HAL Id: tel-01618196
https://tel.archives-ouvertes.fr/tel-01618196
Submitted on 17 Oct 2017HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Synthèse et évaluation pharmacologique d’analogues
préactivés de l’ifosfamide : prodrogues et nanoparticules
à visée antitumorale
Charles Skarbek
To cite this version:
Charles Skarbek. Synthèse et évaluation pharmacologique d’analogues préactivés de l’ifosfamide : prodrogues et nanoparticules à visée antitumorale. Pharmacie galénique. Université Paris Saclay (COmUE), 2017. Français. �NNT : 2017SACLS247�. �tel-01618196�
3
REMERCIEMENTS
Je voudrais exprimer mes remerciements aux membres du jury, aux rapporteurs : les Maitres de Conférences Joseph Ciccolini et Sylvie Bégu, ainsi qu’aux examinateurs : les professeurs Elias Fattal, Thierry Martens et Nathalie Chaput ainsi que les docteurs Alain Herrera et Emilie Roger, d’avoir accepté de juger mon travail de thèse.
Je souhaite remercier le Dr Lluis Mir, directeur de l’unité UMR CNRS 8203 pour m’avoir accueilli dans son laboratoire « vectorologie et thérapeutiques anticancéreuses » et de m’avoir permis de réaliser mes travaux de thèse après mon stage de Master 2, au cours desquels j’ai pu développer la formulation des dérivés préactivés de l’ifosfamide et réaliser leur évaluation pharmacologique. Je tiens à exprimer ma profonde gratitude au Pr Angelo Paci, mon directeur de thèse, pour m’avoir accueilli dans l’équipe « vectorologie chimique », pour m’avoir offert l’opportunité de travailler sur un projet de recherche scientifique intéressant et pour m’avoir guidé pendant ces années de thèse. Je le remercie de m’avoir transmis ses connaissances en pharmacologie avec pédagogie ainsi que pour m’avoir encouragé pour présenter mes travaux lors de plusieurs congrès internationaux. J’adresse mes remerciements au Pr Erwan Le Gall pour son accueil au sein de son équipe « électrochimie et synthèse organique » au sein de l’institut de chimie des matériaux Paris Est (UMR CNRS 7182) et de m’avoir permis de réaliser la synthèse des analogues préactivés de l’ifosfamide durant mon stage de master 2 et mes années de thèse.
Je veux adresser un grand merci au Dr Michaël Rivard, maitre de conférences, pour m’avoir transmis tout son savoir dans le domaine de la chimie organique. Je le remercie pour sa grande disponibilité pendant mes expériences de laboratoire. Je le remercie aussi pour sa contribution lors de la rédaction de mon manuscrit.
Je souhaite remercier le Pr Jean-Pierre Benoit ainsi que le Dr Emilie Roger pour leur collaboration et pour la mise en œuvre des travaux d’encapsulation de certains analogues préactivés de l’ifosfamide.
Je voudrais aussi remercier le Pr Nathalie Chaput et les Dr Mélanie Desbois et Dr Sophie Viaud (Laboratoire d’Immunomonitoring en Oncologie, Gustave Roussy) qui m’ont permis d’acquérir une expérience en immunologie lors des travaux sur l’étude de l’effet immunomodulateur de l’ifosfamide. Je remercie aussi plus particulièrement Julia Delahousse qui a participé grandement à cette étude. Je remercie vivement Alain Deroussent, ingénieur de recherche, qui m’a apporté ses connaissances en chimie analytique et son expertise en spectrométrie de masse. Je le remercie aussi pour sa disponibilité lors de la correction et la rédaction de mes articles ainsi que de mon manuscrit.
4
J’exprime ma sincère gratitude à l’ensemble de mes collègues de l’équipe « électrochimie et synthèse organique » et plus particulièrement au Dr Anthony Olivier ainsi qu’à Patrice Renevret qui ont tous les deux participé à la réussite de mon projet de recherche.
Je tiens à remercier le Dr Eric Le Cam, Catherine Durieu et Sonia Baconnais (UMR 8126) pour leur collaboration lors de la réalisation de l’étude des nanosystèmes par microscopie électronique à transmission.
Je remercie aussi le Dr Jean-Rémi Bertrand ingénieur de recherche pour avoir obtenu l’analyseur des nanoparticules par diffusion de la lumière.
Je remercie également l’équipe de la « plateforme d’évaluation préclinique » de Gustave Roussy, et plus particulièrement, le Dr Karine Ser Le Roux et Mélanie Polrot pour leur contribution à plusieurs études précliniques chez la souris.
Je remercie tout particulièrement le Dr Estelle Daudigeos-Dubus, Léa Lesueur, Julia Delahousse, Ludivine Le Dret, Antoine Azan, Alexandre Plessier, Atmane Seck pour leur présence, leur écoute, leur aide au quotidien dans le déroulement de mon projet de recherche ainsi que pour leur participation à mon épanouissement au sein du laboratoire.
Je remercie toutes les stagiaires (Aurore, Sandra, Valentine, Déborah, Naïma...) que j’ai pu encadrer lors de mon doctorat et qui ont participé à l’avancement de ce projet de recherche.
Je remercie enfin l’ensemble de l’unité « vectorologie et thérapeutiques anticancéreuses » de Gustave Roussy pour l’ambiance cordiale au sein du laboratoire.
5
TABLE DES MATIERES
REMERCIEMENTS ... 3
ABREVIATIONS ... 9
LISTE DES ILLUSTRATIONS ... 11
1 INTRODUCTION ... 15
1.1 L
ES OXAZAPHOSPHORINES,
AGENTS ALKYLANTS DE L’ADN ... 15
1.1.1 Historique du développement des agents alkylants ... 15
1.1.2
Mécanisme d’action des agents alkylants ... 16
1.1.3 Généralités sur les oxazaphosphorines ... 17
1.1.4
L’ifosfamide ... 18
1.2 L
ES NANOTECHNOLOGIES... 24
1.2.1 Généralités ... 24
1.2.2 Historique ... 25
1.2.3 Classification des nanomédecines ... 25
1.2.4 Classification selon leur composition ... 28
1.2.5 Utilisation des nanomédicaments en cancérologie ... 34
1.2.6 Caractérisation des nanomédicaments ... 38
1.2.7 Propriétés des nanomédicaments ... 41
1.2.8 Mécanismes de ciblage ... 45
1.2.9 La nanotoxicologie ... 48
2 OBJECTIFS DES TRAVAUX DE THÈSE ... 53
3
ETAT DE L’ART ... 57
3.1 S
TRATÉGIES DE PHARMACOMODULATION... 57
3.1.1 Modification du cycle oxazaphosphorine ... 57
6
3.1.3 Modification de la moutarde alkylante ... 62
3.2 I
MMUNITÉ ET CANCER... 67
3.2.1 Généralités ... 67
3.2.2
L’immunoediting ... 70
3.2.3 Association chimiothérapie/immunothérapie ... 72
3.2.4 Effet immunomodulateur des agents cytototoxiques ... 73
4 RESULTATS ... 77
4.1 A
NALOGUES D’
IFOSFAMIDE PREACTIVES PERMETTANT LA LIBERATION DE LA MOUTARDE ISOPHOSPHORAMIDEE:
SYNTHESE ET PREUVE DE CONCEPT... 77
4.1.1
Introduction de l’article ... 77
4.1.2
Discussion de l’article ... 80
4.2 A
NALOGUES D’
IFOSFAMIDE POLY-
ISOPRENIQUES:
AGENTS ANTITUMORAUX PREACTIVES SOUS LA FORME LIBRE OU NANOPARTICULAIRE... 82
4.2.1
Introduction de l’article ... 82
4.2.2
Discussion de l’article. ... 95
4.3 E
NCAPSULATION D’
ANALOGUES PREACTIVES D’
IFOSFAMIDE AU SEIN DEN
ANOC
APSULESL
IPIDIQUES(NCL
S). ... 97
4.3.1 Introduction ... 97
4.3.2
Etat de l’art ... 97
4.3.3 Formulation de NCLs de pentanyloxy-ifosfamide (P-IFO) ... 99
4.3.4 Matériel et méthodes ... 101
4.3.5 Résultats ... 103
4.3.6 Discussion... 105
4.3.7 Conclusion et perspectives ... 106
4.4 E
FFET IMMUNOMODULATEUR DOSE-
DEPENDANT DES OXAZAPHOSPHORINES:
OPTIMISATION DE LA COMBINAISON DES EFFETS IMMUNOMODULATEUR ET CYTOTOXIQUE... 109
4.4.1
Etat de l’art ... 109
7
4.4.3 Introduction ... 114
4.4.4 Materials and Methods ... 116
4.4.5 Results ... 118
