HAL Id: tel-00207959
https://tel.archives-ouvertes.fr/tel-00207959
Submitted on 18 Jan 2008HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
et de l’agitation en mélangeur Triaxe®
Jean-François Demeyre
To cite this version:
Jean-François Demeyre. Caractérisation de l’homogénéité de mélange de poudres et de l’agitation en mélangeur Triaxe®. Génie des procédés. Institut National Polytechnique de Toulouse - INPT, 2007. Français. �tel-00207959�
Nod’ordre : 2490 2007
Thèse
présentée pour obtenir le titre de
Docteur de l’I
NSTITUTN
ATIONALP
OLYTECHNIQUE DET
OULOUSEÉcole doctorale : Sciences des Procédés
Spécialité : Génie des procédés et de l’environnement par
M. Jean-François D
EMEYREC
ARACTÉRISATION DE L
’
HOMOGÉNÉITÉ DE
MÉLANGE DE POUDRES ET DE L
’
AGITATION
EN MÉLANGEUR
T
RIAXE
®
Soutenue le 22 juin 2007, devant le jury composé de :
Jamal CHAOUKI Rapporteur
Pierre GUIGON Rapporteur
Henri BERTHIAUX Examinateur
Guillaume DELAPLACE Examinateur
Cendrine GATUMEL Examinateur
Michel GRANDJEAN Examinateur
Catherine XUEREB Examinateur
Thèse préparée au Centre de Recherche d’Albi en Génie des Procédés des Solides Divisés, de l’Énergie et de l’Environnement, UMR CNRS 2392, École des Mines d’Albi-Carmaux
« On croit qu’on emmène son chien pisser matin et soir. Grave erreur : ce sont les chiens qui nous invitent deux fois par jour à la méditation. » Daniel Pennac, La fée carabine
Remerciements
Un laboratoire, une équipe scientifique, une famille, des copains, plein de VTT, des
runnings. . . voilà comment décrire grossièrement cette thèse !
Merci pour ton soutien pendant ces trois années, Anne-C, malgré une rédaction « in-tensive ». Tes sourires, ta bonne humeur m’ont souvent encouragés ! Merci aussi à mes parents pour leur soutien pendant vingt sept ans, et oui, déjà. . .
J’ai travaillé au sein d’une équipe super, tant sur le plan humain que scientifique : merci à Cendrine et Henri mais aussi Séverine, Sylvie, Laurent, Olivier et Philippe ainsi qu’à tous les thésards. . . Merci particulier à Jennifer pour m’avoir aidé à obtenir une partie des résultats expérimentaux dans la chaleur de la cabine !
Je sais ! Si je continue en disant que le laboratoire était super, vous allez pensez que c’était la thèse idéale : c’est pas faux ! J’ai apprécié d’être entouré de personnes prêtes à donner un coup de main. Merci donc au sevrice info & al., j’ai nommé Cathy, Emmanuel, les Jean-Jacques, Olivier, Paul, Thomas, notamment pour m’avoir fait découvrir LATEX. Merci aussi aux secrétaires Anne-Marie, Elisabeth, Eve, Maryse et à l’ensemble du personnel de l’école. . . Merci à Yann pour les quelques conseils et les nombreuses bonnes bouffes, j’ai failli ne pas rentré dans mon costume de marié !
Les petits footing sur les splendides parcours proposés par les aficionados me resteront longtemps en tête ainsi que la splendide victoire de l’équipe 2 lors d’un magnifique relais en 2005 ! Désolé JJ, tu n’auras plus beaucoup l’occasion de courir plus vite que moi, mais il ne fallait pas me donner l’envie ! Révérence, Maître Jacques, tes temps restent infranchis-sables ! Mais bon, comme toujours, la loi du thésard s’est encore vérifiée : l’endurance est inversement proportionnelle au temps passé à écrire sa thèse. . . Il faudra que je revienne faire un Ekiden !
Oh là, j’ai failli oublier Guillaume, qui m’a permis de découvrir les aspects positifs de la
recherche et Michel, sans qui je ne serai pas là aujourd’hui pour parler du Triaxe®. . . Merci à tous.
Juin 2007 Jean-François Demeyre
Table des matières
Remerciements v
Table des matières vii
Table des figures xi
Liste des tableaux xv
Liste des symboles xvii
Introduction générale 1
1 État de l’art 5
1.1 Introduction . . . 5
1.2 L’état de mélange : un concept à plusieurs échelles . . . 6
1.3 Échantillonnage : estimation de la qualité du mélange . . . 9
1.3.1 Estimation et statistiques . . . 9
1.3.2 Méthodes d’échantillonnage . . . 12
1.4 Les méthodes de mesures en ligne de l’homogénéité de mélange . . . 16
1.4.1 Les méthodes capacitives . . . 16
1.4.2 Les méthodes optiques . . . 18
1.4.3 Les méthodes proche infrarouge . . . 19
1.4.4 Les méthodes par analyse d’images . . . 23
1.5 Propriétés des poudres et du milieu influençant l’écoulement . . . 27
1.6 Mécanismes de mélange et de ségrégation . . . 29
1.6.1 Mécanismes de mélange . . . 29
1.6.2 Mécanismes de ségrégation . . . 30
1.6.3 Comment éviter la ségrégation . . . 33
1.7 Les différents types de mélangeurs . . . 34
1.7.1 Les cuves tournantes . . . 34
1.7.2 Les mélangeurs convectifs . . . 35
1.7.3 Les mélangeurs à haut cisaillement . . . 37
1.7.4 Les mélangeurs statiques . . . 38
1.7.6 Les combinaisons de mélangeurs . . . 39
1.7.7 Quelques pistes pour le dimensionnement des mélangeurs . . . 39
1.8 Conclusion . . . 43
2 Développements méthodologiques et expérimentaux 45 2.1 Systèmes particulaires . . . 45
2.1.1 Caractéristiques des poudres . . . 45
2.1.2 Caractéristiques de l’écoulement . . . 47
2.1.3 Ségrégabilité des mélanges . . . 47
2.2 Le Triaxe®, un nouveau type de mélangeur . . . 49
2.2.1 Les caractéristiques mécaniques du Triaxe® . . . 50
2.2.2 Le Triaxe®et l’attrition . . . 53
2.3 La plate-forme expérimentale . . . 53
2.4 Méthode de pilotage du Triaxe® . . . 55
2.5 Mesure de la qualité du mélange . . . 57
2.5.1 Acquisition de l’image . . . 57
2.5.2 Traitement de l’image . . . 60
2.5.3 Analyse de la qualité du mélange . . . 61
2.5.4 Protocole expérimental pour la mesure de la qualité du mélange . . . 67
3 Étude de l’agitation 69 3.1 Mesure des couples . . . 69
3.1.1 Préchauffage des moteurs . . . 69
3.1.2 Couple développé par les moteurs . . . 70
3.1.3 Couple et puissance reçus par les produits . . . 70
3.2 Les trajectoires et les vitesses du Triaxe® . . . . 71
3.2.1 Les trajectoires des pales . . . 71
3.2.2 La vitesse linéaire de bout de pale . . . 71
3.2.3 L’angle de pénétration des pales dans le produit . . . 74
3.3 Analyse des résultats bruts . . . 78
3.3.1 Poudres à écoulement libre . . . 78
3.3.2 Poudres cohésives . . . 80
3.3.3 Mélanges . . . 82
3.3.4 Le Triaxe®. . . un outil de rhéologie ? . . . 84
3.4 Essai d’adimensionnalisation . . . 86
3.5 Conclusion sur les régimes d’écoulement . . . 91
4 Étude des temps de mélange : lien avec l’homogénéité des mélanges 93 4.1 Conditions étudiées . . . 93
4.2 Cinétiques de mélange . . . 94
4.2.1 Qualité du mélange . . . 95
4.2.2 Répétabilité des essais . . . 99
4.2.3 Stabilité des mélanges . . . 99
4.2.4 Conclusion . . . 100
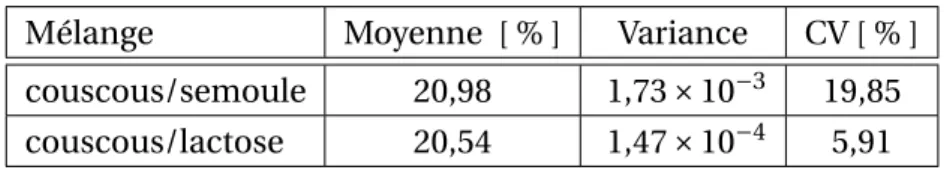
4.3 Analyse des résultats des mélanges couscous/semoule . . . 100
4.3.1 Influence des différents paramètres sur le temps de mélange . . . 100
TABLE DES MATIÈRES ix
4.4 Analyse des résultats des mélanges couscous/lactose . . . 104
4.4.1 Influence des différents paramètres sur le temps de mélange . . . 104
4.4.2 Puissance dissipée et travail . . . 106
4.5 Conclusion sur l’homogénéité des mélanges . . . 110
Conclusion générale 111 Bibliographie 115 A Tableau de synthèse des caractéristiques de différents types de mélangeurs 119 B Distributions granulométriques des différents produits testés 121 C Dispostif de mesure de la masse volumique tassée 123 D Les vitesses et les trajectoires mises en jeu dans le Triaxe® 125 D.1 Les trajectoires . . . 127
D.2 Les vitesses . . . 129
E Les cinétiques de mélange 133 E.1 Les mélanges couscous/semoule . . . 133
