HAL Id: jpa-00233850
https://hal.archives-ouvertes.fr/jpa-00233850
Submitted on 1 Jan 1943
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Solutions. Solubilité. II
E. Darmois
To cite this version:
E. Darmois. Solutions. Solubilité. II. J. Phys. Radium, 1943, 4 (11), pp.233-245.
�10.1051/jphys-rad:01943004011023300�. �jpa-00233850�
LE
JOURNAL
DE
PHYSIQUE
ET
LE
RADIUM
SOLUTIONS.
SOLUBILITÉ.
II.Par E. DARMOIS.
Sommaire. 2014 Un
premier exposé a donné l’essentiel de la thermodynamique des solutions. On montre dans le présent exposé que la solubilité d’un gaz et celle d’un solide sont prévisibles quand la solution obéit à la loi de Raoult; celle d’un liquide est totale dans le même cas. On établit pour un solide l’équation de Schröder-Le Chatelier, qui permet de tracer la courbe de congélation d’un mélange, de calculer la
composition et le point de fusion d’un eutectique. On traite le cas d’un corps dissous dissociable, des cristaux mixtes.
L’expérience n’est pas toujours d’accord avec la théorie; c’est parce que la loi de Raoult n’est qu’une loi-limite. Si l’on connaît la courbe des pressions de vapeur d’un composant, on peut prévoir sa solubilité. Les déviations par rapport à la loi de Raoult introduisent la notion d’activité selon Lewis. A cause de
l’importance de cette notion et de la difficulté où l’on est actuellement de se procurer le livre de Lewis et Randall, nous croyons utile d’indiquer succinctement les procédés de calcul de l’activité.
Le coefficient d’activité varie avec la concentration de la solution; quand la formule est simple, on
peut prévoir certaines singularités, en particulier la démixtion, formation de deux couches liquides.
SRc VIII, - TOME IV. N° 11. NOVEMBRE 1943.
_-- - - -
--Cornlne nous l’avons vu dans un article
précé-dent
(1),
la loi de Raoult relie laproportion
d’une substance dans laphase
gazeuse à saproportion
dans la
phase liquide;
ellepermet
donc de calculer la solubilité d’une vapeur dans une solution. Cettesolubilité « idéale »
est,
dans certains cas, voisinede la solubilité réelle. Nous commencerons donc
par la
calculer,
quitte
à voir ensuite les différences entre le calcul etl’expérience
et les raisons de cesécarts.
A. Solubilité des gaz. --
Quand
le gaz es[au-dessous de sa
température
critique,
c’est unevapeur et ce que nous avons dit
précédemment
s’applique.
Pour certains gaz dont lespressions
de saturation sont
élevées,
des correctionss’impo-seraient pour tenir
compte
du fait que les lois desgaz
parfaits
nes’appliquent plus.
Onpeut
géné-ralement s’en
dispenser
car, en mêmetemps,
la loi deRaoult n’est
plus qu’approchée.
Au-dessus de la
température
critique,
on nepeut
(1) J. de Physique, ~g43, 4, 129. Cette référence est notée (Exp. I) dans ce qui suit. Nous renvoyons à cet article pour les notations.plus
parler
depression
de vapeur,mais,
dans legraphique
delog
p en fonction deT,
onpeut
extra-poler
jusqu’à
latempérature
del’expérience.
Lafigure
ireprésente
cetteextrapolation
pour leméthane à la
température
de25° C;
latempérature
critique
est -â3~C;. On trouve à 250 po =
37o
atm.Si la
pression
du gaz est i atm au-dessus de lasolu-tion,
lerapport
molaire du corps dissous dans lasolution est
x _ ,
soit -3 l ===
0,0027. Ce nombrepu 7
représente
la solubilitéthéorique
du méthane dansun
liquide
quelconque
sous lapression
ordinaire.En
dépit
desapproximations
etextrapolations
faites,
c’est bien l’ordre degrandeur.
Dans l’hexane,
oùla loi de Raoult
s’applique, l’expérience
donne0,0031;
dans le
xylène,
on trouveo,0026.
Théoriquement, exprimée
enrapport
molaire,
la solubilité est la même dans tous lesliquides
donnant des solutions idéales.Exprimée
en concentrationvolumique,
la solubilité estplus
grande
dans les solvants de faible volume molaire.Si l’on
change
lapression
du gaz au-dessus de lasolution,
x2 varieproportionnellement
à cettepression,
c’est la loi deHenry.
Si l’on
augmente
T,
poaugmente,
la solubilitédiminue;
sil’ex1 rapolation
esttoujours
permis,
la variation de po
peut
être calculée par la formule deClapeyron.
Fig. 1.
Enfin,
sous despressions
extérieureségaies,
le gaz leplus
soluble sera celui dont latempérature
critique
est laplus
élevée,
Po étant alorsplus
faible. Cette dernièrerègle
est assez bienvérifiée,
commele montre le Tableau I donnant
quelques
solubilités dans le benzène à 200 C.TABLEAU I.
Nous reviendrons
plus
tard sur les solubilités réelles des gaz.B. Solubilité des
liquides.
- Comme le corpsdissous est à l’état
liquide
dans lasolution,
si celle-ciest
idéale,
elle est unesimple juxtaposition
des deuxespèces
moléculaires;
les deuxliquides
sont doncmiscibles en toute
proportion
et la solubilité estillimitée. On sait que, ce n’est pas
toujours
le cas,comme nous le reverrons
également.
Parexemple,
on obtient souvent deux couchesliquides
super-posées ;
c’estqu’alors
lemélange
ne suit pas la loi de Raoult. Eneffet,
lapression
de vapeurpartielle
du no 1 est forcément la même dans les deux couchespuisque
c’est cellequi
existe dans la vapeur. Sila loi de Raoult
s’applique,
lerapport
molairedu no 1 est le même dans les deux
couches;
elles n’en forment doncqu’une.
Nous reviendrons
plus
loin sur la solubilité limitéedes
liquides;
dès maintenant il est certain que, entrela solution idéale et la démixtion
visible,
il existera évidemment tous les intermédiaires.C. Solubilité des
solides;
courbe de solubilitéidéale. - A la
saturation,
la solution est en contactavec le solide. La
pression
partielle
de celui-ci est doncégale
à lapression
desublimation,
sinon iln’y
aurait paséquilibre;
donc p, =p,. D’autre
part
lapression
de vapeur est aussip~ . ~, où p°
estla
pression
de vapeur du no 2 pur,supposé
à l’étatliquide.