4.4.6 Discussion... 128
4.4.7
Discussion de l’article ... 130
5 DISCUSSION GENERALE ... 132
6 CONCLUSION ... 136
7 PERSPECTIVES ... 144
8 REFERENCES ... 148
9
ABREVIATIONS
ACR : Acroléine
ADC : Antibody-drug conjugates ADH : Aldéhyde déshydrogénase ADN : Acide désoxyribonucléique
Br-IPM : Moutarde bromo-isophosphoramidée CAA : Chloroacétaldéhyde
CD : Cellules dendritiques
CPA : Cellules présentatrice de l’antigène CPM : Cyclophosphamide
CYP : Cytochrome P450
DDL : Diffusion dynamique de la lumière EPR : Enhanced permeability retention FDA : Food and drug administration GSH : Glutathion
HO-IFO : 4-hydroxy-ifosfamide IC50 : Concentration inhibitrice à 50%
IPM : Moutarde isophosphoramidée
LC-MS/MS : Liquid chromatography-tandem mass spectrometry MEB : Microscopie électronique à balayage
Mesna : 2-mercaptoéthane sulfonate de sodium MET : Microscopie électronique à transmission
MTS : (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) NCLs : Nanocapsules lipidiques
NICs : Nano-immuno combos NK : Natural killer
NPs : Nanoparticules
10 PEG : Polyéthylène glycol
PLA : Acide polylactique
PLGA : Acide polylactique-co-glycolique ROS : Reactive oxygen species
SD: Système de délivrance SLN : Solid lipid nanocapsules
11
LISTE DES ILLUSTRATIONS
Figure 1: Structure chimique du gaz moutarde (sulfure de 2,2'-dichlorodiéthyle) ... 15
Figure 2 : Structure chimique de la chlormétine
(2-chloro-N-(2-chloroéthyl)-N-méthyl-éthanamine) ... 15
Figure 3 : Structure chimique des oxazaphosphorines ... 15
Figure 4 : Structure chimique du 2-mercaptoéthane sulfonate de sodium ou mesna ... 22
Figure 5 : Liste non-exhaustive d’applications médicales des nanomédecines (adaptée de
[100] ... 25
Figure 6 : Représentation des trois générations de nanosystèmes [119] ... 27
Figure 7 : Illustration et exemples de structures 0D, 1D, 2D and 3D (adaptée de [72]) ... 28
Figure 8 : Représentation de nanosystèmes à base de polymères, (A) nanosphères, (B)
micelles polymériques et (C) dendrimères. ... 29
Figure 9: Liposomes unilamellaires [129] ... 30
Figure 10 : Liste des techniques courantes utilisées pour synthétiser des nanosystèmes à
base de silice [158] ... 33
Figure 11: Proportions de nanomédicaments en cours d’essais cliniques en 2016 [188] .. 36
Figure 12 : Schéma des principales voies d'internalisation cellulaire : (A) la phagocytose et
les différentes voies d’endocytose (B) (C) (D) et (E) [206] ... 41
Figure 13 : Les étapes de phagocytose d’un nanosystème [206] ... 42
Figure 14: Représentation du ciblage actif [235] ... 47
Figure 15 : Evolution du nombre de publications relatives aux nanotechnologies et à la
toxicité des nanotechnologies [242] ... 48
Figure 16 : Structure chimique d’analogues soufrés du CPM ... 57
Figure 17 : Numérotation du cycle oxazaphosphorinine ... 58
Figure 18 : Structure chimique de l’analogue 4-méthyle-cyclophosphamide ... 58
Figure 19 : Structure chimique du 4-hydropéroxy-ifosfamide (A) et du 4-hydroxy-ifosfamide
(B) ... 59
12
Figure 21 : Exemples de substitutions en position 5 ou 6 ... 59
Figure 22 : Structure chimique d’analogues polycyclique à jonction [5,6] ... 60
Figure 23 : Structure chimique d’analogues par remplacement d'halogène(s) ... 60
Figure 24 : Structure chimique du 7,9-diméthyle-ifosfamide (7,9-diMe-IFO)... 61
Figure 25 : Structure chimique de l'estramustine ... 62
Figure 26 : Structure chimique de la bendamustine ... 63
Figure 27 : Structure chimique du melphalan ... 63
Figure 28 : Structure chimique du glufosfamide ... 64
Figure 29 : Structure chimique du sel de trométhamine-palifosfamide ... 64
Figure 30 : Structure chimique du sel de lysine-palifosfamide ... 64
Figure 31 : Structure chimique du l’évofosfamide ... 65
Figure 32 : Cellules de l’immunité innée et adaptative [368] ... 67
Figure 33 : Représentation d’une nanocapsules lipidique (adaptée de [86]... 98
Figure 34 : Courbes de survie du P-IFO sous la forme libre (●), encapsulé dans les NCLs
(■) ou des NCL non chargées (▲) sur les lignées A673 (A) ou RMS-1 (B) après 72h
d’incubation ... 104
Figure 35 : Cinétique de libération du P-IFO et du HO-IFO à partir des NCLs chargées (n=2)
... 105
Figure 36: Représentation chimique du cyclophosphamide... 109
Schéma 1 : Mécanisme de formation de l’aziridinium ... 16
Schéma 2 : Mécanisme d’alkylation de la moutarde azotée ... 16
Schéma 3: Métabolisme de l’IFO ... 20
Schéma 4 : Disciplines entrants en jeux lors du développement de nanomedecament .... 37
Schéma 5 : Principe de la technique de diffusion dynamique de la lumière [200] ... 39
Schéma 6 : Méthodologie de détermination du potentiel zêta (HORIBA Scientific) ... 40
Schéma 7 : Les voies d’endocytose [206]... 43
13
Schéma 9 : Caractéristiques du CPM et de l'IFO ... 53
Schéma 10: Représentation schématique du projet relatif à la préactivation de l’IFO. ... 55
Schéma 11 : Représentation schématique du projet
relatif à l’immunomodulation par les
oxazaphosphorines ... 56
Schéma 12 : Métabolisme des analogues 7,9-diMe-IFO ... 61
Schéma 13 : Représentation de la théorie d' « immunoediting » [387] ... 70
Schéma 14 : Représentation schématique du projet relatif à la préactivation de l’IFO. ... 79
Schéma 15: Représentation schématique du projet relatif à la préactivation de l’IFO. ... 94
Schéma 16 : Représentation schématique de la méthode d’inversion de phase (adaptée de
[414])... 99
Schéma 17: Représentation schématique du projet relatif à la préactivation. ... 107
Schéma 18 : Etude de l’effet immunomodulateur de l’IFO comparativement au CPM .... 111
Schéma 19 : Mise en évidence de l'effet immunodulateur de l'IFO ... 130
Schéma 20 : Synthèse d’analogues préactivés de l’ifosfamide ... 136
Schéma 21 : Synthèse et évaluation pharmacologique in vitro des analogues X-IFO : ... 137
Schéma 22 : Evaluation pharmacologique in vivo de l’analogues G-IFO ... 139
Schéma 23 : Preuve de concept de l'effet immunomodulateur de l'IFO ... 141
Schéma 24 : Pégylation et évaluation pharmacologiques des nano-X-IFO ... 144
Schéma 25 : Combinaison de la méthylation et de la préactivation de l’IFO donnant lieu à
de nouveaux analogues de l’IFO ... 145
Schéma 26 : Stratégies de combinaison de la préactivation et de l’effet immunomodulateur
des oxazaphosphorines ... 146
15
1 INTRODUCTION
1.1
Les oxazaphosphorines, agents alkylants de l’ADN
Historique du développement des agents alkylants
Lors de la première guerre mondiale (1914-1918), l’armée allemande a mis au point en 1917 un nouveau gaz de combat, le gaz « moutarde », nom caractéristique lié à sa couleur et à son odeur irritante (Figure 1).
Figure 1: Structure chimique du gaz moutarde (sulfure de 2,2'-dichlorodiéthyle)
Les victimes de ce gaz moutarde présentaient, outre les brûlures des voies respiratoires et digestives supérieures, une diminution des éléments figurés du sang, ou leucopénie, pouvant aller jusqu’à des aplasies fatales [1, 2]. Puis, l’utilisation clinique de ces composés cytotoxiques dans les années 1940 marque les débuts des traitements de chimiothérapie anticancéreuse avec la modification structurale du gaz moutarde en chlorméthine (Ledaga™) [3-5] (Figure 2).