Table des figures
1 Illustration des différentes échelles définissant l’homogénéité des mélanges 1
2 Un exemple de trajectoire du Triaxe® . . . 3
1.1 Différents états d’un mélange binaire . . . 6
1.2 Notion d’intensité de ségrégation . . . 7
1.3 Mélanges de même intensité de ségrégation et de structures différentes . . . 8
1.4 Notion d’échelle de ségrégation . . . 8
1.5 Allure typique d’un autocorrélogramme . . . 9
1.6 Évolution des valeurs des bornes d’un intervalle de confiance . . . 11
1.7 Exemple de mélange en accord avec les standards industriels . . . 11
1.8 Différentes étapes d’un prélèvement dans un mélange . . . 12
1.9 Schéma d’une loi d’intégration au hasard . . . 13
1.10 Schéma d’une loi d’intégration systématique avec implantation au hasard . 13 1.11 Schéma d’une loi d’intégration stratifiée au hasard . . . 13
1.12 Différents types de sondes utilisées pour l’échantillonnage des mélanges . . 14
1.13 Perturbation induite par une sonde de prélèvement . . . 14
1.14 Schéma de systèmes industriels de prélèvement . . . 15
1.15 Croquis de l’installation expérimentale de la mesure de capacité électrique 17 1.16 Schéma du principe de mesure de la capacité électrique . . . 17
1.17 Schéma de conception du capteur optique . . . 18
1.18 Signaux de réflexion relative . . . 19
1.19 Courbes de calibration d’une méthode par fibre optique . . . 19
1.20 Schéma du mélangeur Nauta muni de l’interface PIR . . . 20
1.21 Comparaison des performances de mélanges pour différents batches . . . . 21
1.22 Schéma du montage expérimental utilisant un spectrophotomètre PIR . . . 22
1.23 Comparaison de résultats obtenus par tamisage et par proche infrarouge . . 22
1.24 Dispositif expérimental de KEHLENBECK(2007) . . . 23
1.25 Comparaison du modèle et des expériences de KEHLENBECK(2007) . . . 23
1.26 Schéma d’un système de vision . . . 24
1.27 Comparaison des concentrations prédites par le système de vision et le PIR 25 1.28 Dispositif expérimental et méthodologie de traitement du film . . . 26
1.29 Évolution de l’échelle de ségrégation lors d’une perturbation . . . 26
1.30 Relation entre la tension superficielle et la taille des particules . . . 27
1.32 Ségrégation due aux trajectoires . . . 31
1.33 Ségrégation par percolation des fines . . . 32
1.34 Ségrégation par élutriation . . . 32
1.35 Courbe caractéristique d’un procédé de mélange . . . 33
1.36 Mise en évidence des étapes ségrégeantes . . . 34
1.37 Quelques exemples de mélangeurs à cuve tournante en « V » . . . 35
1.38 Mélangeur à rubans hélicoïdaux . . . 36
1.39 Mélangeur à socs . . . 36
1.40 Mélangeur orbital à vis . . . 36
1.41 Mélangeur à turbine à cuve biconique . . . 37
1.42 Turbosphère Moritz . . . 37
1.43 Mélangeurs à haut cisaillement . . . 37
1.44 Broyeur à marteaux . . . 38
1.45 Silo-mélangeur . . . 38
1.46 Installation de mélange en lit fluidisé . . . 39
1.47 Éléments de dimensionnement d’un mélangeur à tambour horizontal . . . 40
1.48 Éléments de dimensionnement d’un mélangeur à double cuve en V . . . 41
1.49 Corrélation entre puissance et vitesse d’un mélangeur orbital à vis . . . 43
1.50 Temps de mélange en fonction des dimensions du mélangeur . . . 43
2.1 Dispositif du test de ségrégation . . . 48
2.2 Résultats des tests de ségrégabilité des mélanges . . . 48
2.3 Le Triaxe®utilisé . . . 50
2.4 Schéma du Triaxe® . . . 51
2.5 Un exemple de trajectoire du Triaxe® . . . 52
2.6 Différents types de pales disponibles pour le Triaxe® . . . . 52
2.7 Distributions granulométriques des tests d’attrition du Triaxe® . . . 53
2.8 Schéma général de la plate-forme expérimentale . . . 54
2.9 Distributions granulométriques des tests d’attrition du tamisage . . . 54
2.10 Schéma du dispositif de tamisage par vibration à 3 niveaux . . . 54
2.11 Relation entre la tension du variateur et la vitesse du moteur . . . 55
2.12 Face avant du logiciel développé pour piloter le Triaxe® . . . . 56
2.13 La caméra linéaire . . . 58
2.14 Comportement de la caméra linéaire en fonction de sa vitesse d’acquisition 59 2.15 Définition de la notion d’image et d’échantillon . . . 60
2.16 Face avant du logiciel développé pour l’analyse d’image en ligne . . . 62
2.17 Étalonnage liant la proportion de pixels noirs à la composition massique . . 62
2.18 Échantillonnages complets de tailles différentes . . . 63
2.19 Échantillonnages de même taille complet ou incomplet . . . 64
2.20 Échantillonnage successif de même taille incomplet ordonné ou aléatoire . 64 2.21 Échantillonnage suivant une implantation stratifiée au hasard . . . 65
2.22 Schéma des différents plans d’échantillonnage . . . 65
2.23 Moyennes et intervalles de confiance pour différents échantillonnages . . . 66
2.24 Variances et intervalles de confiance pour différents échantillonnages . . . 66
2.25 Protocole suivi pour obtenir un point d’une cinétique de mélange . . . 67
TABLE DES FIGURES xiii
3.1 Couples mesurés sur les deux moteurs sans préchauffage . . . 69
3.2 Couples mesurés sur les deux moteurs lors d’un essai à vide . . . 70
3.3 Exemple de trajectoire de deux pales du Triaxe® . . . 71
3.4 Exemple de vitesse linéaire de bout de pale . . . 72
3.5 Vitesse linéaire maximale en fonction de la vitesse de l’axe de rotation . . . 72
3.6 Coefficient de variation de la vitesse linéaire . . . 74
3.7 Schéma de la pénétration de la pale dans le lit de poudre . . . 74
3.8 Angle de pénétration de la pale dans le produit . . . 75
3.9 Angle maximal de pénétration en fonction de la vitesse de giration . . . 76
3.10 Angle maximal de pénétration en fonction du rapport des vitesses . . . 76
3.11 Coefficient de variation de l’angle de pénétration de la pale dans le produit 77 3.12 CVθen fonction de θmax . . . 77
3.13 Couple effectif mesuré pour les poudres à écoulement libre . . . 78
3.14 Puissance effective pour les poudres à écoulement libre . . . 79
3.15 Couple effectif mesuré pour les poudres cohésives . . . 80
3.16 Puissance effective absorbée par les poudres cohésives . . . 81
3.17 Couple effectif pour les mélanges . . . 82
3.18 Puissance effective pour les mélanges . . . 83
3.19 Couple effectif en fonction de θmax . . . 83
3.20 Puissance effective en fonction de θmax . . . 83
3.21 Comparaison des puissances consommées par les quatre produits . . . 84
3.22 Puissance effective développée par les moteurs en fonction de leur vitesse . 85 3.23 Puissance spécifique maximale en fonction d’indices d’écoulement . . . 86
3.24 Ne en fonction de F rmaxpour les différents produits « bruts » . . . 87
3.25 Ne en fonction F rmaxpour les différents mélanges . . . 88
3.26 Ne en fonction F rmaxpour les différents produits « bruts » . . . 89
3.27 Influence de l’angle de pénétration sur le nombre de Newton Ne . . . . 90
3.28 Npmaxen fonction de vmaxpour les différents mélanges . . . 90
3.29 Npmaxen fonction de vmaxpour les différents produits . . . 91
4.1 Exemples de cinétiques pour les mélanges couscous/semoule . . . 94
4.2 Exemples de cinétiques pour les mélanges couscous/lactose . . . 95
4.3 Variance minimale pour l’ensemble des cinétiques couscous/semoule . . . 96
4.4 Variance minimale pour l’ensemble des cinétiques couscous/lactose . . . . 96
4.5 Influence de divers paramètres sur la variance minimale . . . 97
4.6 Variance minimale pour l’ensemble des cinétiques . . . 98
4.7 Les mélanges obtenus face aux standards industriels . . . 98
4.8 Répétabilité du protocole opératoire . . . 99