Celiquide
ne serait pas stable à latempé-rature de
l’expérience puisque
la forme stable estle solide. Donc p, > ps et la
présence
du solvant aprécisément
pour effet d’abaisser lapression
de vapeur duliquide
au niveau de celle du solide. Lacomparaison
des deux relationsprécédentes
donne de suiteCette relation très
simple s’applique
àl’appa-rition des cristaux du no 2
quand
on refroidit unmélange homogène
de(2)
et de(1);
x2représente
la solubilité du no 2 dans le no 1.
Pour connaître la
solubilité,
il faut donc calculerle
rapport
despressions
de saturation du solideet du
liquide
à la mêmetempérature.
Ce calcul estpossible
en utilisant la formule deClapeyron.
Nousemploierons
d’abord les notationsclassiques
enFrance.
Pour le
liquide,
on aoù
L,,
est la chaleur devaporisation
molaire;
on asupposé
commed’usage
le volumespécifique
duliquide
négligeable
vis-à-vis de celui de la vapeur,celle-ci suivant
l’équation
des gazparfaits.
Pour lesolide,
on a de mêmeoù
L,
est la chaleur desublimation,
avec les mêmesapproximations.
En retranchant les deux
équations précédentes,
ilvient
a T, ..
où
Lj
est la chaleur de fusion.Cette
équation
peut
aussi s’écrire avec lesnota-tions
employées
précédemment (Exp. I).
L’équa-tion
(14)
(Exp. 1) réglait
la variation de lafugacité
partielle
d’uncomposant;
c’est lagénéralisation
de la formule deClapeyron.
En retranchant l’une del’autre les deux
équations analogues
pour leliquide
et le
solide,
on obtientoù ~H est la chaleur différentielle de dissolution. Comme la solution est
idéale,
et que le corps dissousest à l’état
liquide,
c’est donc aussi la chaleur de fusion du corps dissoussupposé
solide.L’équation
(2)
a d’abord été obtenue parSchrôder
[ 1 ],
puis
indépendamment
par LeCha-telier
[2].
Pour utiliser cette
équation,
il est nécessaire del’intégrer.
Pour cela onpeut
supposer d’abord que la chaleur de fusion est constante etindépendante
de T. La constante
d’intégration
se détermine enfaisant T =
T f~ (2);
on a alors x2 = 1; c’estl’équi-libre du
liquide
no 2 enprésence
du solide. On trouve de suiteSi l’on veut tenir
compte
de la variation deLj.
avec
T,
on écriral’équation
de Kirchhoffoù les
(C)
sont les chaleursspécifiques
molaires duliquide
et du solide. En lessupposant
de nouveauindépendantes
deT,
on trouve finalementLI’
f est la chaleur de fusion aupoint
de fusionnormal. Les deux formules
(3)
et(4) représentent
avec deux
approximations
différentes
la courbe desolubilité du no 2 dans le solvant no 1 ou encore la
courbe de
congélation
dumélange
homogène
des deuxcomposants.
Schrôder a
déjà indiqué
des vérifications de cesformules pour la solubilité de la
naphtaline
et descorps
analogues.
PourC1oHs,
on aCI
=56,6;
CS
=5 I,8
àTIf.
PourT}-
=298,1
1(230 C)
et T =3~3o,r~,
on trouve x2 =o,31
parl’équa-tion
(3)
eto,32
par l’équation (4). C’est la Solubilité idéale deC10Hs
dansn’importe quel
solvant.Schrôder a vérifié que
C10Hs
se dissout à peuprès
de même dansC,H,, C,H,CI, CCI,,
Enportant
enordonnée:!
et en abscisselog
x2, on doit avoir unedroite. La
figure 2 représente (3)
les courbes decongélation
deC10Hs
enprésence
de diversesimpu-retés. La droite idéale est effectivement suivie
(2) Plus exactement, l’ est égale à la température du point
triple où p~ = po.
(3) D’après HILDEBRAND, Solubilité, New-York , 1936.
pour
C6H5CI
et ladiphénylamine;
son coefficientangulaire
est4~7.
Dans lesliquides
qui
luiressem-blent
chimiquement,
comme lebenzène,
lenaph-talène suit assez convenablement la loi de
Raoult;
Fig. 2.
la solubilité dans
C6H6
est o,2g, très voisine decelle calculée. La flèche
indique
lepoint
de fusiondu
naphtalène.
Schrôder a aussi vérifié par
exemple
que la courbe de solubilité(ou
decongélation)
idéale dep.C6H4Br2
est à peuprès
la même dansC 6H 6, CS2, C 6H5Br,
mais pas dans l’éthanol.
TABLEAU J 1.
Les chaleurs de fusion des corps normaux sont peu
variables;
elles sont de l’ordre de 5ooocal/mol;
mais lespoints
de fusion sont trèsvariables;
dans la formule(3),
c’est doncTj qui
détermÍneTLl lesgrosses variations de la solubilité. Le Tableau Il donne par
exemple
les solubilitésthéoriques
pour des substances dont les P. F. varient de 5oo à ioooocet pour deux chaleurs de fusion :
4ooo
et jooo
cal. Certaines solubilités suivent bien larègle soulignée
ci-dessus.Exemple
m. o. et dansLa loi idéale
(3)
donne la solubilité par lerapport
molaire du corps dissous. Pour passer durapport x2
au
titre,
il faut connaître lepoids
moléculaire desdeux
composants
et enparticulier
celui du solvant. Pour les solvantsorganiques
comme lebenzène,
l’emploi
du P. M.chimique
donne des résultatscorrects,
comme on l’a vu par les nombres ci-dessus.Quand
ils’agit
d’un corpsanormal,
commel’eau,
la formule
(3)
donne des résultats inexacts enprenant
18 comme P. M. de l’eau. Parexemple,
la solubilité de lanaphtaline
dans l’eau estenvi-ron o,1
g/1,
cequi
donneo , 1 1000
_
M > ,
i ei ° ,_ ° ’ ,, ,_ ,=
etI’ ’
’x, exp. est donc seulement Certains auteurs ont vu dans cette discordance une preuve
de la
polymérisation
de l’eau. Onpeut
en effet raisonner comme suit.Supposons
l’eaupolymérisée
dans lerapport
q;t . 0,1 1 . J 000
E ,
t
on a
touj ours n,
=_8’
mais n
=-8.
