HAL Id: tel-02180614
https://tel.archives-ouvertes.fr/tel-02180614
Submitted on 11 Jul 2019
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Macrophages, inflammation et réparation cardiaque
Ivana Zlatanova
To cite this version:
Ivana Zlatanova. Macrophages, inflammation et réparation cardiaque. Cardiologie et système cardio-vasculaire. Université Sorbonne Paris Cité, 2017. Français. �NNT : 2017USPCB047�. �tel-02180614�
Pour l’obtention du diplôme de : Docteur de l’université Paris V Ecole doctorale : Hématologie Oncogenèse et Biothérapie (HOB) Spécialité : Pathologie cardio-vasculaire Titre : Présentée et soutenue publiquement par : IVANA ZLATANOVA Le 20 Octobre 2017 Thèse dirigée par Dr JEAN-SEBASTIEN SILVESTRE Jury Dr. François Canonne-Hergaux, rapporteur Dr. Ebba Brakenhielm, rapporteur PU-PH. Jean-Sébastien Hulot, examinateur Dr. Agnès Lehuen, examinatrice Pr. Bernard Levy, examinateur Dr. Jean-Sébastien Silvestre, directeur de thèse Equipe 6 : Thérapies régénératives des pathologies cardiaques et vasculaires –PARCC Inserm U970
Macrophages, Inflammation
et réparation cardiaque
L’inflammation joue un rôle crucial dans les processus de réparation du muscle cardiaque consécutifs à un infarctus du myocarde (IM). En particulier, les monocytes (MO) et les macrophages (Mf) assument une fonction majeure dans la régulation de la taille d’infarctus et de la fibrose du cœur infarci.
Dans un premier travail, nous avons analysé le rôle coordonné de la myeloid-epithelial- reproductive protein tyrosine kinase (Mertk) et du milk fat globule epidermal growth factor (Mfge8) dans la propension des MO et des Mf à éliminer les débris cellulaires. Nous avons notamment émis l'hypothèse que ces médiateurs pourraient jouer un rôle clé dans la synchronisation de l’efférocytose dans le tissu cardiaque infarci. Les souris dépourvues de Mertk et Mfge8 présentaient un phénotype cardiaque défectueux par
rapport aux souris contrôles. Fait intéressant, au 3ème jour après l’IM, le niveau de protéine
cardiaque d’un facteur pro-angiogénique majeur, le VEGF-A, était plus faible chez les souris chimères Mertk-/ -Mfge8-/-par rapport aux contrôles. À partir de ces données, nous avons postulé qu'une altération du taux de la protéine VEGF-A pourrait être liée à une réduction de la capacité des MO/Mf cardiaques à libérer ces facteurs et donc à participer au remodelage bénéfique du cœur infarci. De fait, après un IM, la suppression du VEGF-A dans les cellules myéloïdes réduit la fonction cardiaque. Ces effets étaient associés à une taille d'infarctus supérieure et à un nombre limité de capillaires dans les cœurs infarcis des souris présentant une invalidation conditionnelle du VEGF-A dans les cellules myéloïdes. Ainsi, le VEGF-A dérivé des cellules myéloïdes joue un rôle essentiel dans la régulation de la fonction cardiaque, probablement grâce à l'activation du processus angiogénique dans la zone ischémique.
Dans un deuxième travail, nous nous sommes intéressés au rôle de l’hepcidine dans les fonctions réparatrices des MO et des Mf. L'hepcidine est une hormone qui régule activement le trafic du fer. De façon inattendue, la déficience complète en hepcidine restaure considérablement la fonction cardiaque après un IM. L'hepcidine est exprimée par les Mf cardiaques. En outre, les souris ayant une moelle déficiente en hepcidine (Hamp -/-)
ou présentant une invalidation conditionnelle dans les cellules de la lignée myéloide (LysM-Cre+/Hampf/f) ont une fonction cardiaque fortement améliorée. Cet effet a été associé à une
réduction de la taille de l'infarctus, de la fibrose et à une stimulation imprévue du renouvellement des cardiomyocytes. De même, dans un modèle de régénération cardiaque induite par résection apicale chez des souris nouveaux nés, nous avons révélé que les macrophages déficients en hepcidine favorisent la prolifération des cardiomyocytes. L'impact des Mf déficients en hepcidine repose sur l'altération de leur métabolisme du fer et sur la libération d'IL-4 et d'IL-13. D’ailleurs, la suppression génétique de l'IL-4 et de l'IL-13 dans les Mf dépourvus d'hepcidine annihile leur effet bénéfique sur la fonction et la réparation cardiaque. Ainsi, l'hepcidine commande la réparation et la régénération cardiaque induite par les Mf par une voie liée à l'IL-4 / IL-13.
Ces travaux ont permis de démontrer l’importance des Mf dans la revascularisation et la régénération post-ischémique et de souligner l’efficacité potentielle d’un traitement basé sur la modulation de l’activité des cellules immunitaires dans les pathologies ischémiques cardiaques.
Remerciements
Je tiens tout d’abord à remercier les membres du jury qui ont accepté d’évaluer mon travail de thèse :
Je remercie le Docteur Ebba Brakenhielm et le Docteur François Canonne-Hergaux d’avoir accepté d’être les rapporteurs de cette thèse et d’avoir pris le temps d’évaluer mon manuscrit de thèse. Je remercie également le Professeur Jean-Sébastien Hulot, le professeur
Bernard Levy et le Docteur Agnès Lehuen d’avoir accepté de participer à mon jury de thèse
en qualité d’examinateurs.
Je remercie tout particulièrement le Professeur Bernard Lévy, pour sa présence tout au long de mes études. Votre gentillesse et votre disponibilité durant ces années ont été d’une grande aide, ainsi que votre soutien. Pour cela, je vous exprime toute ma gratitude.
Je tiens également à remercier Alain Tedgui, Directeur du PARCC.
Je tiens à exprimer ma sincère reconnaissance à mon directeur de thèse, le Docteur
Jean-Sébastien Silvestre. Merci de m’avoir accepté au sein de ton équipe depuis mon Master et
de m’avoir supporté aussi longtemps. Merci pour ta disponibilité sans failles, merci de m’avoir consacré tout ce temps pour me former, tous les jours depuis mon M1 j’ai appris énormément à tes côtés, y compris sur le foot, tu as su me guider, m’encourager et me pousser à me dépasser tout au long de ma thèse, merci d’avoir eu confiance en moi. Au-delà de ces aspects professionnels, ta sympathie, ta bonne humeur contribue énormément à la bonne ambiance de l’équipe, c’était une grande joie et un honneur pour moi d’avoir fait partie de ton équipe. J’ai toujours su que j’avais énormément de chance de t’avoir comme directeur de thèse. Merci Jean-Sé, Merci pour tout.
Je remercie également José Vilar, mon cher José je pense qu’il n’y aura pas assez de pages dans cette thèse aux remerciements que tu mérites. Merci 100000 fois José pour tout ce que tu as su être pour moi un ami, un meilleur ami, un super enseignant et parfois même un père. Vous m’avez tout appris avec Jean-Sé, j’espère un jour que mes étudiants auront autant de reconnaissance et d’admiration que j’en ai pour vous. Merci José cette thèse était une magnifique période de ma vie, tous les jours je suis venu au labo depuis 5 ans avec un grand sourire et une envie d’apprendre et ça c’est grâce à toi sans aucun doute Je remercie également les filles de l’équipe…. « Equipe ou pas Equipe » Merci Cristina Pinto, c’était un plaisir de travailler avec toi, merci pour ton amitié sans failles merci pour tous les moments passés ensemble, on fêtera ça bientôt ensemble au Matrimonio….Merci Bella. Merci Mathilde, je suis ravie que tu aies rejoint l’équipe de Jean-Sé, c’était un plaisir de travailler avec toi, merci de m’avoir fait beaucoup rire, j’ai adoré l’ambiance agréable et rigolotes qu’on a instaurée dans le labo.
Merci Kiave, tous les jours depuis ton départ tu me maques notre amitié, nos discussions scientifiques tu as toujours été de très bon conseil, merci pour tout Kiave.
Merci aux nouvelles recrues Angélique et Ingrid, rappelles-toi Angélique tu es là à vie donc il faut assurer pour la bonne ambiance au labo.
