FACULTÉ DES SCIENCES ET DES SCIENCES APPLIQUÉES
DÉPARTEMENT DE PHYSIQUE
MÉMOIRE PRÉPARÉ POUR L‟OBTENTION DU DIPLÔME
DE MASTER EN PHYSIQUE
OPTION
Physique des matériaux
THÈME
Présenté par : BIBI Hamida
Soutenu le : ……/…../2018
Devant le jury :
M. ZERIRGUI Djamel Président MCB Université de Bouira
M. BENAMARA Salem Rapporteur MAA Université de Bouira
M. CHIBANI Moussa Examinateur MAA Université de Bouira
M. KHEFFACHE Sedik Examinateur MAA Université de Bouira
DE LA RECHERCHE SCIENTIFIQUE
UNIVERSITE DE BOUIRA
Étude par la méthode ab initio des
propriétés magnétiques des agrégats
Dédicaces
Je dédie ce travail à :
La plus chère personne dans la vie : ma mère Mon père qui a quitté ce monde
Mon mari
Mes chers frères et sœurs Toute ma famille
Tous les professeurs de l’université de bouira. Mes amis du groupe physique des matériaux.
Remerciements
Je tiens à exprimer ma reconnaissance à mon encadreur M. Benamara Salem de m’avoir encadré et pour la grande patience avec laquelle il a suivi ce travail jusqu’a son achèvement. Je le remercie également pour ses conseils et suggestions. Cet important Monsieur mérite tous mes respects, toute ma confiance à son égard. Je lui dédie ce travail que j'espère appréciable de son coté.
Je tiens à remercier les membres du jury Monsieur CHIBANI Moussa et Monsieur KHEFFACHE Sedik d'avoir accepté de faire partie du jury en qualité d'examinateur.
Merci à Monsieur ZERIRGUI Djamel pour avoir accepté de présider ce jury.
Je remercie tous mes chers amis pour leurs soutiens et pour les moments agréables qu’on a partagé ensemble. Enfin je remercie toute ma famille qui a toujours été avec moi. Un grand merci pour ma mère, mes frères, mon mari pour leurs encouragements et le soutien morale qu'ils m’ont offert.
J’exprime enfin mes vifs remerciement à tous mes amis et enseignants de graduation.
Tables de matières
Table des matières
Table des matières
Introduction générale………..1
Chapitre1 : les outils théoriques ………...3
1.1 Problème à N corps ……….3
1.2 L‟équation de Schrödinger………...4
1.3 Les approximations fondamentales.………4
1.3.1 Approximation de Born-Oppenheimer...………...4
1.3.2 Approximation de Hartree………...6
1.3.3 Approximation de Hartree-Fock………...7
1.3.4 Approximation de Hartree-Fock-Slater (méthode 𝑋𝛼) .………..8
1.4 Théorie de la fonctionnelle de la densité (DFT).………...9
1.4.1 Approche de Thomas et Fermi……….………...9
1.4.2 Les théorèmes de Hohenberg et Kohn ………10
1.4.3 Les équations de Kohn et Sham .………...11
1.4.4 Les approximations de la DFT………..13
1.4.5 Mise en œuvre de la DFT………..15
1.5 Les base de projection………...16
1.5.1 Base d‟ondes planes………..16
1.5.2 Les bases d‟orbitales atomiques (LCAO)……….18
1.6 Algorithme de la DFT dans une version Pseudo………...20
Chapitre2 : Pseudopotentiels et code de calcul : SIESTA……….22
2.1 Pseudopotentiels………...22
2.2 Les pseudopotentiels à norme conservée………...24
2.2.1 Méthode de Kerker………..25
2.2.2 Méthode de Troullier-Martins………..27
2.2.3 Correction non linéaire de cœur………...28
2.2.4 La séparation de Kleinman-Bylander………..30
2.2.5 Dynamique moléculaire………...31
2.3 Les algorithmes d‟ordre N et d‟ordre N3………...34
2.3.1 Code SIESTA ………35
Tables de matières
3.1 Introduction………...39
3.2 Les agrégats de Fen………..………. ...41
3.2.1 Pseudopotentiel du Fer………...42
3.2.2 Base associée au Fer………...45
3.2.3 Optimisation de l‟énergie Shift……….46
3.2.4 Le dimer de fer (Fe2)………....48
3.2.5 L‟agrégat de Fe3 ..………...49 3.2.6 L‟agrégat de Fe4 ………...50 3.2.7 L‟agrégat de Fe5.………...50 3.2.8 L‟agrégat de Fe6………...51 3.2.9 L‟agrégat de Fe7.………...52 3.2.10 L‟agrégat de Fe8………...53 3.2.11 L‟agrégat de Fe9…..………54 3.2.12 L‟agrégat de Fe10………...54 3.3 Les agrégats de FenP (n=1,...,10)………...55 3.3.1 Pseudopotentiel du Phosphore………..55
3.3.2 Base associée au Phosphore.……….………...56
3.3.3 L‟agrégat de Fe1P………...56 3.3.4 L‟agrégat de Fe2P…...………...57 3.3.5 L‟agrégat de Fe3P...………...57 3.3.6 L‟agrégat de Fe4P………...58 3.3.7 L‟agrégat de Fe5P………...59 3.3.8 L‟agrégat de Fe6P………60 3.3.9 L‟agrégat de Fe7P………61 3.3.10 L‟agrégat de Fe8P..………....62 3.3.11L‟agrégat de Fe9P………...63 3.3.12 L‟agrégat de Fe10P.………...64
3.3.13 Discussion des résultats...………...66
Conclusion générale………...68 Bibliographie
Introduction générale
1
Introduction générale
Depuis la naissance de nouvelles technologies à l‟échelle du nanomètre dans les années 80, l‟intérêt porté aux systèmes de faible dimensionnalité ne cesse de croître. En effet, les recherches menées sur les propriétés physiques et/ou chimiques des nanomatériaux est un enjeu majeur de l‟industrie actuelle, et ce quelque soient les domaines d‟applications considérés (matériaux émergents, micro-électronique, environnement, biomatériau, énergie etc.).
L‟objectif des nanotechnologies consiste à produire des objets ou matériaux de taille inférieur à 100 nanomètres. Ces nanomatériaux sont composés de nanostructures, ou agrégats dont la taille peut aller de quelques atomes à quelques milliers d‟atomes. Leur caractérisation très précise, à l'état de dimension réduite a mis en évidence des propriétés structurales, et magnétiques surprenantes par rapport à celles de l‟état massif.
Les méthodes de simulation ont joué un rôle très important pour la détermination des différentes propriétés; car elles ont en effet donné une nouvelle dimension à l‟investigation scientifique de nombreux phénomènes physiques et chimiques, et en particulier les méthodes ab initio. Ces méthodes sont devenues aujourd'hui un outil indispensable et permettent non seulement de mieux comprendre les différentes propriétés des matériaux et des systèmes les plus complexes mais de les prédire, parfois elles ont pu remplacer des expériences très coûteuses ou même irréalisables en laboratoire.
Ainsi le développement des méthodes de simulation avec l'augmentation de la puissance des ordinateurs ont permis l‟étude d‟une grande gamme de matériaux. Parmi lesquels nous citons les matériaux de transitions. Ces derniers occupent actuellement une position privilégiée dans le domaine des sciences des matériaux. Ces derniers présentent des propriétés magnétiques et structurales intéressantes dans l‟industrie des technologiques. Ce qui motive un intérêt de plus en plus croissant et de nombreuses études aussi bien théoriques qu‟expérimentales leurs sont consacrées.
Le but de ce mémoire est d‟apporter une contribution à la compréhension des propriétés géométriques et magnétiques des petits agrégats de fer (𝐹𝑒𝑛) en première étape et des agrégats
binaires de fer-phosphore (𝐹𝑒𝑛𝑃) en seconde étape, avec n varie de 1 a 10. Ces calcules ont
Introduction générale
2
Electronic Simulations with Thousands of Atoms), Ce code est basé sur la théorie de la
fonctionnelle de la densité, et permet une optimisation des structures par dynamique moléculaire. Il utilise des bases de fonctions localisées de type LCAO (Linear Combination of
Atomic Orbitals) et des pseudo-potentiels non locaux. Les problèmes d‟échange et de
corrélation entre les électrons sont traités dans l‟approximation du gradient généralisé GGA. Dans le premier chapitre, nous avons présenté le cadre théorique qui est à la base des méthodes modernes de calcul de structures électroniques. Les différentes approximations associées à la théorie de la fonctionnelle de la densité (DFT) seront exposées. Ainsi, nous avons donné un aperçu sur les principales bases de projection qui sont utilisées dans les codes de calcul.
Le second chapitre portera sur le formalisme de la méthode LCAO combinée à des pseudo-potentiels ab initio. Notre intérêt, c‟est porté sur les pseudo-potentiels à norme conservée et
aux étapes à suivre pour générer des pseudo-potentiels. A la fin de ce chapitre nous parlerons du dynamique moléculaire ab initio, nous discuterons du code de calcul SIESTA et comment générer une base de pseudo-orbitales atomiques (PAO). Nous terminons par la présentation du code de calcul.
Les résultats et leurs discussions font l‟objet de chapitre 3. Après un bref rappel sur les détails de calcul, nous présentons les résultats théoriques obtenus concernant les calculs des structures géométriques et magnétiques des agrégats de fer et de fer-phosphore dont la taille n‟excède pas les 10 atomes de fer.
