Publisher’s version / Version de l'éditeur:
Virology, 368, November 1, pp. 32-40, 2007
READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE. https://nrc-publications.canada.ca/eng/copyright
Vous avez des questions? Nous pouvons vous aider. Pour communiquer directement avec un auteur, consultez la première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées. Si vous n’arrivez pas à les repérer, communiquez avec nous à [email protected].
Questions? Contact the NRC Publications Archive team at
[email protected]. If you wish to email the authors directly, please see the first page of the publication for their contact information.
NRC Publications Archive
Archives des publications du CNRC
This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.
For the publisher’s version, please access the DOI link below./ Pour consulter la version de l’éditeur, utilisez le lien DOI ci-dessous.
https://doi.org/10.1016/j.virol.2007.06.019
Access and use of this website and the material on it are subject to the Terms and Conditions set forth at
Tropism of Tanapox virus infection in primary human cells.
Nazarian, Steven H.; Barrett, John W.; Stanford, Marianne M.; Johnston,
James; Essani, Karim; McFadden, Grant
https://publications-cnrc.canada.ca/fra/droits
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
NRC Publications Record / Notice d'Archives des publications de CNRC:
https://nrc-publications.canada.ca/eng/view/object/?id=2b563b96-9ca0-49a0-892d-0718b155fba7 https://publications-cnrc.canada.ca/fra/voir/objet/?id=2b563b96-9ca0-49a0-892d-0718b155fba7Tropism of Tanapox virus infection in primary human cells
Steven H. Nazarian
a, John W. Barrett
a, Marianne M. Stanford
a, James B. Johnston
a,1,
Karim Essani
b, Grant McFadden
a,⁎
aBiotherapeutics Research Group, Robarts Research Institute, and Department of Microbiology and Immunology,
University of Western Ontario, London, Ontario, Canada N6G 2V4
bLaboratory of Virology, Department of Biological Science, Western Michigan University, Kalamazoo, MI 49008, USA
Received 2 March 2007; returned to author for revision 11 April 2007; accepted 20 June 2007 Available online 16 July 2007
Abstract
Tanapox virus (TPV) belongs to the genus Yatapoxvirus and causes a relatively benign zoonotic disease in man, with symptoms that resemble a mild version of human monkeypox. In order to investigate the underlying mechanisms of TPV pathogenesis, the tropism and replication characteristics of TPV were examined in a variety of primary human cells. A GFP expressing TPV (TPV-GFP) was constructed and used to infect primary human dermal fibroblasts (pHDFs) and peripheral blood mononuclear cells (PBMCs), both of which are believed to be major in vivo targets of poxvirus infection. pHDFs fully supported productive replication and cell–cell spread of TPV-GFP. However, induction of cell cycle arrest in pHDFs by contact mediated inhibition or rapamycin treatment eliminated the ability of TPV to fully stimulate cell cycle progression and dramatically reduced viral replication. TPV-GFP-infected human PBMCs were screened for permissiveness by FACS analysis. CD14+ cells (monocytes) were the primary cellular target for TPV infection. A small proportion of CD3+ cells (T cells) were positive for GFP expression, yet TPV was not able to replicate and spread in cultured peripheral blood lymphocytes, regardless of their state of activation. Primary human monocytes, however, demonstrated robust TPV replication, yet these cells no longer supported replication of TPV once they differentiated into macrophages. This unique ex vivo tropism of TPV gives key insights into the basis for the self-limiting pathogenicity of TPV in man. © 2007 Elsevier Inc. All rights reserved.
Keywords: Poxvirus; Tanapox; Pathogenesis; Primary human cells; Replication
Introduction
Tanapox virus (TPV) is a member of the Yatapoxvirus genus of poxviruses that is characterized by its ability to cause relatively mild monkeypox-like infections of primates, includ-ing man (Damon, 2007; Knight et al., 1989). TPV is endemic to equatorial Africa and causes occasional zoonotic infections in humans, likely transmitted through biting arthropod vectors, but direct human-to-human transmission of TPV has never been observed. TPV infection begins with brief febrile illness,
followed by the development of one or few inflamed nodule-like lesions and concurrent lymphadenopathy. Maximum lesion size is reached by approximately 2 weeks and resolution of the lesion usually occurs within 6 weeks (Jezek et al., 1985).
This self-limiting poxvirus infection is contrasted with Variola virus (VARV), which causes smallpox and is a systemic disease of comparatively short duration with significant morbidity and mortality (Damon, 2007). From the study of other orthopoxviruses, it is likely that VARV, like other orthopoxviruses, spreads from the initial site of infection (usually the skin or respiratory route), through infected dendritic cells that traffic to the draining lymph node (Damon, 2007). Primary viremia is caused by viral replication within the lymph node and causes subsequent infection of secondary sites through the blood, including the spleen, liver, and bone marrow. Secondary viremia from these sites causes other localized infections, which leads to the characteristic pock lesions or rash (Damon, 2007). Studies with Vaccinia Virology 368 (2007) 32 – 40
www.elsevier.com/locate/yviro
⁎ Corresponding author. University of Florida, 1600 SW Archer Road, ARB Room R4-295, PO Box 100332, Gainesville, FL 32610, USA. Fax: +1 352 273 6849.
