Identification de variants génétiques rares aux loci 1p13
et 8q22 dans la maladie osseuse de Paget
Les gènes CTHRC1 et TM7SF4 associés à la maladie osseuse de
Paget
Mémoire
Mariejka Beauregard
Maîtrise en physiologie-endocrinologie
Maître ès sciences (M. Sc.)
Québec, Canada
© Mariejka Beauregard, 2013
Résumé
CONTEXTE:La maladie osseuse de Paget (MOP) est transmise sur un mode autosomique dominant dans 30% des cas. Les mutations de SQSTM-1 expliquent 37% des formes familiales, ce qui suggère la présence d’autres loci.
OBJECTIFS:
Identifier des variants rares (VR) de gènes candidats situés aux nouveaux loci 1p13 et 8q22.
Rechercher une association génétique de la MOP avec ces gènes candidats dans la population canadienne-française.
MÉTHODES:
Certains gènes candidats à la MOP aux loci 1p13 et 8q22 ont été sélectionnés, puis séquencés dans un échantillon de découverte. La fréquence de l’allèle mineur d’un VR devait être inférieure à 0,05. 4 VR ont été génotypés chez 240 cas et 297 témoins.
RÉSULTATS:
74 VR ont été identifiés. Un VR (TM7SF4; rs62620995; Leu397Phe) était prédit dommageable par 2 outils d’analyse in silico. rs35500845 (CTHRC1) et rs62620995 (TM7SF4) étaient associés à la MOP de façon statistiquement significative.
Mot clés : Maladie osseuse de Paget, Remodelage osseux, Variants rares, Polymorphismes nucléotidiques simples (SNPs), CTHRC1, TM7SF4 (DC-STAMP), Population canadienne-française, Population à effet fondateur.
Abstract
BACKGROUND:Paget's disease of bone (PDB) is transmitted through autosomal dominant mode of inheritance in 30 percent of cases. Mutations of the SQSTM-1 gene account for 37 percent of familial forms of PDB, suggesting the involvement of other loci.
PURPOSE:
Identify rare variants (RV) of candidate genes located on new loci 1p13 and 8q22.
Search for a genetic association of PDB with these candidate genes in the French-Canadian population. METHODS:
We selected candidate genes on 1p13 and 8q22 loci and sequenced them in a discovery sample. RV was defined by a minor allele frequency less than 0.05. 4 RV were genotyped in 240 PDB patients and 297 healthy individuals.
RESULTS:
74 RV were identified. One RV (TM7SF4; rs62620995; Leu397Phe) was predicted to be damaging by two in silico analysis tools. rs35500845 (CTHRC1) and rs62620995 (TM7SF4) were statistically associated with PDB.
KEY WORDS: Paget’s disease of bone, bone remodelling, rare variants, polymorphisms, SNP, CTHRC1, TM7SF4, DC-STAMP, French Canadian population, founder effect population
Remerciements
Je tiens d’abord à remercier ma directrice de recherche, Dre Laëtitia Michou, qui m’a transmis sa passion pour la recherche. Elle a su me laisser suffisamment d’autonomie, tout en me fournissant l’encadrement requis à ma progression. Elle m’a fourni plusieurs opportunités d’apprentissage uniques et inestimables, tant aux plans clinique que scientifique (et parfois même culturel!), et m’a ainsi montré la réalité d’une chercheure-clinicienne. L’enseignement reçu de sa part dépasse largement les limites de mon projet de recherche.
Je remercie Dr Jacques P Brown et Dr Jean Morissette, fondateurs du laboratoire au sein duquel j’ai réalisé mes travaux, de m’avoir fait bénéficier de leur expertise et de m’avoir prodigué de judicieux conseils.
Je remercie mes évaluateurs qui ont accepté de lire ce mémoire et de contribuer à mes apprentissages en me partageant leurs commentaires.
Je remercie Édith Gagnon de m’avoir montré comment réaliser les différentes manipulations nécessaires à mon projet et de m’avoir grandement aidée dans la prise en charge de mes nombreux «boulets»!
Je remercie Julie Parrot d’avoir répondu, toujours aussi efficacement et agréablement, à mes multiples questions et demandes d’ordre administratif.
Je remercie tous les membres de l’équipe du séquençage pour votre travail efficace et professionnel.
Je remercie Claudia, Davy, Iris et Sabrina sans qui je n’aurais pu apprécier autant mes journées au sous-sol du bloc S. Nous avons bien ri ensemble! Vous me manquez déjà. Je vous souhaite bonne chance dans vos projets actuels et futurs.
Merci papa pour ton aide précieuse à plusieurs étapes de la rédaction du mémoire et ta confiance inébranlable en mes capacités. Merci maman de m’avoir accompagnée en congrès, de m’avoir aidée à prendre des bonnes décisions, et d’avoir enduré nos nombreuses discussions scientifiques père-fille! Merci Ben d’avoir partagé de nombreuses heures de travail avec moi et de m’encourager à me réaliser. Merci à vous trois de m’avoir démontré votre soutien par toutes vos attentions pendant mes périodes de travail et de m’avoir distraite aux moments opportuns!
Finalement, j’aimerais remercier les organismes suivants sans lesquels ce projet n’aurait pu être réalisé : le Fonds de recherche du Québec – Santé, les Instituts de recherche en santé du Canada, la fondation du CHUQ, le Groupe de Recherche en Maladies Osseuses, la Fondation canadienne pour l’innovation, l’université Laval et le Centre de Recherche du CHUQ (CHUL).
Table des matières
Résumé ... iii
Abstract ... v
Remerciement ... vii
Table des matières ... ix
Liste des tableau ... xiii
Liste des figures ... xv
Liste des annexes ... xvii
1. MISE EN CONTEXTE ET REVUE DE LA LITTÉRATURE ... 1
GÉNÉTIQUE DES MALADIES COMPLEXES; Un défi imposant ... 1
1.1. Composition et diversité du génome ... 1
1.1.1. Variations génétiques ... 2 1.1.2. Maladies monogéniques ... 3 1.1.3. Maladies complexes ... 3 1.1.4. Maladies communes – Variants communs ... 3
1.1.4.1. Maladies communes – Variants rares ... 4
1.1.4.2. Influence de la sélection naturelle sur la fréquence des variants génétiques ... 4
1.1.4.2.1. Impact fonctionnel des variants rares ... 5
1.1.4.2.2. Méthodes de détection des variants rares ... 6
1.1.4.2.3. Maladies à début tardif et variants rares... 8
1.1.4.2.4. Population canadienne-française et variants rares ... 9
1.1.4.2.5. MALADIE DE PAGET; Une maladie du remodelage osseux ... 9
1.2. Tissu osseux normal ... 9
1.2.1. Aperçu général de la maladie osseuse de Paget ... 11
1.2.2. Épidémiologie ... 11
1.2.3. Présentation clinique ... 13 1.2.4.
Étiologie ... 17 1.2.6. Composante environnementale ... 17 1.2.6.1. Hypothèse virale ... 17 1.2.6.1.1. Alimentation ... 19 1.2.6.1.2. Substances toxiques ... 20 1.2.6.1.3. Composante génétique ... 20 1.2.6.2.
Régions chromosomiques liées à la MOP ... 20 1.2.6.2.1.
Mutations du gène SQSTM1 ... 21 1.2.6.2.2.
Gènes impliqués dans les maladies apparentées à la MOP ... 23 1.2.6.2.3.
2. HYPOTHÈSES ET OBJECTIFS ... 27 3. INDIVIDUS ET MÉTHODES ... 29 INDIVIDUS ... 29 3.1.
RÉPLICATION DE L’ASSOCIATION GÉNÉTIQUE DES LOCI 1p13 ET 8q22 AVEC LA MOP DANS 3.2.
LA POPULATION CANADIENNE-FRANÇAISE ... 29 SÉLECTION DES GÈNES CANDIDATS ... 29 3.3.
RECHERCHE DE VARIANTS RARES DANS LES GÈNES CANDIDATS SÉLECTIONNÉS ... 30 3.4.
CARACTÉRISATION IN SILICO DES VARIANTS GÉNÉTIQUES ... 31 3.5.
ÉTUDE DE SÉGRÉGATION INTRA-FAMILIALE ... 32 3.6.
ÉTUDE D’ASSOCIATION GÉNÉTIQUE CAS-TÉMOINS ... 32 3.7.
ANALYSES STATISTIQUES ... 33 3.8.