En écrivant12, 1
que le
rapport
molaire dunaphtalène
estégal
aurapport
idéalo,31,
ontrouve q
= 3 2 ooo. On saitqu’on
préfère
admettre maintenant une structurequasi
réticulaire pour l’eau. A larigueur
le résultatci-dessus est
compatible
avec cette structure.Le nombre « idéal » pour la solubilité de CINa
est x2 =
0,00015
environ(L~~=
7220 etT j = I077°K).
Ce nombre est nettement inférieur à la solubilité mesurée de CINa dans l’eau : 361 g pour 1000 gd’eau;
ici il est
impossible
de rendrecompte
de ladiscor-dance en
invoquant
unepolymérisation
del’eau.
Les
électrolytes
sedistinguent
nettement des nonélectrolytes.
Dans. la formule
(3),
on voit de suite que lasolu-bilité
augmente
avecT;
c’est,
comme onsait,
lecas pour tous les corps normaux.
CALCUL DE LA TEMPÉRATURE D’EUTEXIE. -
Quand
on connaît la chaleur de fusion d’un corps, on
peut
prévoir
sa courbe decongéiation
idéale,
c’est-à-diresa solubilité dans un solvant idéal. Si l’on connaît (4) Cette variation de la solubilité avec le P. F. du corps
dissous avait déjà été remarquée par LAVOISIER, Traité
élémentaire de Chimie,
la chaleur de fusion des deux constituants d’une
solution,
on doitpouvoir
calculer leur solubilitéréciproque,
c’est-à-direprévoir
latempérature
d’eu-texie. C’est ce
qui
a été fait par Washburn et Read~3].
On a
Pour une valeur de T
donnée,
chaque
f ormulcdonne x1
et x~ et en x2 est différent del’unité. Au
point
d’eutexie,
la même valeur de Tdoit
correspondre
à deux valeurs dont la sommeest T . En
posant I
= 0 et~’
_1,
la condition es tT
Le calcul de 0 s’effectue par
approximations
successives;
il est d’ailleurs aussisimple d’opérer
directement sur les formules enlog
et de comparer les valeurs des(x).
Pour
C10Hs
etC6H6,
les valeurs des(L)
sontQ570
et
237°,
celles des(l)
T o0oet 5 I g,
celles des(1’)
353,3
et
z~8,~,
celles des(0)
0,00283
eto,oo35g.
Le calcul donne 0=0,00371,
T= 26g,7,
c’est-à-diret=-~a,5~~.
L’observation donne -
3~,48.
On a des concordancesdu même ordre pour les deux
couples
suivants :COURBE DE SOLUBILITÉ D’UN CORPS DISSOUS DISSOCIABLE. - Pour établir la formule
(3),
nous avonssupposé que
le corps solide de fond estiden-tique
auliquide
no2;
cela exclut la formation de cristaux mixtes ou le cas où le corps fondu se dissocie.Nous traiterons d’abord ce dernier cas; il est assez
répandu (5);
ilcorrespond
aux courbes decongé-lation avec maximum. La courbe de solubilité du
corps
présente
un maximum pourlequel
lacompo-sition du
liquide
et celle du solide de fond sontles mêmes. La
figure
3 serapporte
aumélange
C2H2
+
(6).
Lesystème
estbinaire,
maisune
partie
des constituants est combinée sous laforme
A . pB.
Lesystème
peut
être considéré comme(5) Nous avons par exemple relevé dans Timmermans
(Solutions concentrées, Massoii, i g36) la présence de courbes de congélation avec maximum pour les mélanges
aldéhyde + éthanol; aniline -~- phénol, etc. C’est aussi le
cas des nombreux hydrates (ou solvates) tels que C12Ca.6 H.0.
(s) Si la combinaison était complète, la courbe du milieu serait composée de deux droites se coupant à angle vif
au-dessus du maximum de la courbe réelle. On peut dire déjà que ce sont les produits de décomposition qui abaissent le P. F. du corps pur.
formé à l’aide de ni moles de
A,
n2 deB,
n3 ducom-posé
ApB.
Fig. 3.
Au
point
de fusion de cecomposé,
on a n2 =pn~
Supposons qu’on
ajoute dni
moles de A. La formulede
Duhem-Margtdes
s’écritElle donne ici
Comme les trois substances sont en
équilibre,
En
comparant
(5)
et(6),
on aLa
fugacité
ducomposé
nechange
donc pas paraddition d’une
petite
quantité
d’un de ses composants;
lecomposé
reste enéquilibre
avec le solideà
température
constante.Le calcul
précédent
ne donne aucunrenseignement
sur l’étendue de la dissociation en solution. Deuxméthodes ont été
proposées
pour évaluer cettedisso-ciation,
MÉTHODE DE KREMANN
’41.
- Elle utilisel’abais-sement
cryoscopique
dans la combinaisonaddi-tionnelle
fondue;
si l’on dissout dans celle-ci uncorps
qui
n’est pas l’un desconstituants,
l’abais-sement observé donne la constante
cryoscopique.
Si l’onajoute
un desconstituants,
A parexemple,
l’équilibre
A ~
pB ,-= A Bp
sedéplace;
unepartie
de A secombine,
donc neproduit
pas d’abaissement.On admet que
l’équilibre
obéit à la loi d’action demasses
et,
moyennant
un certain nombred’hypo-lhèses,
onpeut
calculer ledegré
de dissociation.Kremann étudie par
exemple
la combinaison additionnelle aniline-m-crésol. Sonpoint
de fusionà l’état pur est -
~C~~,2.
En y dissolvant du benzèneou de l’acétate
d’éthyle,
il détermine l’abaissementcryoscopique
normal;
on trouve ainsi un abais-sement A =oa,z8
pour la dissolution de 1 moldans 100 au total. On
ajoute
successivement lesdeux
composants
de lacombinaison,
à savoir le rn-crésol et l’aniline. On constate d’abord quel’abaissement est le
même,
quel
que soit lecomposant
ajouté,
pourvu que sa fraction molaire soit lamême. On obtient ainsi les nombres suivants :
La dissociation du
composé
est dutype AB~ A-[-B.