E-mail address:[email protected](G. McFadden).
1Present address: Institute for Nutrisciences and Health, National Research
Council of Canada, 550 University Avenue, NRC-INH Building, Charlottetown, PE, Canada C1A 4P3.
0042-6822/$ - see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.virol.2007.06.019
virus (VACV) suggest that monocytes and activated T-cells are the main targets of orthopoxvirus replication within the blood and may be primarily responsible for systemic spread of the highly pathogenic poxviruses (Chahroudi et al., 2005;
Sanchez-Puig et al., 2004; Yu et al., 2006). Both the dermal and
epidermal layers of the skin have been implicated in pock formation, with viral replication within the dermal layer being important for systemic spread of the virus from the periphery (Damon, 2007).
At the other end of the pathogenic spectrum of poxviruses in man lies Molluscum contagiosum virus (MOCV), for which viral replication is highly restricted to differentiating keratino-cytes, resulting in small benign lesions as the only clinical feature in immunocompetent hosts (Gottlieb and Myskowski,
1994; Lewis et al., 1997). Lesions may persist for months
without any immune cell infiltrate and, as a result, are noted for a lack of inflammation at sites of viral infection (Heng et al., 1989).
Findings from these viruses suggests that cellular tropism of poxviruses that infect man may be closely linked to its pathogenicity. Poxvirus tropism of cultured cells is largely mediated at the intracellular level and it is generally accepted that poxviruses, through receptor non-specific interactions, are able to bind and enter most cell types and express at least some early gene products (Moss, 2006). In fact, resting human naive T-cells are one of the few cell types which have been reported to be refractory to the binding and/or entry of vaccinia virus (Chahroudi et al., 2005). Permissiveness of poxviruses is therefore thought to be controlled at the level of the intracellular environment, which varies based on cell type, cellular activation state, and various induced anti-viral signaling cascades (McFadden, 2005). A successful poxvirus infection regulates the intracellular signaling environment of the infected cell to ensure both the efficient transcription and translation of viral genes that lead to the production of progeny virions, and also to mitigate the various anti-viral cellular responses.
One of the most intricately regulated cell processes is the cell cycle, in which chromosomes are replicated (S) and then are segregated into two daughter cells through mitosis (M). Regulatory checkpoints at G0, G1, and G2, allow for rigorous control of replication through the cell cycle (reviewed by
Murray, 2004). Many viruses favor cells in a certain phase of the cell cycle in order to provide an adequate environment for viral replication. As such, stimulating progression through the cell cycle is a key adaptive strategy for many viruses, inclu-ding cytomegalovirus (Castillo and Kowalik, 2004), adeno-virus (Berk, 2005), and poxviruses (Johnston et al., 2005;
Wali and Strayer, 1999). For example, VACV infection can
increase transit of cells out of G1 into S phase through a modulation of cyclin and cyclin-dependent kinase levels (Wali
and Strayer, 1999). Similarly, the Myxoma virus (MYXV)
M-T5 host range protein has been shown to interact with host cell cullin-1, and a MYXV construct deficient in M-T5 was shown to be unable to transition infected cells out of cell cycle arrest, correlating with a reduction in MYXV replication (Johnston et al., 2005).
The current study was initiated to analyze TPV infection in primary human cells to correlate its cellular tropism in human cells ex vivo to its pathogenicity profile in man. A TPV expressing EGFP from a synthetic early/late poxvirus pro-moter was constructed to aid in the identification of TPV-infected cells, which was used to assess replication efficiencies in primary human cells in vitro. Various modulations of cell states were also tested, including differentiation, activation, and cell cycle arrest induced by treatment with cytostatic drug rapamycin, to evaluate their role in TPV replication. The cellular tropism profile of TPV in primary human cells is discussed as a potential ex vivo surrogate system to ratio-nalize the restricted spread of TPV throughout the infected host and better understand its self-limiting pathogenesis pro-file in man.