Merci à tous les stagiaires qui m’ont accompagné pendant cette thèse Raphael Cohen, Gabi,
Thomas, Ange, Antoine, Vincent, Sehar, Mattieu merci pour votre aide, j’ai pris beaucoup
de plaisir à vous faire partager ma science.
Merci Marie Vandestienne, merci d’être mon amie, j’ai eu une chance incroyable de t’avoir rencontré, heureusement que tu es partie de Bretagne et moi de Macédoine … on a passé des superbes moments ensemble et on passera pleins d’autres sans aucun doute je te souhaite beaucoup du courage pour ta thèse et sache que je serai toujours là pour toi. Merci Andréas Giraud, merci mon pote, tu étais une des plus belles rencontres qui m’aient arrivé pendant cette thèse, je n’oublierai jamais toutes nos aventures innombrables de rire, de danse de fête et aussi de science. Merci à Bettina, un véritable bonheur de t’avoir rencontré, merci à Marouane Kheloufi Merci pour tous les moments passés ensemble. Merci Adèl pour ta gentillesse et ta bonne humeur. Merci Yacine, et c’est fini je te donnerai plus d’anticorps de FACS. Merci Johanne de ta présence et ta gentillesse sans failles. Merci Camille, merci pour toutes les tries, rires, merci d’être resté des soirées entières pour moi, merci pour ton énergie. Merci Cécile pour ne pas m’avoir engueulé de pas nettoyer le L2 et merci pour ta gentillesse. Merci Marion de passer toujours me dire bonjour. Merci Pierre Nivoit, merci pour tous les bons moments passés ensemble et merci pour toute tes conseils. Merci à Yann, Merci Maria Chavez, Merci Martin Steinmetz, Merci Safa Mellak ….
Merci à tous les amis pour tous les soirées tous les congrès, le ski le Balard…. Merci les copains.
Merci Xavier Loyer ohhh pardon Docteur Loyer, merci pour tous ces moments passés à travailler avec toi, j’espère qu’on va publier, merci pour toutes tes blagues matinales c’était un plaisir de te croiser tous les jours.
Merci Professeur Hafid Ait-Oufella, merci pour tous tes conseils merci pour ta bonne humeur et ton exigence « montre-moi des datas » ne t’inquiète pas ça arrive, là aussi j’espère qu’on va bien publier ensemble.
Merci au Professeur Ziad Mallat de m’avoir encouragé à la soumission de mon papier à Nature Medecin et surtout d’avoir surveillé mon alcoolémie tous les vendredis soir au Balard et bien sûr merci Stéphane merci pour tout le service et pour les cacahuètes. Merci
Je tiens à remercier Docteur Pierre-Louis Tharaux, pour toutes les danses partagées. Merci Carole pour ta douceur au quotidien, tu as toujours été de très bon conseil.
Merci mon cher Philippe Bonin, merci pour tout le travail que tu as fait merci pour les journées de rigolades passées à Lariboisière, merci pour ta bonne humeur et pour ton amitié.
Merci à toutes les personnes de l’animalerie tout particulièrement Nicolas et Dominique, merci pour tout.
Merci au sécréteur général Phillipe Coudol, merci pour et surtout pour toutes ces super soirées mixées par Dj Coudol.
Merci tout particulièrement à toutes les filles de l’administration Véronique la maman,
Annette la star et Martine Jolibis, merci d’avoir été là et merci pour cette superbe
ambiance.
Merci le PARCC et les souris aussi.
Je remercie également Thi Tran pour sa disponibilité et sa gentillesse, je remercie aussi la plateforme de l’HEGP d’histologie, Merci Florence et courage pour ta thèse.
Je tiens tout particulièrement à remercier mes amis d’enfance et toutes les copines Mélanie,
Lara, Débo, Juliana, Emmeline…. Merci les filles
Je tiens également à remercier mes parents pour tout, vous êtes des parents formidables, c’est grâce à vous que j’en suis là aujourd’hui, je remercie ma grande sœur pour son soutien sans failles mais je remercie tout particulièrement ma petite sœur Nevena merci de m’avoir supporté, merci énormément pour ton soutient pour tous les cafés que tu m’as apportés pour m’avoir écouté te raconter toute ma science, merci bébé.
Et enfin je remercie Jérémie de partager ma vie et de me faire rêver au quotidien, merci mon amour.
Liste des abréviations Ag : Antigène Alpha-SMA : alpha Smooth Muscle cell Actin APC : Cellule Présentatrice de l’Ag Arg1 : Arginase 1 AVC : Accident Vasculaire Cérébral AV : Auriculo-Ventriculaire BAFF : Récepteur du facteur d'activation des cellules B BMS : Bear Metal Stent CML : Cellule Musculaire Lisse CMH : Complexe Majeur d'Histocompatibilité CLR : Lectine de type C CPI : Cardiopathie Ischémique Cx43 : Connexine 43 DAMP : Danger-Associated Molecular Patterns DC : Cellule Dendritique DES : Drug Eluting Stent ou Stents médicamenteux DMT-1 : Transporteur de Métal Divalent EC : Cellule endothéliale ECA : Enzyme de Conversion de l’Angiotensine ECG: Electrocardiogramme EGF: Epidermal Growth Factor EMT : Transition épithélio-mésenchymateuse ERM : l'ézrine, la radixine et la moesine FEVG : Fraction d’Ejection du Ventricule Gauche FA : Ataxie de Friedreich FT : Facteur Tissulaire FPN : Ferroportine Fn : Fibronectine FR : Fraction de raccourcissement GFP: Green Fluorescent Protein G-CSF: Granulocyte Colony Stimulating Factor HH: Hémochromatose héréditaire HMOX-1: Hémoxygénase-1 HIF-1 : Hypoxia Inducible Factor 1 HGF: Hepatocyte growth factor IDO: Indoléamine 2,3-dioxygénase ICAM-1 : InterCellular Adhesion Molecule -1 IEC : Inhibiteur de l’Enzyme de Conversion de l’Angiotensine Ig : Immunoglobuline IM : Infarctus du Myocarde iNOS : inducible Nitric Oxide Synthase IRE : Iron Response Element IREBP1: Iron-responsive element-binding protein 1 IRM: Imagerie par Résonance Magnétique KO : Knock Out
KIR : Killer-immunoglobulin-like KYN : Kynurénine LVID : Diamètre Interne Systolique du Ventricule Gauche LVPW : Epaisseur de la Paroi Postérieure du Ventricule Gauche MC : Mastocytes MMP : Métalloprotéinases MC : Monocytes Classiques MNC: Monocytes Non Classiques Mer TK: Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase MFG-E8: Milk Fat Globule Epidermal Growth Factor 8 MO/Mφ : Monocytes/ Macrophages MEC : Matrice Extracellulaire MyBPC: Myosin-binding protein C NK: Natural Killers NO : Monoxyde d’azote LPS : Lipopolysaccharide bactérien OMS : Organisation Mondiale de la Santé PAI-1 : Activateur du Plasminogène 1 PAMPs : Motifs Moléculaires Associés aux Pathogènes PCI: Percutaneous Coronary Intervention PMN: Polymorphonuclear leukocytes PS : PhosphatidylSérine PTP1B : Protein-Tyrosine Phosphatase 1B ROS : Reactive Oxygen Species ou Espèces Radicalaires de l’Oxygène RM : Récepteur au Mannose SCA : Syndrome Coronarien Aigu SCF: Stem Cell factor SDF-1: Stroma Derived Factor-1 TCR : Récepteur des cellules T TG2 : Transglutaminase tissulaire 2 TfR : Récepteur de la Transferrine TLR : Toll-Like Recepteur TNF-α : Tissue Necrosis Factor- α TIMP: Tissue Inhibitor of Metalloproteinase TNc : Troponine C cardiaque TPH : la thrombolyse pré-hospitalière TSP-1: Thrombospondine-1 VG: Ventricule Gauche VEGF: Vascular endothelial growth factor VCAM-1 : Vascular cell adhesion protein 1
