The Core Mammalian Pluripotency Network in Induced Pluripotent Stem Cell (iPSC) Formation: Models for Genetic and Epigenetic
Reprogramming by Hussein Abdallah
S.B., Electrical Engineering, MIT, 2016 Submitted to the
Department of Electrical Engineering and Computer Science in Partial Fulfillment of the Requirements for the Degree of
Masters of Engineering in Electrical Engineering at the
Massachusetts Institute of Technology February 2018
The author hereby grants to M.I.T. permission to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole and in part in
any medium now known or hereafter created.
Author:
Department of Electrical Engineering and Computer Science February 2, 2018
Certified by:
Domitilla Del Vecchio, Associate Professor, Thesis Supervisor February 2, 2018
Accepted by:
Abstract
In 2006, history was made in a seminal experiment that converted mouse fibroblasts to a pluripotent phenotype coined the ‘induced pluripotent stem cell’ (iPSC) state. Unhindered by ethical or immunogenic constraints, iPSCs potentially hold the keys to tremendous applications in therapeutic and regen-erative medicine. Furthermore, on-demand iPSC generation has the capacity to revolutionize basic research in disease modeling and drug discovery. These promises notwithstanding, the economics of iPSC formation—which remains a slow, inefficient, expensive, and laborious process—still stand in the way of fully making use of this extraordinary technology. In this thesis, I present mathematical models aimed at understanding the theoretical reprogrammabil-ity of the core pluripotency gene regulatory network being awakened in iPSC reprogramming. Using these modeling insights, I discuss the merits of current reprogramming strategies, which can be viewed as open-loop perturbations in control theoretic terms. I then discuss an alternative paradigm of closed-loop reprogramming, which is theoretically shown to be far superior when it comes to the reprogrammability of the pluripotency network. Finally, I propose a re-programming model that incorporates the e↵ect of DNA demethylation on the activation of the network, with attention given to the relationship between this epigenetic transformation and the cell proliferation barrier that somatic cells seemingly face on the road to pluripotency.
Contents
I Introduction 4
II The Waddington Landscape and the Core Pluripotency GRN 6
III The iPS Reprogramming Process 9
IV The Biology of Epigenetics 13
V Epigenetics and iPSC Reprogramming 16
List of Figures
1 Waddington Landscape of Cell Di↵erentiation 39
2 The OSN Triad as the Core Pluripotency Network 40
3 Steady State Landscape of Reduced ODE Model 41
4 Visual Depiction of Open-Loop Reprogramming 42
5 Open Loop Overexpression 43
6 Feedback Controller Gain 44
7 Reprogramming from TR to PL Using Closed Loop
Overex-pression on Oct4 45
8 Steering a Golf Cart Analogy: Comparing Closed-Loop and
Open-Loop Control 46
9 Realizing Feedback Overexpression with a Synthetic Genetic
Circuit Feedback 47
10 An Overview of Chromatin Remodeling 48
11 The Cell Proliferation Barrier as a Gatekeeper to Pluripotency 49 12 An Overview of DNA (de)Methylation as an Epigenetic
Regu-latory Mechanism 50
13 Simplified Motifs of the Core Pluripotency Network, With and
Without Demethylation 51
14 The E↵ect of Cell Proliferation Rates on the Temporal
Re-sponse and Steady State Levels of Oct4 52
I
Introduction
The notion of reprogramming cell fate is a direct challenge to what was once a traditionally-held view in developmental biology that a cell’s phenotypic identity is sealed after undergoing di↵erentiation. As early as 1938, Hans Spemann laid ground-work for the earliest experiments that would begin to chip away at the permanence of cell fate [1]. Spemann introduced the idea of somatic cell nuclear transfer (SCNT), in which a di↵erentiated cell’s nucleus could be inserted into a de-nucleated egg cell with hopes that the fused cell would give rise to an embryo. In 1952, the first major SCNT experiment [2] successfully took place when a SCNT-fused cell, made from a blastula cell’s nucleus and an egg cell from the frog species Rana pipiens, gave rise to normal tadpoles. Following this breakthrough, nuclear transfer technologies ad-vanced remarkably in the second half the twentieth century [3], in some cases drawing remarkable public attention as in the cloning of the popularized sheep ‘Dolly.’ Nonetheless, though nuclear transfer hinted at the plasticity of developmental poten-tial even after di↵erentiation, at the start of the 21st century it was still widely be-lieved that cell di↵erentiation was permanent and irreversible. This notion was finally completely shattered in 2006, when a team of Japanese researchers reverted mouse embryonic and adult fibroblast cells (skin cells) back to the pluripotent state, a pheno-type they coined the induced pluripotent stem cell (iPSC) state [4]. These iPSCs pos-sessed the major features of pluripotent embryonic stem cells (ESCs): they expressed ESC marker genes, showed ESC epigenetic properties, displayed ESC morphology and growth properties, and could give rise to all three germ layers [5, 6, 7, 8, 9]. Even more remarkable was that the fibroblast to iPSC transformation was achieved by there mere overexpression of four transcription factors (TFs) in somatic fibroblasts placed in ESC culture conditions: Oct3/4, Sox2, Klf4, and c-Myc, (also known as ’OSKM factors’).
Yamanaka‘s work and the variations that have followed [10, 11] have been remarkable achievements in their own right, and many mechanisms of iPS reprogramming have been elucidated thus far [12, 13]. But over a decade later, there remains many more questions than answers about the the biochemical and biomolecular changes that ac-company the reprogramming process [?, 14]. Chief among these questions are those surrounding the low efficiencies, variable quality, and high latency of iPSC formation [15, 16, 17] that persist in the laboratory today. Yamanaka himself, when describing the first clinical trial with iPSCs in 2016 [18], remarked: “what we learned from the first patient, in which we performed autologous cell transplantation, is that the entire process is too expensive. It also took almost a year to make iPSCs from the patients own cells, to transfer the original cells, and to perform all the rigorous quality control tests. It was too expensive, and it took long. ”
Needless to say, iPSC reprogramming remains an important area of intense research. And though it is, after all, an experimental technique, significant ground can poten-tially be gained by studying the gene regulatory networks (GRNs) that give rise to stem cell identity at a theoretical level. It has become clear that TF-mediated re-programming of somatic cells with OSKM factors, if successful, artificially awakens a naturally silenced endogenous network that then gives rise to pluripotency. This core endogenous pluripotency network (henceforth referred to as the ‘core pluripotency network’) consists of the TFs Oct4, Sox2, and Nanog. By studying the ways in which OSKM or other factors artificially perturb the core pluripotency network during re-programming, the hope is that we can potentially uncover theoretical insights about the intrinsic properties of the network that can in turn be used to identify problems or gaps with current reprogramming strategies. Then, by synthesizing these findings, the ultimate goal is to inform improved reprogramming strategies that address the low efficiency, quality, and latency of iPSC formation. Solving these issues will move us closer to realizing the full potential of iPSCs in clinical applications and basic research without hindrance by the current economics of the process.
In this thesis, I present the findings of theoretical analysis of the core pluripotency net-work that considers both genetic and epigenetic changes to this GRN during iPSC re-programming. The approach I will use is epistemologically rooted in an idea proposed by the British developmental biologist Conrad Waddington in the mid-20th century. Waddington introduced the notion of a ‘landscape’ in which cell development and the capacity for di↵erentiation is akin to a marble’s potential energy as it rolls down a hill with several possible paths to the bottom. I will review the Waddington Landscape in Section II, which will establish the connection between cell phenotypes and their un-derlying GRNs. In that section, I will also introduce an ordinary di↵erential equation (ODE) model for the concentration of the TFs in the core pluripotency network. This ODE model allows us to move the iPSC reprogramming problem to the state-space domain in Section III. By making this transition, we can zoom out from experimental views of reprogramming and understand the network and the phenotypes and it gives rise to at a theoretical level. Then, it becomes possible to characterize the admissible steady state landscapes of the system and determine which state transitions are pos-sible under the influence of external perturbations. This section will address these questions, and then portray the contrast between open and closed-loop control for reprogramming the core pluripotency network. Since iPSC reprogramming is more than just a genetic process, in Section IV I review the relevant epigenetic mechanisms needed to study the epigenetic changes for reprogramming in Section V. In that sec-tion, I will study an even further simplified model for the pluripotency network that assesses the impact of methylation on the timing and levels of concentrations achieved during reprogramming. All in all, the purpose of this work is to garner insight about the theoretical reprogrammability of the network with current ‘open-loop’ strategies,
propose an alternate ‘closed-loop strategy,’ and begin to understand the epigenetic transformations that need to take place for the core pluripotency GRN to be properly activated during iPSC reprogramming.
II
The Waddington Landscape and the Core
Pluripo-tency GRN
A common conceptualization for cell fate reprogramming, of which iPSC reprogram-ming is a type, is the Waddington view of cellular di↵erentiation [20]. In this metaphor, a ball rolls down a hilly landscape with di↵erent valleys representing the multiple paths that undi↵erentiated cells may take in acquiring their di↵erentiated identity (Figure 1). Along the way to di↵erentiation, these cells are often lumped under the umbrella term stem cells, though they have di↵erent potencies depending on their degree of di↵erentiation [21]. Zygotes and embryonic stem cells (ESCs) in the first few divisions after fertilization are totipotent, and can give rise to an entire organism including the placenta and umbilical cord. When ESCs become pluripotent, they can continue to give rise to all three germ layers, and thus an entire organism [22]. Multipotent cells such as haematopoietic stem cells, neural stem cells, and mesenchymal stem cells are adult stem cells that retain some potential for further di↵erentiation that is typically limited to a particular tissue type.