Figure 2 : Structure chimique de la chlormétine (2-chloro-N-(2-chloroéthyl)-N-méthyléthanamine)
La chlorméthine est la première moutarde à l’azote à être utilisée dans le traitement de la maladie de Hodgkin [5]. Comme toutes les moutardes à l’azote, la chlorméthine est un agent bisalkylant, porteur de deux chaînes chloroéthyles responsable de la propriété alkylante.
Cependant, sa très grande instabilité en milieu aqueux et sa faible spécificité vis-à-vis des cellules tumorales est un frein à son utilisation. Pour pallier ces inconvénients, il a été envisagé une cyclisation de la moutarde à l’azote, ayant pour finalité la formation d’une prodrogue stable et permettant une libération de l’entité active uniquement après métabolisation. Cette stratégie a servi au développement de la synthèse des oxazaphosphorines dont les principaux représentants sont : le cyclophosphamide (CPM) et l’ifosfamide (IFO) porteurs de deux chaînes, et chloroéthyles ainsi que le trofosfamide porteur de trois chaînes chloroéthyles (Figure 3).
16
Ces composés anticancéreux représentent un groupe important d’agents alkylants, ayant une activité antinéoplasique, accompagnée d’une activité immunomodulatrice dans le cas du CPM. Ils sont encore très utilisés en clinique, aussi bien en oncologie qu’en hématologie dans le cas du CPM et de l’IFO [6-8], qu’en médecine interne et en rhumatologie pour ses propriétés immunosuppressives dans le cas du CPM [9, 10].
Mécanisme d’action des agents alkylants
Les agents alkylants sont des molécules qui possèdent la propriété d’interagir directement avec les bases de l’ADN. Cette interaction mène à la formation de liaisons covalentes avec généralement l’azote en position 7 de la guanine via la formation d’une substance électrophile. Dans ces composés, ce sont les chaînes chloroéthyles qui sont responsables de l’alkylation via la formation de l’ion aziridinium (Schéma 1).
Schéma 1 : Mécanisme de formation de l’aziridinium
Le mécanisme d’alkylation correspond à une réaction de greffage d’une chaîne alkyle sur un radical accepteur par une liaison covalente correspondant à une réaction de substitution nucléophile d’ordre 2. Une première chaîne chloroéthyle subit une cyclisation avec le départ de l’ion chlorure permettant la formation de l’ion aziridinium (intermédiaire électrophile). Puis, le doublet non-liant de l’azote en position 7 de la guanine réagit ensuite avec l’arizidinium permettant la formation d’une liaison covalente entre l’agent alkylant et l’ADN. La seconde chaîne chloroéthyle subit la même réaction permettant la formation d’une seconde liaison covalente avec l’ADN (Schéma 2).
17
Ces deux liaisons peuvent être inter-caténaires (entre deux brins complémentaires d’ADN) ou intra-caténaires (dans le même brin d’ADN). La formation de ces liaisons covalentes engendre une rigidité de l’ADN modifiant ainsi sa structure. Ceci a pour conséquence le blocage des processus normaux du fonctionnement de l’ADN, tels que sa réplication et sa transcription. L’alkylation conduit finalement à la mort cellulaire par apoptose [11].
Généralités sur les oxazaphosphorines
Parmi les principaux représentants des oxazaphosphorines (Figure 3), l’IFO et le CPM sont des isomères de position liés à la présence des deux groupements chloroéthyles se trouvant, soit sur les azotes endocyclique et exocyclique dans le cas de l’IFO, soit exclusivement sur l’azote exocyclique dans le cas du CPM, (Figure 3). La première synthèse du CPM a été décrite en 1954 et suivie de sa commercialisation en 1959 par les Laboratoires Asta Medica puis par Baxter Oncology sous le nom d’Endoxan® [12, 13]. Concernant l’IFO, sa première synthèse a été décrite
dans les années 1960 suivie de sa commercialisation en France depuis 1977 par les Laboratoires Asta Medica puis Baxter Oncology sous le nom d’Holoxan® [14, 15]. Ces deux composés, en
mélange racémique, sont administrés par voie parentérale. De plus, le CPM est aussi administré par voie orale à faible dose (Endoxan® 50mg). Bien qu’il s’agisse d’isomères, ces deux composés
présentent des activités différentes liées à leurs métabolismes distincts (Tableau 1).
18
L’effet cytotoxique des oxazaphosphorines dépend de leur capacité d’alkylation de l’ADN. Comme prodrogues, elles sont métabolisées principalement dans le foie par les cytochromes P450 (CYP) menant à la formation de la moutarde azotée après oxydation du carbone en position 4 du cycle oxazaphosphorine. Cette moutarde azotée entre ensuite dans le noyau afin de réagir avec les bases de l’ADN, et plus particulièrement l’azote en position 7 de la guanine, par liaison covalente du groupe actif à l’azote de la guanine aboutissant à la formation de ponts inter- ou intra-brins provoquant une toxicité tissulaire et la mort cellulaire [16, 17]. D’autres bases de l’ADN peuvent aussi être ciblées par l’action de la moutarde azotée comme l’azote N-1 et N-3 de l’adénine, l’azote N-3 de la cytosine, et l’oxygène O-6 de la guanine mais elles sont plus rares [18].
L’ifosfamide
1.1.4.1 Présentation
L’IFO possède deux groupements chloroéthyles, l’un sur l’azote exocyclique et l’autre sur l’azote endocyclique (Figure 3). Il est administré par voie parentérale uniquement et est indiqué dans le traitement de divers types de cancers : lymphomes non Hodgkiniens, maladie de Hodgkin en rechute, cancer de l’ovaire, du col utérin et des testicules en rechute, les cancers pulmonaires et des voies aéro-digestives supérieures dont les cancers bronchiques à petites cellules ou non, les sarcomes des tissus mous, les cancers osseux chez l’homme et l’enfant, ainsi que les cancers du sein métastatiques. L’administration s’effectue principalement par voie intraveineuse en perfusion lente (1,5 g/m2/h) en perfusion continue sur 24, 72, 96 heures, et jusqu’à 7 jours. Selon le traitement
et l’indication thérapeutique, la posologie est réalisée suivant quatre schémas d’administration, d’après Baxter sur Holoxan_RCP :
1) en association, 1 à 3 g/m²/j par cycles courts de 3 à 5 jours, renouvelables toutes les 3 à 4 semaines avec une dose totale / cycle de 3 à 15 g/m² ;
2) en perfusion continue, sur 24 heures, 5 à 8 g/m², à renouveler toutes les 3 à 4 semaines ; 3) à fortes doses, en perfusion continue, sur 5 jours, 10 g/m², à renouveler toutes les 3 à 4 semaines ;
4) ou à hautes doses, en perfusions répétées sur 6 à 7 jours, la dose maximale tolérée est de 24 g/m², à renouveler toutes les 3 à 4 semaines.
Du fait de la relation entre l’efficacité cytotoxique et la dose administrée, des protocoles dits « hautes doses » ont été développés permettant de surmonter les résistances tumorales [19]. La posologie de ces protocoles peut ainsi atteindre des doses totales de plus de 20 g/m², administrées en 6 à 7 jours, répétées toutes les 3 à 4 semaines.
19
1.1.4.2 Métabolisme
L’IFO est intrinsèquement inactif : il s’agit d’une prodrogue qui, pour libérer l’entité active, doit subir une activation enzymatique principalement dans le foie par les CYP. Ces enzymes intracellulaires sont situées au sein du réticulum endoplasmique des hépatocytes ainsi que dans toutes les cellules détenant ce type de matériel enzymatique (cellules rénales, cellules du tube digestif…). Cette activation permet la libération de la moutarde isophosphoramidée (IPM) qui constitue la forme active responsable de l’activité alkylante. On distingue deux voies de métabolisme pour l'IFO (Schéma 3). 1) La première, la voie activatrice dite "anabolique", consiste en l’oxydation enzymatique par l’isoforme CYP3A4 en position 4 du cycle de l'IFO conduisant au 4-hydroxy-ifosfamide (HO-IFO), qui est en équilibre avec sa forme tautomère, 4-aldo-Ifosfamide (aldo-IFO). Cependant, cette voie activatrice permet la libération du HO-IFO à hauteur d’environ 10 à 20 % [20]. Deux types de réactions peuvent ensuite avoir lieu :
a) une réaction chimique de type « retro-Michael », à l’origine de la libération de la moutarde isophosphoramidée, correspondant à l’entité active ayant l’activité bisalkylante, accompagnée d’acroléine (ACR), un aldéhyde hautement réactif et responsable d’urotoxicité.
b) une oxydation de l’aldo-IFO par l’aldéhyde déshydrogénase humaine de type 1 (ADH-1) menant au carboxy-ifosfamide (carboxy-IFO) inactif et sans activité alkylante.