4.9 Absence de démélange dans le Triaxe® . . . 100
4.10 Temps de mélange tmpour l’ensemble des cinétiques couscous/semoule . 100 4.11 tmet vitesses d’agitation pour les mélanges couscous/semoule . . . 101
4.12 tmen fonction de vmaxet de θmaxpour les mélanges couscous/semoule . . 102
4.13 tmen fonction de la puissance effective Peff . . . 103
4.14 tmen fonction de la puissance effective Weff . . . 103
4.15 Temps de mélange tmpour l’ensemble des cinétiques couscous/lactose . . 105
4.17 tmen fonction de vmaxet de θmaxpour les mélanges couscous/lactose . . . 106
4.18 tmen fonction de la puissance effective Peff . . . 107
4.19 tmen fonction de la puissance effective Weff . . . 107
4.20 Peffet Weffen fonction du temps de mélange tm . . . 108
4.21 Utilisation de la corrélation d’ENTROPdans le cas du Triaxe® . . . 109
4.22 Temps de mélange en fonction du produit des vitesses des axes NAR× NAG . 109 B.1 Distribution granulométrique des systèmes particulaires utilisés . . . 121
B.2 Distribution granulométrique des mélanges . . . 122
C.1 Dispositif de mesure de la masse volumique tassée . . . 123
D.1 Schéma introduisant les variables utilisées dans les trajectoires du Triaxe® . 125 D.2 Exemple de vitesse normale de bout de pale . . . 130
Liste des tableaux
1.1 Caractéristiques des mélanges de REALPE& VELAZQUEZ(2003) . . . 24
1.2 Coefficients utilisés pour dimensionner des mélangeurs à ruban ou à pales 42 2.1 Différents diamètres caractéristiques des produits . . . 46
2.2 Différentes masses volumiques des produits . . . 46
2.3 Différents indices caractérisant l’écoulement des produits . . . 47
2.4 Résultats des tests de ségrégabilité des mélanges . . . 49
2.5 Caractéristiques de la caméra linéaire Lord DVL 5000 T . . . 57
2.6 Caractéristiques de l’échantillonnage utilisé pour les mélanges . . . 68
3.1 Vitesse linéaire maximale en fonction des vitesses des moteurs . . . 73
3.2 Coefficient de variation de la vitesse linéaire de bout de pale . . . 73
3.3 Influence de θmaxet de vmaxsur le couple effectif . . . 92
3.4 Les différentes relations intégrant les nombres sans dimension définis . . . 92
4.1 Récapitulatif des données pour les 14 expériences choisies . . . 94
4.2 Récapitulatif des temps de mélange pour les mélanges couscous/semoule . 101 4.3 Récapitulatif des temps de mélange observés dans le Triaxe® . . . 104
4.4 Conditions qui permettent le temps de mélange le plus court . . . 108
4.5 Comment mélanger vite et bien en fonction des produits mélangés . . . 110
Liste des symboles
Lettres latines
Aλi absorbance d’une substance i qui absorbe à la longueur d’onde λ
−
Ceff couple effectif reçu par le produit N · m
c concentration des molécules de l’espèce i qui absorbent à la longueur d’onde λ
mol · m−3
C couple N · m
CV coefficient de variation −
dp diamètre de la particule m
F rmax nombre de Froude calculé avec vmax −
g accélération de la pesanteur m · s−2
Iλ0 intensité du rayon incident −
It intensité du rayon réfléchi ou transmis −
j valeur de niveaux de gris −
l trajet optique de la cellule m
Lpale longueur des pales m
M masse de produit kg
MGVi valeur moyenne de niveaux de gris du
com-posant i
−
N vitesse de rotation de la cuve tr · min−1
Z nombre total d’échantillons −
z nombre d’échantillons prélevés −
N vitesse de révolution tr · min−1
Ne nombre de Newton −
Ni vitesse de révolution dont l’axe est défini en
indice
tr · min−1
Np nombre de Puissance −
Npmax nombre de Puissance calculé avec vmax −
Peff puissance effective reçue par le produit W
pi proportion volumique de produit i −
P puissance effective absorbée par le produit W
R rayon de la cuve m
Re nombre de Reynolds −
Remax nombre de Reynolds calculé avec vmaxet Ceff −
s écart-type observé −
s2 variance estimée −
Uf tension aux bornes du variateur de fréquence V
vmax vitesse linéaire maximale de bout de pale m · s−1
v0 vitesse initiale horizontale m · s−1
xi composition des échantillons en constituant
clé
−
xm teneur moyenne estimée −
Lettres grecques
α risque lié à l’estimation −
χ2inf valeur inférieure de la fonction discriminante −
χ2sup valeur supérieure de la fonction discrimi-nante
−
ελi coefficient spécifique d’absorbance molaire de l’espèce i
mol−1· m2
εi permittivité diélectrique à l’état dense du
ma-tériau i
−
εm permittivité diélectrique du mélange des n
constituants de proportion pi
−
µ teneur moyenne −
η viscosité dynamique du fluide Pa · s
φj nombre de pixels dans une image avec un
niveau de gris égal à j (fréquence de chaque niveau de gris)
−
ρ masse volumique aérée du produit kg · m−3
ρs masse volumique du solide kg · m−3
σ écart-type vrai −
σ2 variance vraie −
Indices
0 se rapporte à un essai sans produit −
A se rapporte à un axe de révolution −
à vide se rapporte à un essai sans produit − en charge se rapporte à un essai avec 48 L de produit −
G se rapporte au mouvement de giration −
M se rapporte à un moteur −
Introduction générale
Le mélange des solides divisés, de poudres ou de milieux granulaires, est une opéra-tion clé pour de nombreux domaines industriels — aussi variés que la pharmacie, l’agro-alimentaire, l’industrie des ciments, des matières plastiques. Cette opération permet d’at-teindre les spécifications et propriétés d’usage des produits formulés car elle est respon-sable en grande partie de l’homogénéité du produit à l’échelle requise — souvent celle du conditionnement. C’est donc une étape incontournable au cours de laquelle de nom-breuses difficultés peuvent survenir. La notion d’homogénéité d’un mélange de solides, indissociable de celles d’échelles d’observation et de ségrégation, est difficile à atteindre par la mesure. La figure 1 illustre ces notions d’échelles de ségrégation et d’observation. En effet, suivant la région de l’image analysée — l’échelle d’observation — la taille minimale de l’échantillon à prélever pour avoir une répartition homogène en « nombre de poisson » — l’échelle de ségrégation, la taille maximale des régions ségrégées — est différente.
FIG. 1: Oeuvre de Maurits Cornelis ESCHER— 17 juin 1898 – 27 mars 1972 — pouvant
illustrer les différentes échelles qui définissent l’homogénéité des mélanges de poudres, les carrés noirs représentent les échelles de ségrégation de deux régions différentes,
l’échelle de ségrégation de la figure est la plus grande.
prélèvement d’échantillons, ce qui pose des problèmes techniques et statistiques. Le dé-veloppement actuel de méthodes de mesure en ligne, non intrusives, devrait toutefois contribuer à une meilleure définition, et par la suite à un meilleur contrôle, de l’homogé-néité dans un avenir plus ou moins proche.
A l’inverse du mélange des fluides, les phénomènes réglant le mélange des pulvérulents sont encore mal connus. Ceci étant probablement le fait de la nature mésoscopique de ces milieux et du manque de modèles les décrivant à cette échelle. Il est cependant possible d’utiliser certaines méthodologies classiques en « génie des procédés », comme l’étude de cinétique des mélanges, pour modéliser globalement l’opération. Enfin, s’il est clair que l’opération de mélange a une grande importance dans l’atteinte de l’homogénéité, il est indispensable de considérer l’ensemble du procédé pour atteindre la qualité du mélange. En effet, il existe un grand nombre d’étapes du procédé où la ségrégation peut faire évoluer l’homogénéité comme les étapes de vidange, de transport, de stockage, etc.