In an energetic landscape sense, di↵erentiation is represented as a spontaneous down-hill process associated with typical development during an organism‘s periods of growth and maturation. By contrast, iPSC reprogramming is tantamount to an up-hill movement from a somatic state (e.g. fibroblast) to the pluripotent state. We assume that there is a gene regulatory network (GRN) that gives rise to these valleys (phenotypes) in the landscape, and that by perturbing this network in artificial ways we can induce transitions between phenotypes that would not otherwise occur natu-rally. Viewed in this vein, when Yamanaka and colleagues first created iPSCs in 2006, they must have successfully hit the mark on perturbing the correct GRN needed to induce pluripotency. What is that GRN?
Master Regulators in the Core Pluripotency Network
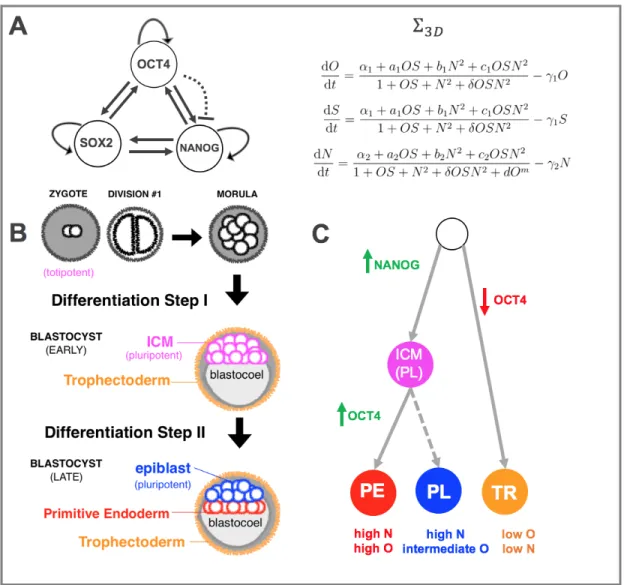
Shortly before the Yamanaka experiments, Boyer and colleagues identified a GRN in which the TFs Oct4, Sox2, and Nanog (the ‘OSN triad’) interact with one another as a fully-connected-triad (FCT) wherein all three TFs mutually and auto-activate one another (Figure 2A). Since then, these TFs have emerged as the ‘master’ pluripotency regulators [23, 24, 25, 26, 27, 28, 29, 30], owing to their essential role in establishing stem cell identity. It is worth noting that from a mathematical point of view, FCTs are a class of GRNs that are known to give rise to multi-stability [31], and that in cell fate
modeling it is common to draw a correspondence between the mathematical attractor states of multi-stable GRNs and experimental phenotypes [32, 33, 34]. It is also worth noting that the OSN triad itself is embedded in an extended regulation network involving various other TFs, signaling factors, di↵erentiation genes, and chromatin remodelers [35, 26, 36, 37, 38].
The Phenotypes of the OSN Triad Given its position at the very top of the di↵erentiation hierarchy in developing mammalian embryos, the OSN triad is impli-cated in the earliest di↵erentiation events following zygote fertilization. Figure 2B shows that in the first few steps after fertilization, three rounds of cell division form an 8-cell mass known as the morula. The first di↵erentiation event occurs when an epithelial layer known as the trophectoderm (TR) forms around the perimeter of the mass, leaving the rest of the cells in an inner cell mass (ICM, from which pluripotent ESCs are derived: [44, 45]), pushed against the blastocoel cavity. This step is marked by an up-regulation of Nanog in ICM cells and a downregulation of Oct4 in TR cells. The second di↵erentiation event occurs when ICM cells adjacent to the blastocoel form an epithelium known as the primitive endoderm (PE), leaving the rest of the cells to the epiblast lineage. This event is accompanied by an up-regulation of Oct4 in PE cells. As shown in (Figure 2C), this leaves three phenotypes characterized by the following concentration levels of Oct4 and Nanog: a pluripotent state (PL) with high N and intermediate O and two relatively di↵erentiated states (PE, TR) with high O, highN and low O, low N, respectively. Based on experimental evidence [40, 41], the relative concentration of Oct4 is a crucial marker of whether or not a cell will be in the TR, PL, or PE lineage. More generally, Oct4 levels are crucial markers of whether or not a cell is pluripotent or di↵erentiated [42, 46, 47, 48, 49, 40, 50]. For modeling purposes, it will be crucial to keep in mind that the pluripotent state is characterized by an intermediate Oct4 concentration level.
Ordinary Di↵erential Equation Model of the Core Pluripotency Network Based on the topology of the GRN in Figure 2A, the following ordinary di↵erential equation model for the evolution of the concentrations of Oct4, Sox2, and Nanog (O,S,N respectively) over time can be formed:
⌃3D dO dt = ↵1+ a1OS + b1N2+ c1OSN2 1 + OS + N2+ OSN2 1O = H1(O, S, N ) 1O dS dt = ↵1+ a1OS + b1N2+ c1OSN2 1 + OS + N2+ OSN2 1S = H2(O, S, N ) 1S dN dt = ↵2+ a2OS + b2N2+ c2OSN2 1 + OS + N2+ OSN2+ dOm 2N = H3(O, S, N ) 2N,
where the linear terms are due to dilution and degradation, and the nonlinear terms Hi(O, S, N )(i2 1, 2, 3) are regulatory functions referred to as Hill functions [51, 52]
and model the species’ interaction with one another. In particular, the Hill functions incorporate activation by the heterodimer OS, the homodimer N2, and the molecule
OSN2. The assumptions of this model are:
• Oct4 and Sox2 act together as a heterodimer, and have the same dynamics. Given that Oct4 and Sox2 are known to work together [53] and have been considered as the same species in previous models [54], I assume that their dynamics are the same. This simplifies the analysis without a↵ecting the main conclusions.
• Nanog acts as a homodimer. In contrast to the models of [55, 56], which consider Nanog as a monomer, it is treated as a homodimer (N2) in ⌃
3D. This
is based on strong evidence suggesting Nanog only binds to other pluripotency factors when dimerized [57, 58]. Moreover, Nanog acts as an activator whether or not it is bound to the heterodimer OS, though it activates much more strongly in the latter scenario.
• Repression of Nanog by Oct4. At higher levels, Oct4 has been shown to correlate with lower Nanog levels [54]. Although no regulatory link has been demonstrated, this empirical observation is modeled using the term d · Om,
which is included as a higher order repressive term (m 2). In addition, since Oct4 and Sox2 function as a heterodimer, the order of repression is assumed to be an even number.
To study the location and stability of the steady states of ⌃3D, I will study a reduced
order system given by:
⌃2D dO dt = ↵1+ a1O2+ b1N2+ c1O2N2 1 + O2 + N2+ O2N2 1O = H1(O, S) 1O dN dt = ↵2+ a2O2+ b2N2+ c2O2N2 1 + O2+ N2+ O2N2+ dOm 2N = H2(O, N ) 2N,
which is obtained by substituting S = O in the equations of ⌃3D. This reduction
is mathematically justified in [59]. Now that a model for the endogenous pluripo-tency network has been established, in the next section I discuss the modeling of reprogramming as the perturbation of this endogenous network.
III
The iPS Reprogramming Process
Induced pluripotent stem cells (iPSCs) Although the 2006 discovery of iPSCs was a culmination of several decades of mounting evidence for the plasticity of cell phenotypes [60], it remains a watershed event in the history of cell fate reprogram-ming. And despite several technical challenges still standing in the way of bringing iPSCs to the clinic [61], there are high hopes for their use in therapeutic and regenera-tive medicine (in theory, iPSCs can be personalized from any patient’s somatic cells). The autologous transplantation that iPSCs could enable would be far superior to using ESCs from other organisms, which are hindered by ethical controversy [62, 63] and immunogenic barriers [64]. In addition to the tremendous curative potential that iPSC technologies hold [65], the ability to efficiently produce on-demand iPSC lines already has, and would continue to have, enormous benefit to basic research in disease modeling and drug discovery [66, 12, 67, 68, 69].
Timeline of molecular events during reprogramming When Yamanka and colleagues first discovered that the small cocktail of OSKM factors was sufficient to induce pluripotency in murine fibroblasts (replicated shortly thereafter in human fi-broblasts [9]), they did it using an informed trial-and-error approach that began with 24 candidate factors. Since then, several molecular and morphological events along the timeline from the fibroblast to iPSC state have been identified [70, 71, 72, 73, 74]. In these studies, many arbitrary divisions of the process into various stages and phases have been proposed, though many events are common regardless of nomenclature. Briefly, it is understood that one of the earliest steps towards successful iPSC re-programming is loss of the fibroblast signature as fibroblast specific surface markers become down-regulated, causing the cells to undergo a morphological change through a mesenchymal to epithelial transition (MET) [75]. In addition, the early phases of reprogramming see an uptick in proliferation rates [76] in the cells that progress forward in iPSC transformation. Following this initial phase, pluripotency genes gradually begin to get activated, including the embryonic marker SSEA. Moreover, DNA demethylation gradually starts increasing [77, 78]. In the late stages of repro-gramming, cells that pass on to the iPSC state overcome an epigenetic barrier and undergo complete loss of methylation and chromatin remodeling at crucial loci. Modeling TF-mediated reprogramming It is evident from these studies that the transition in cell identity from the somatic state to the iPSC state is complex and multi-faceted. At the same time it is also clear that activation of the master pluripo-tency regulators governs the entrance into pluripopluripo-tency [41, 42, 35, 39, 40, 42, 79]. Hence, activation of the pluripotency GRN can be the starting point for a first-pass description of the process. To this end, I start by treating the reprogramming process as a transition between stables steady states (SSSs) in the state space of the model for the endogenous network in ⌃2D. In particular, reprogramming from the fibroblast
to iPSC state is treated as a transition from a SSS with low concentrations of Nanog and Oct4 (as in the TR state of Figure 3B) to a SSS with intermediate levels of Oct4 and high levels of Nanog (as in the PL state shown in Figure 3B).