1 Introduction ... 1 A. L’infarctus du myocarde ... 1 1. Nosologie ... 1 2. Epidémiologie de la maladie coronarienne ... 4 3. Physiopathologie de l’athérosclérose coronaire ... 5 4. Physiopathologie du syndrome coronaire aigu et de l’infarctus du myocarde ... 8 5. Prise en charge de l’infarctus du myocarde à la phase aigue ... 11 Traitements médicamenteux actuels et perspectives de recherche ... 11 a) Méthodes de reperfusion coronaire ... 12 b) (1) Thrombolyse Médicamenteuse ... 13 (2) Angioplastie percutanée ... 13 (3) Chirurgie ... 14 6. Remodelage ventriculaire et cardiopathie ischémique ... 15 B. Réponse immuno-inflammatoire lors d’un infarctus du myocarde ... 18 1. Effecteurs cellulaires de la réponse immunitaire locale et systémique post-infarctus ... 18 Rôle de l’immunité innée ... 19 a) (1) Neutrophiles ... 19 (2) Les Lymphocytes Natural Killers (NK) ... 23 (3) Les Cellules Dendritiques ... 24 (4) Les Mastocytes ... 26 (5) Les Basophiles ... 28 (6) Les Eosinophiles ... 28 Rôle de l’immunité adaptative ... 30 b) (1) Les Lymphocytes T CD4+ ... 31 (2) Les Lymphocytes T régulateurs (Tregs) ... 32 (3) Les Lymphocytes T CD8+ ... 33 (4) Les autres Lymphocytes T ... 33 (5) Les Lymphocytes B ... 35 2. Déterminants moléculaires de la réponse immuno-inflammatoire post infarctus ... 37 Cytokines pro-inflammatoires ... 37 a) (1) TNF-α ... 37 (2) IL-6 ... 38 (3) IL-1-β et IL-18 ... 39 Cytokines anti-inflammatoires ... 40 b) (1) IL-10 ... 40 (2) Il-4/IL-13 ... 40 C. Monocytes/Macrophages ... 43 1. Monocytes ... 43 Les monocytes humains ... 44 a) Les monocytes murins ... 44 b) 2. Influence des monocytes dans l’infarctus du myocarde ... 45 3. Macrophages ... 46 Ontogénèse et rôle primaire des macrophages ... 46 a) Efférocytose ... 47 b) Diversité des macrophages ... 48 c) Implication des macrophages dans l’infarctus du myocarde ... 52 d) Les macrophages résidents. ... 54 e) 4. Macrophages et métabolisme du fer ... 55 Homéostasie et rôle du fer ... 57 a) Hepcidine ... 59 b) M1-macrophages et leur phénotype séquestrant le fer ... 60 c) M2-macrophages – un phénotype de libération du fer ... 63 d) La régulation de l’hepcidine ... 64 e) L’axe Hepcidine-Ferroportine ... 64 f)
5. Pathophysiologie de la régulation de l’hepcidine et du fer ... 65 L’hémochromatose héréditaire ... 65 a) L’hémochromatose secondaire ... 66 b) Anémie inflammatoire ... 66 c) 6. Fer et pathologies cardiaques chez l’homme ... 68 Anémie et maladies cardiovasculaires ... 68 a) L’Ataxie de Friedreich (FA) ... 69 b) Surcharge en fer et maladies cardiovasculaires ... 70 c) (1) Rôle du fer dans l’athérosclérose ... 70 (2) Cardiomyopathies secondaires à une surcharge en fer ... 74 D. Remodelage et Régénération tissulaire post-ischémique ... 77 1. Résolution de l’inflammation ... 77 2. Modifications de la matrice extracellulaire ... 80 3. L’activation des myofibroblastes et la formation de la cicatrice ... 82 4. Conséquences cliniques du remodelage : l’insuffisance cardiaque ... 85 5. Stratégie pharmacologique dans l’insuffisance cardiaque et conséquences sur le remodelage ... 87 Blocage du système rénine-angiotensine- aldostérone ... 88 a) Les effets des bétabloquants ... 88 b) 6. Régénération cardiaque ... 89 Au stade embryonnaire ... 89 a) Au stade néonatal ... 90 b) Au stade adulte ... 91 c) 7. Role du système neurohormonal dans la régénération myocardique ... 93 8. Rôles des cellules inflammatoires, Monocytes/Macrophages dans le processus de régénération cardiaque. ... 94 9. Implication des macrophages dans la contractilité des cardiomyocytes ... 96 2 Hypothèses ... 97 3 Résultats ... 99 A. Article I: Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase and Milk Fat Globule Epidermal Growth Factor 8 Coordinately Improve Remodeling After Myocardial Infarction via Local Delivery of Vascular Endothelial Growth Factor ... 99 B. Article II: The iron regulator hepcidin impairs macrophage dependent cardiac repair after injury ... 132 4 Discussion et Perspectives ... 170 5 Conclusion ... 176 6 Références ... 177 7 Annexe ... 209 A. Article III: Immune Modulation of Cardiac Repair and Regeneration: The Art of Mending Broken Hearts ... 209 B. Article IV: Genetic and Pharmacological Inhibition of TREM-1 Limits the Development of Experimental Atherosclerosis. ... 217 C. Article V: MicroRNA-21 coordinates human multipotent cardiovascular progenitors therapeutic potential. ... 235
L’infarctus du myocarde
1
Introduction
A. L’infarctus du myocarde
1. Nosologie L'infarctus du myocarde (IM) se traduit par une mort massive des cellules myocardiques suite à une ischémie significative et prolongée. Le plus souvent, il s’agit d’une complication aigue d’une maladie coronarienne préexistante liée à l'athérosclérose. D’autres mécanismes plus rares comme le vasospasme, l’embolie coronaire sur artères saines, la dissection ou encore les artérites inflammatoires non athéromateuses (maladie de Kawasaki, syphilis) peuvent aussi être impliqués.La très grande majorité des évènements cliniques est due à une rupture de plaque d’athérome qui va engendrer un processus athéro-thrombotique, provoquant l’occlusion de l’artère cible et la nécrose ischémique de la zone tissulaire en aval. L'athérosclérose se transcrit par des remaniements de l'intima des artères de gros et moyen calibres consistant en une accumulation focale de lipides, de glucides complexes, de sang et de produits sanguins, de tissu fibreux et de dépôts calcaires ; le tout s'accompagnant de modifications de la media. Les plaques d’athérome les plus à risque de rupture, dites instables, sont caractérisées par une chape fibreuse fine avec une perte des cellules musculaires lisses (CML), une destruction de la matrice extracellulaire et la présence d’un infiltrat lymphocytaire T et macrophagique important, constituant un centre nécrotique de grande taille. Des mécanismes, autres que la rupture, peuvent aboutir à une occlusion thrombotique. Ainsi, une érosion ou une fissuration de plaque peuvent être responsables de la même symptomatologie et des mêmes conséquences tissulaires (Figure 1).
La présentation clinique de l'IM est variable et couvre un spectre allant d’un évènement mineur asymptomatique, à des tableaux bruyants de choc cardiogénique voire de mort subite. Aussi, le recueil de tous ces évènements de gravité et de présentation variable est difficile. Afin d’avoir une approche épidémiologique exhaustive, l'Organisation Mondiale de la Santé (OMS) a, dès les années 1970, promulgué des critères standardisés pour le
diagnostic de la maladie coronarienne et de l’infarctus du myocarde 1. Aussi, dans les années
1970, la définition de cas d'IM utilisée dans les projets collaboratifs internationaux était
L’infarctus du myocarde rapport conjoint avec la Société Internationale et la Fédération de cardiologie en 1979. Les critères du registre européen de l’IM de l'OMS étaient alors basés sur l'histoire clinique, les résultats de l'électrocardiogramme (ECG), les mesures enzymatiques dans le sang et, en cas de mort subite, des constatations post-mortem. La définition moderne de l’IM se caractérisait alors par :
(1) La détection d’une augmentation ou d’une diminution d’au moins un des
biomarqueurs cardiaques, de préférence la troponine C, atteignant à minima le 99e
percentile de la limite de référence supérieure, ainsi que des signes d’ischémie myocardique; avec au moins l’un des symptômes suivants :
(a) Les symptômes de l'ischémie (les symptômes ischémiques incluent diverses combinaisons de douleurs à la poitrine, aux extrémités supérieures, à la mâchoire ou un malaise épigastrique pendant un effort ou au repos ; le malaise est habituellement inférieur à 20 minutes, souvent diffus, non localisé, pas positionnel, pas affecté par le mouvement de la région, et peut être accompagné d'une dyspnée, d'une diaphorèse, d'une nausée ou d'une syncope).
(b) Le symptôme d’ischémie déterminé par évaluation des modifications des ondes S, T ou Q au niveau de l’ECG.