Sur ioo moles ducomposé,
soit oc la fractiondécom-posée
pour le corps pur; il reste moleset il
apparaît 9-oo
oc moles de A etB;
on a doncen tout Ioo
+
100 oc moles. Si l’onajoute
enplus
a moles deA,
le nombre total des molespeut
s’écrire Ioo -~- x --E- a, où x est le nombre de molesdécomposées
dans les ioo molesprimitives.
Si K est la constante dedissociation,
la loi d’action demasses s’écrit :
On ne connaît ni x, ni
K;
onprocède
doncpar tâtonnements. On se fixe un
degré
dedissociation,
soit oc = o,10. Pour a = o, on aalors x~ =
K~I04-
x2),
avecx = Io, d’où .K= 0,0101.
Le nombre r~ de moles
produisant
l’abaissement,
dans 10o moles autotal,
estalors 9o
=18,18.
On110
donnera ensuite à a des valeurs
croissantes,
enadmettant que K reste
constant,
cequi
donne parexemple
l’ensemble de nombres suivants. ~ estOn construit la courbe des 6 en fonction de a
ou du
rapport
molaire de l’addition et l’on cherchela valeur de ac
qui
assure la meilleure concordanceentre la courbe calculée et la courbe
expérimentale.
Van Laar a d’ailleurs
proposé
[5]
une formulequi
tient
compte
de la dissociation du solvantervoseo-pique ;
elle s’écrit :ao est le
degré
de dissociation à l’état pur, a le mêmeen
présence
d’une fraction molaire x du deuxièmecomposant.
AvecLr
=3goo
calories,
on obtientdes ao et oc voisins de o,10. C’est à peu
près
cequ’avait
donné le calcul de Kremann.
Les calculs
cryoscopiques
sont en réalitéplus
compliqués
que ceux de Kremann et ils nepeuvent
donner que l’activité de la substance
dissoute,
dès que la solution devient tant soit peu concentrée.Les calculs de Kremann sont donc basés sur des
hypothèses
assez douteuses.MÉTHODE DE JOB
[6].
- Ellerepose aussi, sur un certain nombre
d’hypothèses;
mais elle al’avan-tage
derenseigner
sur la f ormule ducomposé
eten même
temps
sur la constante dedissociation,
c’est-à-dire sur la stabilité ducomposé.
Ungrand
nombre de travaux effectués dans mon laboratoire
ont utilisé cette méthode de Job. Il se trouve
qu’elle
a été
jusqu’ici
presque exclusivementemployée
pour les solutions
d’électrolytes
dans l’eau. Nous enreparlerons
plus
tard.SOLUBILITÉ
(CONGÉLATION)
DANS LE CAS OU ILSE FORME DES CRISTAUX MIXTES. - On rencontre
ce cas très
fréquemment
à hautetempérature;
lecorps
dissous est soluble dans laphase
solide dusolvant.
Soient xl
et x2 lesrapports
molaires dans lasolution,
X’I
etxg
dans la solution solide. Cette fois lafugacité
du solidedépend
de T et des(x’).
L’équilibre exige que/, (solide) - Il (liquide).
Quand
onajoute
dusoluté,
d f i
=d/1;
cela donneOn suppose les deux
solutions,
liquide
etsolide,
infiniment
diluées en le nO 2.D’après
les formules de l’article sur lathermody-namique
dessolutions,
on aet
l’analogue
pour x2.L’hypothèse soulignée
permet
d’écrire la loi de,
Raoult
= -il
» Il reste alorsi/71
c’est-à-dire
On utilise ensuite la loi de
répartition
Ce
qui
donne enfinHl - Hi
concerne le passage du solide à la solutionet si celle-ci est diluée
(idéale),
c’estl’enthalpie
de fusion du solvant pur.La
thermodynamique
ne donne aucunrensei-gnement
sur le domaine de validité de la formule(7).
II. Discordances
entre la théorie et
l’expérience.
A.Remarques qualitatives.
- S’ily a
quelques
solubilités
qui
coïncident avec la solubilitéidéale,
il y a aussi
beaucoup
de discordances entre la théorieFig. 4.
et
l’expérience.
Elless’expliquent simplement
par le fait que la courbe despressions partielles
n’est pas engénéral
la droiteexigée par la loi
de Raoult. Lapression
de vapeurpeut
êtresupérieure
à l’ordonnéede cette
droite;
la déviation est ditepositive;
c’estle cas du
mélange
CS2-CH2(OCH3)2’
Lapression
déviation est dite
négative;
c’est le cas dumélange
acétone-chloroforme. Enfin, dans tout le domaine
étudié,
la déviationpeut
changer
designe;
c’est le cas pour lapyridine
dans sesmélanges
avec l’eau. Il faut mettre tout à fait àpart
les solutionsélectrolytiques,
qui
ne suivent pas la loi de Raoult. Une fois la courbe despressions partielles
connue,on
peut
toujours
utiliser la relation P2 - p,qui
est
toujours
valable;
on arrive ainsi aux résultatsde la
figure ~[
où l’indice ~désigne
le corps dissous.On a
représenté
sur cettefigure
la droitethéorique
en
pointillé,
les deux courbespositive
etnégative
en trait
plein.
L’ordonnée de hauteur p, coupe lescourbes en trois
points
dont les abscissesx’,
xl et x"représentent
lerapport
molaire des corps dissous dans les trois cas. On voit que les déviationsposi-tives
correspondent
à des solubilités inférieures àla solubilité
idéale,
lesnégatives
à des solubilitéssupérieures.
En tout cas, la courbe despressions
partielles
de vapeur étant connue, la solubilité s’endéduit de suite. Il faut maintenant se
préoccuper :
Iode chiffrer lesdéviations;
5,,o d’en proposer uneexplication.
Dans cetarticle,
nous traiteronsseule-ment le
premier point,
nous réservant de traiter ledeuxième
plus
tard.B. Grandeur des déviations de la loi de
Raoult. -
D’après
la définition que nous avonsdonnée au
premier
article de cettesérie,
pour lecoefficient
d’activité,
la relation y= f
montre.r
que y est
précisément
la mesure des déviations.L’expression
RTlny
est lechangement
d’énergie
libre dans le transfert d’une mole du
composant,
d’une
quantité
considérable de solution idéale à la solution réelle de mêmecomposition.