In Figure 4A, this reprogramming concept is depicted visually as a transition between wells in a valley landscape. In this landscape, the TR, PL, and PE phenotypes correspond to the valley-like depressions shown, and reprogramming is akin to moving a ball from a starting well to a final well by altering the landscape according to the type of transition desired. For instance, to transition from TR (characterized by the lowest relative Oct4 levels) to states with higher concentrations of Oct4, the landscape can be adjusted using overexpression values u in a manner that changes the landscape such that the TR state disappears first, forcing the ball to ‘roll’ into states characterized by higher Oct4 levels.
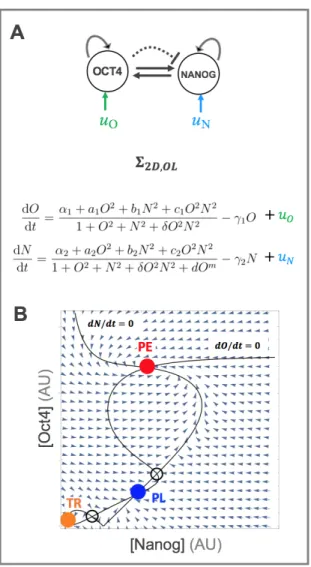
Open-Loop Reprogramming In the original Yamanaka experiments, iPS repro-gramming from fibroblasts was mediated by the ectopic overexpression of the OSKM TFs. Since then, iPS cells have been created from a wide variety of starting phe-notypes [80, 81, 82, 83]. Moreover, other cocktails have been discovered, as well as small molecule modulators of pluripotency signaling pathways and miRNA molecules, that can also induce pluripotency [84, 85, 86, 15, 87, 88, 89, 90, 91, 92, 93]. As a start, I model reprogramming as a TF-mediated process using overexpression of the TFs Oct4 and Nanog in the simplified pluripotency network of Figure 3A. By adding constant overexpression terms to the ODEs in ⌃2D, we get:
⌃2D,OL dO dt = ↵1+ a1O2+ b1N2+ c1O2N2 1 + O2+ N2+ O2N2 1O + uO dN dt = ↵2 + a2O2+ b2N2+ c2O2N2 1 + O2+ N2+ O2N2+ dOm 2N + uN
where the terms uO and uN represent ectopic overexpression of Oct4 and Nanog,
re-spectively.
In current experimental paradigms, the extent of ectopic overexpression (uO and uN)
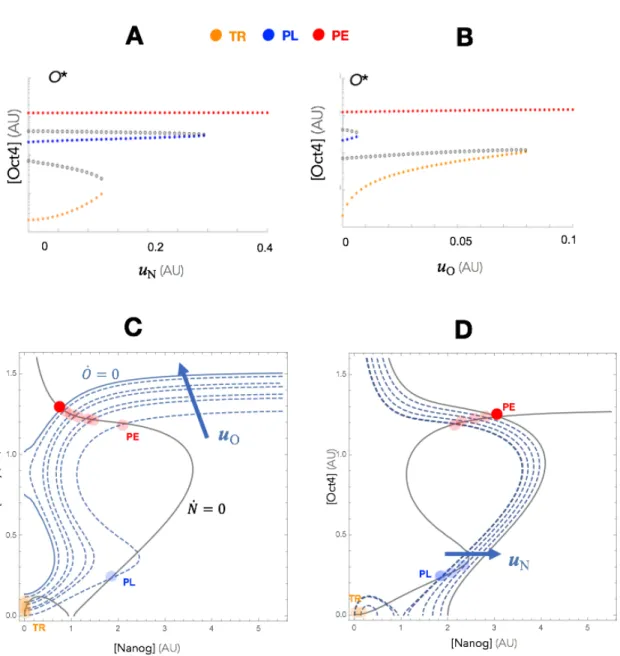
is ‘preset,’ i.e. determined at the beginning of a reprogramming experiment and not iteratively adjusted. From a control-theoretic point of view, this is a type of open-loop control (Figure 4B) since the overexpression levels are not adjusted based on measurements of the state (N, O) during the experiment. Figure 5 show the e↵ect of increasing values of uN and uO on the steady state landscape of ⌃2D. As seen in
in which, starting from the TR state, the system would be pushed to the PL state. Moreover, since the PL state disappears first as uO is increased, there are no values
of uO alone that could induce a TR ! PL transition.
This model makes potentially useful predictions about possible reprogramming strate-gies using open-loop overexpression. In particular, it indicates that there may be a fundamental flaw in these types of perturbations for certain transitions, such as the one from TR to PL. Beyond the narrow intermediate range of overexpression values that could be used to reprogram to PL, the system eventually becomes monostable at the state with maximal levels of whichever TF(s) is (are) being overexpressed (an intuitive outcome).
Perhaps more importantly, the model gives some insight into what we cannot do when it comes to its reprogrammability (i.e. it’s compatibility with the types of state transitions we may want to induce). It turns out there is actually a mathemati-cal reason why certain transitions from states characterized by lower concentrations (TR) to states characterized by higher, but not maximal, concentrations (PL) cannot be guaranteed with open-loop overexpression. It is because our GRN is predomi-nately composed of positive interactions, which means that both Oct4 and Nanog TFs are constantly upregulating themselves as well as each other. Hence, stimulat-ing these nodes artificially (via open-loop ectopic overexpression) very easily sets o↵ an upregulation positive-feedback cascade that pushes concentrations of both TFs to higher and higher levels so that the system becomes monostable at maximal (i.e. non-intermediate) values. In fact, reprogramming GRNs to intermediate states is a difficult task to achieve in an even more general class of GRNs that can be described as cooperative monotone systems [94], of which our example is a type.
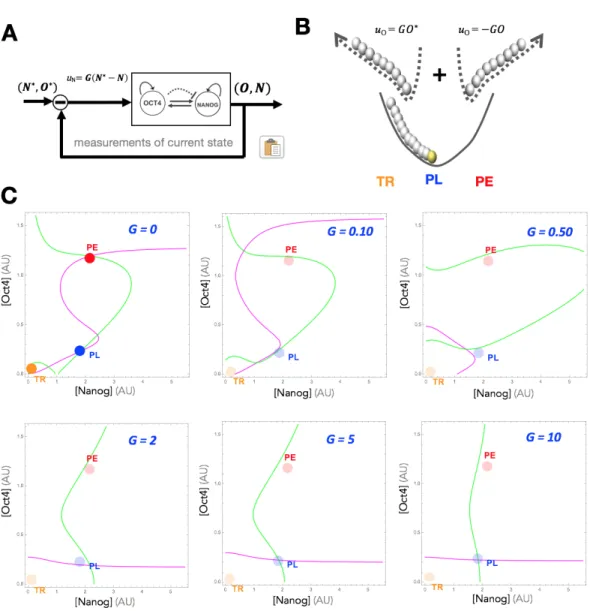
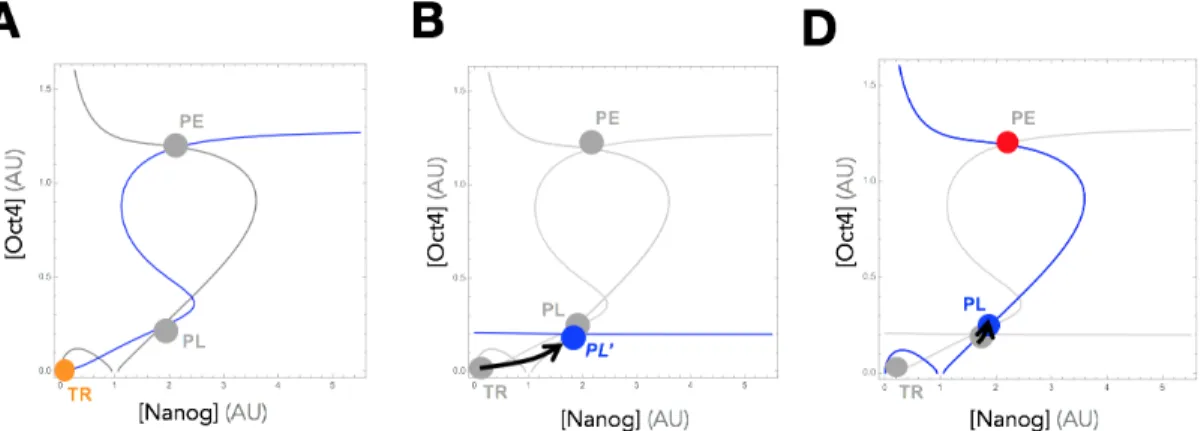
Feedback Controlled (Closed-Loop) Overexpression Based on this analysis, we see that the pluripotency GRN is in a class of GRNs that are not reprogrammable to arbitrary states using preset overexpression. To this end, we have proposed [94] a paradigm of feedback controlled overexpression (Figure 6A), in which the rate of ectopic overexpression is set multiple times throughout an experiment in propor-tion to the distance between the current state (N, O) and a target state (N⇤, O⇤):
uO = G(O⇤ O) and uN = G(N⇤ N ). Figure 6B extends our visual depiction
of reprogramming to this feedback controlled case, and shows that a feedback law on Oct4, for instance, is akin to simultaneously applying two forces that direct the system towards the maximal (+GO⇤) and minimal ( GO) states. By balancing these
two forces, reprogramming to states characterized by arbitrary concentrations can be guaranteed without dependence on the system’s dynamics or parameters. We can see this mathematically by demonstrating the e↵ect of increasing G on the nullclines of the ODE model (Figure 6C). With this new control law, our dynamics from ⌃2D
⌃2D,CL dO dt = ↵1+ a1O2+ b1N2 + c1O2N2 1 + O2+ N2+ O2N2 1O + G(O ⇤ O) dN dt = ↵2+ a2O2+ b2N2 + c2O2N2 1 + O2+ N2+ O2N2+ dOm 2N + G(N ⇤ N )
where G is the gain of the controller. As shown in the figure, for a target state (N⇤, O⇤), increasing values of G transforms the nullclines into approximately straight lines intersecting at at that exact target state. In Figure 7, a reprogramming experi-ment is simulated in which the system starts at the TR state (Fig. 7A). In Fig. 7B, feedback overexpression is applied on Oct4, making the nullcline ˙O = 0 a straight line which intersects the nullcline ˙N = 0 at a point that is in the basin of attraction of the original PL state. Following removal of the input (Figure 7C), the system autonomously converges back to the PL state, and the reprogramming experiment is complete.