Nous terminons notre mémoire par une conclusion générale et les perspectives que notre travail a ouvert.
3
Chapitre 1
Les outils théoriques
Dans ce chapitre, nous présenterons les éléments de la théorie quantique non relativiste sur lesquels sont basées les méthodes de calcul des structures électroniques.
1.1 Problème à N particules
Les propriétés physiques et chimiques des matériaux sont fortement liées à la compréhension des systèmes d‟électrons et des noyaux en interaction, ces propriétés sont rarement décrites avec exactitude à cause de la complexité que présentent ces systèmes à plusieurs particules dits à N corps, La compréhension de ces propriétés passe par la détermination et le calcul des structures électroniques.
Plusieurs techniques de calcul de structure électronique ont été mises au point. Nous distinguons pour ce faire deux catégories principales: les méthodes semi-empiriques qui font appel à des résultats expérimentaux et les méthodes de premiers principes dites ab initio qui ne requièrent aucun paramètre ajustable.
Avec l‟important développement de l‟outil informatique, Les méthodes de premiers principes permettent de simuler et de calculer les propriétés de systèmes de plus en plus complexes. Ces méthodes donnent de très bons résultats et sont d‟une grande fiabilité et ceci sans avoir à introduire de paramètres expérimentaux
4
1.2 Équation de Schrödinger
Dans un système matériel constitué par 𝑁𝑒 électrons de coordonnées d‟espace 𝑟 , 1 𝑟 … 2
𝑟𝑁𝑒
et de masse 𝑚𝑒 en interaction avec 𝑁𝑛 noyaux de charge Z, de masse 𝑀𝑛 et de coordonnées
d‟espace 𝑅 1 , 𝑅 2 …𝑅 𝑁𝑛. Un tel système, est décrit par la fonction d‟onde ψ, en principe, il
peut être déterminé à partir des lois de la mécanique quantique à l‟aide de la résolution de l‟équation de Schrödinger [1] indépendante du temps qui s‟écrit sous la forme (1.1):
Hψ = Eψ (1.1) Où H est l‟opérateur Hamiltonien du système, dont les valeurs propres E sont les valeurs de l‟observable de l‟énergie totale.
L'Hamiltonien exact d„un système non relativiste résulte de la présence des forces d'interaction d‟origine électrostatique et s‟écrit comme :
H =−ℏ 2 2𝑚𝑒 𝛻 𝑖2 𝑁𝑒 𝑖=1 − ℏ 2 2𝑀𝑛 𝛻 𝑎2 𝑁𝑛 𝑎=1 + 𝑒 2 ǀ𝑟 − 𝑟𝑖 ǀ𝑗 − 𝑁𝑒 𝑗 >𝑖 𝑁𝑒 𝑖=1 𝑍𝑎𝑒 2 ǀ𝑟 − 𝑅𝑖 ǀ𝑎 𝑁𝑛 𝑎=1 𝑁𝑒 𝑖=1 + 𝑍𝑎𝑍𝑏𝑒 2 ǀ𝑅 − 𝑅𝑎 ǀ𝑏 𝑁𝑛 𝑏>𝑎 𝑁𝑛 𝑎=1 (1.2)
Le premier terme de l‟équation (1.2), désignant l‟énergie cinétique des électrons, le second celui des noyaux et les trois derniers représentant les énergies potentielles d‟interaction entre électron-électron, électron-noyau et noyau-noyau.
Étant donné la complexité de l‟équation de Schrödinger à 𝑁𝑒+ 𝑁𝑛 particules, la résolution
analytique et rigoureuse est impossible. Il est indispensable de réduire la complexité cette équation et ceci ne peut se faire sans le recours à des approximations.
1.3 Les approximations fondamentales
1.3.1- Approximation de Born-Oppenheimer
Selon Born-Oppenheimer [2] le traitement des électrons et des noyaux d‟une façon séparée permettra de réduire La complexité en ramenant le problème de Ne+Nn particules à celui de
Ne électrons. Cette approximation exploite la grande différence de masse entre les électrons et les noyaux. Car ces derniers étant à plus 1836 fois plus lourdes que les électrons, ils se déplacent beaucoup moins vites que les électrons. Ainsi, les électrons suivent quasi instantanément le mouvement des noyaux.
5
La fonction d‟onde 𝜓(𝑟 ,..,𝑟1 ,𝑅𝑁𝑒 1,.., 𝑅 𝑁𝑛) du système peut s‟écrire comme le produit de deux
fonctions d‟ondes :
𝜓 𝑟 , . . , 𝑟1 , 𝑅𝑁𝑒 , . . . , 𝑅1 = 𝜓𝑁𝑛 𝑒𝑙 𝑟 , . . , 𝑟1 𝛩𝑁𝑒 𝑛𝑜𝑦 𝑅 , . . . , 𝑅1 1.3 𝑁𝑛
Où 𝛩𝑛𝑜𝑦(𝑅 1 ,.., 𝑅𝑁𝑛) est la fonction d‟onde nucléaire et 𝜓𝑅(𝑟 ,..,𝑟1 ) la fonction d‟onde 𝑁𝑒
électronique correspondante à une configuration des noyaux figés dans la position R ≡ 𝑅 1 ,
𝑅2
… 𝑅𝑁 𝑛 .
Dans l‟approximation de Born-Oppenheimer, l‟Hamiltonien du système donné par l‟équation (1.2), peut s‟écrire comme la somme de deux termes :
H =𝐻𝑒𝑙+ 𝐻𝑁 (1.4)
Où 𝐻𝑒𝑙 et 𝐻𝑁 désignent respectivement les Hamiltoniens électronique et nucléaire. Ils sont
donnés par les expressions suivantes :
𝐻𝑒𝑙 = 𝑒2 ǀ𝑟 − 𝑟𝑖 ǀ𝑗 − 𝑁𝑒 𝑗 >𝑖 𝑍𝑎𝑒 2 ǀ𝑟 − 𝑅𝑖 ǀ𝑎 𝑁𝑛 𝑎=1 𝑁𝑒 𝑖=1 𝑁𝑒 𝑖=1 (1.5) 𝐻𝑁= −ℏ2 2𝑀𝑛 𝛻 𝑎2 𝑁𝑛 𝑎=1 + 𝑍𝑎𝑍𝑏𝑒 2 ǀ𝑅 − 𝑅𝑎 ǀ𝑏 𝑁𝑛 𝑏>𝑎 𝑁𝑛 𝑎=1 1.6
Le mouvement des électrons est découplé de celui des noyaux et l‟équation de Schrödinger du système se sépare en deux équations :
1- Équation de Schrödinger nucléaire :
𝐻𝑁Θ(𝑅 1 ,.., 𝑅 𝑁𝑛) = 𝐸𝑁Θ(𝑅 1 ,.., 𝑅 𝑁𝑛) (1.7)
2- Équation de Schrödinger électronique:
𝐻𝑒𝜓𝑅(𝑟 ,..,𝑟1 ) = 𝐸𝑁𝑒 𝑒𝜓𝑅(𝑟 ,..,𝑟1 ) (1.8) 𝑁𝑒 A présent, le problème consiste à résoudre l‟équation de Schrödinger électronique :
𝐻𝑒 𝜓𝑅 𝑟 , . . , 𝑟1 = 𝐸𝑁𝑒 𝑒𝜓𝑅 𝑟 , . . , 𝑟1 (1.9) 𝑁𝑒
6
Cette approximation réduit d‟une façon significative le degré de complexité mais la présence du terme biélectronique associé à l‟interaction électron-électron rend la résolution analytique de cette équation impossible. Alors que d‟autres approximations supplémentaires sont requises pour pouvoir résoudre effectivement cette équation.
1.3.2- Approximation de Hartree
En 1927, Hartree [3] propose une méthode permettant de calculer des fonctions d‟ondes poly-électroniques approchées en les écrivant sous la forme de produits de fonctions d‟ondes mono électronique. Cette approximation, dite de champ moyen, permet de ramener le problème d‟interaction à N-corps à celui d‟un électron indépendant se mouvant dans un champ moyen produit par le restant des électrons. Mathématiquement, on parle de séparation des variables électroniques. Dans ce cas, la fonction d‟onde électronique du système est alors le produit directe des fonctions d‟ondes mono électronique ф𝑖(𝑟 ) : 𝑖
𝜓𝑅 𝑟 , . . , 𝑟1 = ф𝑁𝑒 𝑖 𝑟 𝑖
𝑁𝑒
𝑖=1
1.10 L‟Hamiltonien électronique donné par l‟équation (1.5), s‟écrit comme la somme des Hamiltoniens à un électron : 𝐻𝑒𝑙 = −ℏ2 2𝑚𝑒 𝛻𝑖2 + 𝑒2 ǀ𝑟 − 𝑟𝑖 ǀ𝑗 − 𝑍𝑎𝑒 2 ǀ𝑟 − 𝑅𝑖 ǀ𝑎 𝑁𝑛 𝑎=1 𝑁𝑒 𝑗 >𝑖 1.11 𝑁𝑒 𝑖=1
Le problème de la résolution de l‟équation de Schrödinger d‟un système à 𝑁𝑒 électrons est
réduit à celui de la résolution de l‟équation de Schrödinger à un seul électron suivante : −ℏ 2 2𝑚𝑒 𝛻𝑖2 + 𝑈𝑖 𝑟 + 𝑉𝐻𝑖 𝑟 ф 𝑖 𝑟 = 𝜀𝑖 𝑖ф𝑖 𝑟 1.12 𝑖 𝑈𝑖 𝑟 = − 𝑍𝑎𝑒2 ǀ𝑟 − 𝑅𝑖 ǀ𝑎 . 𝑁𝑛 𝑎=1 1.13 𝑉𝐻𝑖 𝑟 = 𝜌 𝑟 ′ 𝑒2 ǀ𝑟 − 𝑟 ′ǀ 𝑁𝑒 𝑗 >𝑖 𝑑𝑟′. 1.14 𝜌𝑖 𝑟 = ф𝑗 𝑟 2 . 𝑁𝑒 𝑗 =1𝑗 ≠𝑖 (1.15)
7
𝑈𝑖 𝑟 est le potentiel extérieur des noyaux, 𝑉𝐻𝑖 𝑟 est le champ moyen des autres électrons
appelé potentiel de Hartree et 𝜌𝑖 𝑟 représente la densité électronique.