Tous les
procédés
dérivés de la loi de Raoult(tonométrie, ébullioscopie, cryoscopie,
etc.) peuvent
être
employés
enprincipe
pour évaluer le coefficientd’activité. Les méthodes de calcul sont dues à
Lewis;
nous donnerons seulement iciquelques
indications,
renvoyant
le lecteur à lathermody-namique
de Lewis et Randall pour les détails. ACTIVITÉ MESURÉE A PARTIR DE LA PRESSION DEVAPEUR DU SOLVANT. - C’est le
cas le
plus simple.
Si la vapeur est un gaz
parfait,
on asimplement
a = C’est ainsi par exemple que Hildebrand etPI 1
Eastman
[7]
évaluent l’activité du mercure dansles
amalgames
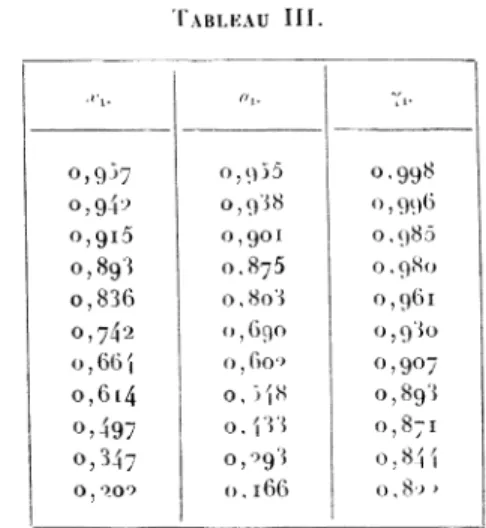
de thallium à Le Tableau IIIdonne,
d’après
cesauteurs,
l’activité et le coefflcientd’activité de
Hg.
ACTIVITÉ DU CORPS DISSOUS A PARTIR DE SA PRESSION DE VAPEUR. -
L’exemple
estemprunté
à Lewis et Storch
[8]
et concerne les solutions debrome dans C
C14,
Les auteursprennent
comme étatstandard du brome celui en solution infiniment
diluée;
dans cetétat,
d’après
la loi deHenrv,
‘’ est1>1
constant, soit k. Comme dans cet état, par
défini-tion,
ag =T~,
à une concentrationquelconque,
onaura
donc a2
=p~. Les auteurs ont ainsi étudié les solutions entre les concentrations .r~ =
et
0,025
et ils ont trouvé unrapport /]/
2 constant etégal
ào,~3g
(p
enatm)
(P.
M. du bromepris
égal
à160).
Dans tout cetintervalle,
la solutionest donc idéale. Le brome
n’y
est d’ailleurs pas dansle même état que dans le
liquide
pur, car sapression
de vapeur est seulement0,280
atm à l’état pur;l’extrapolation
donnerait au contraireo,539
atm.Si la vapeur du corps dissous ne suit pas les lois des gaz
parfaits,
on introduira facilement lafugacité.
TABLEAU III.
ACTIVITÉ A PARTIR DES FORCES ÉLECTROlVIOTRICES.
- Le
procédé
a étébeaucoup
employé
pour lesamalgames.
Parexemple,
deux électrodes constituéespar des
amalgames
dethallium,
decompositions
différentes,
plongent
dans une même solutionélectrolytique
d’un sel thalleux.Quand
lapile
fonctionne,
si Tl est le métalélectrométriquement
actif,
l’un desamalgames
s’enrichit auxdépens
de l’autre. La f. é. m. de la
pile
est donnée par laformule
Les
(a)
sont les activités de Tl dans les deuxélec-trodes,
N est la valence dumétal, 5’
leFaraday.
Avec les valeurs desconstantes,
faisant N = i,il vient
Richards et Daniels
[9]
ont effectué des mesures240
On
peut
convenir que l’accentdésigne
l’état deréférence.
L’expérience
donne une liste de valeursde E en fonction de ~~. On écrit alors
(9)
sous la formeOn
porte
alors en ordonnée laparenthèse
dudeuxième
membre,
connueexpérimentalement,
enFig. 5.
fonction
de x2
enabscisse,
commel’indique
lafigure 5
et l’onextrapole jusqu’à x~
= o. Si l’onremarque
qu’en
solution infinimentdiluée,
a2 -+ x2ou
log
-
o, l’ordonnée àl’origine
estsim-/
plement -- log
a2, cequi
détermine l’activité dans , Bl’amalgame
de référence. Connaissant cettevaleur,
on calcule
facilement a2
et y 2 pour toute valeurde x2. Le Tableau
IV,
où est laconcen-tration de
référence,
donne les valeurs ainsi déduites par Lewis et Randall des mesures de Richards.TABLEAU IV.
On remarquera dans le tableau comment Y2
s’éloigne rapidement
de l’unité même pour desconcentrations très faibles.
La méthode donne ici l’activité du, corps
dissous ;
lapression
de vapeur nous donne au contraire directement celle du solvant. Onpeut
passer del’une à l’autre par la méthode que nous avons
indiquée.
La formule c(e Gibbss’applique toujours
en effet sous la forme ..
Supposons
qu’on
veuille parexemple
obtenir l’activité du mercure dans lesamalgames
précé-dents. On remarque que
dXl
=-dx2,
donc(10)
peut
s’écrire :’2,
ou finalement
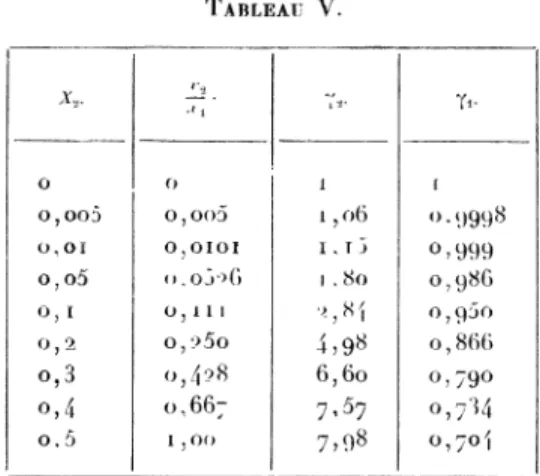
Pour calculer y,, on construira la courbe
de x2
enionction de In Y2 et l’on déterminera l’aire
comprise
sous la courbe à
partir
de ~y2 = I icorrespondant
à la dilution infinie. On obtient ainsi les nombres
du Tableau V.