4. RÉSULTATS ... 37 ÉTUDE DE RÉPLICATION DES ASSOCIATIONS GÉNÉTIQUES ENTRE LA MOP ET LES LOCI 4.1.
1p13 ET 8q22 DANS LA POPULATION CANADIENNE-FRANÇAISE ET RECHERCHE
D‘ASSOCIATION GÉNOTYPE - PHÉNOTYPE ... 37 IDENTIFICATION DE VARIANTS GÉNÉTIQUES AU SEIN DES MEILLEURS GÈNES CANDIDATS 4.2.
À LA MOP ... 41 Sélection de 10 gènes candidats ... 41 4.2.1.
Répartition des variants rares identifiés chez des individus atteints par la MOP selon leurs 4.2.3.
localisations chromosomique et génique ... 47 Description détaillée des 74 variants rares identifiés ... 48 4.2.4.
Gène ALX3 (Homeobox protein aristaless-like 3) ... 48 4.2.4.1.
Gène AMPD2 (Adenosine monophosphate deaminase 2) ... 48 4.2.4.2.
Gène CSF1 (Colony stimulating factor 1) ... 49 4.2.4.3.
Gène CTHRC1 (Collagen triple helix repeat containing 1) ... 49 4.2.4.4.
Gène EPS8L3 (Epidermal growth factor receptor kinase substrate 8-like protein 3) ... 50 4.2.4.5.
Gène GSTM3 (Glutathione s-transferase mu 3 (brain)) ... 51 4.2.4.6.
Gène GSTM4 (Glutathione s-transferase mu 4) ... 52 4.2.4.7.
Gène LRP12 (Low density lipoprotein receptor-related protein 12) ... 52 4.2.4.8.
Gène PSMA5 (Proteasome subunit, alpha type 5) ... 53 4.2.4.9.
Gène TM7SF4 (DC-STAMP) (Transmembrane 7 superfamily member 4, (Dendritic cell 4.2.4.10.
specific transmembrane protein)) ... 53 ÉTUDE DE LA SÉGRÉGATION INTRA-FAMILIALE ... 54 4.3.
ÉTUDE D’ASSOCIATION GÉNÉTIQUE ... 56 4.4.
5. DISCUSSION ... 61 ASSOCIATION GÉNÉTIQUE ENTRE LA MOP ET LES LOCI 1P13 ET 8Q22 DANS LA
5.1.
POPULATION CANADIENNE-FRANÇAISE ... 61 IDENTIFICATION DE VARIANTS GÉNÉTIQUES RARES AU SEIN DES GÈNES CANDIDATS À 5.2.
LA MOP SITUÉS AUX LOCI 1p13 ET 8q22. ... 63 ASSOCIATION GÉNÉTIQUE ENTRE LA MOP ET DEUX VARIANTS GÉNÉTIQUES DU LOCUS 5.3.
8q22 ... 63 Association allélique entre la MOP et le gène CTHRC1, gène codant pour une protéine
5.3.1.
impliquée dans la prolifération et la différenciation de cellules précurseurs d’ostéoblastes ... 63 Association génotypique entre la MOP et le gène TM7SF4, codant pour une protéine impliquée 5.3.2.
dans la multinucléation des ostéoclastes ... 65 RETOUR SUR LA MÉTHODE UTILISÉE POUR EXPLORER LE RÔLE DE VARIANTS
Limites de l’étude et améliorations potentielles ... 66 5.4.1. Forces de l’étude ... 70 5.4.2. DÉMARCHES COMPLÉMENTAIRES ... 70 5.5. INTÉRÊTS ET APPORTS DE L’ÉTUDE ... 71
5.6. 6. CONCLUSION ... 73
Bibliographie ... 75
Bases de données ... 89
Outils de prédiction in silico ... 89
Liste des tableaux
Tableau 1. Loci associés à la MOP via deux études pangénomiques d’association ... 24
Tableau 2. Réplication de l’association génétique des loci 1p13 et 8q22 avec la MOP dans la population canadienne-française ... 38
Tableau 3. Sélection des gènes candidats potentiels à la MOP ... 42
Tableau 4. Nouveaux variants rares identifiés ... 44
Tableau 5. Variants rares sélectionnés pour l’étude de la ségrégation intra- familiale ... 55
Tableau 6. Résultats de l’étude d’association génétique par variant rare ou par groupe de variants rares ... 57
Tableau 7. Comparaison des fréquences de l’allèle mineur des quatre variants communs associés avec la MOP chez des individus non mutés appartenant à différentes populations ... 62
Liste des figures
Figure 1. Structure d’un gène ... 1
Figure 2. Variations génétiques ... 2
Figure 3. Distributions possibles des variants rares ... 6
Figure 4. Fonctions des diverses cellules osseuses au sein de l’unité de remodelage osseux ... 10
Figure 5. Système RANK RANKL OPG ... 11
Figure 6. Déformations osseuses dans la maladie osseuse de Paget ... 13
Figure 7. Phases d’évolution du remodelage osseux dans la maladie osseuse de Paget ... 15
Figure 8. Architecture en microscopie optique de l’os sain et de l’os pagétique ... 16
Figure 9. Architecture en microscopie à balayage de l’os sain et de l’os pagétique ... 16
Figure 10. Ostéoclaste pagétique hypermultinucléé ... 17
Figure 11. Domaines de la protéine SQSTM1 ... 21
Figure 12. Étapes décisionnelles pour la sélection des variants génétiques à étudier ... 34
Figure 13. Nombre de variants génétiques identifiés en fonction de la fréquence de l'allèle mineur du variant génétique selon EntrezSNP ... 45
Figure 14. Répartition des variants génétiques rares identifiés selon leur localisation génique ... 47
Figure 15. Conservation de la méthionine (M) en position 35 de la protéine EPS8L3 ... 50
Figure 16. Conservation de l’acide glutamique (E) en position 240 de la protéine EPS8L3 ... 51
Figure 17. Conservation de la valine (V) en position 650 de la protéine LRP12 ... 53
Figure 18. Conservation de l’acide aminé leucine (L) en position 397 de la protéine TM7SF4 ... 54
Liste des annexes
Annexe A : Amplification et séquençage ... 91 Annexe B : Tissu osseux ... 97 Annexe C : Étude des associations génotype-phénotype entre la MOP et les variants communs
associés dans la population canadienne-française ... 99 Annexe D : Gènes situés dans les intervalles génomiques étudiés ... 103 Annexe E : Variants génétiques identifiés au cours de l’étude ... 105
1. MISE EN CONTEXTE ET REVUE DE LA LITTÉRATURE
GÉNÉTIQUE DES MALADIES COMPLEXES; Un défi imposant
1.1.
Composition et diversité du génome
1.1.1.
Le génome contient plus de trois milliards de paires de bases azotées. Les quatre différentes bases azotées soit l’adénine, la cytosine, la guanine et la thymine s’enchaînent dans un ordre qui est précisément défini. La molécule d’ADN qui en résulte mesure environ deux mètres. Elle se replie sur elle-même afin de pouvoir être contenue dans le noyau de chaque cellule. Environ cinq pour cent de cette longue molécule forment les quelque 20 000 gènes du génome, constitués de régions régulatrices, d’exons et d’introns. (Figure 1) Les exons, soit les segments de gènes qui encodent les protéines, représentent environ 1 % du génome. (Marian, 2012) D’autres segments du génome, comptant pour environ 2 % du génome ont été conservés à travers les espèces, ce qui suggère qu’ils sont importants pour la fonction de la molécule d’ADN. (Wright, 2005) La fonction de plus de 95 % du génome demeure donc inconnue à ce jour.
Figure 1. Structure d’un gène
Figure adaptée de Alexa Plateform, http://www.alexaplatform.org Environ 0,1 % du génome diffère entre deux individus. (Marian, 2012) Un changement de nucléotide se produit à environ toutes les 1000 à 1250 paires de bases (pb), ce qui fait que chaque individu possède entre trois et quatre millions de variations dans sa séquence d’ADN par rapport à la séquence standard. (Marian, 2012; Wright, 2005) Il est estimé qu’environ 3,5 millions de ces variants sont des polymorphismes nucléotidiques simples (SNPs). De ce nombre, entre 10 000 et 50 000 entraînent un changement d’acide aminé. Plus de deux tiers de ces variants sont considérés comme délétères, pour la fonction des protéines encodées. (Marian, 2012; Wright, 2005) Cette diversité du génome, responsable de la diversité des phénotypes observés dans l’espèce humaine, a pour objectif de permettre des adaptations en présence de conditions difficiles telles que les infections, la famine ou les climats extrêmes. (Wright, 2005) Toutefois, certaines variations génétiques s’avèrent nocives et peuvent causer ou favoriser le développement de certaines maladies. (Iyengar et Elston, 2007)
Variations génétiques
1.1.2.