La solution donnée par l‟approximation de Hartree ne correspond pas tout à fait à la réalité. En effet, les électrons sont des particules identiques indiscernables et obéissent au principe d‟exclusion de Pauli [4]. De ce fait, la fonction d‟onde totale du système électronique doit être antisymétrique par rapport à la permutation de deux électrons.
1.3.3- Approximation de Hartree-Fock (HF)
La théorie de Hartree-Fock ne tient pas compte de tous les effets de corrélation entre les mouvements des électrons dans un système. La corrélation entre deux électrons de spins parallèles (dite de Fermi) est décrite par la méthode Hartree-Fock. Par ailleurs, la corrélation de Coulomb est due à la répulsion électrostatique entre les électrons. L‟énergie de corrélation représente la différence entre les résultats obtenus en HF et ceux de la solution exacte de l‟équation de Schrödinger.
On peut considérer que les électrons sont souvent proches les uns des autres, donc le terme de répulsion inter-électronique est trop grand et l‟énergie de HF est supérieure à E0 (l‟énergie
exacte). Cette différence, peut être importante, est prise en compte dans les méthodes post-Hartree-fock. Ces méthodes permettent de traiter les effets de corrélation qui ne sont pas traité par la méthode HF et se partagent en deux catégories.
Dans l‟approximation de Hartree, le mouvement des électrons est supposé non corrélé. En 1930, Fock [5] a montré que la fonction d‟onde de Hartree (1.10) viole le principe d‟exclusion de Pauli parce qu‟elle n‟est pas antisymétrique par rapport à l‟échange de deux particules quelconques. Il a proposé de corriger ce défaut en ajoutant un terme supplémentaire non-local d‟échange qui complique considérablement les calculs. La fonction d‟onde totale et alors remplacée par un déterminant dit de Slater construit sur les fonctions d‟ondes monoélectroniques ф𝑖 𝑟, 𝜍 . 𝜓 𝑟 , . . , 𝑟1 =𝑁 1 𝑁! 𝜓1 𝑟1 ⋯ 𝜓𝑁 𝑟1 ⋮ ⋮ ⋮ 𝜓1 𝑟𝑁 ⋯ 𝜓𝑁 𝑟𝑁 1.16 Où 𝑟 𝑒𝑡 𝜍 sont les variables d‟espace et de spin.
On obtient ainsi les équations de Hartree-Fock : −1
2 𝛻𝑖
2
8
Où 𝑉𝑥 𝑟 est le potentiel non local d‟échange introduit par Fock, il est définit par son action
sur ф𝑖 𝑟 : 𝑖 𝑉𝑥 𝑟 ф𝑖 𝑟 = 𝑑𝑟 𝑖 ′ 𝑁𝑒 𝑗 ≠𝑖 ф𝑗∗ 𝑟 ′ ф𝑖 𝑟 ǀ𝑟 − 𝑟 ′ǀ ф𝑗 𝑟 1.18
Le système d‟équations (1.12) se résout de manière auto-cohérente dans la mesure où le potentiel dépend de la fonction d‟onde.
En principe l‟approximation de Hartree Fock pose un problème du fait du caractère non local du potentiel d‟échange. De plus, cette méthode ne tient pas compte des effets de corrélation entre électrons de spins antiparallèles.
Les équations de Hartree-Fock sont, de ce fait, très difficiles à résoudre notamment pour les systèmes contenant un grand nombre d‟électrons.
1.3.4- Approximation de Hartree-Fock-Slater (méthode𝑋
𝛼)
Slater [6] approxime le terme d‟échange en supposant qu‟il possède un caractère local
contrairement à l‟AHF. Ce potentiel d‟échange 𝑉𝑥 pour un gaz d‟électron homogène de
densité𝜌 𝑟 s‟écrit sous la forme:
𝑉𝑥 𝑟 = −6𝛼
3𝜌 𝑟 8𝜋
1 3
. 1.19 Où 𝛼 est un paramètre sans dimension.
Cette méthode proposée par Slater et connue sous le non de 𝑋𝛼 soulève deux points
essentiels:
- Premièrement la simplicité de ce potentiel par rapport à l‟AHF (due au fait qu‟il est local). - Deuxièmement, il donne une forme simple du terme d‟échange-corrélation. Toutefois le choix de ce potentiel pratiquement intuitif conduit à des résultats pas toujours satisfaisants. La méthode 𝑋𝛼 ignore les corrélations électroniques, car en réalité ces derniers
interagissent mutuellement de manière beaucoup plus complexe. Car selon Wigner [7], les interactions électron-électron produisent un terme d‟énergie supplémentaire en plus du terme d‟échange introduit par Fock, c‟est le terme d‟énergie de corrélation 𝐸𝑐𝑜𝑟 (énergie négative).
9
On définit l‟énergie d‟échange et corrélation𝐸𝑥𝑐 comme étant la somme du terme d‟échange
𝐸𝑥 introduit par Fock et de l‟énergie de corrélation 𝐸𝑐𝑜𝑟 . En effet, l‟énergie totale exacte est donnée par :
𝐸𝑥𝑐 = 𝐸𝑥 + 𝐸𝑐𝑜𝑟 1.20
𝐸𝑒𝑥 = 𝐸𝐻+ 𝐸𝑥𝑐 1.21
1.4 Théorie de la fonctionnelle de la densité (DFT) :
Les bases de la théorie de la fonctionnelle de la densité (DFT) ont été élaborées en 1927 par Thomas [8] et Fermi [9] qui stipule que les propriétés électroniques peuvent être décrites en terme de fonctionnelles de la densité électronique. En 1928, Dirac [10] introduit le terme d‟échange prédit par Hartree mais il n‟y a toujours aucune prise en compte de la corrélation électronique qui fût finalement ajoutée par Wigner.
Néanmoins, il faudra attendre le milieu des années 1960 où les contributions de Hohenberg et Kohn [11] sont établies le formalisme théorique sur lequel repose la méthode actuelle.
1.4.1- Approche de Thomas et Fermi
En 1920, Thomas [8] et Fermi [9] ont proposés une approche qui repose sur l‟hypothèse qui stipule que les électrons sont distribués uniformément autour des noyaux. La densité électronique 𝜌 𝑟 est utilisée comme la variable en fonction de laquelle s‟exprime l‟énergie cinétique des électrons. Cette énergie est décrite par une approximation locale basée sur le modèle de gaz d‟électron sans interaction 𝜌 𝑟 13 .Un peu plus tard, Dirac [10] a proposé que les effets d‟échange soient pris en compte en incorporant un terme venant de la densité d‟énergie d‟échange dans un gaz homogène d‟électrons, de densité constante 𝜌 𝑟 et sans interaction dans un potentiel effectif 𝑉𝑒𝑓𝑓 𝑟 donné par l‟équation suivante :
𝑉𝑒𝑓𝑓 𝑟 = 𝑉𝑒𝑥𝑡 𝑟 + 𝑑𝑟′
𝜌 𝑟 ′
ǀ𝑟 − 𝑟 ′ǀ 1.22
Enfin, les contributions à l‟énergie électronique totale sont exprimées en fonction de la densité électronique :
10 𝐸𝑇𝐹 𝜌 = 3 5 3𝜋 2 23 𝑑𝑟 𝜌53 𝑟 + 𝑑𝑟 𝑉 𝑒𝑥𝑡 𝑟 𝜌 𝑟 + 1 2 𝑑𝑟 𝑑𝑟 ′𝜌 𝑟 𝜌 𝑟 ′ ǀ𝑟 − 𝑟 ′ǀ 1,23
Le premier terme représente l‟énergie cinétique d‟un système d‟électrons sans interaction, de densité 𝜌 𝑟 , le second terme décrit l‟énergie d‟une densité électronique 𝜌 𝑟 dans un potentiel électrostatique externe𝑉𝑒𝑥𝑡, et le troisième terme correspond à l‟énergie d‟interaction
coulombienne électron-électron.
Afin de déterminé la densité introduite par 𝐸𝑇𝐹 𝜌 , Thomas [8] et Fermi [9] ont employés le
principe variationnel. A noter que l‟énergie de Thomas et Fermi ne contient aucun terme d‟échange et de corrélations.
C‟est un pas de plus par rapport à l‟approche de Hartree, mais elle reste incomplète car elle ne prend pas en considération les effets d‟échange et de corrélation.
1.4.2- Théorèmes de Hohenberg et Kohn
En 1964, Hohenberg et Kohn [11] ont repris le postulat de Thomas et Fermi, selon lequel les propriétés électroniques d‟un système en interaction sont déterminées par la densité électronique et ils ont montré qu‟il existe une fonctionnelle de l‟énergie E[ρ(r)] associée au principe variationnel.