Results
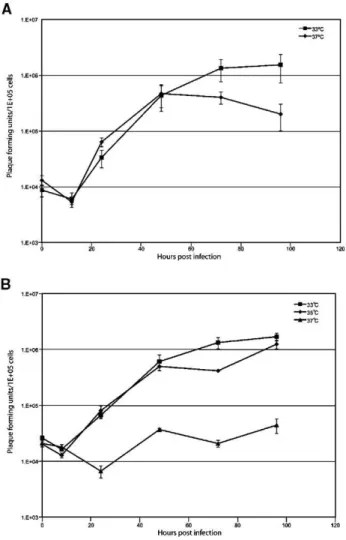
Optimal replication temperature of TPV-GFP is 33–35 °C TPV was engineered to express EGFP from a synthetic early/late promoter. An expression cassette containing the synthetic early/late promoter and EGFP open reading frame was inserted between 5L and 6L in the TPV genome. TPV 5L is predicted to encode a ubiquitin ligase and TPV 6L is of unknown function. TPV-GFP was plaque purified and verified to be pure by PCR analysis of the viral genomic DNA (data not shown). It has been previously reported that TPV grows equally well at both 33 °C and 37 °C, reaching a maximum titer at 96 h post-infection (hpi), in OMK cells (Mediratta and Essani, 1999). However, when tested in OMK cells, single step replication curves of TPV at 33 °C and 37 °C show a decrease in virus progeny at late time points only at the higher temperature (Fig. 1A). Single step replication curves were repeated for TPV-GFP and were carried out at three temperatures: 33 °C, 35 °C, and 37 °C. TPV-GFP grows poorly, if at all, at 37 °C; however, at both 33 °C and 35 °C, TPV-GFP replicates productively with an approximate 2-log increase in infectious progeny (Fig. 1B). Since TPV-GFP replication appears to be highly sensitive to temperature, subsequent experiments with primary human cells were performed at 33 °C.
Cycling, but not growth arrested, primary human dermal fibroblasts are permissive for TPV-GFP
We observed that primary human dermal fibroblasts (pHDFs) that had reached confluence and were in a quiescent state supported only low levels of TPV-GFP replication, accompanied by reduced GFP expression. To investigate this further, pHDFs, either growing exponentially or quiescent, were infected with TPV-GFP at an MOI of 3 and TPV replication kinetics were analyzed over 4 days. Virus from the indicated time points was collected and titrated on OMK cells
(Fig. 2). A 40-fold decrease in progeny virus yield was
observed from the quiescent HDFs as compared to exponen-tially growing pHDFs. To further investigate the idea that 33 S.H. Nazarian et al. / Virology 368 (2007) 32–40
cycling pHDFs are required for optimal TPV replication, pHDFs treated with the drug rapamycin were used. Rapamycin is a highly specific inhibitor of mTOR and treatment with this drug causes arrest in the G1 stage of cell cycle in many types of cells, including pHDFs (Inoki and Guan, 2006). Virus replication in cells treated with rapamycin was found to be particularly restricted, especially at early time points, but reaches a final level similar to quiescent pHDFs by 4 days post-infection (Fig. 2). Interestingly, rapamycin has recently been shown to have the opposite effect on the virus replication of another poxvirus, myxoma virus, in human cancer cells that have altered intracellular signaling pathways (Stanford et al., 2007). In contrast, TPV is non-permissive in these particular human cancer cell lines (data not shown).
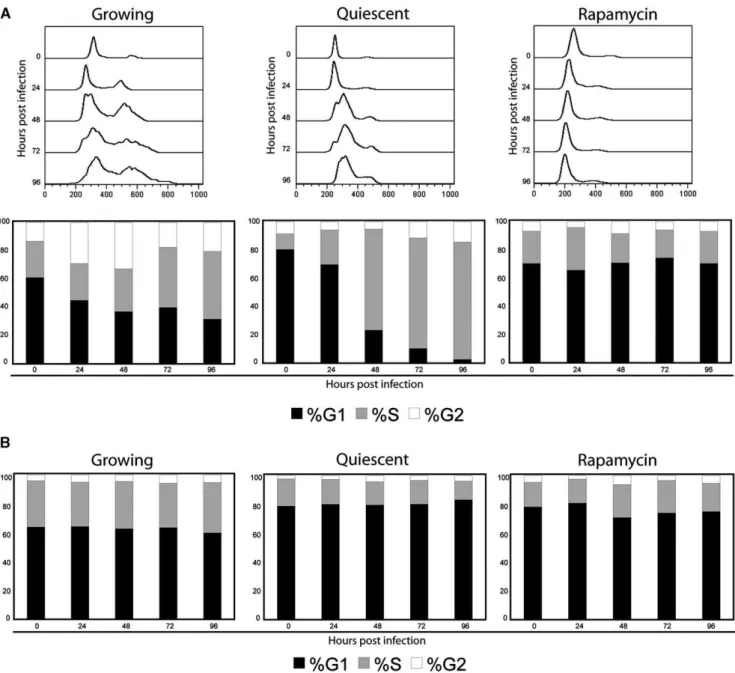
In order to quantify the cell cycle changes caused by these treatments, cells were collected, genomic DNA was stained with propidium iodide, and cells were analyzed by FACS (Fig. 3A). Prior to infection, growing pHDFs showed a typical distribution in the cell cycle, with G1 being the majority, and the rest of the cells in S or G2. Infection of cycling pHDFs caused an increase in the number of cells in the S and G2
phases of the cell cycle, implying that TPV infection stimulates cell cycle progression. In contrast, both quiescent cells and pHDFs treated with rapamycin accumulated in the G1 phase prior to infection. After TPV infection, quiescent cells accumulated in early S phase but did not proceed through the cell cycle to accumulate in G2. Cells treated with rapamycin, however, did not seem to experience any significant change over the time of the experiment, a consequence of their drug induced arrest in G1 that the virus is unable to reverse (Fig. 3A). Growth phase distribution of uninfected cells did not vary over the course of the experiment (Fig. 3B).
