcriptional activity of PPAR-γ which might polarize microglia phenotype toward the anti-inflammatory one (Huang et al. 1999).
The fact that PPAR-γ is involved in the modulation of macrophage differentiation and activation in peripheral tissues led to studying of the role of PPAR-γ in resident microglia in CNS. Several investigations indicate that PPAR-γ endogenous ligand and synthetic agonists might influence brain inflammation by inhibiting different functions related to microglial activation, such as production of inflammatory cytokines, chemokines, nitric oxide and prostaglandins (Bernardo and Minghetti 2006).