Open-Loop vs. Closed-Loop Control Though the open-loop model ⌃2D,OL
made several (evidence-based) assumptions about both form and parameter values, one conclusion is clear that supersedes any choice functional form or parameter set: Open-loop control of the pluripotency GRN, which is pre-dominantly composed of positive interactions, cannot be guaranteed even at a theoretical level. And, while achieving real-world experiments will always be di↵erent and more difficult than even the best models, the fact that the model cannot even guarantee success may be a sign that the experimental paradigm may not be the best for the type of reprogramming we are trying to achieve with iPSC formation.
Realizing Feedback Overexpression using a Synthetic Genetic Feedback Controller In [94], we have proposed a blueprint for a synthetic genetic circuit that could realize the feedback overexpression paradigm presented here. A schematic of the synthetic genetic feedback controller is summarized in Figure 9. Briefly, to control the overexpression of some endogenous gene Xe, the synthetic circuit contains
both a synthetic gene and and a short-interfering RNA that targets the endogenous mRNA mX. If both of these constructs are inducible using small molecules, then an
experimenter could balance the overexpression and degradation as needed to realize the feedback control law.
Now that we have discussed the genetic transformations that take place during repro-gramming, in the next section I will review the relevant biology needed to understand some of the important epigenetic transformations that take place during iPSC repro-gramming.
IV
The Biology of Epigenetics
The models for open-loop and closed-loop control of the core pluripotency network discussed thus far have been the first step towards understanding TF-mediated re-programming from a genetic point of view. However, it is important to consider that genes inside nuclei hardly ever exist in the ‘ideal’ unwound state that is nec-essary for TFs to bind their promoter targets and for the transcriptional machinery to pass through. This is particularly noteworthy for the Oct4 and Nanog genes in the pluripotency network being reprogrammed, which need to be ‘awakened’ from a transcriptionally inactive to active state. As shown in Figure 10, DNA in most nucleated cells is wrapped around a core globular octamer of histones (made up of hi-stone protein classes H2A, H2B, H3, H4) to form nucleosomes that are connected by linker DNA and linker histones (histone class H1). This ‘beads on a string’ configura-tion is called chromatin, which can exist in a relaxed and transcripconfigura-tionally-permissive ‘euchromatin’ state or a tightly compressed and transcriptionally prohibitive ‘hete-rochromatin’ state [95, 96].
Consequently, the status of chromatin provides an additional layer of transcriptional regulation. Switching between these states, a process known as chromatin remodeling, is one of many mechanisms that fall under the umbrella of epigenetic modifications, which are heritable changes to the genome that do not involve changes to the actual base-pair sequence of genes. Epigenetics encompasses an enormous variety of mecha-nisms, including DNA methylation, histone variation, RNA interference, nucleosome remodeling, trans e↵ectors, and non-covalent ATP-dependent chromatin remodeling [95, 96]. As a first pass in modeling the epigenetic changes currently understood to take place during iPS reprogramming, I will review the definitions and mechanisms of DNA methylation and covalent PTMs of histone tails in chromatin, which have emerged as some of the most important changes during reprogramming.
Epigenetic Regulation Point: Covalent Post Translational Modifications (PTMs) at N-Terminal Histone Tails As shown in Figure 10, one important point of epigenetic regulation (i.e. points at which chromatin remodeling can oc-cur) is in the covalent post-translational modification (PTM) of amino acid residues (most commonly lysine (K), arginine (R), serine(S), and threonine(T)) at the N-terminal tails of histones in the core globular octamer within nucleosomes. PTMs include methylation, acetylation, phosphorylation, and ubiquitylation (amongst oth-ers), though methylation and acetylation have emerged as the most important and well-studied thus far. The type and location of these PTMs is specified by a histone code of the form ‘H[C][AA][#][PTM]’ where
• C is the class of histone e.g. H3, H4.
• # is the location on the peptide of the amino acid reside in question.
• PTM is the actual PTM e.g. a triple methylation is represented as ’Me3’, acetylation is represented by ’Ac,’ etc.
Specific histone codes (i.e. specific PTMs at specific residues on specific tails) induce the euchromatin or heterochromatin configuration through a variety of mechanisms that often varies by the histone code in question. For instance, ‘H3K4Me3’ typically gives rise to the euchromatic state while ‘H3K9Me2/3’ typically gives rise to the hete-rochromatic state via recruitment of heterochromatin protein 1 (HP1) [97, 95]. There are several databases that list the observed functions of known PTMs ([98, 95, 99]) and the list is constantly growing as new PTMs are identified and their e↵ects deciphered.
Histone Writers & Erasers
The molecular machinery which is responsible for adding and removing these PTMs are known as histone writers and erasers, respectively. Briefly:
• Writers: depending on the type of PTM, specific classes of enzymes ‘write‘ the covalent modification onto specific residues. There are vast numbers of such writers (see Chapter 3 in [95]). For acetyl and methyl groups, the writers are histone acetylases (HATs) and histone methyltransferases (HMTs), respectively. • Erasers: these classes of enzymes ‘remove’ the covalent PTM in question. For acetyl and methyl groups, the respective erasers are histone deacetylases (HDACs) and histone demethylases (HDMs).
Epigenetic Regulation Point: Cytosine Methylation As depicted in Figure 10, another important epigenetic regulation point is the methylation of cytosine bases at and around the promoters of genes. In general, methylation induces the heterochro-matin state, making it a marker of transcriptional silencing [95]. In Figure 12, this process is explained at the chemical, biochemical, and transcriptional levels. I briefly review these perspectives, as an understanding of them will be crucial to construction of an iPS reprogramming model that includes the aspect of (de)methylation.
The Chemistry of Cytosine Methylation
Cytosine Variants and CpG Sites: At the root of the complex transcriptional e↵ects that methylation gives rise to are very simple organic modifications to cytosine, one of the four base pairs of DNA. As shown in the top panel of Figure 12, C is a pyrimi-dine derivative that can be modified with a variety of organic functional groups at its fifth carbon position. It is currently understood that methylation of cytosine to form
methylated-cytosine is one of the most important such modifications. Since methy-lation only occurs at ‘CpG sites,’ which are adjacent cytosine and guanine base pairs in the 5‘ to 3‘ direction (the p refers to the phosphate connecting these bases in the backbone of the DNA strand), this form of cytosine is typically denoted as 5mC. As shown in the figure, a vast majority of CpG sites are methylated in mammals [100], and when 5mC is found in the vicinity of promoters, it typically has a repressive e↵ect on those genes.
CpG Islands: A related notion is that of ‘CpG Islands,’ which are roughly 1 kb length CpG-rich regions of DNA (the definitions of ‘CpG’-rich varies, but typically means > 50% CpG content [100, 101, 102]) that are (perhaps counter-intuitively) not usually methylated. Since these islands are found at virtually all constitutively-expressed promoters and only 40% of tissue-suppressed genes, it appears that their methyla-tion status is related to the expressiveness of the genes in their proximity. Moreover, CpG islands overlap with the promoters of 60-70% genes in humans, underscoring the prevalence of methylation-based control of transcription.
The Biochemistry of Cytosine (de)Methylation
In mammals, three di↵erent types of DNA methyltransferases (DNMTs) are predom-inantly responsible for establishing methylation patterns ‘from scratch’ as well as replicating them across cell division cycles.
De-Novo Methylation In early development, DNMT3A and DNMT3B are involved in establishing 5mC as various genes in the embryonic genome are silenced [95]. Maintenance Methylation When DNA is being replicated during cell division, the maintenance methylation enzyme DNMT1 is tasked with replicating the methylation patterns on the cytosine bases being copied through semi-conservative replication. A key part of this maintenance methylation process is that DNMT1’s preferred sub-strate is hemi-methylated double-stranded DNA. Hemi-methylated means only one strand in a double strand is methylated, which is the state of DNA following com-plementary transcription before methylation patterns of have been copied (the term hemi-methylated should not be confused with hydroxy-methylated cytosine, a com-pletely di↵erent covalent modification).
Active & Passive Demethylation While the mechanisms for methylation of cy-tosine bases are fairly straightforward and enzymatically driven, the mechanisms for demethylation are a bit more complex. The current consensus points to de-methylation beginning enzymatically with a family of 5mC oxidases known as the TET proteins. TET enzymes can convert 5mC into oxidized versions that include 5-hydroxymethyl-cytosine (5hmC) [103], 5-formylcytosine (5fC), and 5-carboxylcytosine.
For our purposes and without loss of generality later on in modeling, we can consider only 5 hmC and use it as a proxy for all ‘oxidized-but-not-fully demethylated’ versions of cytosine.
The transcriptional e↵ect of 5 hmC is not entirely clear. For instance in [104], one of these intermediates, 5-hydroxymethylcytosine (5-hmC) is shown to be present in both transcriptionally active and repressed promoters. In any case, the most relevant piece here is that it is not de-methylated, which is necessary for full activation of a gene (and hence relevant for reprogramming). It turns out that nature has a clever way of fully demethylating 5-hmC anyways through dilution. At the root of this process is the DNMT1/UHRF1 and 5 hmC recognition problem (UHRF1 is a cofactor of DNMT1). Molecular evidence shows that 5 hmC is not a good substrate of DNMT1 [105, 106, 107, 108]. As a result, once 5mC has been oxidized to 5-hmC, methylation patterns cease to be replicated and eventually disappear at the speed of cell dilution through division. This notion of ‘passive reprogramming’ will be at the core of a model I provide in the next section.