Pour qu’un mélange soit homogène, il faut que les différents constituants soient en mouvement. Cette mobilité du mélange est essentiellement due aux mécanismes de convection, de cisaillement et de diffusion, qui sont le résultat des propriétés des par-ticules, du milieu, mais aussi des possibilités technologiques des appareils de mélange. L’opération de mélange peut être réalisée aussi bien par des procédés en continu qu’en discontinu, ce dernier choix étant souvent le résultat de diverses contraintes de production ou d’habitude des différents secteurs d’activité. Il existe un grand nombre de mélangeurs dont les trois principaux types utilisés dans l’industrie sont classés selon que l’agitation est le résultat d’un mobile interne — mélangeurs convectifs — par la mise en rotation de la cuve — mélangeurs à tambour — ou bien encore par le propre écoulement du matériau — mélangeurs statiques. Les mélangeurs actuels sont souvent basés sur un mouvement d’agitation très systématique et ne balayant pas tout le volume à mélanger. Ceci génère des zones mortes et des surfaces libres importantes qui sont propices aux phénomènes de ségrégation. De plus, ils ont en général été développés pour convenir à un cas particulier de mélange. Nous sommes loin du mélangeur universel dont la polyvalence permettrait de mélanger la plupart des produits. Par exemple un industriel de l’agro-alimentaire peut souhaiter mélanger différentes formulation de céréales ayant des caractéristiques d’écou-lement très différentes dans un même mélangeur.
Cette thèse s’inscrit dans la lignée de divers travaux réalisés au laboratoire, qui visaient à caractériser la qualité de mélange, certainement l’aspect le plus abordé dans la littéra-ture, ou à étudier les mécanismes de mélange dans différents types de mélangeurs. Ici nous avons voulu étudier l’agitation et l’homogénéité produites par un nouveau type de mélangeur encore peu répandu dans l’industrie.
Le Triaxe® est un mélangeur batch dont le mouvement — illustré sur la figure 2 — est basé sur la combinaison de deux axes de révolution pour lesquels les vitesses sont quasiment indépendantes. La combinaison des vitesses permettent au mobile d’agitation de balayer l’intégralité du volume d’une cuve sphérique.
Dans un premier chapitre, nous ferons un point sur les aspects étudiés dans la littéra-ture sur le mélange de poudres. Nous aborderons notamment les différentes échelles qui qualifient l’état de mélange, les points clés de l’échantillonnage, les différents méthodes de mesures en ligne de l’homogénéité de mélange. Nous étudierons les différentes propriétés des poudres et de leur milieu qui influent sur l’écoulement, les mécanismes de mélange et de ségrégation et enfin les différents types de mélangeurs.
INTRODUCTION GÉNÉRALE 3 Distance au centre de la cuve suivant x [ m ] Distance au centre de la cuve suivant y [ m ] D is ta n ce au ce n tr e d e la cu ve su iv an tz [m ] -0,2 -0,1 0 0,1 0,2 -0,2 0 0,2 -0,2 -0,1 0 0,1 0,2
FIG. 2: Un exemple de trajectoire du Triaxe®au bout de 10 secondes de mélange à des
vitesses d’agitation relativement faibles (14% de la vitesse maximale de giration et 32% de la vitesse maximale de rotation)
Le deuxième chapitre nous permettra de présenter les dispositifs mis au point et les méthodes expérimentales développées. Après avoir détaillé les systèmes particulaires utilisés, nous présenterons le mélangeur Triaxe®et l’unité pilote complète — intégrant un dispositif de vidange, de transport et d’analyse de la qualité du mélange — mise en place pour cette étude avec notamment l’élaboration d’un logiciel de pilotage du mélangeur et d’analyse d’image en temps réel permettant de mesurer la qualité du mélange.
Dans le troisième chapitre, nous discuterons des résultats obtenus sur l’agitation de différents milieux à écoulement libre ou cohésifs grâce aux mesures de couples en temps réel qui permettent de connaître la puissance consommée par le mélangeur en fonction des différents paramètres opératoires. Ensuite, nous proposerons une représentation adi-mensionnelle des résultats.
Le quatrième chapitre détaillera enfin l’étude des temps de mélange et de l’homogé-néité de différents mélanges binaires réalisés. Nous essaierons de mettre en évidence les mécanismes mis en jeu dans l’appareil étudié et de relier ces résultats à l’étude sur l’agita-tion afin de déterminer les paramètres opératoires qui permettent un mélange efficace en termes de temps de mélange et de puissance consommée.
C
HAPITRE
1
État de l’art
Dans ce premier chapitre, nous ferons un point sur les aspects étudiés dans la littérature sur le mélange de poudres. Nous aborderons notam-ment les différents mécanismes de mélanges, les propriétés des poudres qui influent sur le mélange, les différents types de mélangeurs et enfin les différentes façons de mesurer l’état de mélange.
1.1 Introduction
D’après MASSOL-CHAUDEUR(2000), le mélange des poudres diffère du mélange des
liquides par trois aspects :
– Il n’y a pas de mouvement relatif des particules solides sans apport d’énergie comme pour les liquides ou les gaz. La vitesse d’homogénéisation des poudres ne dépend que des propriétés d’écoulement des particules, des conditions opératoires et des contraintes mécaniques imposées par le dispositif d’agitation.
– Bien que les molécules d’un système liquide monophasé puissent être différentes et diffuser à des vitesses différentes, elles atteindront toujours un état de mélange par-fait dans un temps plus ou moins long. L’homogénéisation de solides est, en revanche, souvent accompagnée d’un processus de démélange, qui ne permet souvent pas l’obten-tion d’un mélange parfait. L’état final d’un mélange est un équilibre entre un processus d’homogénéisation et un processus de démélange.
– Enfin, la taille d’une particule solide est toujours largement plus élevée que celle de n’importe quelle molécule de liquide ou de gaz. Ceci ajoute un ensemble de phénomènes au niveau mésoscopique que l’on ne sait actuellement pas encore bien décrire.
Une fois les particules mises en mouvement, elles peuvent aussi bien se mélanger que ségréger selon le mouvement imposé au système et les caractéristiques des constituants. Ce dernier point est primordial dans toute manutention de mélange de poudre. Ainsi, pour des solides, l’attention ne doit pas seulement porter sur le dimensionnement du mélangeur mais aussi sur la chaîne de production entière, les étapes de transport, de chargement, de vidange et de stockage comprises, afin de minimiser les risques de ségrégation du mélange.
Ainsi, le phénomène de ségrégation est extrêmement lié aux propriétés d’écoulement des particules : une poudre qui ne s’écoule pas bien ne ségrège pas. Par contre, un mélange
de solides différents qui s’écoulent très bien aura tendance à ségréger. Ces considérations sont essentielles dans le choix et le dimensionnement des mélangeurs.
Si le nombre de particules de chaque composant est identique, l’objectif de l’opération de mélange est l’obtention d’une distribution dans laquelle chaque particule d’un consti-tuant est adjacente d’une particule de l’autre composant. Cette configuration du mélange est appelé mélange parfait. Ceci semble peu réalisable dans un contexte industriel et la qualité du mélange que nous cherchons souvent à atteindre est le mélange aléatoire. Les particules qui constituent le mélange sont distribuées de façon aléatoire, si la probabilité de trouver une particule de l’un des constituants est la même en tout point du mélange, et ceci pour une taille d’échantillon donnée. Pour obtenir cet état de mélange, il faut que les propriétés des poudres soient relativement proches afin d’éviter tout démélange.
Lorsqu’il existe des forces d’attraction interparticulaires pour des particules de nature différentes, on peut obtenir un état de mélange ordonné. Par exemple, dans le cas où les particules seraient de tailles très différentes, grâce aux forces de Van der Waals, les fines particules peuvent venir adhérer sur les plus grandes particules formant un certain ordre lors du mélange.
Si les composants sont complètement séparés on parle de mélange ségrégé. Au cours d’une action de mélange, on tend à s’éloigner de ce type de mélange pour se rappro-cher d’un mélange aléatoire considéré comme étant un état où l’on obtient la meilleure distribution des composants dans le mélange.
La figure 1.1 montre les différents états de mélange de solides divisés théoriques.
(e) (b)
(a) (a)
(a) (c) (d) (f) (g)
FIG. 1.1: Différents états d’un mélange binaire : (a) complètement ségrégé ; (b) aléatoire ;
(c) parfait ; (d) partiellement ségrégé ; (e) cohésif ségrégé ; (f) partiellement ordonné ; (g) parfaitement ordonné d’après HERSEY(1975)
1.2 L’état de mélange : un concept à plusieurs échelles
D’après BERTHIAUX(2002), la qualité d’un mélange se définit par l’usage vers lequel
le produit est destiné. Cette notion d’usage est intimement liée à l’échelle d’observation qui fixe la taille de l’échantillon que l’on doit prélever et qui sert à étudier la qualité du mélange. En effet, la qualité du mélange exigée pour un comprimé pharmaceutique peut être la même que pour un sac d’engrais mais pour des échelles d’observation différentes. Il est donc inutile de réaliser un mélange intime à l’échelle des particules mais plutôt à l’échelle liée aux conditions d’utilisation que l’on appelle « échelle d’observation ».