La théorie de la fonctionnelle de la densité est fondée sur deux théorèmes mathématiques rigoureux, le premier dit d‟existence et le second dit variationnel.
1.4.2.1- Théorème 1
Hohenberg et Kohn ont prouvé que les propriétés du système tel que l‟énergie
fondamentale E0 sont déterminées à partir de la connaissance de la densité électronique en
chaque point.
Pour tout système électronique non dégénéré en interaction dans un potentiel externe Vext(r), l'énergie totale de l'état fondamental est une fonctionnelle de la densité électronique
11
Il existe une seule densité électronique 𝜌0(r) pour l‟état fondamentale, d‟où l‟unicité de
l‟énergie totale E. Autrement dit, deux potentiels extérieurs différents ne peuvent conduire à la même densité pour l‟état fondamentale.
𝐸 𝜌 𝑟 = 𝐹 𝜌 𝑟 + 𝑉𝑒𝑥𝑡 𝑟 𝜌 𝑟 𝑑3𝑟 1,24
𝐹 𝜌 𝑟 = 𝑇 𝜌 𝑟 + 𝐸𝑒𝑒 𝜌 𝑟 1.25
Où 𝐹 𝜌 𝑟 est la fonctionnelle universelle donnée par la somme de l‟énergie cinétique T 𝜌 𝑟 du système d‟électrons en interaction et du terme d‟interaction électron-électron 𝐸𝑒𝑒 𝜌 𝑟 .
1.4.2.2- Théorème 2
Le deuxième théorème stipule que 𝐹 𝜌 𝑟 la fonctionnelle qui permet d‟accéder à l‟énergie de l‟état fondamental non dégénéré 𝐸 𝜌 𝑟 , donne la plus basse énergie si et seulement si la densité électronique correspond à la densité exacte de l‟état fondamental 𝜌0 𝑟 ,
ce qui revient au principe variationnel.
𝐸 𝜌0 = 𝑚𝑖𝑛 𝐸 𝜌 ↔
𝛿𝛦
𝛿𝜌 = 𝜇 1.26 Où 𝜇 est une constante qui impose la conservation du nombre de particules :
𝜌 𝑟 𝑑𝑟 = 𝑁𝑒 1.27
On obtient l‟équation fondamentale de la DFT suivante :
𝜇 =𝛿𝛦 𝜌 𝑟
𝛿𝜌 = 𝑉𝑒𝑥𝑡 𝜌 𝑟 +
𝛿𝐹 𝜌 𝑟
𝛿𝜌 1.28 Si la fonctionnelle 𝐹 𝜌 𝑟 est connue, alors, il sera relativement facile d‟utiliser le principe variationnel pour déterminer l‟énergie totale et la densité électronique de l‟état fondamental pour un potentiel extérieur donné. Malheureusement, la fonctionnelle 𝐹 𝜌 𝑟 n‟est pas connue et les équations correspondantes ne peuvent pas être résolues.
1.4.3- Les équations de Kohn et Sham
En 1965 Kohn et Sham [12] ont développé une approche dans laquelle ils utilisent un système fictif d‟électrons sans interaction baignant dans un potentiel extérieure 𝑉𝑒𝑥𝑡 tel que le
12
système sans interaction a la même densité à l'état fondamental que celle du système réel en interaction. En appliquons les théorèmes de Hohenberg et Kohn, l„énergie totale du système réel dans un potentiel extérieur 𝑉𝑒𝑥𝑡s‟écrit :
𝛦 𝜌 𝑟 = 𝑇0 𝜌 𝑟 + 𝑉𝑒𝑥𝑡 𝑟 𝜌 𝑟 𝑑𝑟 +
1 2
𝜌 𝑟 𝜌 𝑟 ′
ǀ𝑟 − 𝑟 ′ǀ 𝑑𝑟 𝑑𝑟 ′+ 𝛦𝑥𝑐 𝜌 𝑟 1.29
𝑇0 𝜌 𝑟 est la fonctionnelle d'énergie cinétique d'un système de 𝑁𝑒 électrons sans
interaction, et par conséquent elle est différente de la fonction 𝑇 𝜌 𝑟 qui fait partie de 𝐹 𝜌 𝑟 dans l'équation (1.25). 𝛦𝑥𝑐 𝜌 𝑟 est appelée fonctionnelle d‟échange et corrélation.
La différence entre l‟énergie cinétique réelle 𝑇 𝜌 𝑟 et celle des électrons sans interaction 𝑇0 𝜌 𝑟 ainsi que la différence entre l‟énergie d‟interaction réelle et celle de Hartree sont
prises en compte dans l‟énergie d‟échange- corrélation : 𝛦𝑥𝑐 𝜌 𝑟 = 𝑇 𝜌 𝑟 − 𝑇0 𝜌 𝑟 + 𝐸𝑒𝑒 − 1 2 𝜌 𝑟 𝜌 𝑟 ′ ǀ𝑟 − 𝑟 ′ǀ 𝑑𝑟 𝑑𝑟 ′ 1.30 On a: 𝛿𝛦 𝜌 𝑟 𝛿𝜌 = 𝛿𝑇0 𝜌 𝑟 𝛿𝜌 𝑟 + 𝑉𝑒𝑥𝑡 𝑟 + 𝜌 𝑟 ′ ǀ𝑟 − 𝑟 ′ǀ𝑑𝑟 ′ +𝛿𝛦𝑥𝑐 𝜌 𝑟 𝛿𝜌 1.31
Le choix de Kohn et sham de se référer à un système fictif de 𝑁𝑒 électrons sans interaction
implique la résolution de 𝑁𝑒 équations de Schrödinger « mono électroniques ». Cela nous
amène à réécrire le problème sous la forme de trois équations interdépendantes. Les équations de Kohn et Sham sont les suivantes :
- La première donne la définition du potentiel effectif ressenti par les électrons 𝑉𝑒𝑓𝑓 𝑟 = 𝑉𝑒𝑥𝑡 𝑟 +
𝜌 𝑟 ′
ǀ𝑟 − 𝑟 ′ǀ𝑑𝑟 ′+
𝛿𝛦𝑥𝑐 𝜌 𝑟
𝛿𝜌 1.32 -La seconde équation utilise le potentiel 𝑉𝑒𝑓𝑓 estimé dans les 𝑁𝑒 équations de Schrödinger
afin d‟obtenir les ф𝑖 𝑟 :
−𝛻 + 𝑉𝑖2 𝑒𝑓𝑓 𝑟 ф𝑖 𝑟 = 𝜀𝑖ф𝑖 𝑟 1.33
- La troisième équation donne l‟expression de la densité électronique en fonction des Nfonctions d‟onde ф𝑖 𝑟 obtenues :
𝜌 𝑟 = ф𝑖 𝑟 2 𝑁𝑒
𝑖=1
13
L‟équation (1.28) peut s‟écrire pour un système d‟électrons sans interactions, se déplaçant dans un potentiel effectif 𝑉𝑒𝑓𝑓 𝑟
𝛿𝛦 𝜌 𝑟
𝛿𝜌 =
𝛿𝑇𝑆 𝜌 𝑟
𝛿𝜌 𝑟 + 𝑉𝑒𝑓𝑓 𝑟 1.35 Pour le cas spin-polarisé, Les équations de Kohn et Sham généralisées aux systèmes magnétiques non relativistes doivent être résolues, pour chaque composante de spin, de manière auto-cohérente
−𝛻 + 𝑉𝑖2 𝑒𝑓𝑓↑↓ 𝑟 ф𝑖↑↓ 𝑟 = 𝜀𝑖↑↓ф𝑖↑↓ 𝑟 1.36
𝜌 𝑟 = 𝜌↑ 𝑟 + 𝜌↓ 𝑟 1.37
Où 𝜌↑ 𝑟 et𝜌↓ 𝑟 , représentent respectivement les densités des électrons de spin up et de
spin down.
La seule ambiguïté dans l‟approche de Kohn et Sham (KS) est le terme de la fonctionnelle d‟échange-corrélation 𝛦𝑥𝑐 𝜌 𝑟 . La complexité formelle de ce dernier rend la résolution des
équations de KS difficile, en effet, Plus la connaissance de ce terme sera précise, plusф sera connue avec précision, plus l'énergie sera proche de l'énergie exacte. Néanmoins cette fonctionnelle peut être soumise à des approximations de l‟ordre local ou proche local de la densité
1.4.4- Les approximations de la DFT
1.4.4.1- Approximation de la densité locale (LDA)
L‟approximation la plus répandue pour calculer le terme d‟échange et corrélation est l‟approximation de la densité locale (Local Density Approximation), ou LDA. Elle présente la continuité de la démarche de Kohn et Sham. L‟idée de LDA est de considérer le potentiel d‟échange-corrélation comme une quantité locale définie en un point r, dépendant faiblement des variations de la densité autour de ce même point r.
L‟approximation LDA consiste à considérer la densité comme étant équivalente à celle d'un gaz d'électrons homogènes possédant la même densité électronique. L‟énergie d‟échange et corrélation s‟exprime alors de la manière suivante :
14
Où ℰ𝑥𝑐 désigne l‟énergie d‟échange-corrélation pour un gaz homogène d‟électrons, de
densité ρ. Elle peut être considérée comme la somme de la contribution d‟échange et de corrélation.