The Transcriptional Output of Cytosine Methylation
At the end of the day, these covalent modifications of cytosine bases are relevant be-cause they can determine the transcriptional status of a gene. As introduced above, un-methylated promoters are a transcriptionally permissive state while methylated promoters are typically transcriptionally prohibitive. As shown in Figure 12, tran-scriptional silencing by methylation is induced in two ways. First, proteins with methyl-binding domains (MBDs) recognize the methylated promoters and recruit co-repressors that eventually give rise to a heterochromatic state, inactivating the locus [109]. Second, when 5mC is found in the major groove of the DNA helix, it physically impedes TF binding [110, 111, 112]. In any case, the end result is that this simple methylation of cytosine changes the transcriptional status of a locus.
In the next section, I use these principles to discuss the relevance of DNA demethy-lation to reactivation of the core pluripotency network.
V
Epigenetics and iPSC Reprogramming
Since iPSC reprogramming was discovered over a decade ago, many studies have showed the importance of both genetic and epigenetic transformations during the process [113, 114, 115, 69, 116, 117]. In the pluripotent ES state to which reprogram-ming is reverting, the promoter regions of Oct4 and Nanog are demethylated (along with several other pluripotency genes), in contrast to the methylated promoters of the fibroblast and other somatic states [118, 119, 25, 7, 4]. During di↵erentiation, Oct4 and Nanog undergo silencing via methylation [120, 70, 121] that very strongly represseses their activation. For instance, in di↵erentiating ES cells it has been shown
[122, 123, 124] that histone deacetylases and the enzyme Euchromatic histone-lysine N-methyltransferase 2 (also known as G9a) become active and also lead to the de novo methylation of the Oct4 promoter by DnmtA/B. Oct4 in particular is completely si-lenced via H3K9 methylation, Hp1 binding, and DNA methylation [125]
These are very powerful repression mechanisms, which makes TF-mediated repro-gramming an epigenetic challenge as much as it is a genetic challenge. Reversal of these methylation states of the promoter regions of the core pluripotency network seems to be an essential pre-requisite for properly activating the network [70, 78]. In fact, incomplete demethylation of Oct4 and Nanog promoter regions is indeed one of the characteristics of only ‘partially’ reprogrammed iPS cells [6, 126, 127], along with incomplete methylation of somatic genes that persist in some iPS cells [128, 129]. In this section, I construct a model that probes the issue of the DNA demethylation barrier to reprogramming. As introduced in the previous section (Figure 12, mid-dle panel), demethylation is understood to be an active and passive process [130]. The former step is enzymatically-driven by TET enzymes, which convert methylated cytosine bases (5mC) to oxidized forms (5hmC, 5fmC, etc.). Since these oxidized ver-sions of cytosine are not well-recognized by the maintenance methylation machinery (DNMT1 and its cofactor UHRF1), they only transiently exist before disappearing in the next replication cycle. This eventually gives way to fully de-methylated cytosine, which is the state conducive to full reactivation of Oct4 and Nanog. The interaction between the core pluripotency network and the de-methylation machinery has been modeled in [131], though that model did not divide de-methylation into active and passive steps.
Connecting the Demethylation & Fast Cycling Barriers As introduced in the brief review of the phases of reprogramming in Section III, one of the early events in the timeline of iPSC formation is an increase in proliferation rate of the cells being reprogrammed. This fact alone may not seem significant, but becomes very insightful when coupled with knowledge of how demethylation is occurring during reprogram-ming. If the rate-limiting step of demethylation is the passive loss of oxidized forms of methylated cytosine across replication cycles, could it be that the demethylation barrier is just a cell proliferation barrier? In, [134], it is suggested that ‘the low efficiency and time necessary for creating iPSCs are consistent with a passive DNA demethylation model.’
Experimental results from several other studies point to this hypothesis as well. In [135], it was shown that activation of the Ink4/Arf locus, which encodes three anti-proliferative tumor suppressors (p16Ink4a, p15Ink4b, Arf), is a major barrier to re-programming. This paper showed that the locus is completely silenced in iPSCs and ESCs (i.e. cell cycling is not being slowed down by tumor suppressors), while it is
reactivated upon di↵erentiation. More importantly, the study also showed that three of the TFs in the original Yamanaka cocktail (Oct4, Klf, and Sox2) repress the locus upon their own reactivation during reprogramming, pointing to a connection between ectopic overexpression and overcoming the supposed demethylation/proliferation bar-rier. The positive link between proliferation rate and iPSC formation has been argued for in other studies as well [76, 136, 137, 138], though it’s worth noting that it has also been challenged as well [139, 140].
Interaction of TFs with Demethylation Machinery To begin studying the association between TF-mediated reprogramming and the cell-cycling barrier, we can use information from several studies showing a mechanistic link between Oct4, Nanog and the demethylation machinery. In [141], strong evidence hinted at the regulation of Tet1 and Tet2 mRNA by Oct4 and Sox2 while in [142] it was shown that Oct4 alone is necessary for Tet2 transcription. Moreover, it has been shown that Tet1 can replace Oct4 in reprogramming [144], as it demethylates Oct4 regulatory regions. In addition, TET has been shown to be indispensable for the MET step during iPS formation [145].
Based on these studies, the e↵ect of demethylation on reactivating the pluripotency network can begin to be understood by comparing the two motifs in Figure 13. Motif #1 shows an even further simplified version of the pluripotency network in which Oct4 only auto-activates itself. This is also true in Motif #2, which additionally includes both active and passive demethylation as well as transcriptional level activation of TET by Oct4. A biochemical reaction model of these motifs can be constructed, as shown in Table 1. From these reactions, ODE models for Motifs 1 and 2 can be constructed:
⌃5D (Motif 1, auto-activation without methylation)
˙ O2 = aoO2 doO2+ r 1Do r1DO2 O2 ˙ O = m ( O+ )O aoO2 + doO2 ˙ D = r 1Do r1DO2 ˙ DO= r 1Do+ r1DO2 ˙ m = ↵0D + ↵1Do ( m+ )m + u
Biochemical Process Mechanistic Representation
OCT4 Dimerization O + O ao
do O2
OCT4 Auto-activation O2+ D rr11 DO
OCT4 Transcription (leaky) D ↵o
! m + D OCT4 Transcription (activ.) DO
↵1
! m + DO
OCT4 Translation m! m + O
OCT4 Dilution O! , O !
TET/DNMT Dilution DNMT ! , TET ! , C1 ! , C2 !
mRNA/Protein Degradation m m ! , O O ! TET/DNMT Degradation DNMT D ! , TET T ! De-novo Methylation D + DNMT a1 d1 C1 k1 ! ¯D + DNMT Active de-Methylation D + TET¯ a2
d2 C2
k2
! Dh+ TET
Passive De-Methylation Dh ! D
DNMT Constitutive Transcription ! DNMTD TET Transcription/Translation (leaky) DT
o
! DT+ TET
TET Transcription/Translation (activ.) DTO
1
! DTO+ TET
TET Translation DT
o
! DT+ TET
OCT4 Activating TET DT+ O2 rr22 DTO
Ectopic Overexpression !u Om
Table 1: Biochemical Reaction Models for Motif 1 and Motif 2 Species: O = Oct4 Protein, O2 = Oct4 Protein Dimer, m = mRNA of Oct4, D = non-methylated promoter
of Oct4,Dh = hydroxymethylated promoter of Oct4, ¯D = methylated promoter of Oct4, DO
= DNA promoter of Oct4 auto-activated by the dimer O2, DNMT = methylation enzyme,
TET = de-methylation enzyme, DT = TET promoter, DTO = TET promoter activated by
˙ O2 = taoO2 doO2+ r 1Do r1DO2 O2 r2pTO2+ r 2pT O ˙ O = m ( O+ )⇤ O aoO2+ doO2 ˙ D = r 1Do r1DO2 a1DN M T D + d1C1+ Dh ˙¯ D = a2T ET ¯D + d2C2+ k1C1 ˙ DO= r 1Do+ r1DO2 ˙ Dh = k2C2 Dh ˙ DN M T = a1DN M T D + (d1+ k1)C1 ( D+ )DN M T + D ˙ T ET = a2⇤ T ET ⇤ ¯D + (d2 + k2)C2 ( T + )T ET + 0pT + 1DT O ˙ C1 = a1DN M T · D (k1+ d1+ )C1 ˙ C2 = a2T ET ¯D (k2+ d2+ )C2 ˙ m = ↵0D + ↵1Do ( m+ )m + u ˙ DT = r 2DT O r2DTO2 ˙ DT O= r 2DT O+ r2DTO2
In Figure 14, the time courses of activated Oct4 promoter (DO) and Oct4
concen-tration (O) after preset overexpression are shown for increasing proliferation rates (decreasing doubling time). In these simulations, Motif #2 is beginning in a state where Oct4’s promoter is completely methylated, and therefore active and passive demethylation (at the rate of cell proliferation) must first take place. As the left plots show, Motif #2 has a slower accrual of activated DNA. This in turn translates to slower time courses for Oct4’s activation, a proxy for pluripotency. Another impor-tant observation is that although increased proliferation rates diminishes the temporal delay, it also reduces the level of Oct4 that can be reached. This suggests that even if cell proliferation were artificially stimulated, there is a fundamental tradeo↵ between diminishing the temporal delay caused by methylation and the steady state level that will be realized.
Wider TF Involvement With the Epigenetic Machinery The model pre-sented above is only a first pass at understanding a much larger interaction network between TFs and the epigenetic machinery during reprogramming. Oct4 in partic-ular is at the heart of many mechanisms that restore the epigenetic state of TFs [93, 146, 147, 148, 149, 150, 151]. Moreover, Oct4, Klf4 and Sox2 have been described as ‘pioneer factors’ that initiate reprogramming by uncoiling chromatin and recruiting the activating and transcriptional machinery [152, 153, 154]. In [155], it was shown that Tet1 binds with Nanog to the Oct4 locus, a relation that may be included in future methylation models involving other TFs in the core network.