En première approche, cette échelle peut correspondre à la quantité de matière pré-sente dans le volume du conditionnement élémentaire d’un mélange (comprimé, gélule, sachet, sac). Mais si l’atteinte d’une propriété d’usage est spécifiée pour une fraction de ce conditionnement, comme c’est le cas des comprimés sécables en pharmacie, l’échelle d’observation devra être définie plus finement.
1.2. L’ÉTAT DE MÉLANGE:UN CONCEPT À PLUSIEURS ÉCHELLES 7
La notion d’intensité de ségrégation (cf. figure 1.2) introduite par DANCKWERTS(1952)
définit un aspect de la qualité du mélange : l’homogénéité du mélange à l’échelle de l’échantillon. Plus l’intensité de ségrégation diminue, plus la composition de chaque échantillon est proche de la composition moyenne du mélange.
Diminution de l’intensité de ségrégation
FIG. 1.2: Notion d’intensité de ségrégation d’après SCHOFIELD(1970)
Cette notion est quantifiée par la variance : si l’ensemble du mélange peut être divisé en Z échantillons de composition en un constituant xi, on définit la variance σ2de ces
teneurs par : σ2= 1 Z Z X i =1 ¡xi− µ¢2 (1.1)
Avec : Z nombre total d’échantillons [ − ]
xi composition des échantillons en constituant clé [ − ]
µ teneur moyenne [ − ]
On peut noter que dans l’industrie pharmaceutique on utilise, en lieu et place de la variance, le coefficient de variation (cf. équation (1.2)). Ce dernier étant l’écart type ramené à la moyenne, il permet de comparer la qualité de mélanges ayant des compositions différentes. Pour caractériser un bon mélange il doit donc être le plus petit possible et inférieur au standard industriel de 6%.
CV =σ
µ (1.2)
La variance est une caractéristique macroscopique de l’homogénéité du mélange. Plus cette valeur est grande, plus les valeurs des compositions sont éloignées les unes des autres, et plus le mélange est hétérogène. Une diminution de la variance correspond donc à une diminution de l’intensité de ségrégation du mélange.
Mais deux mélanges de mêmes variances peuvent avoir des structures totalement différentes, celles-ci pouvant avoir une répercussion sur l’usage recherché du produit. Imaginons par exemple que la figure 1.3 représente la teneur en principe actif de différents comprimés pharmaceutiques. Les variances sont identiques dans les deux cas. Pourtant, dans le cas (a), le patient subira un surdosage en principe actif au début du traitement et un sous dosage à la fin. En revanche, dans le cas (b) la répartition du principe actif semble plus acceptable.
moyenne
Temps ou coordonnée spatiale (a)
moyenne
Temps ou coordonnée spatiale (b) Ten eu r en const it u ant clé Ten eu r en const it u ant clé
σ
2a= σ
2bFIG. 1.3: Deux mélanges ayant la même intensité de ségrégation globale peuvent avoir
des structures radicalement différentes BERTHIAUX(2002)
La notion d’homogénéité de mélange est donc intimement liée à la définition et à la manipulation de deux échelles :
1. celle d’observation, à laquelle on désirerait que des propriétés d’usage soient garan-ties, et
2. celle de ségrégation, qui correspond à ce qu’un procédé de mélange peut effective-ment produire.
Diminution de l’échelle de ségrégation
FIG. 1.4: Notion d’échelle de ségrégation d’après SCHOFIELD(1970)
Sur la figure 1.4 est représenté un mélange ayant une intensité de ségrégation — une variance — constante mais dont l’échelle de ségrégation — la taille maximale des régions ségrégées — diminue. Plus cette échelle de ségrégation diminue, plus la taille des régions de ségrégation du mélange diminue.
Il est donc bien nécessaire de définir un autre critère macroscopique, retranscrivant cette fois la structure d’un mélange. Considérons en effet deux échantillons séparés par un intervalle r (qui peut représenter un temps ou bien une distance) et pris parmi une série de Z données consécutives. Selon la structure du mélange et la distance entre les prélè-vements, les compositions de ces échantillons peuvent être plus ou moins dépendantes les unes des autres. DANCKWERTS(1952) et SCHOFIELD(1970) ont proposé de quantifier
cet effet par l’étude de la fonction d’autocorrélation normalisée par la variance (ou de sa représentation graphique, un autocorrélogramme), définie par :
1.3. ÉCHANTILLONNAGE:ESTIMATION DE LA QUALITÉ DU MÉLANGE 9 R(r ) = Z −r X i =1 ¡xi− µ¢ ¡xi +r− µ¢ Z X i =1 ¡xi− µ¢2 (1.3)
Avec : r « intervalle » entre deux échantillons [ − ]
Lorsque cette fonction s’annule, les compositions des échantillons situés au delà de la « distance » r0 correspondante se compensent et peuvent être considérées indépen-dantes : un écart à la valeur moyenne sur un échantillon n’a pas de répercussion pour tout échantillon situé à une distance plus grande que r0. On peut donc estimer que r0 corres-pond à une longueur de mélange caractéristique de la structure, mais aussi qu’il définit une échelle au-delà de laquelle on peut considérer que les propriétés macroscopiques sont atteintes. Cette échelle, que l’on peut définir mathématiquement comme l’aire sous la courbe avant annulation de la fonction d’autocorrélation (cf. figure 1.5), est appelée « échelle de ségrégation ». r 2ε Echelle de ségrégation R(r ) 1 0
ε correspond à une tolérance de l’équation R(r ) = 0 r0la longueur caractéristique
FIG. 1.5: Allure typique d’un autocorrélogramme montrant les différentes notions
asso-ciées BERTHIAUX(2002)
Optimiser une opération de mélange de solides divisés sur la base de l’homogénéité re-viendra donc à faire coïncider ces deux échelles en agissant sur les actionneurs (conditions opératoires) du mélangeur.
1.3 Échantillonnage : estimation de la qualité du mélange
1.3.1 Estimation et statistiques
Bien souvent, on ne peut pas avoir accès à tout le mélange pour mesurer sa qualité. On a donc recourt à un prélèvement d’échantillons dont l’analyse doit permettre d’estimer la qualité du mélange. Estimer, c’est au fond, considérer comme acceptable des valeurs dont
on sait qu’elles sont entâchées d’erreurs. Tout le problème est donc de limiter ces erreurs, voire de les estimer elles-mêmes !
Tout d’abord, il faut utiliser une technique de prélèvement adaptée et s’assurer de la validité statistique de la mesure. Mais ceci comporte deux points clés que sont la taille des prélèvements et, surtout, le nombre d’échantillons prélevés et la localisation des échantillons.
Le contrôle de l’homogénéité est un poste coûteux en temps et en argent. Mais il ne faut surtout pas le négliger et diminuer le nombre de prélèvements et/ou augmenter la taille de chaque prélèvement. En effet, on obtiendrait une estimation de l’homogénéité à une échelle plus grande que celle d’observation et on prendrait le risque de ne pas atteindre les propriétés recherchées à cette échelle.