Les mutations et les polymorphismes sont deux types de variations génétiques. (Figure 2) Les mutations sont des altérations peu fréquentes de la séquence normale de l’ADN qui ont des répercussions fonctionnelles délétères. Les polymorphismes sont des variations de la séquence d’ADN fréquentes au sein de la population qui ont des effets fonctionnels mineurs ou nuls. Généralement, pour qu’un variant génétique soit considéré comme un polymorphisme, sa fréquence de l’allèle mineur (MAF) dans la population doit être supérieure à 1 %. (Frazer et al., 2009) Dans la littérature, les polymorphismes sont subdivisés en plusieurs catégories en fonction de leur MAF. Généralement, les variants dont la MAF est inférieure à 5 % sont considérés comme ayant une faible MAF. Le seuil empirique de MAF caractérisant les variants rares varie selon les auteurs. Ainsi, Gorlov (2008) le fixe à 5 %, Bodmer (2008) à 3 % et Frazer (2009) à 1 %. Plusieurs autres catégorisations de variants ont été suggérées pour raffiner la description des variants dont les MAFs se situent entre 0,1 % et 5 % (eg. variant rare, variant très rare, variant de faible fréquence, variant non-commun, variant moins commun, variant privé, variant sub-polymorphiques etc.), mais elles sont peu reprises par des auteurs différents de ceux les ayant définies. Comme les MAF des variations génétiques se répartissent selon un continuum, il existe assurément un chevauchement entre les différentes catégories de variants. (Bodmer et Bonilla, 2008) Dans le présent mémoire, les variants génétiques dont la MAF était inférieure à 5% ont été considérés comme des variants rares.
Maladies monogéniques
1.1.3.
Les maladies monogéniques, aussi appelées maladies mendéliennes ou monofactorielles, sont causées par la mutation d’un seul gène. La pénétrance élevée de ces mutations causales fait en sorte que le mode de transmission de ces maladies est généralement facile à élucider en observant le pedigree d’une famille touchée par la maladie. Environ 1 à 2 % de la population est affectée par une maladie monogénique. (Wright, 2005) Ces maladies causées par l’altération d’un seul gène ne représentent donc qu’une faible proportion des maladies pour lesquelles une composante héréditaire est observée. Par exemple, il est estimé qu’environ 5 % des cas de cancers communs sont causés par la dysfonction d’un seul gène, ce qui ne représente qu’une très faible proportion des cancers pour lesquels il existe une composante héréditaire. (Bodmer et Tomlinson, 2010)
Maladies complexes
1.1.4.
Il semble que la plupart des maladies qui manifestent une composante héréditaire résultent plutôt de l’interaction entre plusieurs facteurs génétiques et environnementaux. (Marian, 2012) Ces maladies complexes ou multifactorielles affectent plus de la moitié de la population à un moment de la vie. (Wright, 2005) Puisque les fréquences alléliques et les effets des variants génétiques sont probablement répartis selon un continuum, il est suggéré que des variants génétiques communs et des variants génétiques rares contribuent à la génération des phénotypes complexes. (Marian, 2012)
Maladies communes – Variants communs
1.1.4.1.
L’hypothèse Maladies communes - Variants communs stipule que les maladies complexes ainsi que la diversité des phénotypes observables entre les espèces du règne animal résultent d’un grand nombre de variants communs, qui possèdent chacun un effet modéré sur le développement du phénotype observé. (Marian, 2012) L’association de variants communs avec plusieurs maladies complexes a été recherchée au moyen de grandes études pangénomiques d’association. Malheureusement, les limites de ce type d’étude ont rapidement été constatées. D’abord, les loci détectés par ces études pangénomiques d’association n’expliquent qu’une petite partie de l’héritabilité qui se définit comme la variation phénotypique attribuée aux différences génotypiques. (Gibson, 2012) En effet, la plupart des variants communs associés à des maladies qui ont été identifiés jusqu’à maintenant confèrent un risque relativement faible de développer la maladie, avec des rapports de cote qui varient entre 1,1 et 1,5. (Manolio et al., 2009) Par exemple, une très large étude pangénomique d’association a identifié 18 variants communs associés avec le diabète de type 2 qui, ensemble, n’expliquaient que 6 % de l’héritabilité de cette maladie. (Cirulli et Goldstein, 2010) De même, les variants communs associés aux cancers les plus communs n’expliquent qu’environ 10 % du risque relatif familial de développer le cancer étudié. (Bodmer et Tomlinson, 2010) Les effets mineurs des variants identifiés par ces études limitent leur intérêt clinique. (Marian, 2012) Aussi, les variants communs associés à une
revanche, ils sont souvent en déséquilibre de liaison avec les variants causaux. (Marian, 2012) Malheureusement, les variants causaux responsables de l’association génétique sont rarement identifiés. (Cirulli et Goldstein, 2010) Ensuite, les immenses cohortes nécessaires à la réalisation de telles études font en sorte que le phénotypage rigoureux des individus étudiés est difficile et que des individus d’origines ethniques différentes sont souvent inclus dans les études. (Manolio et al., 2009) De plus, l’étude d’un très grand nombre de marqueurs génétiques nécessite l’utilisation de niveau de signification statistique très contraignant afin de compenser les multiples hypothèses statistiques vérifiées, ce qui réduit la chance de détecter des loci faiblement impliqués dans la pathogenèse d’une maladie. (Bodmer et Bonilla, 2008)
Malgré leurs nombreuses limites, les études pangénomiques d’association ont permis de déceler de nouvelles pistes physiopathologiques impliquées dans certaines maladies. La possibilité de découvrir de nouvelles pistes à explorer constitue un avantage majeur comparativement aux études qui adoptent une approche par «gènes candidats» puisque la sélection des gènes étudiés par ces approches est souvent basée sur des connaissances incomplètes à propos des maladies étudiées. (Manolio et al., 2009)
Maladies communes – Variants rares
1.1.4.2.
Après avoir constaté que les variants communs découverts via des études pangénomiques d’association ne parvenaient pas à expliquer toute l’héritabilité de maladies complexes, il a été suggéré que les variants génétiques rares, les variants structuraux et les interactions gène-gène pouvaient expliquer en partie l’héritabilité non décelée par ce type d’étude. L’hypothèse Maladies communes - Variants rares s’oppose à l’hypothèse Maladies communes - Variants communs et suggère qu’une partie significative de la susceptibilité génétique aux maladies communes est reliée aux effets de multiples variants dont les MAFs se situent entre celles des mutations et celles des variants communs. Ces variants rares seraient indépendants les uns des autres et confèreraient chacun une augmentation significative du risque relatif de développer une maladie. Cette augmentation du risque relatif serait plus élevée que celle attribuable aux variants communs associés à une même maladie. (Bodmer et Bonilla, 2008)
Influence de la sélection naturelle sur la fréquence
1.1.4.2.1.
des variants génétiques
Le modèle Maladies communes – Variants rares est basé sur la prémisse selon laquelle les variants responsables du développement de maladies héréditaires ont un impact négatif sur la fonction de gènes (ou de régions intergéniques) et sont donc partiellement éliminés par la sélection naturelle. De ce fait, ils ne peuvent pas être fréquents au sein de la population. (Gibson, 2012; Gorlov et al., 2011) Les données empiriques obtenues jusqu’à maintenant sur la génétique des populations sont en faveur de cette hypothèse. D’abord, la très grande majorité des variants non synonymes délétères pour la fonction d’un gène ont une
fréquence inférieure à un pour cent. (Wright, 2005) De plus, une relation inverse entre la MAF des variants génétiques et la sévérité du dommage causé à la protéine encodée est observée; les variants non synonymes qui altèrent sévèrement la structure des protéines encodées sont généralement moins fréquents que les variants synonymes ou neutres. (Gorlov et al., 2011) Aussi, le ratio intronique des polymorphismes non synonymes1 augmente radicalement lorsque les polymorphismes étudiés possèdent une MAF inférieure à 5
%. (Gorlov et al., 2008) Finalement, les variants les plus rares ont généralement les effets de taille les plus élevées. Ensemble, ces observations suggèrent que les polymorphismes fonctionnels sont soumis à la sélection. (Gorlov et al., 2011)
Impact fonctionnel des variants rares
1.1.4.2.2.