ℰ𝑥𝑐 𝜌 𝑟 = ℰ𝑥 𝜌 𝑟 + ℰ𝑐 𝜌 𝑟 1.39
D‟où l‟énergie d‟échange 𝐸𝑐𝜌 𝑟 résulte de l‟anti-symétrie de la fonction d‟onde vis- à-vis
de l‟échange des coordonnées électroniques.
Le potentiel d‟échange-corrélation lui correspondant est : 𝑉𝑥𝑐 𝜌 𝑟 =
𝛿𝛦𝑥𝑐 𝜌 𝑟
𝛿𝜌 𝑟 =
𝛿 𝜌 𝑟 ℰ𝑥𝑐 𝜌 𝑟
𝛿𝜌 𝑟 1.40 L‟extension de la LDA en cas la polarisation de spin est prise conduite à la LSDA (Local Spin Density Approximation), ou S désigne le spin. Elle est importante pour la description des systèmes dans un champ magnétique. Introduire le spin consiste à considérer deux états 𝜌↑ 𝑟 et 𝜌↓ 𝑟 dans la matrice de densité, et le terme 𝐸𝑥𝑐 est maintenant fonction de deux
spins 𝐸𝑥𝑐↑↓ 𝜌 𝑟 , par conséquent l‟énergie d‟échange-corrélation est définit de la manière
suivante : 𝐸𝑥𝑐↑↓ 𝜌 𝑟 = 𝜌 𝑟 𝐸𝑥𝑐↑↓ 𝜌 𝑟 𝑑𝑟 1.41 𝜌 𝑟 = 𝜌↑ 𝑟 + 𝜌↓ 𝑟 (1.42) 𝑉𝑥𝑐 𝜌 𝑟 ↑, 𝜌 𝑟 ↓ = 𝛿 𝜌 𝑟 𝐸𝑥𝑐 𝜌 𝑟 ↑, 𝜌 𝑟 ↓ 𝛿𝜌 𝑟 1.43
L'efficacité de cette approximation est apparue à partir des années 1970 avec les travaux de Zunger et Freeman [13], ainsi que ceux de Moruzzi et al. [14]. Il existe à présent différentes expressions du potentiel d‟échange et corrélation d‟un gaz d‟électrons homogène, qui peuvent être utilisée en LDA et LSDA. Entre autre celles de Hedin et Lundqvist [15], Van Barth et Hedin [16], Janack [17], et d‟autres paramétrisations pour l‟énergie de corrélation d‟un gaz d‟électrons homogène dont celles de Lundqvist et Perdew et Wang. [18].
En générale, l‟approximation LDA est applicable pour de nombreux systèmes proches du modèle de gaz électronique (les électrons dans les solides), mais elle présente un sérieux défaut pour des interactions à long distance comme les molécules par exemple, elle conduit souvent à de très mauvaise donnée énergétique telles que l‟énergie de liaisons et de gap faible pour les semi-conducteurs et les composés isolant. Elle prévoit que la structure de Fe à la température ambiante [19, 20]est cubique à faces centrées cfc (γ-Fe), alors qu‟il s‟agit en fait de la structure cubique centré cc (α-Fe).
15
1.4.4.2- Approximation du gradient généralisé (GGA)
La plupart des corrections qui ont été introduites à la (LDA) reposent sur l‟idée qui consiste à tenir en compte des variations locales de la densité. Pour cette, raison le gradient de la densité électronique a été introduit conduisant à l‟approximation du gradient généralisé (GGA, Generalized Gradient Approximations), dans laquelle l‟énergie d‟échange et de corrélation est en fonction de la densité électronique et son gradient :
𝛦𝑥𝑐 𝜌 𝑟 = 𝜌 𝑟 ℰ𝑥𝑐 𝜌 𝑟 , 𝛻𝜌 𝑟 𝑑𝑟 1.44
Où ℰ𝑥𝑐 𝜌 𝑟 , 𝛻𝜌 𝑟 représente l‟énergie d‟échange-corrélation par électron dans un
système d‟électrons en interaction mutuelle de densité non uniforme.
La GGA (Generalized Gradient Approximation) permet d‟introduire une combinaison entre les termes locaux et des termes dépendant du gradient. Elle donne de bons résultats et permet d‟améliorer les énergies de cohésion et les paramètres de maille. Cependant, l‟amélioration par rapport à la LDA n‟est pas toujours systématique car la GGA permet de corriger les insuffisances de la LDA et s‟avère plus efficace dans de nombreux cas, en effet la GGA donne de meilleurs résultats pour les énergies d‟activations des réactions chimiques [21-22]. En revanche, il existe plusieurs paramétrisations pour la (GGA) dont celles de Perdew et al (1991-1996) [23], et les versions les plus utilisées sont celles de Perdew et Wang [24] et Perdew [25]. Langret et Perdew [26], Langret et Mehl [27], Huand et Langret [28]. Dans ce travail, on a systématiquement préféré la formulation Perdew, Burk et Ernzerhof, qui est connue sous le nom de la PBE.
L‟approximation de la GGA-PBE, conduit à une augmentation significative des paramètres de maille de certains matériaux contenant des éléments lourds (matériaux de transition), et offre des résultats plus satisfaisants pour leurs propriétés magnétiques [29, 30, 31].
L‟approche locale est la mère de toutes les approximations, qui utilise les densités de spin sous la forme LSDA, suivant par l‟approximation GGA qui amène une meilleure précision.
1.4.5- Mise en œuvre de la DFT
Les méthodes basées sur la théorie de la fonctionnelle de la densité DFT (Density Functional Theory) sont celles qui dans la pratique sont utilisées en sciences des matériaux.
16
Les nombreux travaux effectués ces dernières années montrent que les calculs DFT donnent de bons résultats pour les états fondamentaux de systèmes très divers (métalliques, ioniques, organométallique, métaux de transition magnétiques...) pour de nombreuses propriétés. Nous nous sommes placés dans le cadre de l'approximation de Born-Oppenheimer et nous avons vu qu'il était nécessaire d'utiliser une forme approchée de la fonctionnelle d'échange corrélation afin de pouvoir appliquer la DFT en pratique.
Il est néanmoins possible de contrôler les approximations que nous présenterons ci-dessus et d'effectuer des études de convergence. Si on désire comparer des résultats relatifs à des phases différentes, il est essentiel de choisir un jeu de paramètres commun.
1.5 Les bases de projection
Pour résoudre numériquement les équations de Kohn-Sham (1.34), il faut choisir une base pour les fonctions d‟onde ф𝑛 𝑟 que l‟on peut prendre comme une combinaison linéaire
d‟orbitales, appelés orbitales de Khon-Sham (KS) : ф𝑛 = 𝐶𝑃𝑛
𝑝
ф𝑝𝑏𝑎𝑠𝑒 1.45
Où les ф𝑝𝑏𝑎𝑠𝑒, sont les fonctions de base et 𝐶𝑃𝑛 les coefficients de développement.
La résolution des équations de Kohn et Sham revient à déterminer les coefficients 𝐶𝑃𝑛 pour
les orbitales occupées qui minimisent l‟énergie totale.
La décomposition exacte des fonctions d‟onde mono-électroniques ф𝑛 𝑟 implique que le nombre de fonctions de base doit être infini. Nous serons donc amenés à limiter le développement en utilisant certains critères pour pouvoir mener le calcul numérique et résoudre le systèmed‟équations suivant :
ф𝑖 𝐻𝑛 ф𝑗 − 𝐶𝑗𝑛𝐸𝑛 ф𝑖 ф𝑗 𝑗
= 0 1.46
1.5.1- Base d’ondes planes
Considérons un système cristallin périodique basé sur la répétition d‟une cellule unité de volume. Chacune des fonctions d‟onde électroniques ф𝑛 peut se développer sur la base des
ondes planes.
ф𝑛 𝐾, 𝑟 = 𝑒𝑖𝐾 𝑟 𝑈𝑛 𝐾, 𝑟 = 𝑒𝑖𝐾 𝑟 𝑒𝑖𝐺 𝑟 𝐺
17 ф𝑛 𝐾, 𝑟 = ф𝑛 𝐾, 𝐺
𝐺
𝑒𝑖 𝐾 −𝐺 𝑟 1.48
Où 𝐾 est un vecteur de la première zone de Brillouin et 𝐺 un vecteur du réseau réciproque. Théoriquement un tel développement nous permet de résoudre les équations de Kohn et Sham, mais en pratique deux considérations s‟interposent. D‟une part; il y a un nombre infini de vecteurs dans le réseau réciproque, d‟autre part les vecteurs 𝐾 appartenant à la ZB sont également en nombre infini, par conséquent il y a également un nombre infini d‟états propres de l‟Hamiltonien.
La procédure de résolution numérique consiste à résoudre les équations de Kohn et Sham pour une grille de points k et une énergie de coupure 𝐸𝑐𝑢𝑡 donnée en fixant un critère de
convergence. Le calcul est supposé convergé si la différence d‟énergie totale entre deux itérations successives est inférieure à une valeur seuil. Autrement dit on se limitera aux vecteurs du réseau réciproques 𝐺 , contenus dans une sphère de rayon 𝐺𝑚𝑎𝑥.
𝐸𝑐𝑢𝑡 =
ℏ2𝐺𝑚 𝑎𝑥2
2𝑚𝑒
1.49 La méthode utilisée pour décomposer la zone de Brillouin et choisir les points k dans ce manuscrit est celle de Monkhorst-Pack [32].