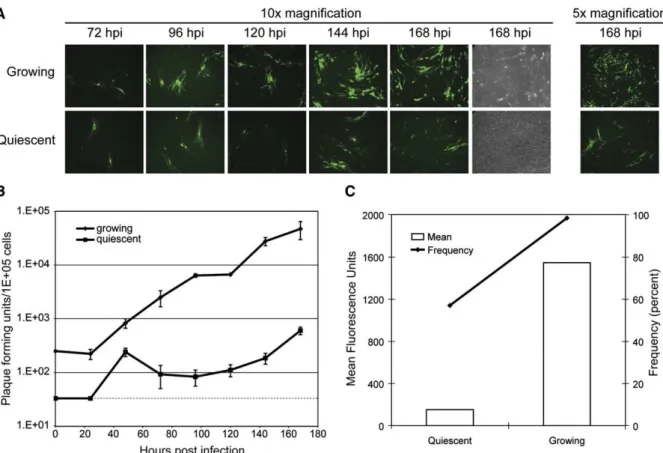
Multistep replication curves were also performed with growing and quiescent pHDFs to examine the effect of cellular cycling on TPV cell-to-cell spread. The inherent infectivity of the virus was not grossly compromised in the quiescent pHDF since a comparable number of GFP positive foci of infection are present in both treatments (Fig. 4A). As in the single step replication curves, TPV propagation in the growing and quiescent pHDFs followed similar early kinetics of growth up to 48 hpi and then a decrease in subsequent progeny titers was detected (Fig. 4B). However, there was reduced fluorescence intensity of the GFP and smaller foci in the quiescent pHDFs as compared to growing pHDFs (Fig. 4A). Additionally, the cytopathic effect of the virus in the quiescent cells was markedly reduced, even at day 7 (Fig. 4A). To quantify the difference in infection and fluorescence intensity, pHDFs at 7 days post-infection were analyzed by FACS. Growing pHDFs were over 98% positive for GFP expression and had a mean fluorescence intensity of over 1500. However, infected quiescent pHDFs were approximately 60% positive for GFP expression, with a mean fluorescence intensity of below 200 (Fig. 4C).
Fig. 2. Cell cycle blockade inhibits TPV-GFP replication in primary HDFs. Primary HDFs were either growing, quiescent through contact-mediated inhibition or pretreated with rapamycin (20 nM) for 16 h when they were infected with TPV-GFP at an MOI of 3. Cells were collected at the indicated time points and samples were titrated in triplicate on OMK cells.
Fig. 1. Replication kinetics of (A) TPV and (B) TPV-GFP in OMK cells at various temperatures. OMK cells were infected at an MOI of 3. After 1 h, the inoculum was removed and the medium was replaced. Cells were collected at the indicated time points and samples were titrated in triplicate on OMK cells.
CD14+ cells are the primary target of TPV-GFP infection in PBMCs
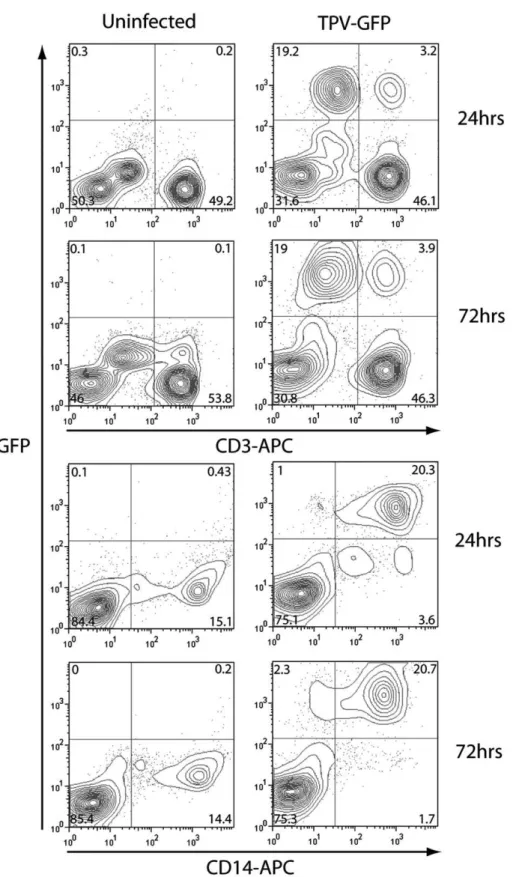
To further this avenue of investigating infection in primary cells, human peripheral blood mononuclear cells (PBMCs) were freshly isolated from donors, purified, and infected with TPV-GFP at an MOI of 3. Cells were harvested at 24 and 72 hpi, stained for CD3 and CD14, markers of T-cells and monocytes, respectively, and analyzed by FACS (Fig. 5). CD14+ cells were the main target of infection for TPV-GFP in PBMCs, showing prolonged high level GFP expression in approximately 20% of PBMCs (84–94% of CD14+ cells). However, CD14lo expres-sing cells were not infected as readily as CD14hi expresexpres-sing cells, but did show some GFP expression by 72 hpi. CD3+ cells were not infected nearly as readily as CD14+ cells; however 3.2
to 3.9% of PBMCs (6–8% of CD3+ cells) were infected and expressed a high level of GFP (Fig. 5).