There are a few notable interactions between Oct4 and chromatin remodeling factors, as depicted in Figure 15. In particular, it has been shown experimentally that H3K9 methylation is a barrier to iPSC formation [156]. G9a is a writer of methylation markers while the H3K9 demethylase genes Jmjd1a and Jmjd2c (‘erasers’) remove this marker. In mouse ESCs, it has been shown [157] that Oct4 activates Jmjd1a and Jmjd2c genes, and that JmJd2c demethylates the Nanog locus. In addition, Oct4 has been linked to the H3K4 methylation writer Wdr5 [?], which is necessary for reprogramming and maintenance of the pluripotent state. These are all interactions that may also be considered in future epigenetic models of reprogramming.
VI
Conclusion
In this thesis, I have introduced a computational model for iPSC reprogramming that is centered around the core pluripotency gene regulatory network. Since their remarkable discovery in 2006, iPSCs have been generated from a variety of somatic cell types and with a variety of reprogramming factor variants. However, the full potential of iPSCs in both clinical and basic research applications has been hindered by the economics and low efficiencies of the process, even over a decade later.
The findings from this work indicate that current reprogramming strategies (open-loop overexpression), in which overexpression levels are ‘preset’ once at the beginning of an experiment and not adjusted throughout the reprogramming experiment, do not guarantee reprogrammability of the type being attempted in iPSC experiments. Fundamentally, this is due to the topology of the core pluripotency GRN, which is composed largely of positive (activating) interactions that make it easy for the sys-tem to cascade into higher and higher concentrations of the TFs involved. Hence, only reprogramming to the state (phenotype) characterized by the maximal concen-trations of TFs can be guaranteed with open-loop overexpression. This makes it an erroneous strategy for iPSC reprogramming, where the final state is characterized by an intermediate level of Oct4 concentrations. Too little overexpression and too much overexpression are both no good; there is a very fine ‘sweet spot’ of Oct4 concen-trations that must be achieved and maintained for pluripotency. In short, current
reprogramming strategies are not even theoretically guaranteed, which suggests a po-tential reason for the low efficiencies and high failure rates of reprogramming still seen in experiments.
As a potential improvement to these strategies, I have presented and summarized a proposal for a new paradigm of ‘closed-loop’ reprogramming in which the concen-tration of TFs is adjusted multiple times throughout the experiment in proportion to the distance between the target state (phenotype) and its current phenotype. In theory, this reprogramming strategy guarantees reprogrammability to arbitrary con-centration levels regardless of network architecture or parameters, including those characterizing the iPSC state.
Finally, I have also presented a model for iPSC reprogramming that takes into account the fact that DNA methylation must first take place on the loci of the genes being reactivated. This model is based on data about the methylation status of Oct4’s pro-moter region before and after reprogramming, and mechanistically takes into account the sequence of active and passive demethylation that takes place during reprogram-ming. This model demonstrates that increased proliferation rates do indeed speed up the reactivation, but at the price of reducing the steady state levels that can be achieved by open loop overexpression.
References
[1] H. Spemann, Embryonic development and induction. New Haven: Yale Univer-sity press; London, H. Milford, Oxford UniverUniver-sity Press, 1938.
[2] R. Briggs and T. J. King, “Transplantation of living nuclei from blastula cells into enucleated frogs’ eggs,” Proceedings of the National Academy of Sciences of the United States of America, vol. 38, pp. 455–463, 05 1952.
[3] J. B. Gurdon and J. A. Byrne, “The first half-century of nuclear transplanta-tion,” Proceedings of the National Academy of Sciences of the United States of America, vol. 100, pp. 8048–8052, 07 2003.
[4] K. Takahashi and S. Yamanaka, “Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors,” Cell, vol. 126, no. 4, pp. 663–676, 2006.
[5] N. Maherali, R. Sridharan, W. Xie, J. Utikal, S. Eminli, K. Arnold, M. Stadt-feld, R. Yachechko, J. Tchieu, R. Jaenisch, K. Plath, and K. Hochedlinger, “Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution,” Cell Stem Cell, vol. 1, no. 1, pp. 55–70, 2007. [6] T. S. Mikkelsen, J. Hanna, X. Zhang, M. Ku, M. Wernig, P. Schorderet, B. E. Bernstein, R. Jaenisch, E. S. Lander, and A. Meissner, “Dissecting direct re-programming through integrative genomic analysis,” Nature, vol. 454, pp. 49 EP –, 05 2008.
[7] K. Okita, T. Ichisaka, and S. Yamanaka, “Generation of germline-competent induced pluripotent stem cells,” Nature, vol. 448, pp. 313 EP –, 06 2007. [8] M. Wernig, A. Meissner, R. Foreman, T. Brambrink, M. Ku, K. Hochedlinger,
B. E. Bernstein, and R. Jaenisch, “In vitro reprogramming of fibroblasts into a pluripotent es-cell-like state,” Nature, vol. 448, pp. 318 EP –, 06 2007.
[9] K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda, and S. Yamanaka, “Induction of pluripotent stem cells from adult human fibroblasts by defined factors,” Cell, vol. 131, no. 5, pp. 861–872, 2007.
[10] F. Gonz´alez, S. Bou´e, and J. C. I. Belmonte, “Methods for making induced pluripotent stem cells: reprogramming `ala carte,” Nature Reviews Genetics, vol. 12, pp. 231 EP –, 02 2011.
[11] S. Chari and S. Mao, “Timeline: ipscs—the first decade,” Cell, vol. 164, no. 3, p. 580, 2016.
[12] M. Stadtfeld and K. Hochedlinger, “Induced pluripotency: history, mechanisms, and applications,” Genes & Development, vol. 24, pp. 2239–2263, 10 2010. [13] K. Plath and W. E. Lowry, “Progress in understanding reprogramming to the
induced pluripotent state,” Nature Reviews. Genetics, vol. 12, pp. 253–265, 04 2011.
[14] L. David and J. M. Polo, “Phases of reprogramming,” Stem Cell Research, vol. 12, no. 3, pp. 754–761, 2014.
[15] N. Malik and M. S. Rao, “A review of the methods for human ipsc derivation,” Methods in molecular biology (Clifton, N.J.), vol. 997, pp. 23–33, 2013.
[16] T. M. Schlaeger and Daheron, “A comparison of non-integrating reprogramming methods,” Nat Biotech, vol. 33, no. 1, pp. 58–63, 2015.
[17] P. A. Goh, S. Caxaria, and Casper, “A systematic evaluation of integration free reprogramming methods for deriving clinically relevant patient specific induced pluripotent stem (ips) cells,” PLoS ONE, vol. 8, no. 11, 2013.
[18] “ips cells 10 years later,” Cell, vol. 166, no. 6, pp. 1356–1359, 2016.
[19] C. Jopling, S. Boue, and J. C. I. Belmonte, “Dedi↵erentiation, transdi↵erentia-tion and reprogramming: three routes to regeneratransdi↵erentia-tion,” Nature Reviews Molec-ular Cell Biology, vol. 12, pp. 79 EP –, 01 2011.
[20] C. H. Waddington, The strategy of the genes; a discussion of some aspects of theoretical biology. London: Allen & Unwin, 1957.
[21] S. Mitalipov and D. Wolf, Totipotency, Pluripotency and Nuclear Reprogram-ming, pp. 185–199. Berlin, Heidelberg: Springer Berlin Heidelberg, 2009. [22] A. G. Smith, “Embryo-derived stem cells: Of mice and men,” Annual Review
of Cell and Developmental Biology, vol. 17, pp. 435–462, 2018/02/03 2001. [23] L. A. Boyer, D. Mathur, and R. Jaenisch, “Molecular control of pluripotency,”
Current Opinion in Genetics & Development, vol. 16, no. 5, pp. 455–462, 2006. [24] H. Niwa, “How is pluripotency determined and maintained?,” Development,
vol. 134, no. 4, pp. 635–646, 2007.
[25] K. Mitsui, Y. Tokuzawa, H. Itoh, K. Segawa, M. Murakami, K. Takahashi, M. Maruyama, M. Maeda, and S. Yamanaka, “The homeoprotein nanog is required for maintenance of pluripotency in mouse epiblast and es cells,” Cell, vol. 113, no. 5, pp. 631–642, 2003.
[26] L. A. Boyer, T. I. Lee, M. F. Cole, S. E. Johnstone, S. S. Levine, J. P. Zucker, M. G. Guenther, R. M. Kumar, H. L. Murray, R. G. Jenner, D. K. Gi↵ord, D. A. Melton, R. Jaenisch, and R. A. Young, “Core transcriptional regulatory circuitry in human embryonic stem cells,” Cell, vol. 122, no. 6, pp. 947–956, 2005.
[27] J. Kim, J. Chu, X. Shen, J. Wang, and S. H. Orkin, “An extended transcrip-tional network for pluripotency of embryonic stem cells,” Cell, vol. 132, no. 6, pp. 1049–1061.
[28] Y.-H. Loh, Q. Wu, J.-L. Chew, V. B. Vega, W. Zhang, X. Chen, G. Bourque, J. George, B. Leong, J. Liu, K.-Y. Wong, K. W. Sung, C. W. H. Lee, X.-D. Zhao, K.-P. Chiu, L. Lipovich, V. A. Kuznetsov, P. Robson, L. W. Stanton, C.-L. Wei, Y. Ruan, B. Lim, and H.-H. Ng, “The oct4 and nanog transcription network regulates pluripotency in mouse embryonic stem cells,” Nat Genet, vol. 38, no. 4, pp. 431–440, 2006.
[29] I. Chambers, D. Colby, M. Robertson, J. Nichols, S. Lee, S. Tweedie, and A. Smith, “Functional expression cloning of nanog, a pluripotency sustaining factor in embryonic stem cells,” Cell, vol. 113, no. 5, pp. 643–655, 2003.