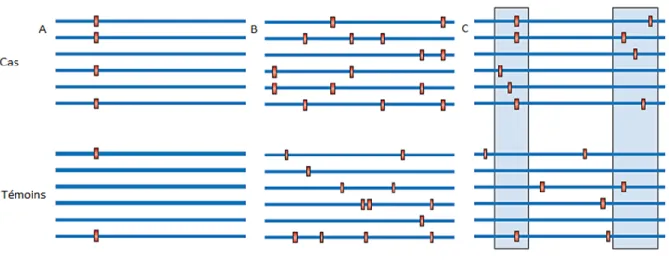
Plusieurs variants génétiques rares interagissent probablement pour entraîner un phénotype. Selon les résultats d’une simulation, 80 variants ayant une MAF entre 0,7 % et 5 % et pour lesquels le risque relatif varie entre 1,6 et 4 seraient nécessaires pour expliquer 40 % de l’héritabilité d’une maladie commune. (Gorlov et al., 2011) Plusieurs modèles de répartition des variants rares ont été créés pour tenter d’élucider leur effet fonctionnel. (Figure 3) Selon le modèle A, un variant causal serait plus fréquent chez les individus atteints que chez les témoins. Selon le modèle B, les variants agiraient en synergie pour contribuer à un phénotype; les individus atteints auraient donc plus de chance de partager des variants fonctionnels que les individus témoins, chez qui les variants sont distribués de façon aléatoire. Selon le modèle C, les individus atteints seraient porteurs d’un plus grand nombre de variants rares que les individus témoins dans certaines régions génomiques précises. (Bansal et al., 2010) Les données acquises jusqu’à maintenant sur les variants rares ne permettent pas de favoriser l’un de ces modèles.
Figure 3. Distributions possibles des variants rares
Figure adaptée de Bansal (2010)
Méthodes de détection des variants rares
1.1.4.2.3.
Il est estimé que chaque gène compte plus de deux polymorphismes délétères dont la MAF est inférieure à 5 %. (Gorlov et al., 2008) Malheureusement, les études pangénomiques d’association ne sont d’aucune aide pour l’étude de tels variants rares puisqu’elles ne permettent pas la détection de nouveaux variants et qu’elles étudient des variants dont la MAF est supérieure à 5% afin d’obtenir une puissance suffisante à la détection d’association. (Manolio et al., 2009) De plus, la pénétrance des variants rares est nettement inférieure à 50 %, ce qui n’est pas suffisant pour causer une concentration de cas dans une même famille. De ce fait, leur détection via des études de liaisons génétiques, utiles pour la détection de régions chromosomiques liées aux maladies mendéliennes, est aussi impossible. (Bodmer et Bonilla, 2008; Bodmer et Tomlinson, 2010)
Une des méthodes fréquemment utilisées aujourd’hui pour identifier des variants rares consiste à séquencer des régions génomiques dans lesquelles des variants communs ont été associés à une maladie au moyen d’études pangénomiques d’association. (Manolio et al., 2009) En effet, il est possible que des variants communs pour lesquels une association a été identifiée soient en déséquilibre de liaison2 avec un ou plusieurs
variants rares responsables de l’association. L’association observée entre une maladie et un variant commun auquel un faible risque relatif est attribué pourrait en fait résulter de quelques variants rares expliquant une partie importante de la susceptibilité à la maladie chez un faible nombre d’individus. (Cirulli et Goldstein, 2010; Gibson, 2012) Autrement dit, le risque attribuable aux variants rares causaux pourrait être très supérieur à celui attribué initialement au variant commun identifié grâce à une étude pangénomique d’association. La dilution de l’effet du variant rare parmi plusieurs individus non porteurs du variant rare qui, par ailleurs,
2 Déséquilibre de liaison : Ségrégation non aléatoire de marqueurs génétiques au sein d’une population. Le déséquilibre de liaison
partagent un même variant commun, pourrait expliquer que le risque attribuable au variant commun associé à la maladie soit inférieur au risque attribuable au variant rare causal. (Dickson et al., 2010)
Il a été suggéré qu’un variant rare responsable d’une association entre une maladie et un variant commun pourrait être situé à des millions de paires de bases du variant commun pour lequel une association a été identifiée en premier lieu. (Dickson et al., 2010) Une autre hypothèse stipule que de multiples variants rares pourraient se retrouver sur le même haplotype et être responsable de l’association significative entre le variant commun et la maladie. La région autour d’un variant commun associé à une maladie qui contient potentiellement un variant rare causal peut donc être assez étendue. Afin de réduire l’ampleur de la tâche de séquençage, il a été suggéré de débuter la recherche de variants rares causaux en séquençant les meilleurs gènes candidats situés dans une région associée à une maladie. Les gènes qui jouent un rôle dans des processus physiopathologiques impliqués dans le développement de la maladie étudiée sont certainement de bons gènes candidats pour la recherche des variants rares causaux. (Bodmer et Bonilla, 2008)
Les variants identifiés lors d’études de reséquençage sont souvent caractérisés puis priorisés selon leur fréquence, leurs effets fonctionnels prédits, la présence d’un autre variant associé à la maladie étudiée à proximité ou la conservation au cours de l’évolution du nucléotide ou de l’acide aminé modifié. (Bodmer et Tomlinson, 2010) Les principaux effets fonctionnels observés sont l’altération de la régulation de l’expression génique via la transformation de sites de liaisons à un facteur de transcription, de microARNs ou de sites d’épissage et la transformation de la structure des protéines encodées. (Bansal et al., 2010; Marian, 2012) Des outils in silico permettent aussi de comparer les séquences de plusieurs organismes afin d’observer quelles régions sont conservées pendant l’évolution. Puisque ces régions ont été conservées grâce à la sélection négative, les variants qui altèrent des nucléotides ou les acides aminés correspondants sont probablement des effets délétères sur la fonction des gènes dans lesquels ils se trouvent. (Cooper et Shendure, 2011)
Les variants génétiques les plus intéressants parmi ceux identifiés peuvent ensuite être génotypés dans un plus grand échantillon afin d’étudier la présence d’une association génétique. Un variant rare dont la MAF diffère de façon statistiquement significative entre les individus atteints par la maladie et les témoins pourrait avoir un effet sur le développement de la maladie étudiée. Par contre, puisque la taille de l’échantillon requise pour démontrer une association entre une maladie et un variant augmente proportionnellement en fonction de 1/MAF et qu’elle augmente abruptement à mesure que le rapport de cote diminue, la taille de l’échantillon nécessaire pour démontrer une association est beaucoup plus élevée que celle requise pour identifier les variants rares. Afin de contrer cette difficulté, il est suggéré de regrouper certains variants pour les analyser
de l’objectif principal qui est d’identifier le variant causal. (Manolio et al., 2009) Pour l’instant, il n’existe pas de consensus sur les méthodes optimales de regroupement de variants. Toutefois, il semble logique de regrouper des variants ayant des MAFs semblables, faisant varier le risque de développer la maladie dans le même sens et appartenant aux mêmes gènes ou aux mêmes voies de signalisation.
Lorsqu’un variant rare est fortement soupçonné d’être impliqué dans le développement d’une maladie après ces différentes étapes, des expérimentations in vitro ou in vivo visant à observer les effets fonctionnels des variants rares identifiés peuvent être effectuées. Des résultats positifs sont de très forts arguments en faveur de la causalité des variants étudiés dans le phénotype étudié. Toutefois, ces expérimentations sont coûteuses et surtout très difficiles à réaliser dans un contexte approprié. En effet, selon le type de cellules utilisées, ainsi que l’organisme sur lequel sont effectuées les expérimentations, les résultats obtenus peuvent s’avérer négatifs malgré l’effet fonctionnel réel du variant étudié. L’action de certains variants peut dépendre de l’état de la chromatine, de l’interaction avec d’autres polymorphismes, de l’interaction avec l’environnement, ou de l’activation par un certain promoteur, ce qui fait qu’une expérimentation qui ne respecte pas certaines conditions précises peut donner des résultats faussement négatifs. De plus, puisque les méthodes d’expérimentations ont chacun leur seuil de sensibilité; un faible impact sur le processus testé peut ne pas être détecté. (Cooper et Shendure, 2011)
Beaucoup de travail est nécessaire dans ce nouveau domaine afin d’élaborer des stratégies efficaces pour déterminer les variants qui sont impliqués dans une maladie. La confirmation de l’implication de variants rares dans certaines maladies permettra d’élucider en partie leurs effets fonctionnels.