2) La deuxième voie de métabolisme, la voie toxicogène dite "catabolique", consiste en l’oxydation des chaînes latérales par les isoformes CYP2B6 et CYP3A4 [21]. Cette voie conduit à la déchloroéthylation de l’IFO en trois métabolites inactifs N-déchloroéthylés, N2
-déchloroéthyl-ifosfamide (N2-DCE-IFO), N2,N3-di-déchloroéthyl-ifosfamide (N2,N3-di-DCE-IFO) et N3
-déchloroéthyl-ifosfamide (N3-DCE-IFO). Cette déchloéthylation est accompagnée de la production
en quantité équimolaire du chloroacétaldéhyde (CAA), qui est responsable de neurotoxicité et de néphrotoxicité [16, 22, 23]. Plus de la moitié de la dose d’IFO administrée subit la voie métabolique de N-déchloroéthylation [20] qui conduit à une libération importante de CAA.
20
Schéma 3: Métabolisme de l’IFO
L’IFO est peu lié aux protéines plasmatiques et sa distribution tissulaire semble être importante. Dans une étude menée chez l’enfant, il a été montré que les concentrations d’IFO dans le plasma et dans le liquide céphalo-rachidien étaient proches, mais ses métabolites les plus polaires franchissaient moins bien la barrière hémato-encéphalique [22]. Les métabolites de l’IFO sont éliminés par voie urinaire ou se lient définitivement à leurs cibles. Au final, moins de 20% de la dose administrée est retrouvée sous forme inchangée dans les urines [20, 24]. Il est noté que le métabolisme de l’IFO peut être affecté par divers paramètres comme la composition du produit (sous la forme de mélange racémique ou d’énantiomères purs) [25], la dose administrée [26, 27] ainsi que les différences physiologiques des patients (sexe, âge, dysfonctionnement hépatique ou rénal). Comme l’IFO est un produit chiral du fait que le phosphore constitue un centre asymétrique, il est administré en tant que mélange racémique. Les deux énantiomères sont ainsi présents (S-IFO et le
R-IFO) avec une distribution égale entre les deux énantiomères. Cependant, la métabolisation des
deux énantiomères est différente et donc leur toxicité l’est aussi. Il existe ainsi une préférence énantiosélective pour l’énantiomère S-IFO ayant ainsi un impact sur le métabolisme, la pharmacocinétique et donc sur l’efficacité des deux énantiomères [25, 28, 29].
21
1.1.4.3 Inconvénients liés au métabolisme de l’IFO
Le métabolisme de l’IFO présente de nombreux inconvénients liés à la variabilité de la voie d’oxydation et aux réactions d’inactivation pouvant influencer son efficacité en clinique.
Faibles hydroxylation et inactivation de l’IFO
Alors même que la réaction d’oxydation du cycle de l’IFO est à l’origine de la libération de l’entité alkylante, le métabolite intermédiaire HO-IFO qui en résulte n’est produit qu’en faible quantité. Ainsi, un taux d’hydroxylation d’environ 10 à 20 % est rapporté, avec une forte variabilité selon les patients, en particulier s’agissant de l’expression des différents isoformes du CYP3A4 [22, 30-33]. Comme indiqué précédemment, après hydroxylation, l’aldo-IFO, peut ensuite libérer le métabolite actif, la moutarde azotée, après avoir subi une réaction de type rétro-Michaël. Dans certains cas, l’aldo-IFO peut, après action de l’ALDH-1, se transformer en un métabolite inactif, le carboxy-IFO [16, 21, 27, 34]. La voie catabolique de l’IFO induit quant à elle une oxydation des chaînes latérales avec production de métabolites déchloroéthylés inactifs. La surexpression de certaines isoformes de CYP favorisant la voie catabolique peut amener à diminuer l’efficacité de l’IFO. Il existe différents isoformes possédant des activités cataboliques plus ou moins fortes et des variations de leurs expressions selon les individus [29, 31]. Cette déchloroéthylation de l’IFO représente 50 à 80 % de la métabolisation de l’IFO et varie selon les individus [11, 23, 30, 35].
Effets indésirables / Toxicités
En plus des effets secondaires inhérents aux traitements utilisant les agents alkylants (nausées, vomissements…), l’IFO possède d’autres toxicités spécifiques liées à son métabolisme :
1) une urotoxicité, due à l’ACR libérée concomitamment à la moutarde azotée, et commune à toutes les oxazaphosphorines [16, 36, 37],
2) une néphrotoxicité et une neurotoxicité dues à la libération de CAA en quantité équimolaire avec les composés déchloroéthylés, caractéristiques de l’IFO [18, 23, 24, 31, 38].
Urotoxicité
L’un des freins au développement de l’IFO dans les années 1980 a été l’urotoxicité, due à l’ACR, un des métabolites hépatiques de l’IFO. Celle-ci est caractérisée par une hématurie et des cystites hémorragiques pouvant menacer le pronostic vital du patient. En raison de cette toxicité sévère, l’utilisation de l’IFO a été limitée jusqu’au début des années 1990 [39]. En effet, l’ACR éliminée par voie rénale, présente une urotoxicité obligeant à l’arrêt du traitement. L’ACR est aujourd’hui
22
neutralisée par la co-administration systématique du 2-mercaptoéthane sulfonate de sodium (mesna, Uromitexan®) (Figure 4) [37, 40].
Figure 4 : Structure chimique du 2-mercaptoéthane sulfonate de sodium ou mesna
Co-administré avec l’IFO, le mesna réagit avec l’ACR conduisant à un composé thio-éther stable et non toxique, facilitant ainsi l’excrétion puis l’élimination de l’ACR tout en neutralisant sa toxicité [39-43]. Une hydratation adaptée du patient ainsi que l’administration systématique de mesna permet ainsi de prévenir ou diminuer cette urotoxicité rendant possible l’utilisation de l’IFO à hautes doses [6, 19, 44, 45].
Néphrotoxicité
La néphrotoxicité de l’IFO, due à la libération de CAA après oxydation des chaînes latérales, est maintenant bien décrite, tant chez l’adulte que chez l’enfant [46-48]. D’importantes variations interindividuelles sont observées concernant la sévérité, la survenue ainsi que le type d’atteinte de cette toxicité. Les tubules rénaux proximaux représentent la principale cible de cette néphrotoxicité. Elle peut toucher l’ensemble du néphron conduisant à un dysfonctionnement, soit glomérulaire, soit tubulaire, et dans certains cas la combinaison de toutes ces atteintes peut être observées [38, 47-53]. Le CAA peut aussi induire un syndrome de Fanconi associé à une acidose tubulaire proximale accompagnée de résorption tubulaire [46]. De ce fait, cette néphrotoxicité est un facteur limitant lors de l’utilisation de protocoles hautes doses, notamment chez l’enfant [54-56]. Lors de la métabolisation de l’IFO par la voie catabolique une forte N-déchloroéthylation est observée et une quantité équimolaire de CAA à l’origine de la néphrotoxicité est libérée [49, 57]. Le mode d'action toxique du CAA semble lié à une atteinte du métabolisme énergétique cellulaire par déplétion de la concentration cérébrale en glutathion (GSH) [58]. Contrairement à l’urotoxicité qui peut être contrée via l’administration de mesna, rien ne permet en revanche de prévenir la néphrotoxicité de l’IFO [59]. D’autres molécules, telles que l’amifostine, ont été proposées pour réduire cette néphrotoxicité [59]. La N-acétylcystéine a aussi montré un effet éventuel contre cette toxicité de l’IFO chez l’enfant [60].
Neurotoxicité
L’IFO ainsi que ses métabolites les plus lipophiles (CAA et N-DCE-IFO) ont la possibilité de pénétrer la barrière hémato-encéphalique et, de ce fait, se trouver dans le liquide céphalorachidien en concentration du même ordre que celle observée dans le sang [33, 61]. La présence de ces métabolites en concentration importante provoque des effets toxiques sur le système nerveux central assimilés à une encéphalopathie, qui se caractérise par divers effets secondaires tels qu’un
23
état épileptique, la dégénération du cortex cérébral, des hallucinations visuelles ou encore une confusion mentale [36, 62, 63]. Cette encéphalopathie peut avoir plusieurs sources, soit un déficit mitochondrial des acides gras menant à l’inhibition du transfert d’électrons aux flavoprotéines [64, 65], soit via l’accumulation du CAA dans le liquide céphalorachidien menant à une neurotoxicité directe caractérisée par une déplétion de GSH au niveau du système nerveux central [36]. Chez certains sujets, cette neurotoxicité centrale impose l’arrêt du traitement et conduit à des échecs thérapeutiques avec très peu d’alternatives. Les conditions précises de survenue de cette neurotoxicité centrale et/ou périphérique d’origine métabolique restent cependant mal définies [23].