(c) L'angine de poitrine typique ou atypique, ainsi que des changements équivoques sur l'ECG et des enzymes myocardique élevées.
(d) L'angine de poitrine typique et des enzymes élevées sans modification de l'ECG ou non disponible.
(e) Une évidence par imagerie de la perte de masse ventriculaire viable ou la détection des régions akinétiques ou encore identification par angiographie ou par autopsie d’un thrombus intracoronarien.
(2) Une mort cardiaque inattendue et soudaine, impliquant un arrêt cardiaque, parfois précédé de symptômes suggérant une ischémie myocardique et accompagnée :
(a) d’une nouvelle élévation du segment ST,
(b) de l’évidence de la formation d’un thrombus frais par angiographie coronaire et/ou à l'autopsie.
Cette définition moderne a assez peu évolué mais la nécessité d’intégrer les éléments diagnostiques nouveaux comme l’angiographie coronaire et l’imagerie myocardique, tout en préservant le caractère universel de cette définition à des fins de comparaisons
L’infarctus du myocarde épidémiologiques a conduit à leur révision. Ainsi en 2012 un consensus international a élaboré la troisième définition universelle de l’infarctus du myocarde. On distingue donc :
-L’IM aigu : Dans ces conditions, l’observation de l’un des critères suivants répond au diagnostic d’IM :
• Détection d’une élévation et/ou baisse d’un biomarqueur cardiaque (de préférence la troponine cardiaque C (TNc), avec au moins une valeur au-dessus du 99e percentile de la limite de référence supérieure, avec au moins l’un des éléments suivants :
Symptômes d’ischémie ; nouvelles (ou présumées nouvelles) modifications significatives du segment ST ou de l’onde T ou un nouveau bloc de branche gauche ; apparition d’ondes Q pathologiques à l’ECG ; à l’imagerie, preuve d’une nouvelle perte de myocarde viable ou d’une nouvelle anomalie de la contractilité régionale ; Identification d'un thrombus intra coronaire par angiographie ou autopsie.
• Décès d’origine cardiaque précédé de symptômes suggérant une ischémie myocardique avec modifications ischémiques à l’ECG (nouvelles ou présumées nouvelles) ou nouveau bloc de branche gauche, mais décès survenant avant le prélèvement sanguin, ou avant l’augmentation des biomarqueurs dans le sang.
• L’IM associé à une intervention coronaire percutanée est arbitrairement défini par une élévation des TNc (> 5 fois) chez les patients présentant des valeurs initiales normales ou une élévation des valeurs de la TNc > 20% si les valeurs initiales sont élevées et stables ou en diminution. En plus, il est requis la présence (i) de symptômes évocateurs d'une ischémie myocardique ou (ii) de nouvelles modifications ischémiques à l'ECG ou (iii) des observations angiographiques compatibles avec une complication procédurale ou (iv) la preuve d’une nouvelle perte de myocarde viable ou d’une nouvelle anomalie de la contractilité régionale myocardique à l’imagerie.
• L’IM associé à une thrombose de stent est défini par la documentation angiographique ou autopsique de la thrombose, dans un contexte d’ischémie myocardique associée à une élévation et/ou diminution des biomarqueurs cardiaques, avec au moins une valeur au-dessus du 99ème percentile.
• L’infarctus du myocarde associé au pontage aorto-coronarien est arbitrairement défini par une élévation de la TNc (>10 fois) chez les patients présentant des valeurs initiales normales. En plus, il est requis la présence (i) de nouvelles ondes Q pathologiques ou d’un nouveau bloc de branche gauche, ou (ii) d’une nouvelle occlusion d’une coronaire native ou
L’infarctus du myocarde d’un greffon documenté angiographiquement ou (iii) d’une nouvelle perte de myocarde viable ou d’une nouvelle anomalie de la contractilité régionale à l’imagerie.
- L’IM ancien : Le diagnostic d’ancien IM est établi en cas de présence d’au
moins un des critères suivants : • Ondes Q pathologiques à l’ECG, avec ou sans symptômes, en l’absence d’une cause non ischémique. • Présence à l’imagerie d’une région de perte de myocarde viable avec amincissement et akinésie pariétale, en l’absence d’une cause non ischémique. • Signes anatomo-pathologiques d’un IM à l’autopsie ou à la virtopsie.
Une telle définition standardisée de l’IM est d'une importance majeure puisqu'il s'agit d’un outil indispensable pour obtenir des données fiables et comparables pour l'évaluation de l'efficacité des stratégies de prévention de traitement dans les pays où les systèmes de santé sont très variés.
2. Epidémiologie de la maladie coronarienne
Les maladies cardiovasculaires sont un problème mondial de santé publique. En effet, elles contribuent à 30% de la mortalité mondiale et représentent environ 10% de la morbidité à l’échelle de la planète 3. En 2012, 17,5 millions de décès sont directement attribuables aux maladies cardiovasculaires soit 31% de la mortalité mondiale totale, parmi eux, 7,4 millions étaient dus à une maladie coronarienne et 6,7 millions à un accident
vasculaire cérébral (AVC) 4. L’IM est l'une des cinq principales manifestations de la maladie
coronarienne, qui incluent également l'angine de poitrine stable, l'angor de poitrine instable, l'insuffisance cardiaque et la mort subite. L'expression « syndromes coronariens aigus » comprend l'angor instable, l'IM sans élévation du segment ST, l'IM avec élévation persistante du segment ST et la mort cardiaque soudaine. Dans les études épidémiologiques, l'incidence de l'IM dans une population peut servir de base à l'estimation de la charge athéromateuse coronarienne.
L’incidence des maladies cardiovasculaires augmente à la fois dans les pays à revenus élevés, en raison du vieillissement de la population mais aussi dans les pays à revenus faibles et intermédiaires. Ce poids épidémiologique est croissant dans les pays en voie de développement en raison de la taille des populations et de l'exposition généralisée à des
L’infarctus du myocarde niveaux critiques de facteurs de risque tels que l'alimentation malsaine, l'inactivité physique, l'obésité, le tabagisme, le diabète, l’hypertension artérielle et les dyslipidémies. Il a été montré que les facteurs de risque de maladies cardiovasculaires sont largement similaires
entre les pays à revenus élevés et ceux en voie de développement 5, alors que les politiques
de prévention dans ce domaine sont quasiment inexistantes. Les conséquences de la
mondialisation et de l'urbanisation sont également des facteurs contributifs 6.
En France, les analyses de certificat de décès rapportent environ 35 000 décès par an lié à un IM. Les enquêtes "Handicap Santé Ménages" et "Handicap Santé Institutions" coordonnées par l’Institut National de Veille Sanitaire ont permis d’estimer la prévalence "déclarée" des patients vivants avec une cardiopathie ischémique (CPI) dans l’ensemble de la population française. La prévalence des CPI est estimée à 2,9 % et celle des patients avec antécédent d’IM à 1,2 %. D’après ces estimations, 1 810 000 patients vivaient avec une cardiopathie ischémique en 2008-2009 et 780 000 patients déclaraient un antécédent d’IM.
3. Physiopathologie de l’athérosclérose coronaire
Considérée longtemps comme une maladie d’accumulation du cholestérol, il est admis actuellement que l'athérogenèse implique une interaction complexe des facteurs de risque avec différents acteurs cellulaires que sont : les cellules de la paroi artérielle, les leucocytes et les plaquettes via une signalisation moléculaire complexe. En particulier, l'inflammation
joue un rôle majeur à chaque stade de son développement 7, dès la phase d’initiation
jusqu’ à la constitution de la plaque mature, puis lors d’évènements aigus engendrant une rupture de plaque.