Maladies à début tardif et variants rares
1.1.4.2.4.
Les maladies qui n’altèrent pas le succès reproducteur, comme la maladie osseuse de Paget (MOP), pourraient être causées par des variants rares ou des variants communs puisque les variants causaux de ces maladies ne devraient pas être confrontés à la sélection naturelle. Or, plusieurs autres maladies à début tardif sont associées à des variants rares. Par exemple, moins de 10 variants communs figurent parmi plus de 1000 variations génétiques des gènes BRCA1 et BRCA2 associées au cancer du sein. (Wright, 2005) Le fait qu’un même variant exerce plusieurs fonctions différentes (effets pléïotropiques) pourrait expliquer que certaines maladies à début tardif soient tout de même associées majoritairement à des variants rares. En effet, les variants associés à des maladies à début tardif peuvent influencer d’autres traits qui diminuent le succès reproducteur. (Manolio et al., 2009) Par exemple, les gènes impliqués dans certains cancers à début tardif sont aussi impliqués dans le contrôle du cycle cellulaire, l’apoptose et l’angiogenèse, qui sont des processus soumis à la sélection négative. (Gorlov et al., 2008)
Population canadienne-française et variants rares
1.1.4.2.5.
Il est suggéré que la plupart des variants rares sont population spécifiques. Les populations à effets fondateurs, comme la population canadienne-française, dans lesquelles la dérive génétique3 a été favorisée
sont donc potentiellement enrichies en variants rares. (Bodmer et Bonilla, 2008; Laberge, 2007; Manolio et al., 2009) Lorsque la MAF d’un variant est plus élevée dans une population à effet fondateur, il est plus facile de détecter une association entre une maladie et ce variant. (Bodmer et Bonilla, 2008) De plus, il est probable que des individus provenant d’une même population à effet fondateur partagent certains variants causaux. Il est aussi probable que ces individus aient été exposés à des facteurs extérieurs semblables, ce qui facilite la détection d’association entre une maladie et des variants qui nécessitent certaines expositions environnementales particulières pour exprimer leur effet. (Marian, 2012) L’utilisation d’une cohorte provenant d’une population à effet fondateur fait donc partie des stratégies pour tenter de surpasser les difficultés énoncées précédemment quant à l’étude des variants rares. (Bodmer et Bonilla, 2008; Manolio et al., 2009) La population canadienne-française est une population de choix pour effectuer la recherche de variants génétiques rares. D’abord, il s’agit d’une population à effet fondateur. En effet, près de 80% de la population québécoise actuelle provient de quelque 8500 colons français venus s’établir en Nouvelle-France. Pour des raisons sociales et linguistiques, cette population est demeurée isolée pendant près de 300 ans. (Laberge, 2007) Aussi, jusqu’à récemment, les familles comportaient de nombreux enfants ce qui constitue un atout pour la recherche en génétique. Finalement, la conservation de registres religieux (naissance, mariage, décès) jusqu’aux premiers arrivants facilite grandement la reconstruction des pedigrees. (Gagnon, 2011)
MALADIE DE PAGET; Une maladie du remodelage osseux
1.2.
Tissu osseux normal
1.2.1.
Le tissu osseux est un tissu dynamique qui s’adapte constamment à son environnement et aux contraintes mécaniques. Les os assurent plusieurs fonctions essentielles comme le soutien des organes et la protection du système nerveux central. Ils forment une réserve de certains minéraux et ils hébergent l’hématopoïèse. Avec les muscles, ils permettent le déplacement et le mouvement. (Clarke, 2008)
Deux types d’os peuvent être distingués d’un point de vue structural : l’os cortical, aussi appelé l’os compact et l’os trabéculaire, aussi appelé l’os spongieux. L’os cortical forme la couche externe des os. Sa grande densité confère à l’os sa force et sa rigidité. L’os trabéculaire est constitué de travées positionnées stratégiquement
pour résister aux contraintes mécaniques. Il assure un support structural sans alourdir inutilement l’os. On le retrouve aux extrémités des os longs (épiphyses) et dans certains os plats. (Clarke, 2008)
Il existe trois types de cellules osseuses; les ostéoblastes, les ostéoclastes et les ostéocytes. (Figure 4) Toutes trois sont impliquées dans la régulation du remodelage osseux qui est nécessaire pour extraire les minéraux de l’os ou de les y emmagasiner selon les besoins, tout en conservant un tissu osseux résistant adapté aux contraintes mécaniques. (Raisz, 1999) Les ostéoblastes sont des cellules d’origine mésenchymateuse qui forment l’os. Ils produisent notamment le collagène de type 1, la principale protéine formant la matrice osseuse. Ils disposent ces fibres de façon lamellaire, chaque lamelle étant orientée de façon perpendiculaire, ce qui confère à l’os sa solidité. (Marks et Popoff, 1988) Les ostéoclastes sont des cellules multinucléées, issues de la fusion de cellules circulantes mononuclées de la lignée monocyte/macrophage, qui résorbent l’os via la sécrétion d’ions hydrogènes et de différentes enzymes. (Raisz, 1999) Les ostéocytes sont des ostéoblastes emmurés dans la matrice osseuse lors de sa synthèse. Ces cellules ont été beaucoup moins étudiées que les ostéoblastes et les ostéoclastes malgré qu’elles représentent environ 90% des cellules osseuses. Elles seraient impliquées dans la régulation du remodelage osseux. (Bonewald, 2011)
Figure 4. Fonctions des diverses cellules osseuses au sein de l’unité de remodelage osseux
Figure adaptée de Longo (2011) La différentiation des ostéoblastes à partir de cellules souches mésenchymateuses est principalement régulée par la voie de signalisation Wnt, alors que le recrutement, la différenciation et l’activation des ostéoclastes sont régulés par la voie de signalisation du NF-κB via l’interaction entre le récepteur RANK porté par les ostéoclastes et le ligand RANK (RANKL) exprimé par les ostéoblastes et les ostéocytes. Le récepteur leurre ostéoprotégérine (OPG), aussi sécrété par les ostéoblastes, inhibe l’interaction entre RANK et RANKL nécessaire à l’activation de la voie de signalisation du NF-κB. (Figure 5) (Caetano-Lopes et al., 2009; Udagawa et al., 2000)
Figure 5. Système RANK RANKL OPG
Figure adaptée de Kumar (2009)
Aperçu général de la maladie osseuse de Paget
1.2.2.
La maladie osseuse de Paget (MOP) est la deuxième maladie osseuse métabolique la plus fréquente après l’ostéoporose. (Roodman et Windle, 2005) Elle se caractérise par une augmentation focale du remodelage osseux qui résulte en la formation de tissu osseux dont l’architecture est anormale. Étrangement, l’atteinte osseuse demeure localisée aux os atteints et ne se propage pas aux os adjacents. (Roodman et Windle, 2005) La MOP se transmet selon un mode autosomique dominant avec un pénétrance incomplète dans environ un tiers des cas. (Morales-Piga et al., 1995; Siris et al., 1991) Actuellement, un seul gène causal de la maladie a été identifié; le gène Sequestosome-1 (SQSTM1). (Chung, 2011) Dans la population canadienne-française, la mutation de SQSTM1 la plus fréquente, soit la mutation P392L, a été identifiée chez 46 % des individus souffrant d’une forme familiale et 16 % des individus souffrant d’une forme sporadique de MOP, ce qui suggère l’implication d’autres gènes dans la maladie. (Laurin, 2002)
Épidémiologie
1.2.3.