Cardiotoxicité
A fortes doses, l’IFO induit une myocardite toxique pouvant aboutir dans certains cas à une nécrose diffuse du cœur. Les observations cliniques peuvent être variables allant de la péricardite à la nécrose du myocarde en passant par l’insuffisance cardiaque [66, 67].
24
1.2 Les nanotechnologies
Généralités
Le terme de nanotechnologie fait référence à une nouvelle discipline visant à concevoir des objets de taille de l’ordre de l’échelle nanométrique (10-9m). Le préfixe "nano", du grec "nanos" signifiant
nain, est associée à diverses disciplines pour faire référence à l’utilisation d’objets dont la taille est de l’ordre du nanomètre, telles que :
1) les nanosciences, qui peuvent être définies comme l'étude des phénomènes et de la manipulation de matériaux à l'échelle atomique et moléculaire ;
2) les nanotechnologies, qui sont liées à la conception, la caractérisation, la production et l'application de structures, dispositifs ou systèmes par contrôle de leur forme et de leur taille au niveau nanométrique ;
3) parmi les nanotechnologies, les nanotechnologies pharmaceutiques et médicales, également appelées nanomédecine, englobent les applications de la nanoscience à la santé sous la forme de nanomatériaux ou nano-objets inertes de taille submicronique ou de dispositifs utilisés pour le traitement, le diagnostic, l'imagerie et l’adressage spécifique au niveau biologique.
A l’échelle du nanomètre, il est possible de concevoir une multitude d’objets tels que : des cristaux, des sphères, des fils et des tubes dont l’utilisation changent le mécanisme d’interaction moléculaire. De nos jours, les nanotechnologies peuvent être employées dans des domaines aussi variés que les matériaux et produits courants (tissus, cosmétiques, etc.) [68-70], l'électronique avec le développement de nano-composites et supports informatiques [71], les applications énergétiques durables (cellules solaires) [72] ainsi que les nanocapteurs [73]. Cependant, le terme de nanotechnologie est largement représenté par ses applications dans le domaine de la santé, l'imagerie médicale, le diagnostic [74, 75] et le traitement du cancer [76-81]. Le développement des nanotechnologies dans le domaine de la pharmacie et pour le traitement de maladies graves est en pleine expansion depuis le début des années 2000 [82, 83]. Ainsi, une multitude de types de nanotechnologies sont mis au point dans le domaine de la médecine, comme le développement de liposomes [84-88], de nanoparticules pour l'administration de médicaments (nanomédicaments) [75, 89-91], ou encore d’agents de contraste pour l’imagerie [92]. Lorsqu'elle est appliquée au domaine de la médecine, plus que la taille de la nanotechnologie, le système de délivrance (SD ou DDS en anglais) du médicament idéal peut être obtenu via l’utilisation de matériaux spécifiques ou d’objets. Ceux-ci ont l’avantage, dans certain cas, d’être biodégradables et également biocompatibles afin de diminuer tout effet indésirable potentiel ou une accumulation toxique dans l’organisme [93].
25
Historique
L'objectif principal d'un système de délivrance est de permettre une libération des agents thérapeutiques au niveau du site d'action souhaité, à une concentration appropriée et pendant la durée souhaitée. Faraday est considéré comme le premier à avoir découvert à la fin du 19e siècle
des objets de taille nanométrique à la suite de ses travaux sur les relations expérimentales de l'or et des autres métaux à la lumière [94]. Puis en 1959, Feynman a donné une conférence à l'American
Physical Society décrivant les immenses possibilités offertes par la miniaturisation. Cette conférence
est maintenant considérée comme la pierre angulaire de l'invention des nanotechnologies. En outre, ses travaux ont été également récompensés par le prix Nobel de physique en 1965 [95], suivis de la conception de systèmes à libération prolongée des médicaments dans les années 1970 [96]. De nos jours, les applications des nanotechnologies au service de la santé sont en plein essor tant dans le domaine du traitement de maladies tels que le cancer, les troubles inflammatoires et les maladies cardiovasculaires, que dans le domaine du diagnostic [75, 95, 97].
Classification des nanomédecines
Le terme de « nanomédecine » correspond à l'application de la nanotechnologie dans le domaine de la santé. Elle conduit à la conception et à l'utilisation de nanomatériaux ou d’objets finement construits permettant le développement de nouvelles thérapies et de nouveaux produits de diagnostic. Leurs applications sont nombreuses (Figure 5), parmi lesquelles l'administration de médicaments, l'imagerie in vivo ou le diagnostic in vitro [98, 99].
26
Le développement de ces nanomédecines permet une collaboration étroite entre la science fondamentale et l’ingénierie complexe qui intègre la chimie, la physique et la biologie des nanostructures. Les nanomédecines englobent ainsi l’élaboration, le développement et la production de nouvelles architectures nanostructurées, de nanomatériaux fonctionnels et de nanocomposants intelligents possédant des propriétés uniques [68, 101]. L’ensemble des laboratoires de recherche privés ou publics ou des entreprises de pointes ont aujourd'hui consacré de nombreux efforts au développement de ces nanomédecines menant à une révolution dans le domaine de la science et plus particulièrement dans la délivrance des médicaments par vectorisation de ces composés au sein d’un système de délivrance adéquat et donnant lieu aux « nanomédicaments » [102, 103]. Certaines de ces nouvelles nanomédecines sont entrées sur le marché ou en développement, quand bien d'autres sont encore au stade de la recherche. Le potentiel de ces innovations est énorme, mais leurs utilisations à long terme et leurs sécurités / toxicités sont encore inconnues.
La médecine conventionnelle se situe dans l’échelle du submicron et son action est souvent limitée en raison de sa faible spécificité. De ce fait, le développement de nanomédicaments, permet une augmentation de la spécificité d’action du composé et conduit à des progrès significatifs en termes de capacité de passage des différentes barrières biologiques [104]. Les nanomédicaments peuvent être classés selon différentes caractéristiques, telles que la dimensionnalité, la morphologie, la composition, l’homogénéité et l’agglomération.
1.2.3.1 Classification selon la complexité
Les nanomédicaments peuvent être classés selon leur complexité en trois grands types de générations (Figure 6).
Selon cette classification, la première génération décrit des systèmes d'administration de médicaments qui ciblent la lésion par un mécanisme passif utilisant l'effet EPR (Enhanced
Permeability and Retention effect) proposé par Maeda [105] . Parmi ceux-ci, nous pouvons citer les
liposomes [84, 88], les conjugués polymériques [106-108] ou lipidiques [86, 109], les micelles [110] ou les dendrimères [111, 112]. Cependant, ces produits de première génération se concentrent principalement au niveau du foie en raison de leur adsorption sur les protéines (opsonines) ce qui conduit ensuite à leur reconnaissance par les macrophages du foie. Comme indiqué précédemment, les opsonines qui s’adsorbent sur les nanosystèmes sont à l’origine de la capture hépatique. Pour y remédier, la « décoration » du nanosystème via l’utilisation de polymères hydrophiles et flexibles, tel que le polyéthylène glycol (PEG), permet d’inhiber l’adsorption des opsonines à la surface du nanosystème. Ces systèmes sont dits de deuxième génération. Ceci a mené au développement de nanosystèmes dits « furtifs » n’étant plus reconnus par le système réticulo-endothélial et menant à de nouvelles propriétés, tel qu’un temps de circulation accru. Cette augmentation du temps de circulation (ou du temps de résidence moyen) va ainsi permettre le ciblage passif de la tumeur par
27
concentration pharmacocinétique [113-115]. Cette deuxième génération peut ensuite évoluer vers des nanosystèmes d’un nouveau type ayant la propriété de cibler spécifiquement un type cellulaire. Cette troisième génération de nanosystèmes, aussi connue sous la dénomination de nanosystème « fonctionnalisé », possède des fonctionnalités avancées telles que :
1) la fixation de fragments de reconnaissance à la surface des nanosystèmes pour permettre une reconnaissance d’un récepteur cible bien identifié [116, 117], ou,
2) la possibilité d'une libération programmée du médicament au niveau de site actif [88, 118].
Figure 6 : Représentation des trois générations de nanosystèmes [119]
Ces générations successives de nanosystèmes sont, pour la plupart, une évolution progressive du nanosystème de première génération associée à une augmentation de sa sophistication.