TABLEAU V.
Nous retrouverons ce genre de calcul dans le
paragraphe
suivant.ACTIVITÉ DÉDUITE DES MESURES CRYOSCOPIQUES.
- Nous tenons enfin à
traiter,
au moinssommai-rement,
cettequestion,
pour bien montrer que,pendant
desannées,
lesphysicochimistes
ontemployé
des méthodes « élémentaires »qui
n’avaient aucunevaleur.
I~ewis, ayant
à traiter del’équilibre solide-liquide,
choisit un état de référence. Pour le
liquide,
onprend
l’état pur; l’activité duliquide
est i dans cetétat;
sonenthalpie
relative est zéro. Le solidepossède
alors une activité a et uneenthalpie
à Hégale
à la chaleur desolidification.
La relation(14)
On
développe
ensuite AN en série dans levoisi-nage du
point
de fusion. Pourcela,
on poseEn
appelant
la chaleur de solidificationau P. F. et
ACP
la différence descapacités
calori-fiques
du solide et duliquide,
on a, à latempé-rature
T,
On substitue
(14)
et(15)
dans(13)
et l’ondéveloppé
parrapport
auxpuissances
de0;
on obtient ainsila série
On
intègre
ensuite àpartir
de 6 = o, enremarquant
que, à cette
température,
il y aéquilibre
entre lesolide et le
liquide,
donc as
= ai = I. Il vientPour la
glace
et l’eau parexemple, gardant
seu-lement le terme en
0,
on aTj -
2 7 3, 1
A H,
=-1 G 3 8
et AC =-
9, ce
qui
donneC’est cette
équation
qui
sert de base aux calculsdes résultats
cryoscopiques.
On
peut
remarquer que, pour unecomposition
donnée d’une solution aqueuse, il existe unabais-sement déterminé du
point
defusion,
de sorte que, dans(18), 0
est fonction de la molarité parexemple.
Aupoint
de fusion dans lasolution,
l’activité del’eau en solution est
égale
à celle de laglace, d’après
la convention ci-dessus.Donc,
si l’indice i serapporte
au
solvant,
on aet en
intégrant
Dans
l’équation (20),
a1 est l’activité de l’eau aupoint d’apparition
de laglace.
Ennégligeant
lavariation de
l’enthalpie
avec laconcentration,
c’est-à-dire en
négligeant
la chaleur dedilution,
onpeut
supposer que a1, pour une concentrationdonnée,
estindépendant
de latempérature.
Celaétant,
on considérera 0 dans leséquations
précé-dentes comme une
propriété
de lasolution,
fonction de la concentrationet,
pour calculer l’activité du corpsdissous,
onemploiera l’équation (10)
ci-dessus.On en tire
Dans cette
équation, quand b - o,
on obtient ’ahaissementcryoscopique
limiteSoit 2, ce nombre. En introduisant ce
nombre,
l’équation (21) prend
la formePour
intégrer
cetteéquation,
Lewis posPon en Lire
et,
en substituant dans(22),
On
intègre
entre les molarités 0 et m; la constanted’intégration
estnulle,
a2 tendant vers met j
verszéro
quand
la dilution est infinie. On afinalement
’
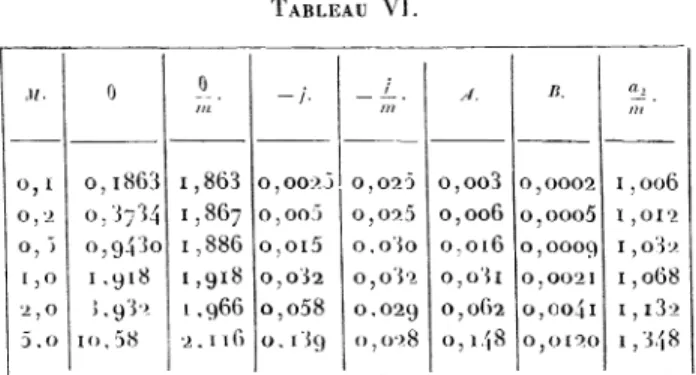
Lewis a par exemple traité à l’aide de cette
équa-tion les résultats relatifs aux solutions aqueuses de
glycérine. L’intégrale
A se déduit de la courbe-
tr
en fonction de m;
l’intégrale B
de lacourbe -
enm
fonction de 0. Cette dernière courbe a été
précisément
considérée par
Raoult,
pour obtenir son « ordonnéeà
l’origine
». Les calculs de l,ewis ontdonné,
pour laglycérine,
les résultats suivants(Tableau VI).
TABLEAU VI.
La dernière colonne donne le coefficient d’activité
(rapporté
à lamolarité).
En solution assez
étendue, ~
’ est à peuprès
constant. Si l’on admet cette
régularité,
on obtientl’équation approchée
qui
sufpltgénéralement
pour traiter les mesuresassez peu
précises
que renferme la littérature dessolutions non
électrolytiques.
On trouvera dansLewis et Randall les
compléments
relatifs àl’ébul-lioscopie.
C. Courbes et formules pour le coefficient
d’activité. - Nous allons supposer de nouveau que nous nous occupons de
mélanges
deliquides
Fig. 6.
Le coefficient d’activité du no 1 est
égal
à 1 pourle
liquide
pur,log y1
est nul pour x2 = o; c’estdonc une fonction
de x2 qui
s’annule pour X2 - o.De même pour log Y2. La
figure
2~t
représente
lesvaleurs de
logyi
en fonction et de mêmepour
log
y,, pour lesmélanges
sulfure decarbone-méthylal.
On voit que ces courbes ont une allureparabolique
suggérant
undéveloppement
suivant lespuissances
croissantes des(x).