Cependant, les coefficients 𝐶𝑛 𝐾, 𝐺 pour les ondes planes ayant de petites énergies
cinétiques ℏ2 2𝑚𝑒 𝐾 + 𝐺 2 sont typiquement plus importants que celles aux plus grandes
énergies. Il est donc nécessaire d‟effectuer un échantillonnage dans le développement en ondes planes, de ce fait seules les ondes planes vérifiant :ℏ2 2𝑚𝑒 𝐾 + 𝐺 2≤ 𝐸𝑐𝑢𝑡 sont
incluses dans l‟ensemble de base. Le nombre d‟ondes planes prises en compte dans le calcul est donc : 𝑁𝑝𝑤 = 𝑁𝐾 𝛺 2𝑚𝑒 𝐸𝑐𝑢𝑡 3 2 1.50
Où 𝑁𝐾 est le nombre de vecteurs 𝐾 à l‟aide desquels la zone de Brillouin est échantillonnée,
et 𝛺 est le volume de la cellule de simulation.
L‟utilisation d‟une base d‟ondes planes combine des avantages ainsi que des inconvénients, parmi les caractéristiques intéressantes on dénombre les suivantes :
La base est non biaisée, ainsi tout l‟espace est traité de la même façon. Elle est complète.
18
Les ondes planes sont mathématiquement simples, et leurs dérivées sont des produits dans l‟espace k.
Les ondes planes sont indépendantes des positions atomiques. Parmi les inconvénients de cette base :
Le nombre d‟ondes planes utilisées est déterminé en fonction de la courbure la plus importante des fonctions d‟ondes.
L‟espace vide est traité sur le même pied d‟égalité (coût) que les régions d‟intérêt.
1.5.2- Les bases d’orbitales atomiques (LCAO)
D'une manière pratique, la résolution des équations Hartree-Fock se fait en réécrivant la partie spatiale des Orbitales Moléculaires (OM) sous la forme d'une combinaison linéaire d'Orbitales Atomiques (OA). Ce développement est appelé méthode de Combinaison Linéaire d'Orbitales Atomiques (LCAO), Elles sont classées en deux catégories qui sont d'un usage courant :
Bases constituées d‟orbitales atomiques de type Slater (STO : Slater type orbital) Bases constituées d‟orbitales atomiques de type gaussiennes (GTO : Gaussian type
orbital)
1.5.2.1- Les fonctions de type
SlaterLes fonctions de type Slater (STO): ce sont des fonctions qui ont un bon comportement à très courte et très longue distance du noyau, elles semblent bien appropriées pour la description d‟un système atomique. Ce sont aussi les meilleures OA qu‟on présente sous une forme analytique simple, on les obtient généralement en minimisant l‟énergie des atomes correspondants. En coordonnées sphériques ces fonctions sont définies par :
𝜒𝑛,𝑙,𝑚 ,𝜎= 𝐶𝑛−1𝑒𝑥𝑝 −𝜎𝑟 𝑌𝑙,𝑚 𝜃, 𝜑 1.51
Où 𝜎 , est un paramètre de décroissance exponentielle et 𝑌𝑙,𝑚 𝜃, 𝜑 l‟harmonique sphérique définie par :
𝑌𝑙,𝑚 𝜃, 𝜑 =
2𝑙 + 1 𝑙 − 1 ! 4𝜋 𝑙 + 𝑚 ! 𝑃𝑚
19 𝑃𝑚𝑙 𝑐𝑜𝑠 𝜃 = −1 𝑙 2𝑙𝑙! 1 − 𝑐𝑜𝑠 𝜃 𝑚 2 𝛿𝑙+𝑚 𝛿𝑐𝑜𝑠𝑙+𝑚 𝜃 𝑐𝑜𝑠2 𝜃 − 1 𝑙 1.53
Cependant, ces fonctions ne sont pas utilisées à grande échelle dans les programmes moléculaires ab initio, cela est dû à la complexité du calcul d‟intégrales moléculaires sur la base STO, excessivement long. Pour cette raison les théoriciens ont exploré d‟autres voies afin de faciliter et d‟accélérer les calculs d‟intégrales. Boys [33] fut le premier à proposer l‟utilisation des fonctions Gaussiennes.
1.5.2.2- Les fonctions de type Gaussiennes
Les fonctions gaussiennes sont largement utilisées, spécialement pour les calculs ab initio.Les fonctions de type G.T.O. (Gaussian type orbitals) sont définies en coordonnées cartésiennes par :
𝜒𝐿,𝛼 = 𝐶𝑥𝑙𝑦𝑚𝑧𝑛𝑒−𝛼𝑟
2
𝑌𝑙,𝑚 1.54
Où 𝑌𝑙,𝑚 est l‟harmonique sphérique correspondant au moment orbital « l » et sa composante
« m », C est une constante de normalisation, 𝛼 est la largeur de diffusion de la fonction et L =
l + m + n est un paramètre, qui détermine le type de symétrie de la fonction :
L = 0 : Ce sont des orbitales s. L = 1 : Trois orbitales p (𝑝𝑥,𝑝𝑦 et 𝑝𝑧).
L = 2 : Cinq orbitales de type d (𝑑𝑥𝑦,𝑑𝑥𝑧, 𝑑𝑦𝑧, 𝑑𝑥2−𝑦2et 𝑑3𝑍2−𝑟2), et la sixième orbitale est de
type s.
Ces fonctions présentent une propriété essentielle : le produit de deux Gaussiennes est aussi une Gaussienne [34], Cette propriété permet de faciliter considérablement le calcul d‟intégrales moléculaires multicentriques. Cependant de telles fonctions n‟ont pas un comportement physique tout à fait satisfaisant. En effet, contrairement aux STO les fonctions GTO reproduisent mal la région proche du noyau, et les grandes distances. Huzinaga [35] a donné une impulsion déterminante aux calculs sur les bases de Gaussiennes, en considérant une combinaison de plusieurs OA gaussiennes (GTO) (comme fonctions de base) pour reproduire une orbitale atomique OA de Slater (STO). Cela veut dire que le nombre de fonctions nécessaires pour atteindre l‟énergie HF sur une base GTO est beaucoup plus élevé sur une base STO. Ces fonctions sont appelées « fonctions gaussiennes contractées » [36].
Le choix, tant en nombre qu‟en qualité, de la base de fonctions représentant les orbitales atomiques est important car il influence autant la précision des résultats obtenues que les
20
temps de calculs. Ceci étant, nous distinguons plusieurs dimensions de bases d‟orbitales atomiques, à savoir i) les bases minimales, ii) les bases étendues, iii) et les bases de valence. Dans les bases minimales, nous ne prenons en compte que les orbitales atomiques qui sont effectivement occupées à l‟état fondamental de l‟atome en y ajoutant toutefois des orbitales inoccupées de la couche de valence. Chaque orbitale est alors décrite par une seule fonction. Les bases étendues, quant à elles, sont construites à partir de la base minimale à laquelle sont ajoutées un certain nombre d‟orbitales excitées des différents atomes. Ces dernières, situées au-delà de la couche de valence, sont appelées orbitales de polarisation. Ceci dit, chaque orbitale est décrite par au moins deux fonctions. Enfin, les bases de valence ne comprennent que les orbitales de la couche de valence de chaque atome et sont décrites en général par une seule fonction de base par orbitale. Les électrons des couches internes (ou électrons de cœur) ne sont pas décrits explicitement dans ce type de base : un potentiel (le pseudo-potentiel) reproduit cependant leurs effets.
1.6 Algorithme de la DFT dans une version Pseudo
Les équations de Kohn Sham sont résolues par une technique itérative dite auto-coherente, visant à diagonaliser la matrice de l‟hamiltonien. Pour cela, on a choisi une densité de charge électronique initiale, un potentiel 𝑉𝑒𝑓𝑓 et des orbitales de Kohn-Sham
Φ
i(r) qui seront injectéspour construire l‟hamiltonien. De nouveaux paramètres (valeurs et vecteurs propres) seront calculés, ils servent à construire une nouvelle densité électronique permettant de remonter au potentiel d‟échange-corrélation ; cette procédure itérative est poursuivie jusqu‟à ce que la convergence d‟énergie ou de charge soit réalisée.
Une fois la convergence atteinte, on accède à l‟énergie de l‟´etat fondamental du système, l‟ensemble de cette procédure est représentée sur la figure (1.1).
21
Oui Début obtenir 𝜌𝑖𝑛𝑖𝑡
Détermination des grandeurs recherchées Teste de convergence
Mélange de 𝜌𝑖𝑛𝑖𝑡et ρ
Calculer la nouvelle
ρ=ΣΦ
i*
Φi
Résoudre les équations Kohn et Sham Utiliser une base d‟orbitales atomiques (LCAO)
Diagonaliser la matrice
Utiliser 𝜌𝑖𝑛𝑖𝑡 pour calculer 𝑉𝑒𝑓𝑓 = 𝑉𝑒𝑥𝑡 + 𝑉𝐻𝑎𝑟𝑡𝑟𝑒𝑒 + 𝑉𝑥𝑐
Figure 1.1- Schéma de résolution du cycle auto-cohérent sur un calcul de convergence
Oui Non
22
Chapitre 2
Pseudo potentiels et code de calcul : SIESTA
Nous commencerons ce chapitre par l‟exposition du formalisme des pseudo-potentiels et les différents schémas permettant de les générer, et nous terminerons par la présentation du code de calcul SIESTA.
