HAL Id: tel-03155152
https://tel.archives-ouvertes.fr/tel-03155152
Submitted on 1 Mar 2021
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Optimisation de molécules complexantes du Ru et du Tc
pour le recyclage des combustibles nucléaires usés
Alexiane Thevenet
To cite this version:
Alexiane Thevenet. Optimisation de molécules complexantes du Ru et du Tc pour le recyclage des combustibles nucléaires usés. Autre. Université Montpellier, 2020. Français. �NNT : 2020MONTS023�. �tel-03155152�
THÈSE POUR OBTENIR LE GRADE DE DOCTEUR
DE L’UNIVERSITÉ DE MONTPELLIER
en Chimie séparative et matériaux
École doctorale Sciences Chimiques Balard (ED 459)
Unité de recherche Département de recherche pour la Mine et le Recyclage du Combustible
Présentée par Alexiane THEVENET
Le 9 octobre 2020
Sous la direction de Philippe GUILBAUD
Devant le jury composé de
Jean AUPIAIS, Directeur de recherche, CEA DAM
Véronique HUBSCHER, Maitre de conférences, Université de Strasbourg Valéria AMENDOLA, Professeur d’université, Université de Pavie Nicolas DACHEUX, Professeur d’université, ICSM
Philippe MOISY, Directeur de recherche, CEA Marcoule Philippe GUILBAUD, Directeur de recherche, CEA Marcoule Isabelle HABLOT, Ingénieur, Orano
Cécile MARIE, Ingénieur, CEA Marcoule Christelle TAMAIN, Ingénieur, CEA Marcoule
Rapporteur Rapporteur Examinateur Président du jury Examinateur Directeur de thèse Invitée Invitée Invitée
Optimisation de molécules complexantes du Ru
et du Tc pour le recyclage des combustibles
Remerciements
Je tiens à remercier…
… les membres du jury d’avoir accepté de juger ce travail : Jean Aupiais et Véronique Hubscher en qualité de rapporteurs et Valeria Amendola, Nicolas Dacheux et Philippe Moisy en tant qu’examinateurs. Merci également à Isabelle Hablot.
… Philippe Guilbaud pour ta disponibilité. Je suis consciente de la chance que j’ai eue d’avoir un directeur de thèse aussi présent. Merci également pour ton soutien et ta bonne humeur tout au long de ces trois années. (« Au fait, tu skies Philippe ? »).
… Cécile Marie qui m’a encadrée pendant trois années et demie. J’ai pris énormément de plaisir à travailler à tes côtés. Merci pour ta disponibilité, ton implication, ton soutien et pour la confiance que tu m’as accordée dès le début du stage. Notre meilleure expérience restera notre expédition à Pavie dans un avion 18 places… Surtout l’arrivée à Marseille par temps de tempête… Merci également d’avoir relu ce manuscrit pendant ton congès maternité ! Je sais que ça a été compliqué… Je te souhaite plein de réussite scientifique et surtout plein de bonheur auprès de ta famille.
… Christelle Tamain qui a accepté de prendre part au projet de thèse. Je te remercie pour ta disponibilité, ton aide pour les études de complexation et tous les bons moments passés au LN1 ! Merci également pour ton soutien en général et pour tes conseils en pâtisserie ! On aura réussi à passer une semaine chez les bonnes sœurs pendant cette thèse… Moment mémorable Merci de m’avoir alimentée en corrections pendant le confinement ! Je te souhaite également beaucoup de réussite scientifique et beaucoup de bonheur dans ta vie personnelle.
… Orano et EDF pour leur implication dans le projet avec les discussions que nous avons pu avoir. Merci à Isabelle Hablot pour son soutient.
… le LCPE puis le LCIS pour la très bonne ambiance de travail ! Tout d’abord, merci à Christian Sorel, chef du laboratoire, de m’avoir accueillie dans ses locaux. Merci pour les discussions et les conseils scientifiques que tu as pu me donner. Travailler dans ce laboratoire a été un réel plaisir.