Onpeut
donc poserNous avons
déjà
dit que la formule deDuhem-Margules
établit une relation entre lespressions
partielles,
donc en définitive entre les coefficientd’activité;
c’est la relationdéjà
vueplus
haut etque nous écrirons
’
Si on limite les
développements (26)
auxquatre
premiers
termes,
on tire de suite de la formules deDuhem-Margules
les relations suivantes :Il est à retenir d’abord de ces relations que les
développements
n’ont pas de terme en ri et x2.Cela prouve que, dans un
mélange
réel,
la loi deRaoult est
vérifiée,
non seulement pour x = o,mais pour une série de valeurs de x voisines de zéro. Ce résultat
correspond
au fait que, dans les courbesde
pression
partielle,
la courbe esttangente
à la (miroite de Raoult vers x = o. Autrement ditencore,
la loi de Raoult est une loi-limite
indépendante
ducorps
dissous,
pour un solvants donné(x2
nefigure
pas au
premier degré
danslog Yl)’
Comme cas
particulier
des formules(26),
nousutiliserons un
développement
du ’ledegré
Ces
développements
semblent convenir pour lessolutions de Tl et de Cd dans
l’étain;
dans ce cas,le
graphique
delog
y en fonctionde X2
est une droite.Van Laar a
proposé
une formule dutype
et
l’expression analogue
pour y 2; les r sont lerapport 11)
et son inverse. Les coefficients(oc)
et()
:1’"2
sont
également
reliés parl’équation
deDuhem-Margules. D’après
Hildebrand,
pour lesamalgames
de Cd et Sn à le coefficient d’activité de
Hg
est bienreprésenté
par deséquations
de cetype;
on aurait
respectivement
oc, ==2013 et 0..22et
131
= 1,90 et0, ’:>.6.
D. Effet de la
température
sur les déviationsde la loi de Raoult. - L’élévation de T a
géné-ralement pour effet de rendre lesmélanges
plus
idéaux. C’est ce que montre la
figure 7
qui
repré-sente,
d’après
Schulze[viol,
lapression
totale pourle
système
chloroforme-acétone à 3 0° etgooc.
L’échelle de
gauche,
en millimètre deHg,
celle dedroite en
kilogramme
par centimètre carré sontrelatives aux deux
températures.
La déviation estnégative
dans les deux cas, mais moins forte àgo°
qu’à
30°.Inversement,
l’abaissement de latempérature
rend lemélange
de moins en moins idéal. Cet effet estparticulièrement marqué
dans lesmélanges
qui
se
séparent
en deux couchesliquides
à bassetempé-rature.
Fig. 7.
E.
Apparition
de deux couchesliquides.
-La discussion de cettequestion
est relativementsimple
si l’on admet que ledéveloppement
delog
,-est réduit au terme en x2. Nous verrons,plus
tardque,
d’après
Hildebrand,
ce cas est assezgénéral
pour les
mélanges
dont lescomposants
ont desvolumes molaires assez voisins. Si =
Ax2,
on obtiendra des déviations croissantes en augmen-tant A. Nous avons
reproduit
le calcul del’activité du corps no 2 pour divers
(x2),
en donnant à A des valeurscroissantes;
on trouve ainsi que lacourbe a2
= forme donnée par lafigure
8pour différentes valeurs de
A ;
A = ocorrespond
au cas idéal. Pour les
grandes
valeurs deA,
la courbea deux bosses et la
partie
médianecorrespond
à un étatinstable;
lemélange
correspondant
sesépare
en deux couches P etQ.
L’importance
de ces deux couchesdépend
de laquantité
deproduit
dispo-nible. Si l’onajoute
au no 1 pur desquantités
crois-santes du no2,
il y a d’abordsimple
dissolution;
lepoint
représentatif
décrit l’arc degauche
de la courbe. Pour lacomposition
P,
apparaît
la deuxième couche dont laquantité
augmente,
le volume de lapremière
couchediminuant;
pour unequantité
suffisante du no
2,
la couche decomposition
Q
subsiste seule et lepoint
décrit ensuite lapartie
de droite de la courbe.Quand
latempérature augmente,
les déviationssont
moindres;
il faut admettre que Adiminue;
on arrive à la courbe
marquée
A =o,g et le
mélange
homogène
devient alorspossible.
Au-dessus d’une certainetempérature,
il y a donc dissolutionhomo-gène
des deuxliquides;
on dit que lecouple
deliquides possède
unetempérature critique
dedisso-lution
(T. C. _S.);
on aurait alors pour les deuxcomposants
2013
= o et~~ , =
o. Ces deuxrelations,
~ du d2 a
écrites pour
logio
y =Ax,
donnent x,
= x2 =0"51
avec A _
9 -
o,868.
Cette valeur «critique
»,
de A est bien
voisine
de A = o,gqui
donne lalongue
inflexion dans la courbe. Comme nous l’avons
Fig. ,,.
remarqué
ci-dessus,
dès l’instantqu’on
arrête ledéveloppement
delog
y au terme enx2,
il y asymétrie
entre les deuxcomposants.
Ce n’est pas tout à fait le cas enpratique,
comme le montre lafigure
gqui
donne lacomposition
desmélanges
enéquilibre
aux diversestempératures
pour lecouple
aniline-hexane
(T.
C. S.6oO).
T. C. S. SUPÉRIEURE ET INFÉRIEURE. - Dans
ce
qui précède,
il a étéquestion
demélanges
semblablesau
mélange
aniline-hexane,
qui
subissent la démixtion au-dessous d’une certainetempérature.
On dit que cesmélanges
possèdent
une T. C. S.supérieure.
C’est le cas aussi pour les
mélanges
eau-phénol,
hexane-nitrobenzène,
pétrole-alcool,
etc. Cesmélanges
ont été
beaucoup
étudiés à toutes sortes depoints
de vue. Ils ofirent en effet uneimage
facilementaccessible des
phénomènes
devaporisation critique
auxquels
ils ressemblentbeaucoup.
Fig. 9.
Quand
latempérature
s’élève,
les deuxliquides
superposés
tendent l’un versl’autre;
l’indice deréfraction
(Mlle Schlegel) [13],
ladensité,
la tensionsuperficielle
(Antonoff)
etc.,
deviennentiden-tiques.
On retrouve mêmel’opalescence critique
qu’on
aégalement
étudiée,
tant aupoint
de vueexpé-rimental
qu’au point
de vuethéorique (Rocard
etAndant
[ 15],
RoussetC r 6]).