2.1- Pseudo-potentiels
A l‟aide des concepts développés jusqu‟à présent, il est déjà possible de définir un schéma de principe afin de déterminer l‟état fondamental électronique d‟un système quelconque. Le problème étant que les calculs deviennent de plus en plus coûteux au fur et à mesure que la taille des atomes augmente, à cause d‟une part, de l‟augmentation du nombre d‟électrons et d‟autre part, du caractère localisé de certaines orbitales, comme par exemple les orbitales d des métaux de transition, Qui vont nécessiter des moyens de calcul extrêmement puissants. Or dans la plupart des cas les électrons de valence sont les seuls à intervenir dans l‟établissement des liaisons chimiques. Les électrons de cœur pourront donc être regroupés avec les noyaux pour constituer des ions rigides : c‟est l‟approximation du cœur gelé [37− 39].
Afin de tenir compte des interactions qui ont perdu leur caractère explicite, le potentiel effectif dans les équations de Kohn-Sham doit être remplacé par un potentiel effectif, beaucoup moins attractif que le potentiel créé par le noyau. Ce potentiel effectif est appelé un pseudo-potentiel[40]
Chapitre 2 : Pseudo-potentiel et code de calcules SIESTA
23
Les fonctions d‟ondes réelles associées aux électrons de valence oscillent très rapidement dans la région de cœur et possèdent beaucoup de nœuds. L‟orthogonalisation et la description de fonctions nécessitent un grand nombre de fonctions de base. Ce qui rend les calculs trop lents.
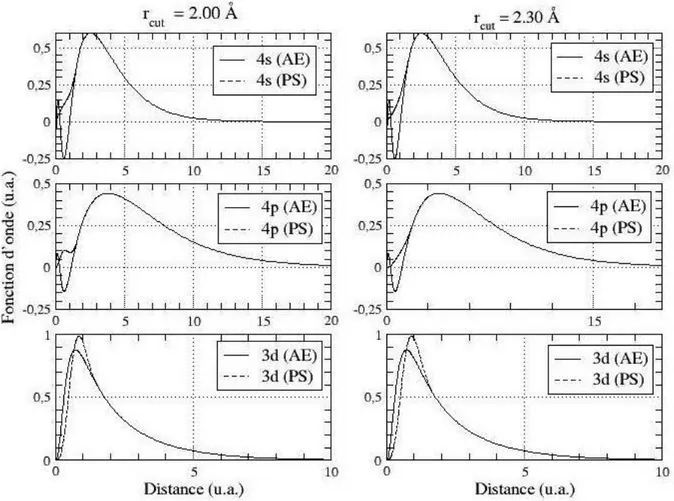
La théorie du pseudo-potentiel à été mise au point pour les solides par Herring en 1940 [41], l'idée de cette approche est de développer les fonctions d‟onde de valence sur un nombre réduit d‟ondes planes. On remplace la fonction d‟ondes réelle (AE) par un pseudo fonction (PS), qui au delà d‟un certain rayon 𝑟𝑐dit rayon de coupure, elles coïncident avec les
fonctions d‟ondes réelles. Ceci de façon à ce que les pseudo-fonctions d'onde n'aient pas de nœud dans la région du cœur figure (2.1) :
Figure 2.1- Fonction d‟onde réelle (AE) et pseudo-fonction d‟onde (PS) pour l‟orbitale 4s du
chrome.
La transférabilité d‟un pseudo-potentiel est un élément essentiel à prendre en compte lors de sa génération. Un pseudo-potentiel doit pouvoir être utilisé dans différents environnements, tout en donnant la meilleure approximation possible du système modélisé.
La transférabilité d‟un pseudo-potentiel est très influencée par le choix de 𝑟𝑐. Plus 𝑟𝑐 est
«petit» plus la capacité de transférabilité du pseudo-potentiel sera assurée.
Donc le pseudopotentiel permet d'une part de limiter le nombre d'électrons et d'autre part de réduire la taille des bases d'ondes planes nécessaires à la représentation de la fonction d‟onde du système, et cela réduirait considérablement les calculs numériques.
Chapitre 2 : Pseudo-potentiel et code de calcules SIESTA
24
Trois grandes familles de pseudo-potentiels ont ainsi été créées : les pseudo-potentiels dits à norme conservée introduits par Hamann et al. [42], les pseudo-potentiels de Vanderbilt appelés ultra-mous ou US [43] et les Pseudo-potentiels “dual-espace gaussian” introduits par Goedecker et al. [44, 45].
Pour satisfaire au mieux du critère de transférabilité et de la conservation de la charge dans la région de cœur, des pseudo-potentiels à norme conservée ont été élaborés.
2.2- Les pseudo-potentiels à norme conservée
Cette famille correspond -à des potentiels dits à norme conservée (la pseudo-fonction d‟onde correspondante est normalisée). Ces pseudo-potentiels modernes sont construits selon une méthode proposée par Hamann [46] en 1979, et systématisée par Bachelet et al. [47].
Pour une configuration électronique de référence de l‟atome isolé, le pseudo-potentiel conduit aux valeurs propres exactes et à des fonctions propres appelées pseudo-fonctions aussi régulières que possible en accord avec les fonctions d‟ondes atomiques au-delà d‟un rayon de coupure 𝑟𝑐.
Chaque état propre de l‟équation de Schrödinger atomique est défini par trois nombres quantiques (n, l, m). Les orbitales atomiques ф𝑛𝑙𝑚 𝑟, 𝜃, 𝜑 et les pseudo-orbitales ф𝑛𝑙𝑚𝑃𝑆 𝑟, 𝜃, 𝜑
s‟écrivent :
ф𝑛𝑙𝑚 𝑟, 𝜃, 𝜑 = 𝑅𝑛𝑙 𝑟 𝑌𝑙,𝑚 𝜃, 𝜑 2.1
ф𝑛𝑙𝑚𝑃𝑆 𝑟, 𝜃, 𝜑 = 𝑅𝑛𝑙𝑃𝑆 𝑟 𝑌𝑙,𝑚 𝜃, 𝜑 2.2
Où 𝑅𝑛𝑙 𝑟 ,est la partie radiale et 𝑌𝑙,𝑚 𝜃, 𝜑 la partie angulaire.
Le pseudo-potentiel à norme conservée respecte les conditions suivantes :
1- égalité des valeurs propres pseudo (PS) de l‟Hamiltonien associées à 𝑅𝑛𝑙𝑃𝑆 𝑟 et réelles (AE)
associées à 𝑅𝑛𝑙 𝑟 pour une configuration électronique de référence donnée :
𝐸𝑛𝑙𝑃𝑆 = 𝐸𝑛𝑙 2.3
2- les fonctions d‟ondes réelles 𝑅𝑛𝑙 𝑟 et pseudo 𝑅𝑛𝑙𝑃𝑆 𝑟 sont égales au-delà du rayon de
Chapitre 2 : Pseudo-potentiel et code de calcules SIESTA
25
𝑅𝑛𝑙𝑃𝑆 𝑟 = 𝑅𝑛𝑙 𝑟 𝑝𝑜𝑢𝑟 𝑟 > 𝑟𝑐 2.4
3- la pseudo-fonction d‟onde 𝑅𝑛𝑙𝑃𝑆 𝑟 est choisie de manière à supprimer les nœuds et les
oscillations dues à l‟orthogonalisation des fonctions d‟onde.
4- les intégrales des densités de charge réelles et pseudo s‟accordent pour chaque état de valence (conservation de la norme):
𝑑𝑟𝑟2 𝑟𝑐 0 𝑅𝑛𝑙𝑃𝑆 𝑟 2 = 𝑑𝑟𝑟2 𝑟𝑐 0 𝑅𝑛𝑙 𝑟 2 2.5
De cette condition découle le fait que les dérivées logarithmiques des fonctions d‟onde réelles et pseudo et leurs premières dérivées par rapport à l‟énergie s‟accordent pour 𝑟 > 𝑟𝑐.
La pseudo-fonction d‟onde 𝑅𝑛𝑙𝑃𝑆 𝑟 , qui satisfait aux quatre conditions précédentes, donnent
une densité de charge identique à la densité de charge réelle.
Ces conditions permettent d‟obtenir des pseudo-potentiels de bonne qualité, mais laissent une grande liberté de choix dans la région de cœur. De nombreuses méthodes pour générer des pseudo-potentiels à norme conservée, ont donc été créées, chacune imposant ses propres conditions supplémentaires. Parmi celles-ci, les plus utilisées sont :
La méthode de Kerker [48] qui utilise une fonction analytique pour représenter les orbitales de valence dans la région de cœur.La méthode de Bachelet Hamann et Schlüter [49], qui la première a souligné l‟importance du concept de conservation de norme. La méthode de Troullier et Martins [50], et la méthode Greenside, et Schlüter [51].