Dans un deuxième temps, je voulais remercier les filles d’Atalante du L16 et du L28 : Marie-Thé, Emilie, Estelle, Jessica mais surtout Justine et Sophie avec qui j’ai partagé le plus de temps sur Atalante. L’ambiance qui régnait dans les labos était juste géniale et travailler dans ces conditions a été un réel plaisir. Merci pour votre aide lors des manips Tc et de m’avoir toujours accompagnée pour manger au réfectoire (parfois très tard) ! Les élutions étaient bien rentabilisées grâce à vous ! Merci pour votre soutien au quotidien et pour tous les bons moments passés ensemble au CEA mais surtout en dehors. MERCI ! Je vous souhaite le meilleur… et vivement le 11 juin 2022…
Merci à Pauline pour ton aide côté modélisation et surtout pour ton implication dans le projet. Nous sommes souvent revenus vers toi pour changer le modèle et c’était à chaque fois un plaisir pour toi de m’aider dans la compréhension de nos données d’extraction. Merci pour ta disponibilité quand j’avais des questions et merci pour la relecture de la modélisation de ce manuscrit. Je suis contente que ce travail aboutisse à une publication.
Merci à Anne pour ta générosité, ta bonne humeur, les discussions Ru au début de la thèse. Merci surtout pour ton aide après le déconfinement, ton PC m’aura fait gagner énormément de temps ! Merci à Franck pour ta bonne humeur et nos moments de rigolade. Merci à Xavier pour nos discussions très enrichissantes. Merci pour ton aide pour la radiolyse et d’avoir fait,
le LCIS... J’ai pu voir tout au long de ces trois années, mais aussi pendant la période de confinement, que l’entraide au sein du laboratoire est remarquable !
Et évidemment merci à Isabelle !
… Valéria Amendola et ses étudiantes : Ana et Sonia. Merci pour la synthèse des molécules cages, pour votre réactivité et les nombreux échanges que nous avons pu avoir.
… à mes différents co-bureaux : Philippe M., Damien et Guillaume. J’ai toujours été bien entouré et je vous en remercie.
… le LILA, ma deuxième maison. Je tenais à remercier Georges et Laurence pour les expériences ESI-MS ; Claude, Laeti et Mathieu pour la RMN ; Thomas et Thomass pour les discussions sur le Ru et Domi pour les calculs de DFT. Merci de m’avoir accepté aussi souvent au LN1. Je remercie également ma copine Nath pour la partie calo, COTmètre et pour toutes les questions que j’ai pu te poser, mais surtout MERCI pour ton soutien pendant ces trois années. Je te souhaite également le meilleur pour la suite.
… aux personnes du free : Seb, Nath, Manon, Marie-T, Justine & Romain ! C’était toujours agréable de manger ensemble pour rigoler et se raconter les derniers potins d’Atalante. … Jackie et Emilie pour m’avoir accueillie au L18 pour la caractérisation des solutions mères de Tc par UV.
… mes amis de Strasbourg. Merci à Zélie, Elo, Boris et Thomas de nous avoir accueillis assez souvent à Strasbourg. Ces week-ends à se retrouver autour de crêpes, à visiter des grottes (ou non…) ou encore à rencontrer des tchèques (parfois de trop près) resteront inoubliables. Merci aux filles d’avoir été aussi présentes pendant cette période particulière du confinement. Merci à Elo, Mathias et mon filleul Léon pour leur soutien et les bons moments passés ensemble à Strasbourg, Bagnols ou dans le Jura. Je vous souhaite beaucoup de bonheur auprès de la petite Adèle.
Merci à Fannie et Anna. Malheureusement, je n’ai pas pu vous voir assez souvent mais se retrouver était un réel plaisir.