MOP est fréquente en Europe de l’Ouest, en Amérique du Nord, en Australie et en Nouvelle-Zélande. (Cooper et al., 1999) Elle est plus rare en Scandinavie, en Irlande, au sud de l’Europe, en Afrique et en Asie. (Cooper et al., 2006; Siris et al., 1991) La prévalence globale la plus élevée a été observée en Grande-Bretagne. (Cooper et al., 1999)
L’incidence et la prévalence de la maladie augmentent avec l’âge. (Laurin et al., 2001) Dans une étude réalisée au Royaume-Uni, Van Staa et coll. (2002) ont observé que l’incidence de la MOP passait de 0,5 cas par 10 000 années-personnes pour les hommes âgés de 55 à 59 ans à 7,6 cas par 10 000 années-personnes chez les hommes de plus de 85 ans. En Grande-Bretagne, Cooper et coll. (1999) ont observé que la prévalence de la MOP s’étalait de 0,3%, pour les hommes âgés de 55 à 59 ans, à près de 7% pour les hommes de plus de 85 ans. Aux États-Unis, les prévalences sont semblables; la MOP touche de 1 à 3% des individus de plus de 40 ans et de 8 à 10% des individus de plus de 80 ans. (Siris et al., 1991)
Étonnamment, l’incidence, la prévalence et la sévérité de la MOP diminuent depuis plusieurs années. Une diminution de la mortalité attribuable directement à la MOP ou à une tumeur osseuse primaire chez des adultes de plus de 40 ans a été observée entre les années 1950 et 1970, suggérant une diminution de la fréquence de la maladie ou de la sévérité de l’atteinte osseuse. (Barker, 1984) Une diminution de 60% de la prévalence de la MOP a été observée en Grande-Bretagne entre 1974 et 1994, alors qu’en Nouvelle-Zélande, la prévalence de la maladie a diminué de moitié entre 1983 et 2002. (Cooper et al., 1999; Cundy, 2006) Aussi, en Angleterre, l’incidence a chuté de près de la moitié entre 1990 et 1997. (Van Staa et al., 2002) Bolland et coll. (2007) ont observé que les porteurs d’une mutation du gène SQSTM1 développaient la MOP plus tardivement que le parent porteur de la mutation et que leur maladie était moins sévère que celle du parent porteur. Aussi, des corrélations négatives ont été observées entre l’année de naissance et le taux sérique des phosphatases alcalines, le degré moyen d’extension de la maladie mesuré par scintigraphie osseuse et le nombre d’os atteints, suggérant que les individus plus jeunes souffrent d’une forme moins sévère de la maladie depuis quelques années. (Cundy, 2006)
Étonnamment, ni la prévalence, ni la sévérité de la maladie ne semblent diminuer en Italie. Au contraire, la prévalence de la MOP observée à Turin s’est élevée entre 1986 et 2002. À Sienne, le degré de sévérité de la maladie, déterminé par le nombre d’os atteints ou le degré d’extension de la maladie, est demeuré constant depuis les dernières années. Aussi, une forme sévère de la maladie caractérisée par un début précoce, une atteinte osseuse extensive et un risque plus élevé de dégénérescence tumorale des os atteints en ostéosarcome ou en tumeur à cellule géante est décrite chez les individus pagétiques originaires de la région de la Campanie. (Gennari et al., 2006; Rendina et al., 2006)
Présentation clinique
1.2.4.
La MOP est souvent diagnostiquée fortuitement lorsque des anomalies radiographiques ou scintigraphiques sont apparentes lors de l’investigation d’une autre condition ou lorsque des valeurs élevées de la phosphatase alcaline sérique sont obtenues lors d’un bilan biochimique de base. (Michou et Brown, 2011a) La MOP peut aussi être diagnostiquée lors d’un dépistage effectué chez un individu ayant des antécédents familiaux de MOP. (Falchetti et al., 2010)
Entre 10 et 30 % des personnes atteintes par la MOP sont symptomatiques. (Chung et Van Hul, 2012) Les manifestations les plus communes de la maladie sont la douleur osseuse et les déformations osseuses qui apparaissent tardivement au cours du processus pathologique. (Figure 6) (Lyles et al., 2001)
Figure 6. Déformations osseuses dans la maladie osseuse de Paget
Figures tirées de la Fondation Paget (www.paget.org) et de la collection personnelle du Dr Jacques P. Brown Les parties du squelette qui sont les plus souvent atteintes sont, par ordre de fréquence, le bassin, le rachis, le crâne, le fémur, le tibia, l’humérus et la clavicule. Rarement, d’autres os, comme ceux formant les extrémités, peuvent être atteints. (Michou et al., 2006) Les complications de la MOP varient en fonction de l’os touché par la maladie. Les déformations osseuses peuvent causer des difficultés à marcher ou favoriser l’apparition précoce d’arthrose. (Michou et al., 2006; Singer, 2009; Theodorou et al., 2011) Malheureusement, les chirurgies impliquant un os pagétique sont particulièrement difficiles dû à l’hypervascularisation de l’os pagétique et aux déformations présentes; les résultats sont souvent décevants en raison de l’accélération de l’ostéolyse suite à l’intervention ou de la survenue d’un descellement de prothèse. (Lyles et al., 2001; Singer, 2009) Certains os, comme le fémur, le tibia et l’humérus sont prédisposés aux fractures pathologiques
2009; Theodorou et al., 2011) Aussi, l’atteinte de certains os du visage peut entraîner la déformation du visage, la perte de dent ou la malocclusion des mâchoires. (Singer, 2009)
Les os pagétiques sont plus à risque de développer certaines tumeurs osseuses comme les ostéosarcomes et les tumeurs à cellules géantes. (Singer, 2009; Theodorou et al., 2011) La dégénérescence de l’os pagétique en ostéosarcome est une complication grave de la MOP qui survient chez moins de 1 % des patients ayant une atteinte mono ou oligo-ostotique et chez 5 à 10 % des patients ayant une atteinte polyostotique. (Theodorou et al., 2011)
Certaines complications extra-osseuses sont possibles dans la MOP. Des troubles neurologiques, comme des céphalées, des problèmes auditifs, des paralysies de nerfs crâniens, des compressions de racines nerveuses ou une sténose spinale peuvent survenir lorsque le crâne ou une vertèbre sont affectés par la MOP. (Michou et Brown, 2011b; Singer, 2009) Aussi, les individus atteints par la MOP peuvent manifester des complications cardiovasculaires secondaires aux calcifications artérielles ou coronariennes importantes chez les individus pagétiques, ou à l’augmentation du débit cardiaque en raison de l’importante vascularisation dont bénéficient les os pagétiques. (Laroche et Delmotte, 2005; Singer, 2009; Strickberger et al., 1987)
Caractéristiques de l’os et des cellules osseuses pagétiques
1.2.5.
La MOP est caractérisée par une augmentation focale et couplée du remodelage osseux; la résorption osseuse est accélérée et la formation osseuse est à la fois accélérée et désorganisée. (Ralston, 2008) Habituellement, les lésions ostéolytiques (lésions en V aux os longs, ostéoporose circonscrite au crâne) précèdent les lésions sclérotiques, l’élargissement de l’os et l’épaississement du cortex. (Figure 7) (Michou et Brown, 2011a) La désorganisation de la formation osseuse est responsable de la présence d’os tissé, moins solide que l’os lamellaire qui compose habituellement le tissu osseux des adultes non atteints par la MOP. (Figure 8 et 9) (Chung et Van Hul, 2012) La moelle osseuse d’un os pagétique est parfois fibrosée et envahie par la vascularisation. Comme mentionné plus tôt, la maladie ne se propage pas aux os adjacents. L’atteinte d’autres os après que le diagnostic ait été posé demeure très rare. (Roodman et Windle, 2005)
Figure 7. Phases d’évolution du remodelage osseux dans la maladie osseuse de Paget
Figure 8. Architecture en microscopie optique de l’os sain et de l’os pagétique
Figures tirées de Roodman (2005) Figure 9. Architecture en microscopie à balayage de l’os sain et de l’os pagétique
Figures tirées de la Fondation Paget (www.paget.org) Les ostéoclastes d’individus pagétiques sont plus nombreux, plus volumineux et possèdent plus de noyaux que des ostéoclastes normaux. En effet, ils peuvent contenir jusqu’à 100 noyaux, alors qu’un ostéoclaste normal en contient entre trois et 20. (Figure 10) (Roodman et Windle, 2005) Leur survie serait augmentée dû à un dérèglement du mécanisme de l’apoptose. (Michou et Brown, 2011a) Ces ostéoclastes contiennent parfois des inclusions nucléaires de nature indéterminée qui pourraient correspondre à des nucléocapsides de paramyxovirus ou à des agrégats de protéines non dégradées. (Mills et Singer, 1976; Ralston, 2008; Rebel et al., 1976)
a) Os sain b) Os pagétique
Figure 10. Ostéoclaste pagétique hypermultinucléé
Figures tirées de Roodman (2005) Les précurseurs d’ostéoclastes pagétiques forment des ostéoclastes en présence de concentrations de 1,25-(OH)2D3 et de RANKL très inférieures à celles nécessaires à la formation d’ostéoclastes à partir de
précurseurs normaux. (Neale et al., 2000) Des niveaux plus élevés de TAF12 (anciennement TAFII-17), un coactivateur du récepteur de la vitamine D (VDR) qui augmente la sensibilité du VDR à la 1,25-(OH)2D3,
expliquent en partie l’hypersensibilité à la 1,25-(OH)2D3 des précurseurs d’ostéoclastes pagétiques qui
expriment la protéine de la nucléocapside du virus de la rougeole selon Kurihara. (Kurihara et al., 2004) Peu d’études ont porté sur le rôle des ostéoblastes dans le développement de la MOP jusqu’à maintenant. Ces ostéoblastes sont morphologiquement normaux, mais plus nombreux et plus actifs que les ostéoblastes sains. (Roodman et Windle, 2005)
Étiologie
1.2.6.