1.2.3.2 Classification selon la dimension et la morphologie
Tout autant que sa complexité, la forme et la morphologie du nanosystème jouent aussi un rôle important sur ses propriétés intrinsèques. Selon la classification la plus récente [120], les nanosystèmes peuvent être classés en nanostructures de dimension zéro (0D), à une dimension (1D), à deux dimensions (2D) et à trois dimensions (3D) (Figure 7). La structure 0D correspond habituellement aux nanosystèmes dirigés dans les trois directions. Leur forme peut être sphérique (nanoparticules ou nanopores), cubique et polygonique. En ce qui concerne leur taille, elle varie de quelques nanomètres à quelques centaines de nanomètres. Les structures 1D sont des nanosystèmes longs, tels que les nanofils ou les nanotubes. Les nanostructures 2D sont quant à elles en dehors de la gamme nanométrique car elles sont principalement représentées par différents types de nanofilms, tels que les films minces multicouches ou encore les nanofeuilles. Les nanosystèmes 3D sont structurées et composées de plusieurs éléments répétés donnant un objet tridimensionnel final [72].
28
Figure 7 : Illustration et exemples de structures 0D, 1D, 2D and 3D (adaptée de [72])
Classification selon leur composition
Une dernière méthode de classification, correspondant aussi à la plus utilisée, se réfère à la composition du nanosystème :
1) les nanosystèmes organiques, à base de polymères ou de lipides ;
2) les nanosystèmes inorganiques, majoritairement constitués de composés métalliques.
1.2.4.1 Nanosystèmes organiques
Les nanosystèmes polymériques
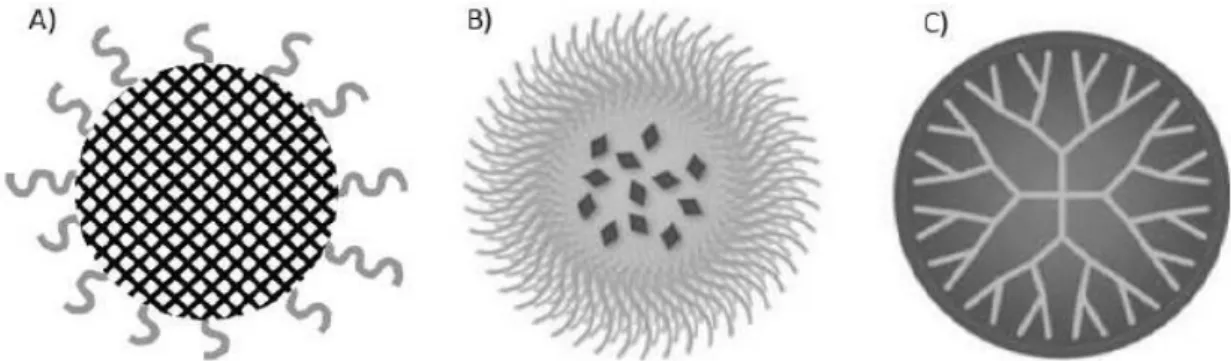
Dans la plupart des cas, ces nanosystèmes sont composés de molécules ou blocks amphiphiles polymériques. Leurs avantages potentiels sont la présence d’une composition chimique plus précise, une multivalence de surface adaptée et la création d'une architecture tridimensionnelle définie soit par une macromolécule hydrosoluble synthétique, soit par création d’une nouvelle structure supramoléculaire [106, 107, 121]. Ces nanosystèmes sont souvent composés d'un noyau dans lequel le médicament est solubilisé et d’une couronne ou coquille qui protège le médicament de la dégradation et ce qui permet l’obtention de leur architecture finale. Les principaux représentants de cette classe sont les nanosphères (A), les micelles (B) et les dendrimères (C) (Figure 8).
29
Figure 8 : Représentation de nanosystèmes à base de polymères, (A) nanosphères, (B) micelles polymériques et (C) dendrimères.
Nanosphères
Ces nanosystèmes sont structurés en deux phases : un noyaux sphérique dans lequel le médicament est physiquement et uniformément dispersé, lui-même entouré d'une enveloppe ou d'un revêtement extérieur [122]. Le noyau est constitué de polymères compacts biocompatibles ou de polyacides tels que l’acide polylactique (PLA) ou l’acide polylactique-co-glycolique (PLGA) [123]. Ce noyau est biodégradable et conduit à des éléments inoffensifs après hydrolyse afin d'éviter l'accumulation de particules ou de molécules toxiques dans l’organisme. L'optimisation de l'utilisation de ces polymères conduit à différents types de propriétés d'encapsulation et de cinétiques de libération [124].
Micelles polymériques
Les micelles polymériques ont un diamètre allant de 10 à 100 nm et sont caractérisées par une structure noyau-enveloppe, formée par l'auto-assemblage de copolymères séquencés amphiphiles dans un environnement aqueux. Le noyau interne est souvent composé de chaînes hydrophobes typiquement composées de polycaprolactone ou d'acide poly-(acide lactique) (PLA) qui créent un espace approprié pour la solubilisation de médicaments lipophiles [125]. En outre, ce noyau interne est entouré d'une couronne composée de blocs hydrophiles. Ceux-ci deviennent très solubles dans l'eau et adoptent une apparence "déployée" qui conduit à divers types de possibilités de conformation [118].
Dendrimères
Les dendrimères sont des macromolécules globulaires de taille monodisperse avec des structures fortement ramifiées. Il existe deux types de structures de base : la première est la structure globulaire avec un noyau central à partir duquel les branches rayonnent. Le deuxième type n'a pas de noyau central et se compose simplement d'une série de polymères fortement ramifiés d'une taille d'environ
30
10 nm [111, 126]. Les dendrimères sont des nanosystèmes formés par des unités multiples avec des branches terminales fonctionnalisables. Ils peuvent incorporer divers types de composés soit par l'intermédiaire des groupes terminaux pouvant être fonctionnalisés, soit par encapsulation au sein de la cavité centrale, qui est composée de multiples monomères parfaitement ramifiés, généralement de polyaminoamine [111].
1.2.4.2 Les nanosystèmes à base de lipides
Liposomes
Les premiers liposomes ont été découverts dans les années 1960 [127] afin d’améliorer l'accumulation sélective du composés d’intérêt dans les tissus et les sites cellulaires souhaités [87, 128]. Ils correspondent à des vésicules sphériques artificielles biologiquement inertes et faiblement immunogènes. Ils sont formés par une ou plusieurs bicouches lipidiques concentriques composées de molécules amphiphiles qui s’auto-assemblent en milieu aqueux. Les liposomes peuvent être composés de phospholipides naturels ou synthétiques, permettant d’encapsuler des composés polaires dans le compartiment aqueux et apolaires dans la bicouche lipidique (Figure 9).
Figure 9: Liposomes unilamellaires [129]
Ils sont facilement formés à partir de molécules amphiphiles (le plus couramment des phospholipides). Ils sont non toxiques, non immunogènes, naturels et biodégradables [85, 87, 130]. Le Doxil®, premier liposome approuvé par la FDA en 1995, est un liposome pégylé encapsulant de
la doxorubicine [131, 132].
La principale difficulté rencontrée lors du développement des liposomes réside dans leur stabilité qui dépend du ratio principe actif / lipide. Idéalement, ce ratio doit être élevé, mais augmenter ce rapport diminue la stabilité des liposomes en milieux aqueux. En général, les liposomes sont lyophilisés pour être conservés sous la forme de poudre, puis solubilisés juste avant utilisation. En
31
plus des problèmes de stabilité menant à la fuite de principes actifs, les liposomes présentent d’autres inconvénients tels que de faibles taux de chargement en principes actifs lipophiles et le besoin d’utiliser un solvant organique pour leur préparation [129].
Les conjugués médicament-lipides
Les conjugués médicament-lipides ont également été développés dans les années 1990 afin d'obtenir une meilleure charge de médicament pour les composés lipophiles [133, 134]. Cependant, de nos jours, ces systèmes sont principalement utilisés pour l'administration de médicaments hydrophobes possédant une faible hydrosolubilité limitant ainsi leur utilisation [135, 136]. Ils sont conçus en liant le médicament à une fraction lipidique par formation de sel ou par liaison covalente qui conduit au complexe médicament-lipide. Pour générer ces nouveaux composés, la liaison de diverses chaînes lipidiques peut être utilisée. Au cours du développement de ces composés, des chaînes lipidiques de types terpéniques ont pu être utilisées [137-139]. Parmi ces terpènes, l'utilisation du squalène a été largement décrite [137, 140, 141]. Le squalène, triterpène précurseur du cholestérol, est un hydrocarbure biocompatible largement répandu dans la nature et couramment utilisé comme excipient dans de nombreuses préparations pharmaceutiques [137, 142]. Lorsque le squalène est lié par liaison covalente à des composés hydrophiles tels que des agents alkylants, des antibiotiques ou des analogues nucléosidiques, le composé obtenu acquiert la propriété de s'auto-assembler spontanément dans des milieux aqueux pour former des structures nanométriques [89, 143-147]. La conjugaison par greffage chimique du squalène conduit à une augmentation de l'entrée cellulaire du composé grâce à la propriété intrinsèque de la molécule d’auto-assemblage sous la forme de nanoparticules (NPs). Basées sur cette technologie, des NPs de monophosphate de squalènoyl didésoxycytidine testées sur des cellules HIV-1 ont montré une augmentation d’un facteur 2 de l'activité anti-HIV in vitro de la molécule mère. De manière plus intéressante, des NPs d'adénosine squalène ont fourni une neuroprotection après un accident vasculaire cérébral engendrant une lésion de la moelle épinière [148]. De plus, la pégylation de ces NPs de squalène a montré une protection contre la dégradation et l'élimination rapide du plasma [149-151].