… Alex-andre ! Merci pour les courses à pied partagées et surtout le marathon d’Annecy ! … ma famille ! Je commence bien évidemment par mes parents. Merci pour votre soutien pendant ces trois ans (et les années d’avant…). Au courant de tout ce qui pouvait se passer au labo et dans mes recherches, les explications pouvaient parfois être longues. Vous vous êtes toujours sacrifiés pour que je réussisse et que je trouve une voie professionnelle qui me plaît. Votre mission est accomplie. Merci à mes frères, Josselin et Dylan. Ces moments en famille sont si important à mes yeux. Je vous souhaite beaucoup de réussite dans tous les projets que vous pouvez entreprendre ! Je voudrais remercier mes grands-parents et ma grand-mère, si contents de venir découvrir, lors de ma soutenance, le monde dans lequel j’ai travaillé pendant trois ans. Merci à ma famille en générale, surtout à JP et Christelle et mes cousins. L’apéro d’après thèse est tout proche… Je voudrai également remercier mes beaux-parents de me soutenir dans ce projet. Vivement les derbys Béziers-Clermont, et le prochain triathlon…
… Romain ! Tu te doutais que ton nom arriverait en dernier. J’ai eu la chance de réaliser cette thèse à tes côtés et tu as été un atout majeur dans cette réussite. Le bonheur que tu m’apportes au quotidien m’a permis de relativiser par rapport au travail quand les résultats n’étaient pas là. Merci !! Vivement 2022
Table des matières
INTRODUCTION GENERALE ... 11
CHAPITRE 1 : REVUE BIBLIOGRAPHIQUE – COMPORTEMENT DU RUTHENIUM ET DU TECHNETIUM DANS LES PROCEDES DE RETRAITEMENT DES COMBUSTIBLES NUCLEAIRES ... 17
1. RETRAITEMENT DU COMBUSTIBLE IRRADIÉ ... 19
1.1. Extraction liquide-liquide ... 19
1.2. Mécanisme d’extraction pour le retraitement des combustibles nucléaires usés ... 21
2. LE PROCÉDÉPUREX ... 22
2.1. Présentation du procédé ... 22
2.1.1. Fonctionnement dans les usines de la Hague ... 22
2.1.2. Avantages et inconvénients ... 24
2.2. Le technétium ... 24
2.2.1. Comportement dans le procédé PUREX ... 25
2.2.1.1. Extraction de HTcO4 par TBP ... 25
2.2.1.2. Extraction du technétium par le TBP en présence d’uranium ... 26
2.2.1.3. Extraction du technétium par le TBP en présence d’autres cations métalliques ... 27
2.2.2. Problématiques liées au technétium dans PUREX ... 29
2.3. Le ruthénium ... 30
2.3.1. Étude du ruthénium dans le procédé PUREX41,42 ... 30
2.3.1.1. Espèces formées en phase aqueuse ... 31
2.3.1.2. Données d’extraction du ruthénium avec le TBP ... 31
2.3.1.3. Espèces formées en phase organique (TBP 30%vol dans le TPH) ... 32
2.3.2. Problématiques du ruthénium dans le procédé PUREX ... 34
3. ÉLABORATION D’UN PROCÉDÉ DE SÉPARATION INNOVANT ... 35
3.1. Co-extraction du technétium avec les monoamides ... 36
3.2. Données sur l’extraction du ruthénium par les monoamides ... 38
4. MÉTHODES POSSIBLES POUR ÉLIMINER LE TECHNÉTIUM ET LE RUTHÉNIUM DE LA PHASE ORGANIQUE ... 39