2.2.1- Méthode de Kerker
En 1980, Kerker [48] introduit une nouvelle classe de pseudo-potentiels à norme conservée dans lesquels les pseudo-fonctions d‟onde 𝑅𝑛𝑙𝑃𝑆 𝑟 s‟écrit sous la forme analytique suivante :
𝑅𝑛𝑙𝑃𝑆 𝑟 = 𝑟
𝑙𝑒 𝑝 𝑟 𝑝𝑜𝑢𝑟 𝑟 < 𝑟 𝑐
𝑅𝑛𝑙 𝑟 𝑝𝑜𝑢𝑟 𝑟 > 𝑟𝑐
2.6 avec pr décrit par un polynôme de 4è𝑚𝑒 degré
Chapitre 2 : Pseudo-potentiel et code de calcules SIESTA
26
Le paramètre 𝜆1, est pris égale à zéro pour que 𝑃′ 𝑟 𝑟et le pseudo-potentiel écranté noté
𝑉𝑠𝑐𝑟𝑙𝑃𝑆 𝜌 𝑟 ne présentent pas de singularité à l‟origine. Kerker a proposé des critères qui
permettent de déterminer les paramètres𝜆0,𝜆2¸𝜆3 et 𝜆4 de façon à satisfaire au critère de
conservation de la norme ainsi qu‟aux critères supplémentaires suivants :
1- La fonction d‟onde réelle 𝑅𝑛𝑙 𝑟 et la pseudo-fonction d‟onde 𝑅𝑛𝑙𝑃𝑆 𝑟 doivent avoir les
mêmes valeurs propres.
2- La pseudo-fonction d‟onde ne doit pas avoir de nœuds et doit être égale à la fonction d‟onde réelle pour 𝑟 > 𝑟𝑐.
3- La pseudo-fonction d‟onde, sa première et sa seconde dérivée doivent être continues en 𝑟 = 𝑟𝑐.
La pseudo-fonction d‟onde obtenue est en suite injectée dans l‟équation radiale de Kohn et Sham : −1 2 𝑑2 𝑑𝑟2+ 𝑙 𝑙 + 1 2𝑟2 + 𝑉𝑠𝑐𝑟𝑙𝑃𝑆 𝜌 𝑟 𝑅𝑛𝑙𝑃𝑆 𝑟 = ℰ𝑛𝑙𝑅𝑛𝑙𝑃𝑆 𝑟 2.8
La méthode utilisée par Kerker consiste à définir une pseudo-fonction d‟onde ф𝑙𝑃𝑆 𝑟 , ayant les propriétés désirées pour chaque atome ensuite à inverser l‟équation de Schrödinger afin de déterminer le potentiel 𝑉𝑠𝑐𝑟𝑙𝑃𝑆 𝜌 𝑟 pour lequel ф𝑙𝑃𝑆 𝑟 est une solution d‟énergie𝐸. La fonction d‟onde égale la vraie fonction au-delà de 𝑟𝑐 et prend la forme d‟une fonction
analytique paramétrée à l‟intérieur de la région du cœur. Le potentiel ainsi obtenu pour chaque l s‟écrit : 𝑉𝑠𝑐𝑟𝑙𝑃𝑆 𝜌 𝑟 = 𝐸𝑛𝑙 − 𝑙 𝑙 + 1 2𝑟2 + 1 2𝑟𝑅𝑙𝑃𝑆 𝑟 𝑑2 𝑑𝑟2 𝑟𝑅𝑛𝑙𝑃𝑆 𝑟 2.9
La forme analytique de la pseudo-fonction d‟onde proposée par Kerker nous donne un pseudopotentiel sous la forme suivante :
𝑉𝑠𝑐𝑟𝑙𝑃𝑆 𝜌 𝑟 = ℰ0+ 𝑙 + 1 𝑟 𝑃 ′ 𝑟 + 𝑃′′ 𝑟 + 𝑃′ 𝑟 2 𝑟 < 𝑟 𝑐 𝑉𝑙 𝑟 𝑟 > 𝑟𝑐 2.10
Où 𝑉𝑙 𝑟 , est le potentiel réel (AE).
Dans cette approche, nous recherchons des pseudo-potentiels en quelque sorte optimisés, c‟est-à-dire qui minimisent le nombre de fonctions de base. Ils doivent en outre être transférables, c‟est-à-dire utilisables dans différents environnements chimiques. Une approche
Chapitre 2 : Pseudo-potentiel et code de calcules SIESTA
27
qui puisse assurer l‟optimisation des pseudo-potentiels consiste à mettre p(r) sous la forme d‟un polynôme du N-ième degré :
𝑝 𝑟 = 𝜆0+ 𝜆0 𝑁
𝑛=1
𝑟𝑛 2.11
avec N pris égale à 4 et ¸1 pris égale à 0.
Afin de générer des pseudo-potentiels locaux de plus en plus élaborés, une amélioration a été proposée par Troullier et Martins [50].
2.2.2- Méthode de Troullier-Martins
Troullier et Martin [50] ont étendu la méthode de Kerker dans l‟optique de rendre la fonction d‟onde aussi douce que possible, en utilisant un polynôme d‟ordre plus élevé et en égalant plus de dérivées de la fonction d‟onde.
𝑃 𝑟 = 𝜆0+ 𝜆2𝛼 6
𝛼=1
𝑟2𝛼 2.12
Où les coefficients𝜆0, 𝜆2, 𝜆4, 𝜆6, 𝜆8, ¸𝜆10 et 𝜆12, doivent être calculés de manière à conserver
la norme.
Dans ce schéma, nous assurons la continuité de la partie radiale de la pseudo-fonction d‟onde et de ses quatre premières dérivées au point 𝑟𝑐 . De plus, nous assurons à la dérivée des
pseudo-potentiels ainsi qu‟à celle des pseudo-fonctions d‟onde d‟être nulles à l‟origine. Les pseudo-potentiels ainsi construits, sont assez lisses et convergent rapidement dans l‟espace réciproque.
A partir de là, il est possible d‟obtenir un pseudo-potentiel intermédiaire « écranté », qui agit sur les pseudo-fonctions d‟onde, comme le potentiel effectif agit sur les fonctions d‟onde de valence. Il suffit pour cela d‟inverser l'équation de Kohn et Sham pour déterminer le potentiel de l‟ion écranté 𝑉𝑠𝑐𝑟𝑙𝑃𝑆 :
𝑉𝑠𝑐𝑟𝑙𝑃𝑆 = 𝐸𝑛𝑙 − 𝑙 𝑙 + 1 2𝑟2 + 1 2𝑟𝑅𝑙𝑃𝑆 𝑟 𝑑2 𝑑𝑟2 𝑟𝑅𝑛𝑙𝑃𝑆 𝑟 2.13
Enfin, le pseudo-potentiel correspondant au moment orbital ℓ est obtenu en soustrayant les potentiels de Hartree 𝑉𝐻 𝜌 𝑟 et celui d‟échange-corrélation 𝑉𝑥𝑐 𝜌 𝑟 des électrons de valence,
dans le pseudo-potentiel écranté :
Chapitre 2 : Pseudo-potentiel et code de calcules SIESTA
28
Maintenant, il reste à choisir une configuration électronique de référence à partir de laquelle, il faut générer des pseudo-potentiels transférables. Comme le montre l‟équation (2.14), le pseudo-potentiel dépend de la valeur de l‟énergie𝐸𝑛𝑙, qui peut être différente d‟une
configuration électronique à une autre.
Pour remédier à ce problème, il est nécessaire de construire plusieurs pseudopotentiels à partir de configurations différentes et effectuer des tests sur les propriétés physiques de systèmes simples. A partir de là, on pourra avoir une idée sur les états de valence à inclure, la configuration correspondante ainsi que les rayons de coupures𝑟𝑐.
2.2.3- Correction non linéaire de cœur
(NLCC):Par ailleurs, dans le formalisme du pseudo-potentiel, on suppose que la contribution des électrons de cœur est bien séparée de celle de valence lors du calcul d‟énergie ou lorsque on génère le pseudo-potentiel. La densité électronique de charge est ainsi séparée en densité de cœur et une densité de valence
𝜌 𝑟 = 𝜌𝐶 𝑟 + 𝜌𝜐 𝑟 2.15 Ceci revient à supposer que le potentiel est linéaire par rapport à la densité :
𝑉𝑥𝑐 𝜌 𝑟 = 𝑉𝑥𝑐 𝜌𝐶 𝑟 ] + 𝑉𝑥𝑐[𝜌𝜐 𝑟 (2.16) Cette approximation est valable lorsque les densités respectives (cœur et valence) sont bien séparées spatialement. Par contre, lorsque ce n‟est pas le cas, cette séparation induit une erreur significative sur l‟énergie. Ainsi, lorsque le recouvrement ne peut être négligé, on doit impérativement prendre en compte du caractère non linéaire du potentiel (énergie) d‟échange-corrélation, dans ce cas précis :
𝑉𝑥𝑐 𝜌 𝑟 = 𝑉𝑥𝑐 𝜌𝐶 𝑟 + 𝜌𝜐 𝑟 ≠ 𝑉𝑥𝑐 𝜌𝐶 𝑟 + 𝑉𝑥𝑐 𝜌𝜐 𝑟 2.17
Or dans la formulation des pseudo-potentiels, le terme du potentiel échange-corrélation correspondant au potentiel ionique contient un terme ayant la forme :
𝑉𝑥𝑐 𝜌𝐶 𝑟 + 𝜌𝜐 𝑟 − 𝑉𝑥𝑐 𝜌𝐶 𝑟 2.18
Autrement dit : la contribution 𝜌𝜐 𝑟 au potentiel est obtenue par simple soustraction du
potentiel total,𝑉𝑥𝑐 𝜌 𝑟 , de la contribution 𝜌𝐶 𝑟 . Dans ce cas précis, la linéarisation de
l‟énergie échange-corrélation réduit la transférabilité du potentiel et engendre des erreurs dans l‟énergie totale. Louie et al. [52] ont développé une expression pour générer des pseudo-potentiels linéaires en définissant le pseudo-potentiel ionique selon la relation :