Les nanoparticules lipidiques
Les NPs lipidiques font fréquemment référence aux NPs lipidiques solides ou nanocapsules lipidiques solides (SLN), et ont été découvertes dans les années 1990 par séchage par pulvérisation et congélation de « micropellets » lipidiques suivi de leur caractérisation par microscopie électronique [152]. Les SLN sont des systèmes colloïdaux de délivrance de médicaments composés de lipides, d’eau et d’émulsifiants. Les lipides qui les constituent ont la particularité d’être solides à température ambiante et liquide à la température physiologique ce qui a pour but d’empêcher une libération immédiate du composé actif, rendant ainsi difficile sa diffusion à la surface [153]. De plus,
32
les lipides utilisés pour former le SLN présentent une faible toxicité aiguë et chronique [154]. Elles permettent d’encapsuler une grande variété de principes actifs, hydrophiles ou hydrophobes, ainsi que des peptides ou des protéines [109, 155]. Les avantages les plus importants des SLN par rapport aux autres nanosystèmes à base de lipides sont la libération contrôlée du médicament et la stabilité physique des préparations. Cependant, elles présentent encore certaines limitations telles que la charge limitée de médicament et l'expulsion du médicament pendant le stockage. Dans la plupart des études, les SLN sont administrées par voie intraveineuse avec des résultats probants mais elles peuvent également être un système de délivrance de médicaments par voie orale très prometteur, puisque cette voie d’administration permet d’éviter l’effet de premier passage et mène à une diffusion lente et continue du principe actif [156].Une des formulations ayant connu un succès commercial est la Mucosolvan retard capsule® permettant la vectorisation de l’ambroxol au sein de
nanocapsules lipidiques dans le cadre du traitement de toux et bronchites chroniques [153]. Le principal inconvénient des SLN concerne leur faible taux de chargement en principe actif. Celui-ci dépend fortement de la pureté des lipides utilisés pour la formulation et de l’étape de cristallisation. De plus, une fois les SLN obtenues, les principes actifs peuvent être expulsés de la maille lors du stockage des NPs.
Au sein de ce groupe, nous trouvons aussi les nanocapsules lipidiques (NCLs) qui font référence à des nanocapsules composés de lipides liquides et qui correspondent à une nouvelle technologie mis au point au sein de l’unité de « micro et nanomedecines translationnelles» (MINT, U1066) de l’Université d’Angers dirigée par le professeur Patrick Saulnier et dont la présentation se fera plus tard.
1.2.4.3 Les nanosystèmes inorganiques
Les NPs à base de silice, les NPs magnétiques guidées, les NPs de métaux nobles ou les nanostructures de carbone [157] constituent la majorité des nanosystèmes inorganiques.
Les nanosystèmes à base de silice
Les nanosystèmes à base de silice sont largement utilisés en raison de la possibilité de moduler leur taille, leur porosité et leur forme, ce qui leur permet d'être utilisés dans les domaines de l’imagerie biomédicale, l'intégration comme agents de contraste d'imagerie ou la délivrance contrôlée de médicaments [158-160]. De nombreuses techniques de préparation sont répertoriées permettant la formulation de nanosystèmes avec une gamme étroite de tailles et une composition presque uniforme [158]. Ces techniques conduisent à de nombreux types de nanosystèmes à base de silice tels que les nanosystèmes basiques de silice ou une architecture plus complexe comme les NPs mésoporeuses, les NPs à coquille centrale ou les NPs gravées. De plus, dans d'autres cas, des formes différentes peuvent être obtenues. Diverses techniques peuvent être utilisées pour synthétiser ces nanosystèmes à base de silice (Figure 10).
33
Figure 10 : Liste des techniques courantes utilisées pour synthétiser des nanosystèmes à base de silice [158]
Les nanosystèmes guidés magnétiques
Louis Néel montre en 1949 qu’en dessous d’une certaine taille, les NPs magnétiques entrent dans un état appelé superparamagnétisme [161]. Ce comportement concerne les NPs d’oxyde de fer possédant un diamètre inférieur à 20 nm et les NPs de fer pur. Lorsqu’elles sont soumises à un champ magnétique extérieur, les NPs peuvent émettre une propriété de magnétisme [162]. Ces nanosystèmes d’oxyde de fer superparamagnétiques (SPION, pour « superparamagnetic iron oxide
nanoparticles ») permettent donc de faire un contraste avec les tissus. Pour ces raisons, les SPION
sont utilisées notamment en imagerie par résonance magnétique (IRM). Ils peuvent également être fonctionnalisés par des ligands de ciblage ou être incorporées dans des nanoparticules NPs encapsulant un principe actif, afin de leur apporter un élément d’imagerie [163].
Les « quantum dots », également connus sous le nom de nanocristaux, sont des semi-conducteurs nanométriques qui, en fonction de leur taille, peuvent émettre de la lumière dans toutes les couleurs de l'arc-en-ciel. Ces nanostructures confinent des électrons de bande de conduction, des trous de bande de valence ou des excitons dans les trois directions spatiales. Certains exemples de « quantum dots » sont les nanocristaux semi-conducteurs et les nanocristaux cœur-enveloppe, où il existe une interface entre différents matériaux semi-conducteurs. Ils ont été appliqués en biotechnologie pour le marquage cellulaire et l'imagerie, en particulier dans les études d'imagerie du cancer [164].
34
Les nanosystèmes à base de carbone
Nous pouvons également trouver des nanosystèmes à base de carbone tels que les fullerènes et les tubes de carbone. Ils se caractérisent par des molécules potentiellement poreuses à base de carbone, en treillis [165]. Les fullerènes sont de forme sphérique tandis que les tubes de carbone sont cylindriques et de forme allongée. Le diamètre d'un tube de carbone peut être de plusieurs nanomètres mais la longueur peut être beaucoup plus grande, jusqu'à plusieurs micromètres, en fonction de son utilisation prévue. Les nanotubes de carbone ont de nombreuses applications en science des matériaux. Cependant, ils ont également trouvé une utilisation dans le domaine de la biomédecine comme porteurs de vaccins, de médicaments et d'autres molécules [166].
Les nanosystèmes à base de métaux nobles
L’utilisation de l’argent ou de l’or dans l’obtention de nanosystèmes est maintenant bien décrite [167, 168]. Elle remonte au 5e siècle avant J.-C., où les NPs d’or, ou « or colloïdal » étaient utilisées pour
donner une couleur rouge aux céramiques et aux verres. C’est au cours du 17e siècle que des
solutions d’or colloïdales ont été utilisées pour le traitement de diverses maladies [169].
Ces nanosystèmes à base d’or peuvent avoir un diamètre allant de 2 à 200 nm et sont utilisés pour diverses applications biologiques, en imagerie et en photothérapie [167, 169]. Diverses méthodes de synthèse sont possibles, mais la méthode de synthèse la plus répandue est la réduction d’un sel de tétrachloroaurate par du citrate de sodium dans l’eau à 100°C. Les ligands citrates stabilisent les nanoparticules d’or. Ils sont ensuite facilement remplaçables par des ligands thiolés. Des ligands hydrophiles sont nécessaires pour apporter de la stabilité aux nanosystèmes et éviter leur agrégation [170].