The adherent cell fraction of PBMCs is highly permissive for TPV-GFP infection
In order to correlate the previous FACS data to viral replication, CD14+ cells and lymphocytes were separated using an adherence step. Additionally, to investigate the roles of activation and differentiation in viral replication, monocytes were cultured for 7 days to allow for differentiation into macrophages (Andreesen et al., 1990) and PBLs were activated with PHA. These cells were infected at an MOI of 3 and virus was collected at various time points (Fig. 6). Neither activated nor naive PBLs were able to support TPV-GFP replication by Fig. 3. Cell cycle analysis of (A) TPV-GFP-infected and (B) uninfected HDFs. Growing, quiescent, or rapamycin treated cells were infected at an MOI of 3. Cells were collected at the indicated time points, stained with PI, and analyzed by flow cytometry. Histograms showing populations of cells in various stages of the cell cycle are shown (A; top row). Curves were analyzed using Flo-Jo software and distribution of cells within the cell cycle is shown (bottom row).
35 S.H. Nazarian et al. / Virology 368 (2007) 32–40
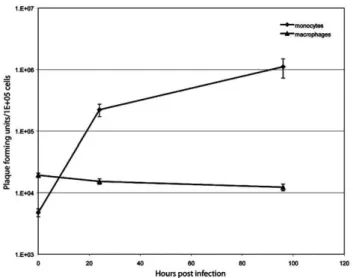
72 hpi (data not shown). However, the TPV-GFP replication kinetics were extremely robust in the freshly isolated monocytes as compared to either OMK or pHDF cells. TPV-GFP grew to yields of approximately 2.5 orders of progeny increase by 24 hpi, which is essentially the maximum since the amount of virus was similar to that obtained at 72 hpi. Interestingly, macrophages failed to support TPV-GFP replica-tion (Fig. 6).
Discussion
It is thought that TPV is primarily transmitted from an unknown animal reservoir to humans by biting insects. Therefore, upon infection, the virus has access to cells of the blood and skin adjacent to the area of the bite. However, the virus rarely causes more than 2 or 3 external skin nodules, and in 80% of infections, there is only a single lesion detected (Jezek
et al., 1985). It is unknown why TPV is unable to cause
clinically significant, systemic infection in contrast to certain other orthopoxvirus counterparts, like monkeypox. This study analyzes TPV tropism and replication efficiency in primary human cells to establish an ex vivo model to study TPV cellular tropism in man. TPV expressing GFP was constructed as a tool to examine the ability of TPV various primary human cell types. At an optimal temperature of 33–35 °C, we found that TPV-GFP was able to replicate efficiently in pHDFs that were in an
exponential growth phase. However, cell cycle blockade either by contact-mediated growth inhibition or treatment with rapamycin, created an intracellular environment that did not allow for efficient TPV replication. Analysis of the permissive cells of the blood suggests that monocytes are the primary cellular vehicle for spread of the virus to secondary sites.
Despite previous reports stating that TPV grows well at 37 °C (Mediratta and Essani, 1999), TPV seems to be somewhat sensitive to temperatures above 35 °C. This sensitivity is more obvious when using the TPV-GFP clones, likely due to overexpression of GFP causing cell stress. This effect is not seen at the lower temperatures tested. The implications of this temperature sensitivity may help explain the self-limiting pathogenesis of the TPV in man. In primates, including humans, TPV causes only a few lesions that tend to regress over several weeks. It is possible that this is primarily due to an inefficiency of TPV spread internally due to a higher core body temperature, especially considering the nature of the typical TPV-associated febrile illness. As a result, the virus may be restricted to replicating efficiently only at the periphery.