On a même retrouvé surles courbes
analogues
à celles de lafigure
g la loidu diamètre
rectiligne
de Cailletet etMathias;
la demi-somme des concentrations(exprimées
entitre)
correspondant
aux solutionsconjuguées
est une fonc-tion à peuprès
linéaire de ~’.Pour
comprendre
complètement
lesphénomènes
dedémixtion,
il faut considérerl’équilibre
des deuxliquides
avec leur vapeur commune, comme l’amontré Kuenen.
Le
système,
comprenant
deuxcomposants
ettrois
phases
estunivariant,
d’après
larègle
desphases,
d’où unecomposition
complètement
déter-minée dès
qu’on
fixe latempérature.
Si celle-civarie,
lescompositions
des deuxphases
liquides
etcelle de la vapeur changent; à la T. C. S. la
phase
vapeur n’a pas la
composition
commune aux deuxliquides.
L’existence d’une T. C. S.
supérieure
est trèsfréquente,
mais pas absolumentgénérale.
Certainsmélanges
(eau-nicotine; glycérine-in-toluidine)
pos-sèdent un domaine de démixtion
complètement
f ermé,
donc à la fois une T. C. S.supérieure
et uneT. C. S.
in f érieure.
Lafigure
1 oreprésente,
dans lediagramme (T,
x),
le domaine de démixtion pour lemélange glycérine-m-toluidine.
Pour certains
couples,
on nepeut
réaliser auxtempératures
accessibles que lapartie
inférieurede la
courbe;
ilspossèdent
seulement une T. C. S.inférieure. Exemples : eau-triéthylamine;
CÛ2-nitro-henzène ;
o-nitrophénolnitrobenzène.
Quand
onaug-mente la
température,
l’une desphases
liquides
sevaporise complètement
et il ne reste enprésence
qu’un
liquide
riche en lecomposant
le moins volatilet une vapeur riche en le
composant
leplus
volatil.
Il existe enfin
quelques couples
où la solubilitémutuelle est
toujours
trop
faible pour donner unmélange
homogène;
iln’y
a aucune T. C. S.(mélange
méthane-alcool).
.Fig, i n.
Les calculs effectués ci-dessus sur
l’équation
-
Ax1
nepeuvent
plus s’appliquer
pour uneT. C. S.
inférieure,
à moins de supposer que Aaugmente
cette foisquand
Taugmente.
Il estpossible
aussi que la formule soitplus compliquée.
On vérifiera
qu’une
formule dutype
log
y=Ar2+Bx3
donne parexemple
des(y)
inférieurs à l’unitéquand
Aest
négatif
et Bpositif,
pour certaines valeurs de Aet B. Pour trouver des
(y)
inférieurs àl’unité,
il fautsupposer A
o; avec un seulterme,
la courbecle a2
n’a dans ce cas aucunesingularité.
Avec deuxmais cette lois au-dessous de la droite de Raoult.
Exemple :
A =-3,
B~+
4;
on obtient une courbeà deux bosses.
Dans un autre
exposé,
nousparlerons
des raisonsinvoquées
pourexpliquer
les discordances avecla loi de Raoult.
,
BIBLIOGRAPHIE.
[1] SCHRÖDER, Zeit. f. phys. Chem., 1893, 11, p. 449.
[2] LE CHATELIER, C. R. Acad. Sc., 1894, 118, p. 638.
[3] WASHBURN et READ, Proc. Nat. Acad. Sc., 1915, 1, p. 191.
[4] KREMANN, Monatsh. f. Chem., 1904, 25, p. 1215 et
1916, 37, p. 59.
[5] VAN LAAR, Kon. Akad. W et. Amst., 1906 711;
[6] P. JOB, Ann. Chim., 1928, 9, p. 113.
[7] HILDEBRAND et EASTMAN, J. Ann. Chem. Soc., 1915, 37, p. 2452.
[8] LEWIS et STORCH, ibid., 1917, 39, p. 2544.
[9] RICHARDS et DANIELS, ibid., 1919, 41, p. 1732.
[10] SCHULZE, Zeits. f. phys. Chem., 1919, 93, p. 368.
[11] E. DARMOIS, L’état liquide de la matière, Albin Michel,
Paris, 1943.
[12] TIMMERMANS, Les solutions concentrées, Masson, Paris,
1936.
[13] Mlle SCHLEGEL, J. Chem. Phys, 1934, 31, 668.
[14] ANTONOFF, Phil. Mag., 1926, 1, 1121 et suivants.
[15] ROCARD et ANDANT, Soc. fr. Phys., 1925, 30. [16] ROUSSET, C. R., 1934, 198, 2152 et 199, 716.
CONTRIBUTION AU SPECTRE ULTRAVIOLET DE LA
MOLÉCULE D’AZOTE
Par Mme RENÉE HERMAN.
Sommaire. 2014 Les mesures plus précises des
longueurs d’onde des têtes de bandes d’un système signale pour la première fois par Kaplan permettent de tracer un schéma de niveaux reliant entre eux plusieurs
états électroniques non identifiés de la molécule d’azote.
Ce schéma fait apparaître une fréquence de 913 cm-1 identique à celle d’une progression trouvée dans le spectre de l’oxygène et située dans l’ultraviolet extrême. L’attribution de cette progression à la molé-cule neutre d’azote et non à celle d’oxygène est compatible avec les anomalies constatées dans les spectres
d’émission ci-dessus.
La
partie
laplus
intense duspectre
ultraviolet de la moléculed’azote,
émis sous faiblepression,
se compose en dehors du second etquatrième
groupes
positifs,
des bandes de Van der Ziel etdu
système
asignalé
pour lapremière
lois parhaplan
(1).
Jusqu’ici,
les niveauxélectroniques
de ces
systèmes
n’ont été rattachés ni entre eux, niautres niveaux connus de
N2
ouNt.
TABLEAU 1. -
Systèiiie
a-p.(1) VAN DER ZIEL,
-Physica,
1, I g34, p. 513. - J. KAPLAN, Phys. ReU., 46,1934,
p.534.
-Mme R. HERMAN et L. HERMAN,
C. R. Acad. Sc., 225, ~ gG z, p. 83; Société trançaise de Physique, Section de Lyon, réunion du 5 mai g~ 3.