4.1. Techniques rapportées dans la littérature ... 39
4.1.1. Séparation du technétium ... 39
4.1.2. Conversion du ruthénium en une espèce non extractible ... 39
4.2. Stratégie développée au cours de cette étude ... 40
4.2.1. Complexation en phase aqueuse ... 40
4.2.2. Étude des complexants dans les conditions du procédé ... 40
4.2.3. Choix des systèmes étudiés pendant la thèse ... 42
CHAPITRE 2 : LE RUTHENIUM – SPECIATION EN MILIEU ACIDE NITRIQUE ET RESULTATS ... 45
1. SPÉCIATION EN MILIEU ACIDE NITRIQUE ... 47
1.1. Le ruthénium nitrosyle ... 47
1.2. Les complexes nitratés ... 48
1.3. Les complexes mixtes nitrés-nitratés ... 50
1.4. Bilan des études de spéciation du ruthénium en solution ... 51
2. LES COMPLEXANTS ET EXTRACTANTS DU RUTHÉNIUM ... 52
2.1. Complexants du Ru(III) en milieu acide nitrique ... 52
2.1.1. La famille des « polyamines » ... 53
2.1.1.1. Études menées par Sasaki ... 53
2.1.1.2. Études menées au CEA sur les polyamines ... 54
2.1.3.2. Cinétique de formation du complexe 1:2 ... 57
2.1.4. Structures trouvées dans la Cambridge Structural Database ... 57
2.2. Extractants du Ru(III) ... 58
2.3. Les molécules envisagées pour la rétention du ruthénium (III) en milieu acide nitrique ... 60
3. COMPLEXATION DU RUTHÉNIUM PAR L’HYDROQUINONE ET SES DÉRIVÉS ... 62
3.1. Méthodologie des tests d’extraction liquide-liquide ... 63
3.2. Étude de l’hydroquinone en extraction ... 65
3.2.1. Tests d’extraction dans les conditions du procédé ... 65
3.2.2. Stabilité de l’hydroquinone en milieu acide nitrique ... 67
3.2.3. Suivi de la complexation du ruthénium avec l’hydroquinone en phase homogène : préparation de la solution aqueuse d’extraction ... 69
3.2.4. Tests d’extraction liquide-liquide à forte concentration de ruthénium ... 70
3.3. Étude de l’hydroquinone en lavage ... 71
3.3.1. Cinétique de désextraction du ruthénium avec l’hydroquinone ... 71
3.3.2. Tests à différentes concentrations d’acide nitrique ... 73
3.3.3. Étude du mode d’extraction de l’hydroquinone en phase organique ... 74
3.4. Bilan des études sur les molécules contenant une fonction OH ... 76
3.4.1. Études en extraction ... 76
3.4.2. Études en lavage... 77
4. COMPLEXATION DU RUTHÉNIUM PAR LA BIPYRIDINE ... 79
4.1. Étude du système ruthénium - bipyridine ... 79
4.1.1. Étude de la bipyridine en extraction ... 79
4.1.2. Étude de la bipyridine en lavage ... 80
4.1.3. Caractérisation du complexe formé en phase aqueuse ... 81
4.1.3.1. Spectroscopie d’absorption Visible ... 81
4.1.3.2. Spectroscopie Raman ... 81
4.1.3.3. DRX monocristal ... 82
4.2. Étude de la sélectivité vis-à-vis de l’uranium ... 83
4.2.1. Tests d’extraction liquide-liquide ... 83
4.2.2. Caractérisation du complexe formé avec l’uranium ... 85
4.2.2.1. Structure DRX ... 85
4.2.2.2. Spectroscopie d’absorption visible ... 86
4.2.2.3. Spectroscopie Raman ... 87
4.3. Bilan sur la bipyridine ... 88
5. CONCLUSIONS ET PERSPECTIVES ... 89
CHAPITRE 3 : LE TECHNETIUM – SPECIATION EN MILIEU ACIDE NITRIQUE ET RESULTATS ... 93