Lack of efficient systemic viral spread notwithstanding, monocytes from the adherent fraction of PBMCs are highly permissive to TPV infection. The observed rapid replication within the primary human monocytes may allow some spread of TPV in naturally occurring infections, and their infection may also be the cause of the pro-inflammatory cytokines that drive Fig. 4. Cell cycle progression is critical for efficient TPV-GFP replication and spread. Quiescent or growing primary HDFs were infected at low MOI (0.01). (A) Fluorescent microscope pictures were acquired at time points indicated. (B) Cells were collected at indicated time points and titrated on OMK cells in triplicate. Dashed line indicates detection limit for the assay. (C) At endpoint (168 hpi), cells were collected and analyzed by flow cytometry. Percent positive cells (line) and mean fluorescence intensity (bars) are shown.
acute febrile illness associated with TPV disease. Additionally, traffic of infected monocytes to the draining lymph node is likely the main culprit in inducing local lymphadenopathy. VACV has been characterized to also primarily infect mono-cytes (Sanchez-Puig et al., 2004). However, VACV was also
found to infect a proportion of T-cells, B-cells, and NK cells (Sanchez-Puig et al., 2004). Interestingly, other groups have demonstrated that activated CD3+ leukocytes are productively infected by VACV but resting naive T-cells are poorly infected due to some restriction of either binding or entry (Chahroudi et
Fig. 5. TPV-GFP primarily infects CD14+ cells in whole PBMC preparations. PBMCs were isolated and infected at an MOI of 3 and harvested at 24 or 72 h. Cells were fixed and stained for either CD3 or CD14. Representative scatter plots are shown including the percentage of cells that lie within each gate.
37 S.H. Nazarian et al. / Virology 368 (2007) 32–40
al., 2005). It is likely that active replication in some population of circulating lymphocytes may be required for the systemic spread of poxviruses in general. In the case of TPV, a small percentage of human CD3+ leukocytes are consistently infected, as measured by GFP expression. However, the failure to support TPV replication as measured by an absence of increase in progeny virus suggests that this is not a productive TPV infection in T-cells, even in lymphocytes activated with PHA. Other subsets of mononuclear cells may be infected but not to any significant degree since CD14-infected cells only represent approximately 3% of cells, similar to the fraction of T-cells that are infected.
pHDFs were used to model TPV infection at the skin. While infection in cycling pHDFs resulted in the production of progeny TPV, cells that were cell cycle arrested only poorly supported viral replication. The effects of cell cycle arrest through contact inhibition or rapamycin treatment, however, were not identical. Either treatment alone caused the accumula-tion of cells in G1; however, upon TPV infecaccumula-tion, cells arrested through contact inhibition progressed into S phase but seemed to be blocked during DNA synthesis, which resulted in accumulation of cells in S phase. This corresponds to the viral replication kinetics since TPV replication paralleled that of the actively dividing cell population until 48 hpi. Once the cells had finished accumulating in S phase, the viral titer remained unchanged for the remainder of the time course experiments. Perhaps even more striking were the detrimental effects on viral gene expression and spread of TPV due to cell cycle arrest. Virus spread to neighboring cells still occurred during these experiments, thus it is likely that the depression of viral gene expression simply slows the replication of the virus and does not block replication at a particular stage.
In contrast, rapamycin treated pHDFs showed evidence of only minimal TPV replication and most cells remained in G1 throughout the experiment indicating effective G1 arrest by the drug. As a consequence, the TPV replication curve was delayed,
before reaching the same low-level of TPV progeny observed with quiescent pHDFs. This is an interesting result, considering the opposite effect that rapamycin has on the replication of myxoma virus in human cancer cell lines (Stanford et al., 2007). This difference is likely due to the altered cell cycle and intracellular signaling milieu in tumor cells, and to the differing cellular requirements of these two poxviruses.
The restricted tropism of TPV to non-quiescent fibroblasts and monocytes gives significant clues into the modest pathogenicity of this virus in man. Without significant amplification of TPV within lymph node T cells, there is little of an internal cellular reservoir of virus to cause significant secondary viremia and delivery of virus to secondary skin sites, resulting in secondary pock formation. In addition, TPV replication in monocytes likely results in the acute febrile illness frequently reported early in TPV infection, and the viral temperature sensitivity may indicate that efficient virus replication occurs only at the periphery. In contrast, the more complex and devastating systemic effects of the more highly pathogenic poxviruses in man, like monkeypox virus and variola virus, are likely tied to their unique ability to replicate and spread within internal lymphocyte reservoirs of the infected host.