La seule alternative possible est donc de prélever un nombre suffisant d’échantillons. Dans ce cas, pour z échantillons considérés à la « taille » de l’échelle d’observation, nous pouvons à nouveau définir la teneur moyenne xmet la variance s2
xm=1 z z X i =1 xi (1.4) s2= 1 z − 1 z X i =1 (xi− xm)2 (1.5)
Avec : z nombre d’échantillons prélevés [ − ]
s écart-type observé [ − ]
En ne prélevant que z échantillons parmi Z , on commet une certaine erreur que l’on peut estimer par le biais de la notion d’inférence statistique. Ceci permet de calculer l’erreur faite pour un certain niveau de confiance à atteindre sur la variance, et selon le nombre de prélèvements effectués. Dans le domaine du mélange et de l’échantillonnage, on utilise habituellement la fonction discriminante χ2, ce qui définit un intervalle de confiance non centré sur la variance
Iαχ= " s2 z − 1 χ2inf(α); s 2 z − 1 χ2sup(α) # (1.6)
Avec : α risque lié à l’estimation [ − ]
χ2inf valeur inférieure de la fonction discriminante [ − ] χ2sup valeur supérieure de la fonction discriminante [ − ]
Dans ce cas, il faut avoir recours à des tables comme celle répertoriées par MOTHES
(1969) pour avoir accès aux valeurs des paramètres χ2infet χ2supselon le risque fixé. Il faut noter que, non seulement l’erreur dans l’estimation de la qualité du mélange peut être très importante, mais aussi qu’on ne peut la réduire significativement qu’en augmentant
z d’une manière drastique. Ainsi comme le montre la figure 1.6, le gain de réduction d’un
1.3. ÉCHANTILLONNAGE:ESTIMATION DE LA QUALITÉ DU MÉLANGE 11 Limite haute Limite basse 0 10 100 1000 0,2 1,0 1,4 0,6 1,8 2,2 2,6 3,0 Dév iat ion stan dar d vr aie Dév iat ion stan dar d estimée Nombre d’échantillons
FIG. 1.6: Évolution selon la loi du χ2 et avec le nombre d’échantillons, des valeurs
des bornes d’un intervalle de confiance (normé par la variance vraie) à 95% d’après SCHOFIELD(1976)
On peut noter qu’il existe des standards donnés par la pharmacopée européenne portant sur le coefficient de variation CV basé sur l’échantillonnage, xi et xm.
xm µ µ− 15% µ+ 15% µ− 30% µ+ 30% CV=5,9487% xi Echantillon 2 4 6 8 10 0,35 0,4 0,45 0,5 0,55 0,6 0,65 0,7
FIG. 1.7: Un exemple de mélange en accord avec les standards industriels de la
pharma-copée européenne ; µ = 0,50, xm= 0,5391, CV = 5,9478%
Un exemple de mélange qui satisfait ces standards est représenté figure 1.7 : pour un mélange donné, le CV estimé doit être inférieur à 6% afin de s’assurer que l’ensemble des échantillons ne diverge pas trop de la composition souhaitée. Chaque échantillon doit avoir une composition xi appartenant à l’intervalle [µ − 30%,µ + 30%] afin de vérifier qu’il
n’y ait pas d’échantillon ayant une composition trop éloignée de celle voulue. Enfin la composition moyenne xm doit appartenir à l’intervalle [µ − 15%,µ + 15%], ce qui d’une
certaine manière « valide » l’échantillonnage. Si ce n’est pas le cas, deux solutions sont possible : soit le régime permanent n’est pas atteint dans un mélangeur continu, soit le
plan d’échantillonnage n’est pas convenable, par exemple parce qu’il existe une zone morte non échantillonnée.
1.3.2 Méthodes d’échantillonnage
Pour échantillonner un mélange de solides divisés, il existe typiquement deux possibi-lités :
– De manière statique, en effectuant des prélèvements à l’intérieur de la chambre de mélange, ce qui n’a de sens que dans le cas d’un procédé discontinu.
– De manière dynamique, en prélevant au sein d’un écoulement survenant au moment de la vidange d’un mélangeur discontinu, ou encore en sortie d’un mélangeur (ou d’une ligne) en continu.
Le prélèvement
Quel que soit l’outil de prélèvement, celui-ci se décompose en trois étapes (cf. fi-gure 1.8) :
– l’intégration qui consiste à sélectionner les points de prélèvement,
– la découpe qui est la matérialisation géométrique de ces points (définissant un prélèvement modèle),
– la prise qui est la matérialisation physique de ces points (définissant le prélèvement réel).
1- lot à évaluer 2- intégration 3- découpe 4- prise
FIG. 1.8: Différentes étapes d’un prélèvement dans un mélange
La nature discrète des solides divisés fait que ces trois phases engendrent des erreurs. En effet, lors de la découpe, il existe une incertitude quant aux particules qui se trouvent sur le tranchant de l’outil de prélèvement.
Les différentes lois d’intégration
Il existe des échantillons de lots à zéro, une, deux ou trois dimensions.
Lot à trois dimensions : Il correspond au type d’échantillon que l’on prélève directement
dans un mélangeur. L’échantillonnage n’est pas représentatif de la qualité du mé-lange.
Lot à deux dimensions : Il correspond aux mélanges qui ont une dimension peu variable
comme les mélangeurs à cuve tournante dont on peut prélever une colonne de section constante qui recoupe toute l’épaisseur du mélange. On choisit, on échan-tillonne, un point de coordonnées (x, y) sur une coupe du mélangeur et on prélèvre sur l’ensemble de l’axe des z. L’échantillonnage n’est toujours pas représentatif de la qualité du mélange et dans ces lots comme dans ceux à trois dimensions, certains éléments ont peu, voire aucune probabilité d’être prélevés.
1.3. ÉCHANTILLONNAGE:ESTIMATION DE LA QUALITÉ DU MÉLANGE 13
Lot à une dimension : La section du lot, perpendiculairement à une direction donnée,
est peu variable. C’est ce type de lot que l’on retrouve dans les veines obtenues en sortie de mélangeur ou sur une bande transporteuse. L’épaisseur et la largeur de la veine sont constantes. On choisit un point de coordonnée x le long d’une veine et on prélève l’ensemble de la coupe suivant y et z. Ce type d’échantillonnage est simple à réaliser, il est représentatif si l’on prend bien en compte toute la section de la veine.
Lot à zéro dimension : Par convention, ce sont des lots divisés en un grand nombre
d’uni-tés de masse peu variables. C’est le cas par exemple des comprimés pharmaceu-tiques et ce cas ne pose en général pas de problème de prélèvement d’échantillon-nage
Pour optimiser l’étape d’intégration, il est recommandé de mettre le mélange sous forme de lot à zéro ou une dimension. Cela peut se faire en vidangeant le contenu à échantillonner sur une bande transporteuse par exemple.
D’après MUZZIOet al. (1997), les deux règles d’or de l’échantillonnage des poudres
sont :
– une poudre ne doit être échantillonnée qu’en mouvement,
– un échantillon doit être pris sur l’intégralité d’une ligne du procédé pendant un laps de temps.
Sachant cela, on peut ensuite utiliser une des lois d’intégration suivantes :
– l’intégration au hasard (cf. figure 1.9) : on réalise n prélèvements sélectionnés au hasard le long de la veine ;
3 4 5
2 n
1
FIG. 1.9: Schéma d’une loi d’intégration au hasard d’après MASSOL-CHAUDEUR(2000)
– l’intégration systématique avec implantation au hasard (cf. figure 1.10) : on choisit au hasard un point M comme origine et on réalise n prélèvements espacés d’une distance régulière du point M ;
θ
2 3 4 5
M n
FIG. 1.10: Schéma d’une loi d’intégration systématique avec implantation au hasard
d’après MASSOL-CHAUDEUR(2000)
– l’intégration stratifiés au hasard (cf. figure 1.11) : on segmente la veine en n segments égaux et on réalise un prélèvement au hasard par segment.
θ
1 2 3 4 5 n
FIG. 1.11: Schéma d’une loi d’intégration stratifiée au hasard d’après
Échantillonnage statique
Lors d’un échantillonnage statique, on a souvent recours à des sondes voleuses (cf. fi-gure 1.12) qui sont introduites dans la masse considérée, et dans lesquelles viennent s’infiltrer (souvent par gravité) les particules. Dans cette famille, on distingue :
– des sondes à prélèvement latéral (ou encore « à logettes »)
– des sondes à prélèvement en tête de canne dont il existe plusieurs types.
(a) (b) (c)
FIG. 1.12: Différents types de sondes utilisées pour
l’échantillon-nage des mélanges d’après MUZZIO et al. (1997) : (a) Sonde
Globe Pharma à prélèvement latéral ; (b) Sonde Slug à prélè-vement en tête ; (c) Sonde Rutgers à prélèprélè-vement en tête
FIG. 1.13: Perturbation induite
par une sonde de prélèvement d’après MUZZIOet al. (1997)
La prise d’un échantillon par une sonde provoque un entraînement des particules se situant sur le trajet de celles-ci (cf. figure 1.13).
Les grosses particules peuvent être éliminées du prélèvement au moment de la prise si elles se trouvent à la limite de l’ouvertue de la sonde, et les sondes ayant des cavités latérales ont souvent un problème supplémentaire : les poudres cohésives ne pénètrent pas facilement dans les cavités, ce qui fausse grandement les résultats de l’échantillonnage. D’après MUZZIOet al. (1997) l’erreur d’échantillonnage peut aller jusqu’à 300 %.
L’utilisation des sondes voleuses reste une technique laborieuse décriée notamment par BERMANet al. (1996). Ils montrent qu’une même sonde voleuse utilisée dans différentes
conditions, ou que deux sondes voleuses utilisées dans des conditions semblables, peuvent extraire des échantillons différents à partir de la même population et donc conduire à des résultats d’analyses complètement différents.