Lorsque l'endothélium artériel est soumis à des produits de dégradation bactériens et/ou des facteurs de risque aussi divers que la dyslipidémie, les hormones vasoconstrictrices, les produits de glycation conséquence d’hyperglycémie chronique, ou les cytokines pro-inflammatoires produites par un tissu adipeux en excès, les cellules endothéliales augmentent l'expression des molécules d'adhésion favorisant l'adhérence des leucocytes sanguins à la surface interne de la paroi artérielle. Puis la transmigration des leucocytes adhérents se fait selon un gradient d'expression de chimiokines attractantes (Figure 2). Une fois infiltrés dans l'intima artérielle, les leucocytes sanguins principalement les phagocytes mononucléaires et les lymphocytes T, communiquent avec les divers types cellules présents
L’infarctus du myocarde dans la paroi artérielle. Les messages échangés entre les différents types de cellules impliqués dans l'athérogenèse dépendent des médiateurs de l'inflammation et de l'immunité, y compris des petites molécules comme les médiateurs lipidiques tels que les prostanoïdes et d'autres dérivés de l'acide arachidonique, par exemple les leucotriènes. D'autres, comme l'histamine, régulent le tonus vasculaire et augmentent la perméabilité vasculaire. Récemment, une attention particulière a été portée sur les médiateurs protéiques de l'inflammation et de l'immunité, comme les cytokines et les composants du complément. Dès les phases précoces de la maladie, les CML migrent de la media vers l'intima et vont changer de phénotype. Elles prolifèrent et élaborent une matrice extracellulaire riche et complexe. En collaboration avec les cellules endothéliales et les monocytes, elles sécrètent des métalloprotéinases matricielles (MMP) en réponse à divers signaux oxydatifs, hémodynamiques, inflammatoires et auto-immuns. Les MMPs, en équilibre avec leurs inhibiteurs tissulaires endogènes, modulent de nombreuses fonctions des cellules vasculaires, y compris l'activation, la prolifération, la migration et la mort cellulaire, ainsi que la formation de nouveaux vaisseaux et le remodelage géométrique de la structure vasculaire. En situation de déséquilibre, l’excès de production de MMPs va provoquer la déstabilisation de la plaque pouvant aboutir à la rupture de la chape fibreuse
et déclencher le processus thrombotique causant l’IM 8.
Pendant de nombreuses années, l’estimation du risque et les stratégies d’angioplastie coronaire étaient guidées essentiellement par le degré de sténose artérielle. Ce point de vue traditionnel a été bouleversé par les connaissances sur la théorie inflammatoire de l’athérosclérose et la compréhension du remodelage artériel. En effet, il est maintenant établi que lors de la progression de la plaque il existe un remodelage excentrique 9. Ainsi, une charge athéromateuse importante ne cause pas systématiquement de sténose luminale significative, alors que le risque thrombotique existe. Les études récentes utilisant une imagerie ultra sonique endovasculaire ont confirmé les données autopsiques plus anciennes : les sténoses coronaires ne représentent que la « pointe de l'iceberg » de l'athérosclérose et ne permettent pas de présager du risque d’évènement thrombotique. Au stade où les lésions ont progressé au point de produire des sténoses significatives, la charge
athéromateuse est habituellement élevée avec une atteinte diffuse et multifocale 10. Mais,
même en l’absence de sténose, on retrouve lors d’échographie endocoronaire des lésions athérosclérotiques, y compris sur des cœurs issus de donneurs d’organes considérés comme
L’infarctus du myocarde sains 11. Ces caractéristiques d’évolution et de distribution de la maladie athéromateuse rendent aujourd’hui difficile une stratification précise du risque d’évènement coronaire à l’échelle individuelle, et représentent une voie de recherche importante pour guider les procédures d’angioplastie préventives.
Figure 1 : Développement de la plaque d’athérome jusqu’à la rupture et l’occlusion coronaire thrombotique. D’après Peter Libby Nature 2002. L'artère coronaire saine a une
structure trilaminaire typique. Les cellules endothéliales en contact avec le sang dans la lumière artérielle reposent sur une membrane basale. La lame élastique interne forme la barrière entre l'intima et la media sous-jacente. La media se compose de plusieurs couches de CML intégrées dans une matrice riche en élastine et en collagène. Dans l'athérosclérose précoce, le recrutement de cellules inflammatoires et l'accumulation de lipides entraînent la formation d'un noyau riche en lipides, car l'artère s'élargit. Si les conditions inflammatoires prévalent et que les facteurs de risque tels que la dyslipidémie persistent, le noyau lipidique peut se développer et les protéinases sécrétées par les leucocytes activés peuvent dégrader la MEC, tandis que les cytokines pro-inflammatoires telles que l’INF-γ peuvent limiter la synthèse du nouveau collagène. Ces changements peuvent diminuer l’épaisseur de la chape fibreuse et la rendre friable et susceptible de se rompre. Lorsque la plaque se rompt, une coagulation s’en suit à cause du sang qui entre en contact avec le FT. Les plaquettes activées
L’infarctus du myocarde par la thrombine générée à partir de la cascade de coagulation et par le contact avec le contenu du centre nécrotique provoquent la formation du thrombus. Si le thrombus occlue de manière persistante, un infarctus aigu du myocarde peut en résulter (la zone bleue dans la paroi antérieure du ventricule gauche, en bas à droite). Le thrombus peut éventuellement se résorber en raison d'une thrombolyse endogène ou thérapeutique. Cependant, une réaction de cicatrisation provoquée par la thrombine générée pendant la coagulation du sang peut stimuler la prolifération des CML. Le facteur de croissance dérivé des plaquettes (PDGF) libéré des plaquettes activées stimule la migration des CML. Le TFG-β également libéré des plaquettes activées stimule la production interstitielle de collagène. Cette augmentation de la migration, de la prolifération et de la synthèse de la MEC par les CML épaissit la chape fibreuse et provoque un remodelage cette fois ci concentrique, entraînant un rétrécissement de la lumière. Les lésions sténotiques produites par la réduction du calibre luminal de la plaque fibrosée peuvent restreindre l'écoulement, en particulier dans des situations d'augmentation de la demande cardiaque, conduisant à une ischémie, provoquant généralement des symptômes tels que l'angine de poitrine. Les plaques sténotiques avancées, plus fibreuses, sont moins sensibles à la rupture et à la thrombose renouvelée.
4. Physiopathologie du syndrome coronaire aigu et de l’infarctus du
myocarde
Au cours des années 1980, une certaine incertitude a prévalu en ce qui concerne le rôle
causal de la thrombose dans le syndrome coronarien aigu (SCA) 12. Les techniques d'imagerie
in vivo appliquées chez l'homme et le succès du traitement anti- thrombotique et fibrinolytique dans le SCA ont formellement incriminé le rôle de la formation du thrombus intra-coronaire. Aussi, les études autopsiques montrent qu’en cas de thrombose létale, il existe le plus souvent une rupture de la chape fibreuse 8, 13, 14. D'autres mécanismes qui représentent une minorité de thromboses coronaires mortelles incluent l'érosion superficielle, l'hémorragie intraplaque, et la protrusion d'un nodule calcifié 15.
La rupture de plaque déclenche un processus thrombotique par plusieurs mécanismes (Figure 2). Tout d'abord, le contact avec le collagène de la matrice extracellulaire (MEC) de la plaque déclenche l'activation des plaquettes. Deuxièmement, le facteur tissulaire (FT) produit par les macrophages (Mφ) et les CML active la cascade de
L’infarctus du myocarde
coagulation 16. La plaque rompue représente donc un stimulus puissant favorisant à la fois la
thrombose et la coagulation. De plus, ces voies s’auto-maintiennent de manière synergique, car la génération de la thrombine augmente l'activation des plaquettes et d'autres cellules dans la lésion. La conversion du fibrinogène en fibrine et la libération du facteur de Von Willebrand à partir de plaquettes activées peuvent fournir des ponts moléculaires entre les plaquettes qui produisent le réseau de plaquettes densifié et piégé dans la fibrine caractéristique du thrombus artériel "blanc". En plus de cette phase solide de la plaque rompue, la « phase fluide » du sang participe aussi à la thrombose coronaire. L'inhibiteur de l'activateur du plasminogène-1 (PAI-1) inhibe le processus fibrinolytique physiologique en situation d’homéostasie et prévient la persistance et l'accumulation du thrombus en inhibant les activateurs du plasminogène de type tissulaire et de l'urokinase. Ainsi, Il a été rapporté que les taux plasmatiques de PAI-1 sont augmentés dans le diabète, l'obésité et que les médiateurs favorisant l'hypertension artérielle comme l’angiotensine II, augmentent l'expression de PAI-1 par divers types de cellules 17, ce qui pourrait favoriser le processus athérothrombotique. En outre, les plaques rompues peuvent élaborer un FT particulier qui
peut augmenter la thrombogénicité du sang 16. Ces changements de phase fluide en lien
avec les facteurs de risques cardiovasculaires ont conduit à définir la notion de « patient vulnérable » ou à « haut risque ». Parallèlement à la thrombose focale, en regard d’une plaque concernant un vaisseau de gros ou moyen calibre, lors d’un SCA il existe une embolisation distale des débris riches en FT provoquant une occlusion microcirculatoire 18, 19. Une telle embolisation distale explique en partie le phénomène de no reflow ("sans reperfusion") qui peut compliquer une procédure de revascularisation en empêchant une reperfusion tissulaire efficace.