1. PROPRIÉTÉS DU TECHNÉTIUM ET BIBLIOGRAPHIE DES COMPLEXANTS ET EXTRACTANTS ... 95
1.1. États d’oxydation stables en milieu acide nitrique ... 95
1.1.1. L’état d’oxydation +VII ... 95
1.1.2. Le rhénium(VII) : un analogue inactif du technétium ... 95
1.2. Les complexants et extractants du Technétium ... 96
1.2.1. Reconnaissance anionique ... 96
1.2.1.1. Les différents types d’interaction ... 96
1.2.1.2. Série de Hofmeister ... 97
1.2.2. Bibliographie des complexants du Tc(VII) en phase aqueuse ... 97
1.2.2.1. Les molécules cages ... 98
1.2.2.2. La famille des « polyamines » ... 100
1.2.3. Autres ligands transposables pour la complexation du Tc(VII) en phase aqueuse ... 101
1.2.4. Bibliographie des extractants du Tc(VII) en phase organique ... 102
1.2.4.1. Un ligand tripodal, analogue du ligand cage ... 103
1.3. Bilan sur les molécules envisagées pour le technétium (VII) ... 107
2. LA FAMILLE DES « POLYAMINES » ... 109
2.1. Méthodologie pour l’évaluation des complexants par extraction liquide-liquide ... 109
2.1.1. Préparation de la solution de complexant ... 109
2.1.2. Tests d’extraction liquide-liquide ... 109
2.2. La famille des « polyamines »... 112
2.2.1. Tests d’extraction liquide-liquide sans uranium ... 113
2.2.2. Tests d’extraction en présence d’uranium en phase aqueuse ... 113
2.3. Bilan ... 115
3. LES MOLÉCULES CAGES... 116
3.1. Influence de la concentration du ligand Lcage sur la co-extraction du Re(VII) ou du Tc(VII) avec U(VI) en phase organique ... 116
3.1.1. Études préliminaires ... 116
3.1.1.1. Solubilité du ligand Lcage ... 116
3.1.1.2. Optimisation du temps d’agitation pour les tests d’extraction ... 117
3.1.2. Extraction de Re(VII) ... 118
3.1.3. Extraction de Tc(VII) ... 120
3.1.3.1. Influence de la concentration d’U(VI) ... 120
3.1.3.2. Influence de la concentration du ligand Lcage ... 122
3.2. Caractérisation des complexes formés ... 123
3.2.1. Étude par microcalorimétrie du complexe Lcage-ReO4- ... 123
3.2.2. Étude du complexe formé avec Re(VII) ... 125
3.2.2.1. Structure du ligand cage ... 125
3.2.2.2. Structure cristalline du complexe formé avec Re(VII) ... 126
3.2.2.3. Comparaison avec la structure de ligand seul ... 128
3.2.3. Spectroscopies infrarouge et Raman des solides ... 128
3.2.4. Conclusion sur la caractérisation des complexes ... 130
3.3. Modélisation des données d’extraction... 131
3.4. Bilan ... 134
4. ÉTUDE DE DERIVES PLUS SOLUBLES DE LA MOLÉCULE LCAGE ... 135
4.1. Solubilité en milieu acide nitrique ... 135
4.2. Test des molécules en extraction en présence d’uranium (VI) ... 136
4.2.1. Études menées sur le rhénium ... 136
4.2.2. Études menées sur le technétium ... 138
4.2.2.1. Influence de la concentration de ligand à HNO3 0,5 M ... 138
4.2.2.2. Influence de la concentration de ligand MOH à HNO3 1 M ... 140
4.2.3. Influence de la concentration d’acide nitrique sur D(Tc) ... 141