Comme on peut le lire dans l’article de STANIFORTH(1982), lors d’un échantillonnage
classique, la variance expérimentale, σ2e, est la somme de la variance vraie résultante du procédé de mélange, σ2m, de la variance introduite par l’erreur d’échantillonnage, σ2s, et de la variance liée à l’analyse de l’échantillon, σ2
a.
1.3. ÉCHANTILLONNAGE:ESTIMATION DE LA QUALITÉ DU MÉLANGE 15
Dans un cas idéal, σ2s et σ2asont négligeables devant σ2m et σ2eest environ égale à σ2m. Malheureusement, une sonde voleuse peut biaiser très fortement la mesure. σ2
sn’est plus
négligeable et on ne peut alors pas estimer correctement la qualité du mélange analysé. De plus, du fait de contraintes industrielles, il est rarement pratique de prendre plus de 10 ou 20 échantillons avec des sondes voleuses dans un équipement industriel, ce qui rend impossible toute caractérisation représentative de l’ensemble des régions du mélangeur. Par exemple, dans le secteur de la pharmacie, la validation industrielle se fait à l’échelle 1/10ème. Ensuite on ne fait que du contrôle de routine sur le mélangeur et du contrôle libératoire sur les comprimés.
Échantillonnage dynamique
Pour éviter l’utilisation de ces sondes, on peut avoir recours à un échantillonnage dynamique. Il existe par exemple des systèmes de prélèvements en sortie de bande trans-porteuse (cf. sous-figure 1.14 (a)) ou par diviseur rotatif (cf. sous-figure 1.14 (b)).
Trémie vibrante
Diviseur Échantillon
(a) prélèvement par bande transporteuse
Souche Échantlillon secondaire Couloir d’alimentation Traitement éventuel Éventuel système de stockage ou ou de transfert de pots Prélever un échantillon représentatif qui correspond à une tranche de toute la section du flux de produit
(b) prélèvement par diviseur rotatif
FIG. 1.14: Schéma de systèmes industriels de prélèvement d’après FORRATECHNIC
(France)
Ces échantillonneurs ne permettent en général pas d’obtenir des échantillons de faibles masses et sont mal adaptés aux lots pulvérulents et cohésifs (MASSOL-CHAUDEUR(2000)).
Pour éviter de perturber le mélange en prenant les échantillons, des méthodes de mesure non intrusives ont été mises au point et feront l’objet de la section suivante qui détaille les méthodes de mesures en ligne de l’homogénéité de mélange. Ces méthodes non intrusives combinent la « prise » des échantillons et leur analyse sans les retirer du reste du mélange.
1.4 Les méthodes de mesures en ligne de l’homogénéité de
mélange
Les méthodes de mesures « en ligne » effectuent des échantillonnages directement sur la chaîne de production.
D’après WEINEKOTTER& REH(1994), les méthodes « en ligne » incluent les méthodes
in-situ et les méthodes in-line — « dans la ligne » en français. Une mesure in-situ définit
une mesure intégrale des propriétés du matériau au niveau d’une coupe d’un réacteur, d’une conduite etc. Les méthodes in-line représentent une mesure locale et rapide où le matériau analysé passe devant une sonde. Ces deux types de méthodes appartiennent aux groupes des méthodes « en ligne » — on-line en anglais — mais évitent l’échantillonnage et la préparation des échantillons nécessaires pour les autres techniques de mesure « en ligne ».
Comme démontré dans la section 1.3 la meilleure façon d’échantillonner est de pré-lever dans l’écoulement et de prendre le maximum d’échantillons possible. Si l’on prend un grand nombre d’échantillons et que l’on utilise une analyse chimique, on perd une grande quantité de produit. D’autre part, si l’on utilise une technique d’analyse intrusive, on perturbe le mélange. Il est donc préférable d’utiliser une méthode non intrusive et non destructive.
Quelques unes de ces différentes méthodes sont décrites dans la suite de cette partie. Elles peuvent permettre, en outre, le calcul en temps réel des grandeurs qui caractérisent l’état de mélange, dans la mesure où la partie sensible de l’analyseur voit un échantillon réellement représentatif de la composition des mélanges étudiés... Il devient alors imagi-nable d’utiliser ces techniques pour contrôler et réguler les propriétés de mélange.
1.4.1 Les méthodes capacitives
Le concept de la mesure capacitive a été utilisé pour la première fois par Abel en 1826, pour un objet de géométrie axi-symétrique. Mais ce n’est que dans le milieu des années 1980 qu’un projet sur la tomographie capacitive électrique pour visualiser des écoulements multi-composants a débuté à l’UMIST — cf. UMIST (2007) — qui n’a de cesse d’améliorer ses méthodes et ses dispositifs d’imagerie depuis le début des années 1990.
Une nouvelle méthode de suivi de mélange par mesure de la capacitance électrique a été mise au point au Laboratoire de Physique des Systèmes Désordonnés de Marseille par DALLOZ-DUBRUJEAUD et al. (2000). Cette méthode ne permet pas de reconstruire
une image du milieu mais a l’avantage de réduire la phase du traitement des données. EHRHARDTet al. (2003) poursuivent le développement de cette méthode à la décharge
d’un mélangeur statique (cf. figure 1.15). Cette méthode, en cours de développement, permet de remonter aux propriétés globales (composition) d’un mélange en écoulement. Le principe consiste à mesurer la capacité électrique du mélange qui passe entre deux électrodes d’un condensateur (cf. figure 1.16).
La mesure ne dépend que de la géométrie du capteur et de la permittivité du milieu entre les deux électrodes et il est possible de connaître la permittivité diélectrique de mélange après étalonnage du capacimètre. L’idée est ensuite d’en déduire la composition du milieu, si toutefois le contraste de permittivité des produits est suffisant.
1.4. LES MÉTHODES DE MESURES EN LIGNE DE L’HOMOGÉNÉITÉ DE MÉLANGE 17 0 0 0 0 0 0 0 1 1 1 1 1 1 1 00 00 00 00 11 11 11 11 0 0 0 0 0 0 0 1 1 1 1 1 1 1 00 00 00 11 11 11 0 0 0 1 1 1 000 000 111 111 00000000000 00000000000 11111111111 11111111111 000000 000000 111111 111111 Capteurs Zéro Gain Mélangeur Capacimètre 1 2 3
FIG. 1.15: Croquis de l’installation expérimentale montrant les sondes (1), le mélangeur
statique (2) et le capacimètre (3) d’après EHRHARDTet al. (2005)
Capacimètre vs Vs= k(Cx+Cp) écran Cp Zéro Gain E2 E1 Cx
FIG. 1.16: Schéma du principe de mesure de la capacité électrique d’après EHRHARDT
et al. (2003)
Il faut noter que cette mesure globale fait une moyenne des caractéristiques diélec-triques du mélange qui s’écoule entre les deux électrodes. Le milieu à échantillonner est considéré comme une structure unidimensionnelle et la proportion de chacun des consti-tuants est identique en tout point d’une section donnée. En d’autres termes, la mesure capacitive considère le milieu comme un milieu homogène de propriétés égales aux pro-priétés moyennes mesurées. De ce fait, l’analyse des résultats se fait en général grâce à la théorie du milieu effectif. Ce milieu possède les même propriétés que le milieu réel, ce qui permet d’utiliser les grandeurs correspondant aux propriétés globales du milieu en s’affranchissant de la complexité réelle du milieu.
En général, on utilise la formule de BRUGGEMAN(1935), symétrisée par Landauer et
n
X
i =1
εm− εi
εi+ 2εm × pi= 0 (1.8)
Avec : pi proportion volumique de produit i [ − ]
εi permittivité diélectrique à l’état dense du matériau i [ − ]
εm permittivité diélectrique du mélange des n constituants deproportion p
i [ − ]
Ces méthodes présentent des résultats très encourageants, moyennant quelques res-trictions. En effet, il est impossible d’analyser les mélanges dont les constituants ont des permittivités proches.
1.4.2 Les méthodes optiques
Récemment, de nombreux auteurs, dont STEINMETZet al. (1996); POUXet al. (1995,
1991); WEINEKOTTER& REH(1994) ont utilisé ces méthodes pour mesurer les
concentra-tions en constituant clé des échantillons analysés.
Le principe est relativement simple : un laser émet de la lumière qui transite par une fibre émettrice et une fibre réceptrice conduit vers un détecteur la lumière réfléchie par les particules (cf. figure 1.17).
FIG. 1.17: Schéma de conception du capteur optique de POUXet al. (1995)
Sur la figure 1.18 on note que les signaux normalisés sont fortement dépendant de la taille des particules. Seuls des particules ayant des distributions de tailles rapprochées peuvent être analysées avec ce type de méthode.