Utilisation des nanomédicaments en cancérologie
Selon l'organisation mondiale de la santé, les maladies les plus meurtrières sont les maladies cardiovasculaires, le cancer et le diabète. Ainsi, le cancer représentant la deuxième maladie la plus mortelle dans les pays développés est en continuelle croissance. Les traitements classiques contre le cancer reposent sur la chirurgie, la radiothérapie et la chimiothérapie [171]. Bien que les agents antitumoraux présentent une grande efficacité dans leur domaine d’action et de traitement, ils doivent encore être administrés à une dose élevée pour obtenir une concentration appropriée au niveau du site d'action et ainsi assurer leur efficacité thérapeutique. Plus radicalement, les traitements de chimiothérapie, réagissent rapidement avec les cellules normales en division de par leur faible spécificité et sont à l'origine de divers effets secondaires toxiques [172-174]. Ce manque de spécificité induit une diminution de la concentration du médicament au niveau de la cible ainsi qu’une faible accumulation au niveau de la tumeur et, par conséquent, la réduction de l'efficacité du
35
traitement. Cette diminution de l'efficacité peut également conduire, dans certains cas, à la récidive de la maladie. Pour prévenir ces toxicités et améliorer l'efficacité et la spécificité de la chimiothérapie, le développement d'agents chimiothérapeutiques ciblés est apparu ces dernières décennies [82, 175, 176]. De nos jours, le développement de nanomédicaments contribue à la renaissance d’une multitude de composés (petites molécules, protéines, acides nucléiques, etc.) dont l’administration n’a pas pu se faire du fait de problème de solubilité (insolubles ou peu solubles dans l'eau) ou d’un manque de spécificité vis-à-vis de leur cible [151, 177-186]. Ainsi, l’utilisation de ces nanosystèmes permet de réduire les toxicités causées par l’administration de doses élevés. Elle permet aussi de diminuer les fluctuations de concentrations plasmatiques en principe actif en contrôlant la libération du principe actif au cours du temps. Ce contrôle permet d’assurer une concentration quasi constante, ce qui, à terme pourrait permettre une réduction ou un fractionnement des doses à administrer, celles-ci étant souvent responsable de la toxicité. Toutefois, parmi ces nanomédicaments, un faible nombre ont réussi à obtenir l'approbation de la FDA, parmi lesquels nous retrouvons les préparations Doxil® ou Myocet®, correspondant à de la doxorubicine encapsulée au sein de liposomes pégylés
ou non, ou encore l’Abraxane® correspondant au couplage du paclitaxel à de l’albumine (Tableau
36
Tableau 2 : Nanomédicaments utilisés en clinique pour le traitement du cancer [100]
De plus, une multitude de nouveaux nanomédicaments sont en cours d’évaluation dans le cadre d’essais cliniques [187, 188] (Figure 11).
37
Ainsi, l’oncologie est devenue l’un des domaines prioritaires de ces nanomédecines [189] mettant en collaboration une diversité disciplines scientifiques [76] (Schéma 4).
Schéma 4 : Disciplines entrants en jeux lors du développement de nanomedecament
Le développement de ces nanomédicaments a pour objectif de réduire la toxicité et d'améliorer la biodisponibilité ainsi que la spécificité permettant d’optimiser l’index thérapeutique du médicament de base et de ce fait la qualité de vie du patient. Cette approche permet également d’augmenter le temps de demi-vie du médicament en réduisant l'immunogénicité et en diminuant le métabolisme du médicament par la vectorisation de celui-ci afin de le protéger. Aujourd'hui, tous les domaines de la recherche, industrielle ou universitaire, ont compris l’importance de ces nouvelles entités, ce qui a conduit au développement de nouvelles formes de nanomédicaments. En outre, ceux-ci peuvent montrer de nombreux avantages apportés par leurs caractéristiques spécifiques :
1) la taille du nanomédicament peut être modulée afin de tirer profit du ciblage passif par l’intermédiaire de l’effet EPR;
2) la surface de celui-ci peut être modifiée afin d’apporter de la spécificité par un ciblage actif ;
3) le contrôle et la durabilité du médicament par le transport vers le site d'action contribuent à sa libération progressive au cours du temps au niveau de la cible, permettant ainsi d’atteindre une efficacité thérapeutique accrue accompagnée d’effets secondaires réduits ;
4) la charge de médicament, correspondant au processus d'incorporation du médicament au sein du nanosystème est optimale. Ainsi, plus cette charge est élevée, plus la dose à administrer peut-être diminuée permettant la réduction des effets secondaires ;
5) le greffage de ligands à la surface du nanosystème peut être effectué afin de permettre un ciblage spécifique d’un récepteur connu, pour assurer le guidage du nanosystème directement vers le site d'action par reconnaissance de récepteur ligand ;
38
6) le nanosystème peut être utilisé pour divers types de voies d'administration, y compris par voie orale et parentérale ;
A ce jour, de nombreux composés anticancéreux ont pu vivre une seconde vie grâce à leur développement sous la forme de nanosystèmes. Cette stratégie a ainsi permis une diminution de la cardiotoxicité de la doxorubicine lors de vectorisation au sein de liposome dans le cas du Caelyx®
[190]. Dans la majorité des cas, ces nouveaux nanosystèmes ont permis d’accroitre considérablement la biodisponibilité ainsi que l’efficacité de la molécule mère, permettant ainsi de diminuer la dose administrée celle-ci étant, pour la plupart des cas, responsable de l’ensemble des effets secondaires toxiques [183, 191, 192].
Si, les nanomédicaments présentent de nombreux avantages, ils ont aussi leurs limitations [193-195]. Ils sont de petite taille et de grande surface, ce qui peut conduire à une fusion ou à une agrégation conduisant à des nanosystèmes plus grands menant à la perte de certaines de leurs propriétés [196].
Caractérisation des nanomédicaments
D’une façon générale, les dimensions de ces nanosystèmes correspondent au diamètre géométrique, mesuré directement par microscopie électronique à transmission ou à balayage (MET ou MEB). En milieu aqueux, le nanosystème est recouvert d’une couche de solvatation formée d’espèces qui agissent comme solvant selon la charge et la taille du nanosystème, mais également selon la nature des solutés [197, 198]. Cette couche influence le mouvement brownien du nanosystème, c’est-à-dire le mouvement aléatoire celui-ci lors de son immersion dans un fluide ; elle est prise en compte dans le calcul du diamètre hydrodynamique du nanosystème. Le diamètre hydrodynamique correspond ainsi au diamètre d'une sphère théorique qui aurait le même coefficient de diffusion que la particule considérée. Il est évalué grâce à la technique de diffusion dynamique de la lumière (DDL, ou Dynamic Light Scattering - DLS en anglais) et correspond à la taille du nanosystème que va appréhender la cellule [199].
1.2.6.1 Caractérisation par diffusion dynamique de la lumière (DDL)
La technique de diffusion dynamique de la lumière permet de mesurer le diamètre de particules en suspension pouvant atteindre quelques centaines de nanomètres (nm) ainsi que leur charge [200]. En effet, en solution colloïdale, les particules rencontrent un faisceau laser et le dispersent dans toutes les directions. L’intensité du faisceau dispersé fluctue en fonction du temps en raison du mouvement brownien des particules. Grâce à un photomultiplicateur, l’appareil détecte les photons et analyse ces variations d’intensité pour donner les coefficients de diffusion des particules. Ces
![Tableau 1: Principales caractéristiques différenciant le CPM de l’IFO [11]](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/18.892.178.714.665.1097/tableau-principales-caractéristiques-différenciant-le-cpm-l-ifo.webp)
![Figure 5 : Liste non-exhaustive d’applications médicales des nanomédecines (adaptée de [100]](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/26.892.272.628.718.1084/figure-liste-exhaustive-applications-médicales-nanomédecines-adaptée.webp)
![Figure 6 : Représentation des trois générations de nanosystèmes [119]](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/28.892.162.804.349.536/figure-représentation-générations-nanosystèmes.webp)
![Figure 7 : Illustration et exemples de structures 0D, 1D, 2D and 3D (adaptée de [72])](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/29.892.98.756.86.519/figure-illustration-exemples-structures-d-d-and-adaptée.webp)
![Figure 9: Liposomes unilamellaires [129]](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/31.892.295.600.562.862/figure-liposomes-unilamellaires.webp)
![Figure 10 : Liste des techniques courantes utilisées pour synthétiser des nanosystèmes à base de silice [158]](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/34.892.273.622.87.491/figure-liste-techniques-courantes-utilisées-synthétiser-nanosystèmes-silice.webp)
![Tableau 2 : Nanomédicaments utilisés en clinique pour le traitement du cancer [100]](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/37.892.89.809.105.614/tableau-nanomédicaments-utilisés-clinique-traitement-cancer.webp)
![Figure 12 : Schéma des principales voies d'internalisation cellulaire : (A) la phagocytose et les différentes voies d’endocytose (B) (C) (D) et (E) [206]](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/42.892.87.813.430.655/figure-schéma-principales-voies-internalisation-cellulaire-phagocytose-endocytose.webp)
![Figure 13 : Les étapes de phagocytose d’un nanosystème [206]](https://thumb-eu.123doks.com/thumbv2/123doknet/14555414.726005/43.892.128.765.156.489/figure-les-étapes-phagocytose-d-un-nanosystème.webp)