4.2.4. Influence de la concentration de nitrates sur l’extraction du technétium ... 142
4.2.5. Influence de la concentration de proton sur l’extraction du technétium ... 144
4.2.6. Étude de la complexation en milieu acide nitrique et nitrate ... 146
4.2.6.1. Études thermodynamiques par microcalorimétrie ... 146
4.2.6.2. Modélisation des données d’extraction ... 148
4.3. Tests des nouveaux dérivés du ligand Lcage en lavage ... 151
4.4. Caractérisation des complexes formés ... 153
4.4.1. Microcalorimétrie avec ReO4- ... 153
4.4.2. Études par spectroscopies infrarouge et RAMAN ... 154
4.4.2.1. Étude spectroscopique avec le rhénium ... 154
4.4.2.2. Étude spectroscopique avec le technétium ... 156
5. STABILITÉ RADIOLYTIQUE DES MOLÉCULES CAGES ... 158
5.1. Stabilité radiolytique - Méthodologie ... 158
5.2.1.2. Tests d’extraction liquide–liquide ... 162
5.2.1.3. Étude radiolytique de Lcage en présence de rhénium... 163
5.2.2. Stabilité de la molécule 1-ARM pour une application en lavage ... 164
5.3. Bilan sur la stabilité radiolytique des molécules cages... 164
... 166
CONCLUSION GENERALE ET PERSPECTIVES... 167
BIBLIOGRAPHIE ... 173
ANNEXES ... 181
ANNEXE 1 : GLOSSAIRE ... 183
ANNEXE 2 : MÉTHODOLOGIE ... 189
1. PRÉPARATION DES SOLUTIONS MÈRES DE TECHNÉTIUM... 189
1.1. Dissolution et caractérisation de la solution ... 189
1.2. Validation de la solution mère de technétium ... 190
2. RUTHÉNIUM : PRÉPARATION DES SOLUTIONS ET DÉSEXTRACTION ... 191
2.1. Préparation des solutions mères ... 191
2.2. Conditions expérimentales pour la désextraction ... 191
3. TESTS D’EXTRACTION LIQUIDE-LIQUIDE ... 192
3.1. Protocole expérimentale ... 192
3.1.1. Pré-équilibrage du solvant ... 192
3.1.2. Tests en mode extraction ... 192
3.1.3. Tests en lavage ... 192
3.1.4. Mesure de l’acidité des phases aqueuses initiales et à l’équilibre ... 193
3.1.5. Mesure des concentrations de Ru(III), Re(VII) et U(VI) ... 193
3.1.6. Calcul des coefficients de distribution du Tc(VII) ... 193
3.2. Appareils utilisés pour la quantification ... 194
3.2.1. ICP-AES ... 194
3.2.2. Spectrométrie gamma ... 195
3.2.3. COTmètre ... 195
3.3. Appareils utilisés pour la caractérisation des complexes ... 196
3.3.1. Diffraction des rayons X ... 196
3.3.2. Spectroscopie Raman ... 196
3.3.3. Spectroscopie Infrarouge ... 197
3.3.4. Spectroscopie RMN ... 198
3.3.5. Microcalorimétrie ... 198
ANNEXE 3 : TESTS COMPLÉMENTAIRES SUR LE RUTHÉNIUM ... 199
1. CARACTÉRISATION DES COMPLEXES DE RUTHÉNIUM ET D’HYDROQUINONE ... 199
1.1. Caractérisation par ESI-MS ... 199
1.2. Caractérisation des phases aqueuses après lavage avec l’hydroquinone ... 199
2. ÉTUDES COMPLÉMENTAIRES SUR LES MOLÉCULES DE LA FAMILLE DE L’HYDROQUINONE ... 201
2.1. Tests de molécules non aromatiques ... 201
2.1.1. Tests en lavage ... 201
2.1.1.1. Tests à concentration fixe de ligands ... 201
2.1.1.2. Test de la molécule TA ... 202
2.1.2. Tests en extraction ... 203
2.2. Tests du phénol... 203