[30] C. Hadjimichael, K. Chanoumidou, N. Papadopoulou, P. Arampatzi, J. Papa-matheakis, and A. Kretsovali, “Common stemness regulators of embryonic and cancer stem cells,” World Journal of Stem Cells, vol. 7, pp. 1150–1184, 10 2015. [31] P. C. Faucon, K. Pardee, R. M. Kumar, H. Li, Y.-H. Loh, and X. Wang, “Gene networks of fully connected triads with complete auto-activation enable multi-stability and stepwise stochastic transitions,” PLoS ONE, vol. 9, no. 7, 2014. [32] S. A. Kau↵man, “Control circuits for determination and transdetermination,”
Science, vol. 181, p. 310, 07 1973.
[33] S. Huang, G. Eichler, Y. Bar-Yam, and D. E. Ingber, “Cell fates as high-dimensional attractor states of a complex gene regulatory network,” Physical Review Letters, vol. 94, pp. 128701–, 04 2005.
[34] S. Huang, “Reprogramming cell fates: reconciling rarity with robustness,” BioEssays, vol. 31, no. 5, pp. 546–560, 2009.
[35] H. Niwa, Y. Toyooka, D. Shimosato, D. Strumpf, K. Takahashi, R. Yagi, and J. Rossant, “Interaction between oct3/4 and cdx2 determines trophectoderm di↵erentiation,” Cell, vol. 123, no. 5, pp. 917–929, 2005.
[36] B. Zhang and P. G. Wolynes, “Stem cell di↵erentiation as a many-body prob-lem,” Proceedings of the National Academy of Sciences, vol. 111, pp. 10185– 10190, 07 2014.
[37] S. H. Orkin, J. Wang, J. Kim, J. Chu, S. Rao, T. W. Theunissen, X. Shen, and D. N. Levasseur, “The transcriptional network controlling pluripotency in es cells,” Cold Spring Harbor Symposia on Quantitative Biology, vol. 73, pp. 195– 202, 01 2008.
[38] E. Bieberich, G. Wang, D. Bhartiya, and N. Lenka, Molecular Mechanisms Underlying Pluripotency, p. Ch. 08. Rijeka: InTech, 2018-01-24 2013-08-28. [39] J. M. Velkey and K. S. O’Shea, “Oct4 rna interference induces trophectoderm
di↵erentiation in mouse embryonic stem cells,” genesis, vol. 37, no. 1, pp. 18–24, 2003.
[40] V. Karwacki-Neisius, J. G¨oke, R. Osorno, F. Halbritter, J. H. Ng, A. Y. Weiße, F. C. K. Wong, A. Gagliardi, N. P. Mullin, N. Festuccia, D. Colby, S. R. Tomlinson, H.-H. Ng, and I. Chambers, “Reduced oct4 expression directs a robust pluripotent state with distinct signaling activity and increased enhancer occupancy by oct4 and nanog,” Cell Stem Cell, vol. 12, no. 5, pp. 531–545, 2013.
[41] H. Niwa, J.-i. Miyazaki, and A. G. Smith, “Quantitative expression of oct-3/4 defines di↵erentiation, dedi↵erentiation or self-renewal of es cells,” Nat Genet, vol. 24, no. 4, pp. 372–376, 2000.
[42] A. Radzisheuskaya, G. Le Bin Chia, R. L. dos Santos, T. W. Theunissen, L. F. C. Castro, J. Nichols, and J. R. Silva, “A defined oct4 level governs cell state transitions of pluripotency entry and di↵erentiation into all embryonic lineages,” Nat Cell Biol, vol. 15, no. 6, pp. 579–590, 2013.
[43] I. Chambers, J. Silva, D. Colby, J. Nichols, B. Nijmeijer, M. Robertson, J. Vrana, K. Jones, L. Grotewold, and A. Smith, “Nanog safeguards pluripo-tency and mediates germline development,” Nature, vol. 450, no. 7173, pp. 1230–1234, 2007.
[44] M. J. Evans and M. H. Kaufman, “Establishment in culture of pluripotential cells from mouse embryos,” Nature, vol. 292, no. 5819, pp. 154–156, 1981. [45] G. R. Martin, “Isolation of a pluripotent cell line from early mouse embryos
cultured in medium conditioned by teratocarcinoma stem cells.,” Proceedings of the National Academy of Sciences of the United States of America, vol. 78, no. 12, pp. 7634–7638, 1981.
[46] G. Shi and Y. Jin, “Role of oct4 in maintaining and regaining stem cell pluripo-tency,” Stem Cell Research & Therapy, vol. 1, no. 5, p. 39, 2010.
[47] G. Zafarana, S. R. Avery, K. Avery, H. D. Moore, and P. W. Andrews, “Specific knockdown of oct4 in human embryonic stem cells by inducible short hairpin rna interference,” STEM CELLS, vol. 27, no. 4, pp. 776–782, 2009.
[48] L. Li, L. Sun, F. Gao, J. Jiang, Y. Yang, C. Li, J. Gu, Z. Wei, A. Yang, R. Lu, Y. Ma, F. Tang, S. Won Kwon, Y. Zhao, J. Li, and Y. Jin, “Stk40 links the pluripotency factor oct4 to the erk/mapk pathway and controls extraembryonic endoderm di↵erentiation,” Proceedings of the National Academy of Sciences, vol. 107, pp. 1402–1407, 01 2010.
[49] D. Zeineddine, E. Papadimou, K. Chebli, M. Gineste, J. Liu, C. Grey, S. Thurig, A. Behfar, V. A. Wallace, I. S. Skerjanc, and M. Puc´eat, “Oct-3/4 dose de-pendently regulates specification of embryonic stem cells toward a cardiac lin-eage and early heart development,” Developmental Cell, vol. 11, pp. 535–546, 2018/01/24.
[50] D. Esch, J. Vahokoski, M. R. Groves, V. Pogenberg, V. Cojocaru, H. vom Bruch, D. Han, H. C. A. Drexler, M. J. Ara´uzo-Bravo, C. K. L. Ng, R. Jauch, M. Wilmanns, and H. R. Sch¨oler, “A unique oct4 interface is crucial for re-programming to pluripotency,” Nature Cell Biology, vol. 15, pp. 295 EP –, 02 2013.
[51] D. Del Vecchio and R. M. Murray, Biomolecular Feedback Systems. Princeton UP, 2014.
[52] M. Santill´an, “On the use of the hill functions in mathematical models of gene regulatory networks,” vol. 3, no. 2, pp. 85–97.
[53] J.-L. Chew, Y.-H. Loh, W. Zhang, X. Chen, W.-L. Tam, L.-S. Yeap, P. Li, Y.-S. Ang, B. Lim, P. Robson, and H.-H. Ng, “Reciprocal transcriptional regulation of pou5f1 and sox2 via the oct4/sox2 complex in embryonic stem cells,” Molecular and Cellular Biology, vol. 25, no. 14, pp. 6031–6046, 2005.
[54] T. Kalmar, C. Lim, P. Hayward, S. Mu˜noz-Descalzo, J. Nichols, J. Garcia-Ojalvo, and A. Martinez Arias, “Regulated fluctuations in nanog expression mediate cell fate decisions in embryonic stem cells,” PLoS Biol, vol. 7, no. 7, 2009.
[55] V. Chickarmane, C. Troein, U. A. Nuber, H. M. Sauro, and C. Peterson, “Tran-scriptional dynamics of the embryonic stem cell switch,” PLoS Comput Biol, vol. 2, no. 9, 2006.
[56] V. Chickarmane and C. Peterson, “A computational model for understanding stem cell, trophectoderm and endoderm lineage determination,” PLoS ONE, vol. 3, no. 10, 2008.
[57] J. Wang, D. N. Levasseur, and S. H. Orkin, “Requirement of nanog dimeriza-tion for stem cell self-renewal and pluripotency,” Proceedings of the Nadimeriza-tional Academy of Sciences, vol. 105, no. 17, pp. 6326–6331, 2008.
[58] N. P. Mullin, A. Yates, A. J. Rowe, B. Nijmeijer, D. Colby, P. N. Barlow, M. D. Walkinshaw, and I. Chambers, “The pluripotency rheostat nanog functions as a dimer,” Biochemical Journal, vol. 411, no. 2, pp. 227–231, 2008.
[59] H. Abdallah, Y. Qian, and D. Del Vecchio, “A dynamical model for the low efficiency of induced pluripotent stem cell reprogramming,” in Submitted to American Control Conference, 2015.
[60] V. K. Singh, M. Kalsan, N. Kumar, A. Saini, and R. Chandra, “Induced pluripo-tent stem cells: applications in regenerative medicine, disease modeling, and drug discovery,” Frontiers in Cell and Developmental Biology, vol. 3, p. 2, 2015. [61] N. Tapia and H. R. Sch¨oler, “Molecular obstacles to clinical translation of ipscs,”
Cell Stem Cell, vol. 19, pp. 298–309, 2018/01/23.
[62] G. d. Wert and C. Mummery, “Human embryonic stem cells: research, ethics and policy,” Human Reproduction, vol. 18, pp. 672–682, 04 2003.
[63] B. Lo and L. Parham, “Ethical issues in stem cell research,” Endocrine Reviews, vol. 30, pp. 204–213, 05 2009.
[64] O. Preynat-Seauve, K.-H. Krause, and J. Villard, The Immune Barriers of Cell Therapy with Allogenic Stem Cells of Embryonic Origin, pp. 181–197. Berlin, Heidelberg: Springer Berlin Heidelberg, 2011.
[65] X. Lu and T. Zhao, “Clinical therapy using ipscs: Hopes and challenges,” Ge-nomics, Proteomics & Bioinformatics, vol. 11, no. 5, pp. 294–298, 2013. [66] Y. S. Chun, P. Chaudhari, and Y.-Y. Jang, “Applications of patient-specific
induced pluripotent stem cells; focused on disease modeling, drug screening and therapeutic potentials for liver disease,” International Journal of Biological Sciences, vol. 6, no. 7, pp. 796–805, 2010.
[67] S.-i. Nishikawa, R. A. Goldstein, and C. R. Nierras, “The promise of human induced pluripotent stem cells for research and therapy,” Nature Reviews Molec-ular Cell Biology, vol. 9, pp. 725 EP –, 08 2008.