Materials and methods Cells and media
Owl monkey kidney (OMK) cells were obtained from ATCC (CRL-1556) and were maintained in Eagle's minimum essential medium with 2 mML-glutamine, 1.5 g/l sodium bicarbonate, 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate, 100 U penicillin/ml, 100 μg streptomycin/ml, and 10% fetal bovine serum. Neonatal primary human dermal fibroblasts (pHDFs) were obtained from Cascade Biologics and were maintained in medium 106 with low serum growth supplement as recommended by the supplier (Cascade Biologics). pHDFs were cultured no more than 16 population doublings. Human peripheral blood mononuclear cells (PBMCs) were isolated by venous puncture of normal healthy donors and separated on Histopaque-1077 (Sigma). The PBMC layer was removed and washed 3 times in PBS and maintained in RPMI 1640 with 2 mML-glutamine, 100 U penicillin/ml, 100 μg streptomycin/ ml, and 20% fetal bovine serum (Complete RPMI). All cells were grown at 37 °C. Quiescence in HDFs was induced by allowing cells to grow to confluence and then cultured at least two more days. Growing cells were infected at approximately 50% confluence to ensure that all cells were actively growing. In some experiments, growing cells were pretreated with 20 mM rapamycin (Sigma) for 16 h. In addition, rapamycin was replaced each time the medium was replaced.
Cell separation and preparation
To isolate monocyte and lymphocyte fractions from PBMCs, freshly isolated cells were allowed to adhere to surface modified cell culture dishes (Becton Dickinson Primaria) for 2 h at 37 °C. Fig. 6. TPV-GFP replicates in adherent monocytes but is unable to replicate in
differentiated macrophages. Monocytes and macrophages were infected at an MOI of 3 and cells were collected at the indicated time points. Samples were titrated on OMK cells in triplicate.
Peripheral blood lymphocytes (PBLs) were gently washed off with PBS and collected by centrifugation at 300×g. Monocytes remained attached to the cell culture dish surface. To activate PBLs, they were treated with 1 μg/ml phytohemagglutinin (Sigma) for 24 h. They were then collected by centrifugation and washed 3 times with complete medium. Monocytes were left in culture for 7 days in complete RPMI media to allow for differentiation into macrophages. The medium was changed every 2 days.
Generation of TPV expressing EGFP
To produce a TPV construct that expressed EGFP under a synthetic early/late promoter, sequences flanking the intergenic region between 5L and 6L were amplified from genomic DNA from TPV-Kenya using the following primer pairs: 5′-GAATT-CCTTTATACAAAAATAAATGAGTG-3′ (EcoRI underlined), 5′-CTCGAGATTAAACGTACGCACCCG-3′ (XhoI underlined), and 5′-CTGCAGTGCTCCGTGTGATAACAATG-3′ (PstI underlined), 5′-CTGCAGGATAAGTTTTTTAACAATTG-3′ (PstI underlined). The PCR products were cloned into pGEM-T easy (Promega) and sequenced. These constructs were amplified and cloned into pBluescript flanking an EGFP cassette under the synthetic early/late promoter to produce pBS:TPV-GFP.
OMK cells were infected with TPV (strain Kenya) at an MOI of 0.1 and then transfected with pBS:TPV-GFP. Infected cells were collected after 72 h and subjected to 3 cycles of freeze/ thaw and sonication. Virus was plaque purified on OMK cells by picking green fluorescent foci over 5 rounds. TPV-GFP was amplified and determined to be pure by PCR of genomic DNA. TPV replication curves
Single step replication curves were performed by infecting cells in serum free medium at an MOI of 3 at 27 °C for 1 h except for infection of PBMCs, which was done at 37 °C. Virus inoculum was removed and replaced with complete medium; this was considered time 0. Multistep replication curves were performed similarly except infecting cells at an MOI of 0.01 instead of 3. Medium was replaced at day 4 of the multistep replication curve to ensure adequate nutrients. Incubators were calibrated immediately before the experiment and temperature was monitored continuously throughout the experiments. Cells were collected as the specified time points and frozen. Virus-infected cells were subjected to 3 cycles of freeze/thaw and were then sonicated to release virus. Cell lysates were used to determine virus titer OMK cells in triplicate. Fluorescent foci were enumerated and graphed using Excel software (Microsoft).
FACS analysis
Cells were harvested either by trypsinization for pHDF cells, or by scraping for PBMCs, and collected by centrifugation at 300×g for 10 min. Cells were washed in PBS. One of each duplicate sample PBMCs was stained with either CD14-APC
(FL-4) or CD3-APC (Caltag Laboratories) to minimize compensation issues due to GFP (FL-1) expression. Isotype control was a mouse IgG2a-APC antibody (ebioscience). Cells were then either fixed in ethanol, to quench GFP expression for cell cycle analysis, or using the cytofix/cytoperm kit as per the manufacturer's instructions (Becton Dickinson). Cells were analyzed on a FACS Caliber (Becton Dickinson), with appro-priate compensation. Data were analyzed using Flo-Jo software (Tree Star).