L’infarctus du myocarde
Figure 2 : Recrutement des cellules monocytaires dans la plaque d’athérosclérose.
D’après Peter Libby Nature 2002. Différentes étapes du recrutement des cellules monocytaires au stade d’initiation de la plaque d’athérosclérose. Suite à une inflammation, les cellules endothéliales subissent une activation, ainsi elles augmentent leur expression de diverses molécules d'adhésion. Dans ce contexte inflammatoire le recrutement des monocytes dans l'athérome par des molécules d’adhésion cellulaire vasculaire-1 (VCAM-1) semble jouer un rôle majeur. Après leur adhésion à la couche endothéliale, les monocytes migrent entre les cellules endothéliales intactes pour pénétrer dans l'intima de la paroi artérielle. Cette migration dirigée nécessite un gradient chimio-attractant. Diverses chimiokines participent à ce processus, en particulier l'interaction de la chimiokine de monocytes, MCP-1 avec son récepteur CCR2. Une fois résident dans l'intima, le monocyte acquiert des caractéristiques des Mφ tissulaires. Dans l'athérome en particulier, les Mφ expriment des récepteurs éboueurs qui lient les particules de lipoprotéines, puis celle-ci sont internalisées, modifiées par oxydation ou glycation. Ces processus donnent lieu à une cellule spumeuse, ainsi nommée en raison de l'apparence mousseuse sous microscope, qui est le résultat de l'accumulation de gouttelettes lipidiques dans le cytoplasme. Les cellules spumeuses peuvent rentrer en apoptose, produisant ainsi le « noyau nécrotique » de la lésion athéromateuse.
L’infarctus du myocarde 5. Prise en charge de l’infarctus du myocarde à la phase aigue
Lors d’un SCA, les symptômes, manifestations électrocardiographiques et les éventuelles perturbations hémodynamiques sont le plus souvent les conséquences d’une thrombose coronaire aigüe occlusive ou sub-occlusive, entraînant une ischémie myocardique. Réversible dès les premières heures, elle se traduit par une séquelle ischémique akinétique myocardique si elle persiste. Aussi, la prise en charge médicamenteuse et interventionnelle doit se faire en urgence selon le paradigme « time is muscle » (Figure 3). La stratégie actuelle de ces situations à risque consiste à associer un traitement médicamenteux destiné à limiter l’extension de la thrombose intra coronaire et, en cas d’occlusion complète à la perméabiliser le plus vite possible. Dans ces situations à haut risque, cette stratégie
agressive permet d’améliorer le pronostic des patients 20.
Traitements médicamenteux actuels et perspectives de recherche a)
Actuellement, une association de traitements anti-agrégants plaquettaires et de traitements anticoagulants est recommandée pour limiter l’extension de la thrombose et faciliter les procédures de recanalisation et d’angioplastie éventuelles.
Il est recommandé lors d’un SCA d’administrer le plus tôt possible un bolus d’aspirine associé à un second anti-agrégant de la famille des inhibiteurs de P2Y12 (clopidogrel, prasugrel, ticagrelor). Chez les patients les plus à risque une inhibition supplémentaire des plaquettes peut être obtenue par adjonction d’un inhibiteur des glycoprotéines Gp2b3a. Un traitement anticoagulant d’activité anti Xa et/ou anti IIa est aussi recommandé (héparine non fractionnée ou héparines de bas poids moléculaire, fondaprinux).
Les futures thérapies anti-plaquettaires pourraient offrir un blocage plus complet des récepteurs P2Y1 et P2Y12, dépendant de l’ADP, et inhiber également le complexe facteur de Von Willebrand, glycoprotéine Ib /IX qui favorise l'adhésion plaquettaire dans des conditions où les forces de cisaillement sont élevées. Cette inhibition de l'activation des plaquettes peut donner des avantages au-delà de la prévention de l'agrégation et de la progression du thrombus, en atténuant la libération par les plaquettes de molécules pro-thrombotiques et pro-inflammatoires puissantes responsables de formation d'agrégats de monocytes-plaquettes, ciblant ainsi le cercle vicieux thrombose-inflammation 21, 22. D’autres agents
L’infarctus du myocarde anticoagulants sont en cours d’évaluation dans le SCA, en particulier des inhibiteurs directs et spécifiques de la thrombine et du facteur Xa, ou encore des inhibiteurs du complexe du
facteur VII-FT qui initie la formation du thrombus 23.
L'application des progrès dans la connaissance de la biologie du SCA et du rôle de l'inflammation offre de nouvelles opportunités pour atténuer la thrombogenicité de la plaque, assurer un contrôle plus rapide du décours de la maladie et prévenir une récidive précoce. La thérapie par statines après SCA améliore probablement les résultats en partie en raison des effets anti-inflammatoires attribuables à la baisse du cholestérol et aux actions
anti-inflammatoires directes 24. Certaines études expérimentales ont montré que l'inhibition
de la cyclooxygénase-2 ou du récepteur thromboxane retarde l'athérosclérose 25. Jusqu'à
présent, seuls quelques essais de phase 2 chez l’homme avec SCA ont testé des agents anti-inflammatoires, sans efficacité concluante. Un essai clinique a testé des bolus de méthylprednisolone par voie intraveineuse pendant 48h au décours d’un SCA sans
efficacité 26. Le Pexelizumab, un anticorps monoclonal contre le facteur C5 du complément,
a également échoué dans 2 essais. Le médicament, cependant, a réduit de façon frappante la mortalité et le choc cardiogénique dans le groupe ayant bénéficié d'angioplastie primaire. Ces résultats suggèrent une nouvelle fois un rôle majeur de l'inflammation et du complément dans la mortalité et de la morbidité associée au SCA. En effet, le Pexelizumab empêche la formation du C5a, une anaphylatoxine puissante associée au recrutement de leucocytes ainsi que l'expression de médiateurs pro-inflammatoires, de cytokines pro inflammatoires, de l’enzyme iNOS (inducible Nitric Oxide Synthase), qui favorisent la
mort cellulaire et l'apoptose via le complexe d'attaque membranaire 27. De plus, le
Pexelizumab, lors d’un SCA, permet de réduire significativement les taux de biomarqueurs
d’inflammation associés à la survenue d’un choc cardiogénique de pronostic péjoratif 7, 28, 29.
Compte tenu de leur rôle dans la déstabilisation de la plaque et dans le remodelage vasculaire et myocardique, les MMPs représentent une autre cible thérapeutique potentielle. Les inhibiteurs des MMP sont en cours d'étude dans l'IM aigu.
Méthodes de reperfusion coronaire b)
En cas de syndrome coronaire aigu avec sus-décalage du segment ST sur l’électrocardiogramme, témoignant d’une occlusion coronaire, l’objectif principal n’est plus
L’infarctus du myocarde de limiter l’extension du thrombus mais de re-perméabiliser au plus vite l’artère en cause. Pour ce faire, il existe deux techniques : l’une médicamenteuse, par thrombolyse intraveineuse, l’autre interventionnelle, par angioplastie transluminale percutanée. Afin de privilégier une des deux options, plusieurs critères sont examinés (Figure 3) : la présence d’une contre-indication à la thrombolyse (un traitement anticoagulant en cours ou un risque important de saignement) doit être recherchée mais l’élément principal aujourd’hui est le délai d’accès à une salle de cathétérisme. En effet, si le temps entre l’évènement ischémique et l’admission prévisible du patient en coronarographie est inférieur à 120 minutes, l’angioplastie primaire est privilégiée. A l’inverse en cas de délais >120minutes, une thrombolyse précoce sera réalisée.