[68] J. T. Dimos, K. T. Rodolfa, K. K. Niakan, L. M. Weisenthal, H. Mitsumoto, W. Chung, G. F. Croft, G. Saphier, R. Leibel, R. Goland, H. Wichterle, C. E. Henderson, and K. Eggan, “Induced pluripotent stem cells generated from pa-tients with als can be di↵erentiated into motor neurons,” Science, vol. 321, p. 1218, 08 2008.
[69] K. Hochedlinger and K. Plath, “Epigenetic reprogramming and induced pluripo-tency,” Development, vol. 136, p. 509, 02 2009.
[70] J. M. Polo, E. Anderssen, R. M. Walsh, B. A. Schwarz, C. M. Nefzger, S. M. Lim, M. Borkent, E. Apostolou, S. Alaei, J. Cloutier, O. Bar-Nur, S. Cheloufi, M. Stadtfeld, M. E. Figueroa, D. Robinton, S. Natesan, A. Melnick, J. Zhu, S. Ramaswamy, and K. Hochedlinger, “A molecular roadmap of reprogramming somatic cells into ips cells,” Cell, vol. 151, pp. 1617–1632, 2017/02/28 2012. [71] L. David and J. M. Polo, “Phases of reprogramming,” Stem Cell Research,
vol. 12, pp. 754–761, 5 2014.
[72] T. Brambrink, R. Foreman, G. G. Welstead, C. J. Lengner, M. Wernig, H. Suh, and R. Jaenisch, “Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells,” Cell Stem Cell, vol. 2, no. 2, pp. 151– 159, 2008.
[73] M. Stadtfeld, N. Maherali, D. T. Breault, and K. Hochedlinger, “Defining molecular cornerstones during fibroblast to ips cell reprogramming in mouse,” Cell Stem Cell, vol. 2, no. 3, pp. 230–240, 2008.
[74] R. Sridharan and K. Plath, “Illuminating the black box of reprogramming,” Cell Stem Cell, vol. 2, no. 4, pp. 295–297, 2008.
[75] B. Ebrahimi, “Reprogramming barriers and enhancers: strategies to enhance the efficiency and kinetics of induced pluripotency,” Cell Regeneration, vol. 4, no. 1, p. 10, 2015.
[76] S. Ruiz, A. D. Panopoulos, A. Herrer´ıas, K.-D. Bissig, M. Lutz, W. T. Berggren, I. M. Verma, and J. C. Izpisua Belmonte, “A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell iden-tity,” Current Biology, vol. 21, pp. 45–52, 1 2011.
[77] E. Apostolou and K. Hochedlinger, “Chromatin dynamics during cellular re-programming,” Nature, vol. 502, pp. 462 EP –, 10 2013.
[78] D.-S. Lee, J.-Y. Shin, P. D. Tonge, M. C. Puri, S. Lee, H. Park, W.-C. Lee, S. M. I. Hussein, T. Bleazard, J.-Y. Yun, J. Kim, M. Li, N. Cloonan, D. Wood, J. L. Clancy, R. Mosbergen, J.-H. Yi, K.-S. Yang, H. Kim, H. Rhee, C. A. Wells, T. Preiss, S. M. Grimmond, I. M. Rogers, A. Nagy, and J.-S. Seo, “An epigenomic roadmap to induced pluripotency reveals dna methylation as a re-programming modulator,” Nature Communications, vol. 5, pp. 5619 EP –, 12 2014.
[79] J. Silva, J. Nichols, T. W. Theunissen, G. Guo, A. L. van Oosten, O. Barrandon, J. Wray, S. Yamanaka, I. Chambers, and A. Smith, “Nanog is the gateway to the pluripotent ground state,” Cell, vol. 138, pp. 722–737, 2018/01/23.
[80] T. Seki, S. Yuasa, M. Oda, T. Egashira, K. Yae, D. Kusumoto, H. Nakata, S. Tohyama, H. Hashimoto, M. Kodaira, Y. Okada, H. Seimiya, N. Fusaki, M. Hasegawa, and K. Fukuda, “Generation of induced pluripotent stem cells from human terminally di↵erentiated circulating t cells,” Cell Stem Cell, vol. 7, pp. 11–14, 2018/01/23.
[81] A. G. Marthaler, U. Tiemann, M. J. Ara´uzo-Bravo, G. Wu, H. Zaehres, J. K. Hyun, D. W. Han, H. R. Sch¨oler, and N. Tapia, “Reprogramming to pluripotency through a somatic stem cell intermediate,” PLOS ONE, vol. 8, pp. e85138–, 12 2013.
[82] T. Aoi, K. Yae, M. Nakagawa, T. Ichisaka, K. Okita, K. Takahashi, T. Chiba, and S. Yamanaka, “Generation of pluripotent stem cells from adult mouse liver and stomach cells,” Science, vol. 321, p. 699, 08 2008.
[83] J. Hanna, S. Markoulaki, P. Schorderet, B. W. Carey, C. Beard, M. Wernig, M. P. Creyghton, E. J. Steine, J. P. Cassady, R. Foreman, C. J. Lengner, J. A. Dausman, and R. Jaenisch, “Direct reprogramming of terminally di↵erentiated mature b lymphocytes to pluripotency,” Cell, vol. 133, pp. 250–264, 2018/01/23. [84] J. Yu, M. A. Vodyanik, K. Smuga-Otto, J. Antosiewicz-Bourget, J. L. Frane, S. Tian, J. Nie, G. A. Jonsdottir, V. Ruotti, R. Stewart, I. I. Slukvin, and J. A. Thomson, “Induced pluripotent stem cell lines derived from human somatic cells,” Science, vol. 318, p. 1917, 12 2007.
[85] C. Yu, K. Liu, S. Tang, and S. Ding, “Chemical approaches to cell reprogram-ming,” Current Opinion in Genetics & Development, vol. 28, pp. 50–56, 2014. [86] K. Liu, C. Yu, M. Xie, K. Li, and S. Ding, “Chemical modulation of cell fate
in stem cell therapeutics and regenerative medicine,” Cell Chemical Biology, vol. 23, no. 8, pp. 893–916, 2016.
[87] J. Liao, Z. Wu, Y. Wang, L. Cheng, C. Cui, Y. Gao, T. Chen, L. Rao, S. Chen, N. Jia, H. Dai, S. Xin, J. Kang, G. Pei, and L. Xiao, “Enhanced efficiency of generating induced pluripotent stem (ips) cells from human somatic cells by a combination of six transcription factors,” Cell Research, vol. 18, pp. 600 EP –, 04 2008.
[88] D. Huangfu, R. Maehr, W. Guo, A. Eijkelenboom, M. Snitow, A. E. Chen, and D. A. Melton, “Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds,” Nature Biotechnology, vol. 26, pp. 795 EP –, 06 2008.
[89] D. Huangfu, K. Osafune, R. Maehr, W. Guo, A. Eijkelenboom, S. Chen, W. Muhlestein, and D. A. Melton, “Induction of pluripotent stem cells from primary human fibroblasts with only oct4 and sox2,” Nature Biotechnology, vol. 26, pp. 1269 EP –, 10 2008.
[90] J. K. Ichida, J. Blanchard, K. Lam, E. Y. Son, J. E. Chung, D. Egli, K. M. Loh, A. C. Carter, F. P. Di Giorgio, K. Koszka, D. Huangfu, H. Akutsu, D. R. Liu, L. L. Rubin, and K. Eggan, “A small-molecule inhibitor of tgf-β signal-ing replaces ¡em¿sox2¡/em¿ in reprogrammsignal-ing by inducsignal-ing ¡em¿nanog¡/em¿,” Cell Stem Cell, vol. 5, pp. 491–503, 2018/01/23.
[91] N. Maherali and K. Hochedlinger, “Tgfβ signal inhibition cooperates in the induction of ipscs and replaces sox2 and cmyc,” Current Biology, vol. 19, pp. 1718–1723, 2018/01/23.
[92] L. Warren, P. D. Manos, T. Ahfeldt, Y.-H. Loh, H. Li, F. Lau, W. Ebina, P. K. Mandal, Z. D. Smith, A. Meissner, G. Q. Daley, A. S. Brack, J. J. Collins, C. Cowan, T. M. Schlaeger, and D. J. Rossi, “Highly efficient reprogramming to pluripotency and directed di↵erentiation of human cells with synthetic modified mrna,” Cell Stem Cell, vol. 7, pp. 618–630, 2018/01/23.
[93] A. Radzisheuskaya and J. R. Silva, “Do all roads lead to oct4? the emerging concepts of induced pluripotency,” Trends in Cell Biology, vol. 24, pp. 275–284, 2018/01/27.
[94] D. Del Vecchio, H. Abdallah, Y. Qian, and J. J. Collins, “A blueprint for a synthetic genetic feedback controller to reprogram cell fate,” Cell Systems, vol. 4, pp. 109–120.e11, 2018/01/23.
[95] D. C. Allis, M.-L. Caparros, D. Reinberg, and M. Lachlan, Epigenetics. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, second ed., 2015.
[96] L. Armstrong, Epigenetics. Garland Science, 1 ed., 2013.
[97] W. Zeng, A. R. Ball, and K. Yokomori, “Hp1: Heterochromatin binding proteins working the genome,” Epigenetics : official journal of the DNA Methylation Society, vol. 5, pp. 287–292, 05 2010.
[98]
[99] T. S. Mikkelsen, M. Ku, D. B. Ja↵e, B. Issac, E. Lieberman, G. Giannoukos, P. Alvarez, W. Brockman, T.-K. Kim, R. P. Koche, W. Lee, E. Menden-hall, A. O’Donovan, A. Presser, C. Russ, X. Xie, A. Meissner, M. Wernig, R. Jaenisch, C. Nusbaum, E. S. Lander, and B. E. Bernstein, “Genome-wide