Composante environnementale 1.2.6.1. Hypothèse virale 1.2.6.1.1.Il a été suggéré qu’une infection virale chronique pouvait contribuer à la pathogenèse de la MOP. (Mills et Singer, 1976) Des inclusions nucléaires et cytoplasmiques semblables aux nucléocapsides des paramyxovirus ont d’abord été observées dans les ostéoclastes de patients pagétiques grâce à la microscopie électronique. (Rebel et al., 1976) Puis, des transcrits, des protéines et des antigènes appartenant à cette classe de virus ont été identifiés dans des ostéoclastes de patients pagétiques à l’aide de techniques comme
l’immunohistochimie, l’hybridation in situ et la réaction en chaîne par polymérase (PCR) in situ. (Roodman, 2010)
Récemment, une équipe a observé que le gène encodant la nucléocapside du virus de la rougeole (MVNP) était exprimé dans la moelle osseuse de huit patients pagétiques sur dix et que tous les précurseurs d’ostéoclastes des patients exprimant le MVNP formaient des ostéoclastes ayant un phénotype pagétique complet. (Kurihara et al., 2011) Étonnamment, les ostéoclastes formés in vivo à partir de précurseurs provenant de patients ayant un phénotype clinique pagétique confirmé, mais dont la moelle n’était pas infectée par le MVNP n’arboraient pas toutes les caractéristiques des ostéoclastes pagétiques (phénotype cellulaire pagétique incomplet). Ces ostéoclastes ne démontraient pas d’hypersensibilité à la 1,25-(OH)2D3, ils n’étaient
pas hypermultinucléés et ils ne produisaient pas de plus grandes quantités d’IL-6 que les ostéoclastes sains. Toutefois, leur activité basale était augmentée dû à une hypersensibilité au RANKL. (Kurihara et al., 2011) Une souris transgénique exprimant l’équivalent de la mutation humaine P392L de la protéine p62 (p62P394LKI)
et exprimant le gène MVNP dans ses cellules de la lignée ostéoclastique a développé des ostéoclastes ayant un phénotype cellulaire pagétique complet. L’hypersensibilité de ces ostéoclastes au RANKL serait attribuable à la mutation, alors que la multinucléation, l’hypersensibilité à la 1,25-(OH)2D3, la surexpression de TAF12 et
la production accrue d’IL-6 seraient attribuables au MVNP. (Kurihara et al., 2011)
Il a été montré que le virus en cause dans la maladie de Carré, le «canine distemper virus», induisait transitoirement l’ostéoclastogénèse dans les cellules précurseurs en culture, grâce à l’activation de la voie de signalisation du NF-κB. Bien qu’un effet transitoire ne puisse expliquer le développement retardé de la MOP, il a été suggéré que ce virus puisse avoir des effets à long terme sur d’autres protéines surexprimées dans la MOP et qu’il pourrait, de cette façon, lui aussi contribuer à la pathogenèse de la maladie. (Michou et Brown, 2011a) Dans cette optique, il est intéressant de noter qu’en Grande Bretagne, l’incidence de l’infection par la maladie de Carré a diminuée drastiquement grâce à la vaccination débutée en 1950 et que l’arrivée de la vaccination concorde avec le déclin de la prévalence de la maladie dans cette région. (Cooper et al., 1999) Une association a aussi été observée entre la MOP et les contacts fréquents avec les animaux. Dans l’étude réalisée par Lopez-Abente, les contacts avec les chats, les chiens et les bovins ont été identifiés comme facteurs de risques de développer la maladie chez les Espagnols. (López-Abente et al., 1997) En Italie, une association significative entre la MOP et les contacts, présents ou antérieurs, avec des animaux, a aussi été observée chez les personnes habitant une région rurale. Dans les régions étudiées, le risque de développer la maladie était accru suite aux contacts avec des cochons, des lapins et du bétail. Ces animaux étant rarement vaccinés contre les infections par un paramyxovirus, il a été suggéré qu’ils pouvaient agir comme vecteur d’infections virales. (Gennari et al., 2006)
Malgré les différentes observations mentionnées plus haut, le rôle de l’infection virale dans la pathogenèse de la MOP demeure controversé. L’hypothèse selon laquelle la détection de nucléocapsides virales dans les ostéoclastes pagétiques serait attribuable à une contamination des produits de PCR a été soulevée. (Ralston et al., 1997) Une autre hypothèse, selon laquelle la défectuosité des systèmes de l’ubiquitine-protéasome ou de l’autophagie dans la MOP entraînerait une mauvaise dégradation des agrégats de protéines cellulaires qui prennent alors la forme d’inclusions cellulaires, a aussi été soulevée en raison de la non-spécificité des inclusions retrouvées dans la MOP. (Chung et Van Hul, 2012; Helfrich et Hocking, 2008; Ralston, 2008) Des inclusions cellulaires semblables ont été observées dans l’ostéopétrose et dans certaines maladies neurodégénératives, comme la maladie d’Alzheimer, la maladie de Parkinson et la démence à corps de Lewy, ou musculodégénératives, comme la myosite à inclusions. (Chung et Van Hul, 2012; Helfrich et Hocking, 2008) Dans plusieurs de ces maladies impliquant des inclusions cellulaires, des composés impliqués dans la voie de dégradation de l’ubiquitine-protéasome semblent former les inclusions, qui seraient, en fait, des agrégats de protéines destinées à être dégradées. L’agrégation des protéines pourrait survenir en cas de surcharge du système de dégradation dans le but de protéger la cellule des effets néfastes des protéines anormales. Des filaments intermédiaires de confinement près du centrosome de la cellule et une abondance de mitochondries ont été observés autour des inclusions présentes dans la MOP, ce qui est cohérent avec l’idée qu’il s’agisse en fait d’aggrésomes4. (Helfrich et Hocking, 2008) L’implication de virus dans la maladie
soulève aussi plusieurs questions. Si ces virus sont en cause, pourquoi la maladie est-elle distribuée de façon aussi restreinte géographiquement alors que ces infections sont communes à travers le monde? Puisque l’infection par ces virus survient habituellement en jeune âge et que la MOP se développe beaucoup plus tardivement, comment expliquer que le virus demeure aussi longtemps dans les ostéoclastes de personnes immunocompétentes? (Cooper et al., 2006) Finalement, de quelle façon l’infection virale peut-elle expliquer l’aspect focal de la maladie? (Roodman, 2010)
Outre l’infection virale, plusieurs facteurs environnementaux pouvant avoir un rôle dans la MOP ont été investigués.
Alimentation
1.2.6.1.2.
Certains facteurs liés à l’alimentation ont déjà été soupçonnés d’influencer le développement de la MOP. Il a déjà été suggéré qu’une faible consommation de calcium provenant de l’alimentation pendant l’enfance pouvait être associée à un risque accru de développer la MOP. (Siris, 1994) Toutefois, il n’a pas été possible de trouver une association entre une déficience en 1,25-(OH)2D3 et le développement de la maladie. (Chung
et Van Hul, 2012) En Espagne, une association entre MOP et la consommation fréquente de viande d’agneau ou de chèvre dont la qualité sanitaire n’était pas évaluée préalablement a été observée. Cette observation a entraîné la suggestion selon laquelle un agent infectieux pourrait être transmis via la consommation de viande contaminée. (Piga et al., 1988)
Substances toxiques
1.2.6.1.3.