(1) Thrombolyse Médicamenteuse
Les premières études concluantes ont été menées au début des années 80, avec l’utilisation intra-coronaire de thrombolytiques. La plus importante d’entre elles a ainsi validé le concept d’une reperfusion précoce de l’artère atteinte avec amélioration de la fonction ventriculaire gauche, réduction de la taille de l’IM et de la nécrose et diminution de la mortalité. La thrombolyse intra veineuse permet une reperfusion coronaire dans la majorité des cas, dans un délai de 30 à 60 minutes, accompagnée par une diminution de la douleur voire une disparition et une baisse du sus-décalage de ST. Cependant, même en cas de succès clinique, la thrombolyse expose à un risque de récidive précoce et tout patient admis pour SCA thrombolysé doit bénéficier d’une coronarographie pour faire le bilan lésionnel coronaire et vérifier angiographiquement la reperfusion. En cas d’échec de reperfusion, une angioplastie de sauvetage doit être pratiquée.
(2) Angioplastie percutanée
L'angioplastie transluminale percutanée est une procédure radio-interventionnelle qui permet, grâce à un cathéter, d’ouvrir les artères occluses et de rétablir le flux sanguin coronaire. Lors d’une angioplastie coronaire, parfois après thrombo-aspiration, un ballon mince et extensible est inséré dans l'artère obstruée et gonflé. Cela permet de restaurer le calibre natif de l'artère en refoulant la plaque sur la paroi artérielle. En 1964, Dotter et Judkins ont été les premiers à tenter l'angioplastie en implantant le premier greffon
L’infarctus du myocarde percutané à l'artère fémorale d'un chien 30. Plus tard, Andreas Gruntzing a développé un ballon à base de polychlorure de vinyle à double lumière cathéter en 1975 et a effectué la première angioplastie chez l'humain en 1977. Depuis, il y a eu de nombreux progrès dans l’intervention coronarienne percutanée (PCI). Dans les années 1990, la révolution a été le
développement d’endoprothèses coronaires appelées stents 31, 32. Différents types de stents
ont été successivement développés : les stents métalliques nus (BMS), les stents « actifs » recouverts de médicament (DES) destinés à limiter la re-sténose intra stent par prolifération de CML dans la néo-intima, DES avec des polymères biodégradables, DES sans revêtement
polymère, et stents auto-expansibles 33. Dès le début des années 90, l’angioplastie primaire,
si elle est réalisée tôt, a montré sa supériorité par rapport à la thrombolyse 34. Plus récemment l’étude Comparison of Angioplastie and Pre-hospital Thrombolysis in Acute Myocardial Infarction (CAPTIM) a montré que l’angioplastie et la thrombolyse pré-hospitalière (TPH), suivie d’angioplastie si nécessaire, sont équivalentes en termes d’efficacité (jugée sur le nombre de décès, de récurrences d’IM et de survenue d’AVC) à
30 jours et à un an 35. Aussi, le choix entre la TPH et l’angioplastie est une question de délai
écoulé depuis l’apparition des symptômes d’une part, et de délai de transport jusqu’à une structure hospitalière d’angiographie coronaire d’autre part. Actuellement, pour les patients pris en charge dans les 2 heures suivant l’apparition des symptômes et ceux pris en charge à plus d’une heure de transport d’un centre de cardiologie interventionelle, une TPH est préférable.
(3) Chirurgie
La revascularisation chirurgicale d’urgence par pontage aorto-coronaire est actuellement indiquée uniquement dans des situations de sauvetage : angor instable subintrant dans le cadre d’une sténose serrée du tronc commun; persistance de l’angor malgré la thrombolyse ou la PCI; infarctus menaçant étendu dans une anatomie défavorable à la PCI; dysfonction ventriculaire sévère sur hibernation; dissection et/ou thrombose coronarienne lors d’angioplastie percutanée ou encore complications mécaniques de l’infarctus : anévrysme avec menace de rupture, rupture pariétale, rupture de pilier mitral, communication interventriculaire massive.
Dans ces situations, malgré une revascularisation, la mortalité reste extrêmement élevée. Suite à un syndrome coronaire aigu, en l’absence de revascularisation, ou même en cas de
L’infarctus du myocarde revascularisation précoce, il se constitue en quelques minutes une nécrose myocardique qui va être le siège d’un processus immuno-inflammatoire complexe à l’origine d’un remodelage tissulaire étendu.
Figure 3 : Remodelage ventriculaire suite aux différentes interventions. D’après Derek
M. Yellon N Engl J Med 2007 Ce schéma montre la forte réduction de la taille de l’IM obtenue par une reperfusion myocardique précoce et réussie après un épisode d'ischémie myocardique aiguë. Cet effet est renforcé par l’adjonction du traitement cardioprotecteur au début de la reperfusion myocardique. Le myocarde infarci est représenté en rose, et le myocarde viable et à risque est rouge. La taille de l'infarctus est exprimée en pourcentage du volume de myocarde infarci.
6. Remodelage ventriculaire et cardiopathie ischémique
Le remodelage cardiaque post infarctus est un processus complexe et dynamique qui associe l’action de l’infiltration myocardique par des cellules inflammatoires, de médiateurs solubles cytokiniques et chimiokiniques, de médiateurs neuro-endocriniens et des conditions de charge, provoquant des modifications transcriptionelles du génome, des changements moléculaires, cellulaires et interstitiels. Macroscopiquement il se traduit par des changements de taille, de forme et de fonction systolique et/ou diastolique.
L’infarctus du myocarde Le cardiomyocyte est la cellule cardiaque cible majeure impliquée dans le processus de remodelage. Les autres composants concernés comprennent des éléments cellulaires comme les fibroblastes et les cellules vasculaires mais également la matrice extracellulaire. Le remodelage cardiaque est un processus physiologique et parfois pathologique qui permet une adaptation à une rupture de l’homéostasie provoquée par différents dommages myocardiques aigus ou chroniques : IM, surcharge de pression (sténose aortique, hypertension), maladie inflammatoire du muscle cardiaque (myocardite), ou encore surcharge volumique (régurgitation valvulaire). Bien que les étiologies de ces maladies soient différentes, elles partagent plusieurs voies de signalisation en termes d'événements moléculaires, biochimiques et mécaniques. Un exemple de remodelage physiologique est observé chez les athlètes, avec une dilatation du ventricule gauche (VG), une hypertrophie ventriculaire et une augmentation des volumes d’éjection systolique.
Dans les modèles post-infarctus, le processus de remodelage du VG commence dès les premières heures et se poursuit pendant plusieurs mois 36, 37. Cependant, le déroulement des événements est influencé par la sévérité de l’atteinte coronaire, les événements secondaires (tels que l'IM récurrent), les conditions de charge et l’activation
neuro endocrine, le génotype et les traitements administrés 38, 39. Les études animales
montrent que l'expansion, la dilatation régionale et l'amincissement de la paroi infarcie peuvent s’observer dès le premier jour post-infarctus 39. L’altération sévère de la fonction ventriculaire globale initiale, qui est un phénomène fonctionnel de sidération transitoire,
doit être différencié clairement du remodelage du VG infarci 40. Bien que la vision intégrative des voies cellulaires et du rôle spécifique des cellules elles-mêmes impliquées dans le remodelage du VG ne soit pas parfaitement établie, le scénario suivant a été proposé au niveau moléculaire. Dès la phase initiale inflammatoire puis lors de la résolution de l’inflammation, l’élévation de la pression de remplissage télédiastolique du VG provoque l’étirement des cardiomyocytes. Cet évènement est le principal mécanisme à l’origine de la reprogrammation de l’expression de leur génome qui conduit à leur hypertrophie et à l’augmentation de la masse du myocarde non nécrosé. Pour maintenir une fonction contractile et un débit cardiaque satisfaisant, les taux tissulaires de norépinephrine locale, d'angiotensine 2 et la libération d'endothéline augmentent. Il en résulte une production accrue de protéines du sarcomère et une hypertrophie compensatrice des zones péri-infarctus. En l’absence de traitement, il se met en place un cercle vicieux avec activation
L’infarctus du myocarde persistante du système sympathique et du système rénine-angiotensine-aldostérone, qui va aboutir à l’insuffisance cardiaque. En outre, l’action de l'aldostérone et des cytokines peut également stimuler la synthèse du collagène, entraînant ainsi une fibrose et un remodelage de la MEC altérant les fonctions diastoliques et systoliques et prédisposant à la survenue de troubles du rythme ventriculaire.