Cell preparation for analysis of cell cycle
Approximately 1 × 106cells were collected in PBS and fixed in cold ethanol (50% final concentration) for a minimum of 24 h at 4 °C. Fixed cells were washed in PBS and incubated covered at room temperature for 30 min in 0.5 ml of PBS containing 50 μg/ml propidium iodide and 50 μg/ml RNase A. Cells were analyzed on a FACS Caliber (Becton Dickinson). Pulse width and area were used to eliminate doublets and debris and curves were produced and analyzed using Flo-Jo software.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (CIHR) and National Cancer Institute of Canada (NCIC). SHN was supported by an Ontario Graduate Scholarship (OGS) and Natural Sciences and Engineering Research Council of Canada (NSERC) post-graduate scholar-ship. MMS is the recipient of a Postdoctoral Fellowship provided by the Pamela Greenaway Kohlmeier Translational Breast Cancer Research Unit of the London Regional Cancer Program. GM is an International Scholar of the Howard Hughes Medical Institute.
References
Andreesen, R., Brugger, W., Scheibenbogen, C., Kreutz, M., Leser, H.G., Rehm, A., Lohr, G.W., 1990. Surface phenotype analysis of human monocyte to macrophage maturation. J. Leukoc. Biol. 47 (6), 490–497. Berk, A.J., 2005. Recent lessons in gene expression, cell cycle control, and cell
biology from adenovirus. Oncogene 24 (52), 7673–7685.
Castillo, J.P., Kowalik, T.F., 2004. HCMV infection: modulating the cell cycle and cell death. Int. Rev. Immunol. 23 (1–2), 113–139.
Chahroudi, A., Chavan, R., Kozyr, N., Waller, E.K., Silvestri, G., Feinberg, M.B., 2005. Vaccinia virus tropism for primary hematolymphoid cells is determined by restricted expression of a unique virus receptor. J. Virol. 79 (16), 10397–10407.
Damon, I.K., 2007. Poxviruses, fifth ed. In: Knipe, D.M., Howley, P.M. (Eds.), Fields Virology, vol. 2. Lippincott, Williams & Wilkins, New York, pp. 2947–2976 (2 vols).
Gottlieb, S.L., Myskowski, P.L., 1994. Molluscum contagiosum. Int. J. Dermatol. 33 (7), 453–461.
Heng, M.C., Steuer, M.E., Levy, A., McMahon, S., Richman, M., Allen, S.G., Blackhart, B., 1989. Lack of host cellular immune response in eruptive molluscum contagiosum. Am. J. Dermatopathol. 11 (3), 248–254. Inoki, K., Guan, K.L., 2006. Complexity of the TOR signaling network. Trends
Cell Biol. 16 (4), 206–212.
Jezek, Z., Arita, I., Szczeniowski, M., Paluku, K.M., Ruti, K., Nakano, J.H., 1985. Human tanapox in Zaire: clinical and epidemiological observations on cases confirmed by laboratory studies. Bull. World Health Organ. 63 (6), 1027–1035.
39 S.H. Nazarian et al. / Virology 368 (2007) 32–40
Johnston, J.B., Wang, G., Barrett, J.W., Nazarian, S.H., Colwill, K., Moran, M., McFadden, G., 2005. Myxoma virus M-T5 protects infected cells from the stress of cell cycle arrest through its interaction with host cell cullin-1. J. Virol. 79 (16), 10750–10763.
Knight, J.C., Novembre, F.J., Brown, D.R., Goldsmith, C.S., Esposito, J.J., 1989. Studies on Tanapox virus. Virology 172 (1), 116–124.
Lewis, E.J., Lam, M., Crutchfield III, C.E., 1997. An update on molluscum contagiosum. Cutis 60 (1), 29–34.
McFadden, G., 2005. Poxvirus tropism. Nat. Rev., Microbiol. 3 (3), 201–213. Mediratta, S., Essani, K., 1999. The replication cycle of tanapox virus in owl
monkey kidney cells. Can. J. Microbiol. 45 (1), 92–96.
Moss, B., 2006. Poxvirus entry and membrane fusion. Virology 344 (1), 48–54. Murray, A.W., 2004. Recycling the cell cycle: cyclins revisited. Cell 116 (2),
221–234.
Sanchez-Puig, J.M., Sanchez, L., Roy, G., Blasco, R., 2004. Susceptibility of different leukocyte cell types to Vaccinia virus infection. Virol. J. 1, 10.
Stanford, M.M., Barrett, J.W., Nazarian, S.H., Werden, S., McFadden, G., 2007. Oncolytic virotherapy synergism with signaling inhibitors: rapamycin increases myxoma virus tropism for human tumor cells. J. Virol. 81 (3), 1251–1260.
Wali, A., Strayer, D.S., 1999. Infection with vaccinia virus alters regulation of cell cycle progression. DNA Cell Biol. 18 (11), 837–843.
Yu, Q., Jones, B., Hu, N., Chang, H., Ahmad, S., Liu, J., Parrington, M., Ostrowski, M., 2006. Comparative analysis of tropism between canarypox (ALVAC) and vaccinia viruses reveals a more restricted and preferential tropism of ALVAC for human cells of the monocytic lineage. Vaccine 24 (40–41), 6376–6391.