Une exposition à des substances toxiques a aussi été suggérée comme facteur de risque de développer la MOP. L’arsenic a été soupçonné de contribuer à la prévalence élevée de la MOP observée dans la région du Lancashire, suite à la détection de concentration élevée d’arsenic dans une source d’eau de cette région. (Bastin et al., 2009; Chung et Van Hul, 2012) Aussi, une association entre la MOP et le tabagisme présent ou antérieur a récemment été observée dans la population française. (Michou et al., 2012) Une surexpression par des cellules épithéliales bronchiques humaines du seul gène dont l’implication dans la MOP est confirmée jusqu’à ce jour, soit le gène SQSTM1, avait déjà été démontrée suite à l’exposition à la fumée de cigarette. (Michou et al., 2012)
Composante génétique
1.2.6.2.
Les caractéristiques épidémiologiques particulières de la MOP suggèrent que des facteurs génétiques et des facteurs environnementaux influencent le développement de la maladie. (Cooper et al., 1999) Dans environ un tiers des cas, la MOP a un mode de transmission autosomique dominant avec une pénétrance incomplète. (Morales-Piga et al., 1995; Siris et al., 1991) Les personnes ayant un parent du premier degré atteint par la maladie ont sept à dix fois plus de risques de souffrir de MOP. (Siris et al., 1991) Aussi, les individus atteints par une MOP familiale souffrent généralement d’une forme plus précoce et plus sévère de la maladie que les individus atteints « sporadiquement » par la maladie. (Cooper et al., 2006; Gennari et al., 2010)
Régions chromosomiques liées à la MOP
1.2.6.2.1.
Jusqu’à maintenant, plusieurs régions chromosomiques potentiellement liées à la MOP (PDB1 à PDB7) ont été décrites, ce qui traduit l’hétérogénéité génétique de la maladie. Le locus 5q35-qter (PDB3) est la seule région contenant un gène dont l’implication dans la MOP est confirmée à ce jour, soit le gène SQSTM1 (Laurin et al., 2002, 2001). Les liaisons entre la MOP et les régions 6p21.3 (PDB1) (Tilyard et al., 1982), 18q22.1 (PDB2) (Cody et al., 1997), 5q31 (PDB4) (Laurin et al., 2001), 2q36 (PDB5) (Hocking et al., 2001), 10p13 (PDB6) (Hocking et al., 2001) et 18q23 (PDB7) (Good et al., 2002), n’ont pas été répliquées lors d’études subséquentes ou n’ont pas permis d’identifier d’autres gènes directement impliqués dans la MOP. (Michou et Brown, 2011b) Certaines de ces liaisons découlent probablement de résultats faussement positifs, alors que d’autres peuvent traduire la présence de gènes modificateurs de l’effet de SQSTM1. (Ralston, 2008)
Mutations du gène SQSTM1
1.2.6.2.2.
La protéine p62, encodée par le gène SQSTM1, est composée de 440 acides aminés et comporte neuf domaines, dont un domaine associé à l’ubiquitine (UBA). (Figure 11) Elle est notamment impliquée dans l’autophagie et l’apoptose. Elle agit aussi en tant que protéine d’échafaudage dans l’activation de la voie de signalisation du NF-κB. (Chung et Van Hul, 2012)
Figure 11. Domaines de la protéine SQSTM1
Figure tirée de Chung (2011) Le rôle du gène SQSTM1 dans la MOP a été découvert grâce à une étude de liaison génétique pangénomique de trois grandes familles québécoises. Un haplotype commun ségréguant avec la maladie, situé au locus 5q35, a été découvert dans deux de ces trois familles. Le génotypage de marqueurs microsatellites a permis de cibler une région de 300 kb, contenant les gènes SQSTM1 et MAPK9. Le séquençage d’un échantillon de découverte composé de quelques cas et contrôles a permis d’identifier la première mutation du gène SQSTM1, soit la mutation P392L, chez tous les individus atteints possédant l’haplotype initialement lié avec la maladie. (Laurin et al., 2001, 2002) Cette mutation entraîne la substitution d’une proline appartenant au domaine UBA, en leucine en position 392 de la protéine (P392L), dû au changement d’un nucléotide cytosine en thymine en position 1215 du gène au sein du huitième exon. (Laurin et al., 2002) Selon la composition des chaînes d’ubiquitine qu’il lie, le domaine UBA permet l’activation de la voie de signalisation du NF-κB, ou la dégradation programmée des protéines par le protéasome 26S. (Chung et Van Hul, 2012) La mutation P392L a été identifiée dans 46 % des cas familiaux et 16 % des cas atteints de MOP non apparentés dans la population canadienne-française. (Laurin et al., 2002) Dans une étude portant principalement sur des individus d’origine britannique, la majorité des porteurs de la mutation P392L possédait un même haplotype dans la région 5q35-qter, et ce indépendamment de la présence d’une histoire familiale positive. Cette observation suggère que la mutation P392L est une mutation ancestrale dans cette population. (Lucas et al., 2005) Plusieurs autres mutations du gène SQSTM1 ont été identifiées chez des individus atteints par la MOP. Jusqu’à maintenant, près de 30 mutations du gène SQSTM1 ont été rapportées dans plusieurs populations; la grande majorité de ces mutations sont situées dans le domaine UBA et induisent une augmentation de l’activité de la voie de signalisation du NF-κB. Près du tiers des patients souffrant d’une forme familiale de MOP et environ 10 % des cas sporadiques sont porteurs d’une mutation de SQSTM1. (Chung et Van Hul,
(Morissette et al., 2006) Il a été observé que les individus porteurs d’une mutation du gène SQSTM1 développaient généralement une forme plus sévère de la maladie. La maladie débute à un plus jeune âge et atteint un plus grand nombre d’os chez ces individus. (Gennari et al., 2010; Michou et al., 2012, 2011) La maladie est aussi plus sévère chez les individus possédant une mutation menant à la formation de protéines tronquées (mutation stop) que chez les individus possédant une mutation entraînant la modification d’un acide aminé (mutation faux-sens). (Chung et Van Hul, 2012) Malgré tout, il existe une grande variabilité de la sévérité du phénotype entre les individus d’une même famille possédant la même mutation. Cette variabilité demeure inexpliquée, mais suggère l’implication d’autres facteurs, génétiques ou environnementaux, dans la pathogenèse de la maladie. (Chung et Van Hul, 2012)
Des études in vitro et in vivo ont été effectuées afin d’élucider le rôle de la protéine p62 dans la pathogenèse de la MOP. Duran et coll. (2004) ont observé que l’ostéoclastogenèse, et l’activation de la voie du NF-κB de souris pour lesquelles le gène SQSTM1 a été invalidé (SQSTM1-/-) étaient altérées, ce qui confirme
l’importance de cette protéine dans le métabolisme osseux. Par contre, le rôle de la mutation P392L dans la MOP n’a pas encore été clairement établi. Kurihara et coll. (2007, 2011) n’ont observé aucune lésion osseuse semblable aux lésions pagétiques au niveau des vertèbres lombaires de souris exprimant l’équivalent murin de la mutation P392L sous le contrôle du promoteur TRAP. Les cellules précurseurs d’ostéoclastes exprimant cette mutation ne parvenaient pas non plus à former des ostéoclastes aux caractéristiques pagétiques complètes. En effet, les précurseurs étaient hypersensibles à RANKL, mais pas à la 1,25-(OH)2D3 et ils ne
sécrétaient pas d’IL-6 ou de TAF12 en grande quantité. Suite à l’obtention de ces résultats, il a été suggéré que cette mutation pouvait prédisposer à développer la maladie en augmentant la sensibilité des précurseurs des ostéoclastes aux cytokines ostéoclastogénique ou au microenvironnement osseux, mais qu’elle n’était pas suffisante pour causer la MOP. Toutefois, Daroszewska et coll. (2011) ont observé des lésions osseuses focales ayant des composantes d’ostéolyse et d’ostéosclérose chez des souris transgéniques possédant la mutation P394L. Les ostéoclastes des souris mutés étaient plus nombreux et possédaient plus de noyaux que ceux des souris sauvages. De plus, certains ostéoclastes présentaient des inclusions semblables à celles retrouvées dans la MOP. Ces résultats contredisent ceux de Kurihara et coll. (2007, 2011) et suggèrent que cette mutation pourrait s’avérer suffisante pour causer le développement de la maladie. Le fait que des os différents aient été étudiés et que des appareils différents aient été utilisés pour déterminer la présence ou l’absence de lésions osseuses pourrait expliquer les conclusions différentes auxquelles sont arrivées les deux études. Des études supplémentaires seront nécessaires pour faire la lumière sur le rôle exact de la mutation P392L dans le développement de la MOP.