La phase initiale de remodelage menant à la réparation de la zone nécrotique et à la formation de cicatrices peut, dans une certaine mesure, être considérée comme nécessaire et bénéfique. Cependant ces réarrangements cellulaires et matriciels de la paroi ventriculaire de la zone infarcie et du reste du myocarde aboutissent, s’ils sont non contrôlés, à une dilatation plus grande et à une augmentation accrue du stress systolique et diastolique, provoquant à terme une altération de la fraction d’éjection 40. Ce risque est d’autant plus important que la taille de l’infarctus est étendue et la revascularisation nulle ou tardive. Ce remodelage pathologique avec dilatation et altération d’ECG est un prédicteur majeur de la mortalité 41, 42.
On distingue classiquement trois phases dans le processus de remodelage : les premiers jours associent nécrose cellulaire, oedème et inflammation tissulaire faisant intervenir les médiateurs solubles (cytokines et chimiokines) ainsi qu’une séquence d’infiltration myocardique par les leucocytes circulants : les polynucléaires neutrophiles et les macrophages (Mφ) envahissent la zone infarcie et phagocytent les myocytes nécrosés jusqu’à la détersion complète du tissu infarci. Les cellules inflammatoires se localisent principalement au niveau de la zone frontière entre tissu nécrosé et tissu sain, qui augmente jusqu’à la fin de la première semaine, puis décroît.
La seconde phase est la réparation fibrotique de la zone nécrosée. Les myofibroblastes jouent un rôle essentiel dans ce processus, dès le troisième jour qui suit l’infarctus. Au
7e jour, on distingue trois zones dans l’infarctus : la zone centrale nécrosée, entourée d’une
zone de Mφ et de myofibroblastes, puis d’une zone périphérique où prédominent les myofibroblastes.
Un mois après l’infarctus, on ne retrouve pratiquement plus de cellules inflammatoires et les myofibroblastes ont envahi la totalité de la zone infarcie, qui est le siège d’un important dépôt de collagène. Parallèlement, le myocarde sain subit un remodelage hypertrophique associant hypertrophie des myocytes, hyperplasie des cellules non musculaires et fibrose
Réponse immuno-inflammatoire lors d’un infarctus du myocarde
B. Réponse immuno-inflammatoire lors d’un infarctus du
myocarde
1. Effecteurs cellulaires de la réponse immunitaire locale et systémique
post-infarctus
Le recrutement des cellules inflammatoires est un processus dynamique et orchestré comprenant une infiltration séquentielle du myocarde lésé par des neutrophiles, des
phagocytes mononucléés, des cellules dendritiques (DC) et des lymphocytes 43-45. Les
neutrophiles migrent vers le myocarde infarci dès les premières heures suivant l'ischémie. Le pic de l’infiltration neutrophilique survient à J1 post-infarctus. Par la suite les monocytes et les Mφ dominent l’infiltration tissulaire et la libération des médiateurs inflammatoires, d’espèces réactives de l'oxygène (ROS) et d’enzymes protéolytiques. Ils contribuent ainsi à l'initiation et à la résolution de l’inflammation, et orchestrent la majorité des processus physiologiques que sont la phagocytose, la protéolyse, l'angiogenèse, le remodelage et la régénération ventriculaire 46. Les rôles spécifiques des monocytes et des Mφ au cours du post infarctus seront détaillés dans un paragraphe suivant. Parallèlement, les DCs et les lymphocytes T sont recrutés dans le myocarde lésé, ce qui contribue à la cicatrisation et au
remodelage ventriculaire 47. Les effets néfastes et le rôle bénéfique de ces cellules
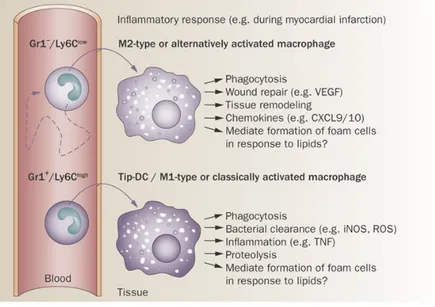
inflammatoires ont été documentés dans la pathophysiologie de l'IM (Figure 4). Leurs rôles divers et même contradictoires peuvent être attribués à l'hétérogénéité des sous-populations et à la diversité fonctionnelle des cellules inflammatoires. 43, 44. Le rôle spécifique de chaque type cellulaire est détaillé ci-après.
Réponse immuno-inflammatoire lors d’un infarctus du myocarde
Figure 4 : L’implication des cellules inflammatoire dans l’infarctus du myocarde.
Zlatanova et al Frontiers Cardiovasc Med 2016
Le nombre, le type et l'état d'activation des différents sous-types de cellules inflammatoires dictent leur impact sur la réparation cardiaque conduisant à un remodelage cardiaque positif ou néfaste post-IM. Rôle de l’immunité innée a) (1) Neutrophiles Les neutrophiles sont indispensables pour la défense contre les microorganismes. Ils sont générés en grand nombre dans la moelle osseuse et circulent dans le sang périphérique avec une demie vie de quelques heures. Si les microorganismes ont réussi à surmonter les barrières physiques fournies par la peau et les muqueuses et ont eu accès aux tissus, les signaux générés par les germes et les Mφ résidents sur les sites d'infection activent les cellules endothéliales locales qui favorisent l’adhésion et le recrutement des neutrophiles et les guident à travers les cellules endothéliales. Les neutrophiles dans les tissus produisent des agents qui amplifient la réponse immune locale et systémique en attirant d’autres neutrophiles, des monocytes/Mφ, des lymphocytes T et régulent leur activité (Figure 5a). La
Réponse immuno-inflammatoire lors d’un infarctus du myocarde production de neutrophiles est quantitativement l'activité principale de la moelle osseuse, où environ deux tiers de l'activité est consacrée à la myélopoïèse, terme utilisé pour la production collective de monocytes et de granulocytes. La production de neutrophiles est
importante à l'état d'équilibre avec 1 à 2x1011 cellules générées par jour chez un adulte. Le
facteur de stimulation des colonies de granulocytes (G-CSF) est essentiel pour accorder la production de neutrophiles ainsi que pour répondre aux besoins accrus pendant les infections, mais le G-CSF n'est pas absolument nécessaire pour la granulocytopoïèse car les souris déficientes pour G-CSF ont approximativement 25% de granulocytopoïèse et génèrent
des neutrophiles complètement matures 48. La production de neutrophiles est largement
régulée par le taux d'apoptose des neutrophiles dans les tissus. Les neutrophiles doivent traverser la paroi vasculaire pour arriver au site d'entrée des microorganismes. La traversée se déroule en grande partie dans les veinules post capillaires. La fixation initiale des neutrophiles aux cellules endothéliales est déterminée par les cellules endothéliales, qui réagissent aux stimuli tels que le TNF-α, l'IL-1β et l'IL-17 qui sont générés lors d'une infection ou d'une inflammation. Une telle stimulation entraîne l'expression de la P-sélectine (également exprimée par les plaquettes activées), la E-selectine, ainsi que les membres de la superfamille des intégrines (les ICAM et VCAM) sur la surface luminale des cellules endothéliales. Ces sélectines se lient à PSGL-1 (ligand de P-sélectine 1) et à la L-sélectine, exprimées de manière constitutive sur les neutrophiles, au niveau des zones de contact intercellulaire organisées notamment par les protéines ézrine, radixine et moesine, (ERM) qui se connectent au cytosquelette d'actine 49-51. La E-sélectine se lie également à l'ESL-1 (ligand de l’E-sélectine 1) présent sur le côté des microfilaments et à CD44, présent sur la membrane cellulaire 51 La liaison de PSGL-1 à la P-sélectine et à la E-sélectine établit le contact initial entre les neutrophiles et les cellules endothéliales activées. E-sélectine et ESL-1 entraînent un roulement plus lent, tandis que la liaison de la E-sélectine à CD44 sert à la médiation d'une redistribution de PSGL-1 et de la L-sélectine pour former des clusters 52 associés à une réduction supplémentaire de la vitesse de roulement. Les molécules clés qui permettre l'adhésion ferme des neutrophiles à l'endothélium activé sont les intégrines β2 LAF-1 (aLb2) et Mac-1 (aMb2), présentes sur les neutrophiles et leurs ligands, membres de la superfamille des immunoglobulines, Intercellular Adhesion Molecule (ICAM)-1 et ICAM-2, présents sur les cellules endothéliales. Lors d’une absence de CD-18, une intégrine de la famille des intégrines de type β, l’adhérence des leucocytes de type I est réduite de part