Contribution à l'étude de l'interaction contraintes-diffusion dans les polymères
149
0
0
Texte intégral
Figure


+7




Outline
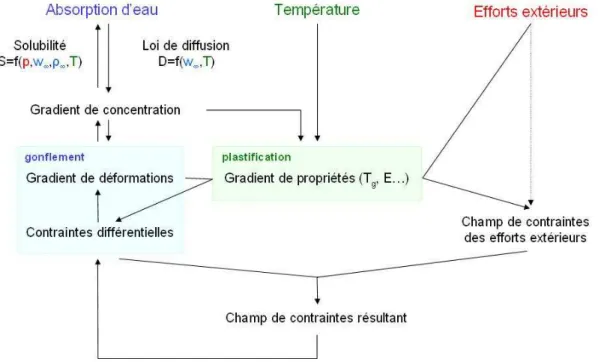
Comparaison entre mod´elisations
Effets de bord
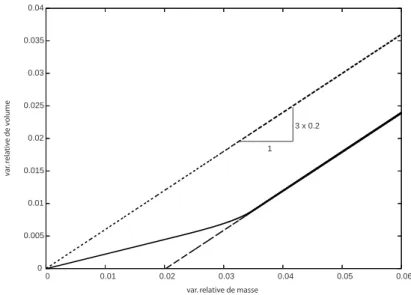
Variation globale de volume et loi locale de gonflement
Pr´eparation des mat´eriaux d’´etude
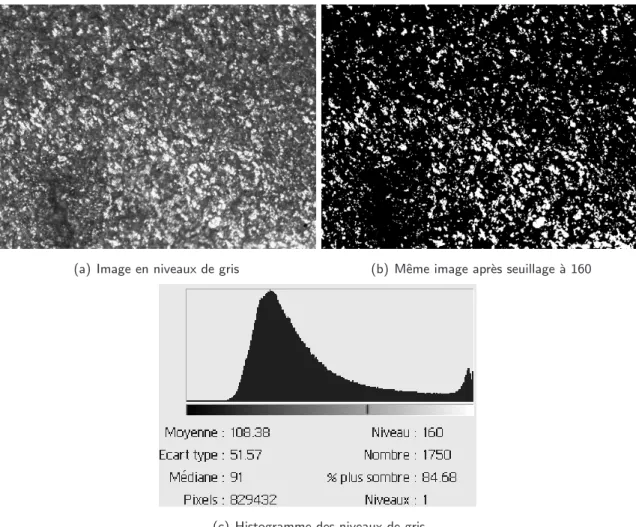
M´ethodes d’analyse des mat´eriaux cuits
M´ethodes d’analyse des mat´eriaux en cours de diffusion
Essais pr´eliminaires
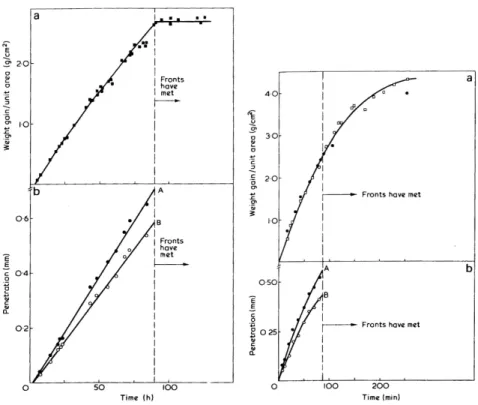
Application `a la diffusion dans une plaque homog`ene
Sur l’influence de l’´epaisseur
Sur la loi de diffusion
Documents relatifs