Par ailleurs, POUXet al. (1995) ont montré que la tension de sortie du détecteur peut
1.4. LES MÉTHODES DE MESURES EN LIGNE DE L’HOMOGÉNÉITÉ DE MÉLANGE 19 30 40 50 60 70 80 0.5 Watt dp3;50£µm¤ R M oy en [− ] Type de sonde : 1) coaxial 2.1) parallèle 2.2) croisée 3,00 2,50 2,00 1,50 1,00 0,50 (a) dp dp 0.5 Watt 0,0 0,8 0 1 2 3 4 5 croisée parallèle R M oy en [− ] Al(OH)3dp 56 − 63 µm 40 − 50 µm 40 − 50 µm 35 − 40 µm
{
{
Concentration SiC£ %massique ¤
SiC dp 1,0 0,6 0,2 0,4 (b)
FIG. 1.18: (a) Signaux de réflexion relative en fonction de la taille des particules d’Al(OH)3 mesurés avec 3 types de sondes d’après WEINEKOTTER& REH(1994) ; (b) Signaux de
ré-flection relative mesurés avec des sondes croisées et parallèles en fonction de la concen-tration en SiC pour différentes tailles de particules de Al(OH)3
fortement quand la concentration du mélange dépasse 40 % (cf. figure 1.19).
FIG. 1.19: Courbes de calibration obtenues par POUXet al. (1995)
Ces méthodes ont de bons résultats, mais ne permettent pas une vision globale du mélange et l’étape du traitement du signal semble assez lourd.
1.4.3 Les méthodes proche infrarouge
La spectroscopie proche infrarouge (PIR) est une technique d’analyse qui permet de re-monter à la composition chimique beaucoup plus rapidement que les dosages chimiques classiques. L’analyse proche infrarouge permet des analyses multicomposants simultanées avec une grande rapidité, une faible quantité de produit qui peut être récupéré si néces-saire, un coût d’analyse faible (généralement pas de solvant et cellule d’analyse robuste, possibilité d’utiliser le verre comme fenêtre). La grande souplesse d’adaptation du matériel
pour le contrôle en ligne permet l’utilisation de cette technique dans de nombreux sites avec l’aide des fibres optiques par exemple.
La bande spectrale dite du proche infrarouge s’étend de 1100 à 2500 nm, on y trouve les harmoniques d’élongation et de vibration des liaisons d’un grand nombre de groupe-ments fonctionnels de molécules organiques. Un spectromètre est constitué d’une source lumineuse couvrant cette bande spectrale, d’un système optique de focalisation,d’un mo-nochromateur, et d’un capteur. Il génère donc un faisceau monochromatique qui vient frapper l’échantillon, dont une partie de l’énergie est absorbée. Cette « absorbance » est mesurée soit dans le faisceau réfléchi « réflectance », soit dans le faisceau ayant traversé l’échantillon. Cette absorbance est proportionnelle à la quantité de liaisons qui absorbent, elle suit une loi de Beer-Lambert
Aλ= log I0 λ Iλt = X i ελilci (1.9) Avec : Aλi
absorbance d’une substance i qui absorbe à la
longueur d’onde λ [ − ]
Iλ0 intensité du rayon incident [ − ]
It intensité du rayon réfléchi ou transmis [ − ]
ελi
coefficient spécifique d’absorbance molaire de
l’espèce i £ mol−1· m2
¤
l trajet optique de la cellule [ m ]
c concentration des molécules de l’espèce i qui
absorbent à la longueur d’onde λ £ mol · m−3 ¤
BAKEEV (2003) présente les applications en proche infra rouge pour les industries
pharmaceutiques. Quelques exemples d’utilisation de la spectroscopie en PIR sont donnés : mesure de l’humidité, suivi de réactions et détermination de polymorphes.
BERNTSSONet al. (2002) ont réalisé un suivi de mélange de poudres dans un mélangeur
Nauta®grâce à la spectroscopie PIR (cf. figure 1.20).
Nauta
Fiber optic interfaces Mixer
NIR
FIG. 1.20: Schéma du mélangeur Nauta muni de l’interface PIR, avec la sonde dans la
position la plus basse des deux possibles (d’après BERNTSSONet al. (2002))
1.4. LES MÉTHODES DE MESURES EN LIGNE DE L’HOMOGÉNÉITÉ DE MÉLANGE 21
et d’une poudre brute (taille des particules supérieure à 300 µm). La différence des tailles de particules rend ce mélange susceptible de ségréger et le suivi du mélange est particu-lièrement intéressant. Après un lourd travail de calibration par la méthode des « spectres moyens » décrite par BERNTSSONet al. (2000), un bon suivi du mélange est possible dans
ce procédé ayant une lente évolution. BERNTSSONet al. (2001) ont montré que les
perfor-mances du spectromètre FT-NIR utilisé pour cette étude permettaient l’analyse de poudres se déplaçant à une vitesse modérée — inférieure à 1 m · s−1. Malgré cette évolution peu rapide de la composition du mélange, un taux d’échantillonnage élevé est nécessaire à l’obtention de résultats cohérents. De plus, cette méthode ne donne qu’un aperçu du procédé de mélange : la surface d’analyse est fixe et ne mesure que 0,12 cm2.
La figure 1.21 montre un exemple de résultats obtenus par BERNTSSONet al. (2002). On
peut noter que l’écart-type se stabilise après environ 30 temps caractéristiques pour un mélangeur à l’échelle du laboratoire et après environ 50 temps caractéristiques pour un mélangeur à l’échelle pilote.
(a) (b)
FIG. 1.21: Comparaison des performances de mélanges pour différents batches :
Écart-type en fonction du temps de mélange (d’après BERNTSSONet al. (2002)).(a) Expériences
avec un mélangeur de laboratoire. (b) Comparaison d’expériences similaires à l’échelle pilote et à l’échelle laboratoire
FRAKEet al. (1997) ont développé une application en ligne de spectroscopie PIR afin de
mesurer l’évolution de la taille des particules dans un procédé de granulation en lit fluidisé dans un batch de 40 kg Glatt GPCG 30/50.
L’analyse in-line à été réalisée grâce à un spectrophotomètre NIR Systems 6500 équipé d’une sonde à fibre optique de 2,54 cm de diamètre externe (NIRSystems, Silver Springs MD), d’un paquet de fibre optique de 210 fibres de la source vers le détecteur et de l’échan-tillon vers le détecteur et du logiciel NSAS (NIR Systems, Silver Springs MD) version 3.30. La sonde était positionnée à une distance définie à l’intérieur du lit au sein du flux descendant dans une zone de forte densité de produit (cf. figure 1.22).
9 12 11 3 2 1 6 4 5 7 8 10
FIG. 1.22: Schéma du montage expérimental de FRAKEet al. (1997) : 1- filtres, 2-
venti-lateur, 3- bras de pulvérisation, 4- buse, 5- direction du mouvement du lit, 6- Chambre d’expansion, 7- bol de production, 8- sonde d’échantillonnage, 9- flux d’air chaud et sec,
10- sonde PIR, 11- monochromato PIR, 12- PC
En choisissant une longueur d’onde unique (2282 nm) et en traçant l’absorbance en fonction du temps (cf. sous-figure 1.23 (b)) un profil comparable à celui produit par analyse par tamisage est obtenu (cf. sous-figure 1.23 (a)), il y a augmentation progressive pendant la pulvérisation de solution puis augmentation plus rapide pendant l’addition de l’eau.
(a) (b)
FIG. 1.23: Comparaison de résultats obtenus par tamisage et par proche infrarouge
(d’après FRAKEet al. (1997)).(a) Croissance des granules déterminée par tamisage ;(b)
Profil d’absorbance PIR d’ordre zéro à 2282 nm
KEHLENBECK(2007) a utilisé une méthode de mesure en ligne en plaçant une sonde PIR
en sortie d’un mélangeur continu — cf. figure 1.24 — pour valider et améliorer le modèle de mélange développé par SOMMER(1994) qui calcule la réduction des fluctuations de la concentration des constituants d’un mélange binaire. Les produits mélangés étaient du carbonate de calcium dont le diamètre moyen est de l’ordre de 2 µm et de l’amidon de maïs dont le diamètre moyen est de l’ordre de 15 µm. Pendant les expériences, la vitesse de rotation de l’agitateur, la fréquence de fluctuation de l’alimentation et le taux de remplissage étaient variables. Les résultats obtenus expérimentalement correspondaient très bien avec les résultats calculés à partir du modèle comme le montre la figure 1.25.