2.2.1. Influence de la concentration de phénol en lavage ... 204
2.3.1. Tests en lavage ... 206
2.3.2. Tests en extraction ... 208
3. ÉTUDE COMPLÉMENTAIRES SUR LA BIPYRIDINE ... 209
3.1. Tests en lavage de la bipyridine ... 209
3.2. Caractérisation des complexes formés ... 209
3.2.1. Spectre Raman des monocristaux Ru-BIPY ... 209
3.2.2. ESI-MS ... 210
ANNEXE 4 : TESTS COMPLÉMENTAIRES SUR LE TECHNÉTIUM ... 213
1. ÉTUDES COMPLEMENTAIRES EN EXTRACTION ET CARACTÉRISATION ... 213
1.1. Test de la molécule TEA ... 213
1.2. Cinétique de désextraction du ligand 1-ARM en lavage ... 214
1.3. Sélectivité du ligand 1-ARM vis-à-vis du plutonium ... 215
1.3.1. Tests réalisés en extraction ... 215
1.3.2. Tests réalisés en lavage ... 216
1.4. Analyse des précipités obtenus pendant les tests d’extraction avec les ligands cages ... 216
1.5. Variation de la concentration de proton avec l’ajout de HCl... 218
1.5.1. Microcalorimétrie avec HCl ... 218
1.5.2. Extraction de l’uranium en milieu chlorure ... 219
2. MODÉLISATION DES DONNÉES D’EXTRACTION ... 221
2.1. Description du modèle et ajustement de ses paramètres ... 221
2.2. Modélisation des données d’extraction en milieu acide nitrique ... 223
3. CARACTÉRISATION STRUCTURALE DES COMPLEXES ... 227
3.1. Synthèse et caractéristiques des monocristaux Re-Lcage ... 227
3.2. Calculs DFT ... 227
3.3. Caractérisation des complexes par spectroscopies Raman et infrarouge ... 229
3.3.1. Caractérisation des complexes formés avec l’anion perrhénate ... 229
3.3.2. Caractérisation des complexes formés avec l’anion pertechnétate ... 231
3.4. Études thermodynamiques ... 234
3.4.1. Comparaison des comportements du ligand MOH en milieu acide nitrique ou nitrate ... 234
3.4.1.1. Complexation en milieu acide nitrique ... 234
3.4.1.2. Complexation en milieu nitrate ... 235
3.4.2. Complexation avec les dérivés du ligand Lcage ... 237
4. ÉTUDES DE RADIOLYSE ET D’HYDROLYSE ... 239
4.1. Irradiation de la molécule Lcage en présence de rhénium ... 239
La demande en électricité ne cesse d’augmenter au niveau mondial. Compétitive et générant
peu de CO2, l’énergie nucléaire est le moyen de production le moins émetteur de gaz à effet
de serre1. En France, près de 70% de l’électricité est produite grâce au nucléaire. Toutefois,
cette énergie se doit de proposer une gestion sûre et durable de ses déchets et de développer des procédés robustes de recyclage des matières énergétiques valorisables (uranium et plutonium) afin de limiter son impact environnemental.
Le parc électronucléaire français compte aujourd’hui 56 réacteurs nucléaires répartis dans 18
centrales nucléaires2. Les réacteurs sont tous alimentés en UOX (oxyde d’uranium enrichi à
hauteur de 3 à 5% en uranium 235) ou en MOX (mélange d’oxydes d’uranium et de plutonium). Environ trois ans après y avoir été introduit, le combustible irradié est déchargé des réacteurs.
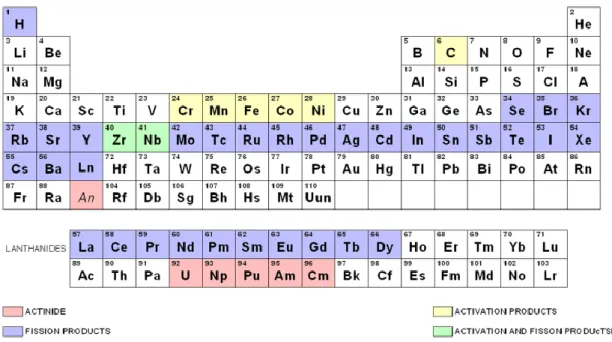
Le combustible nucléaire, dit « usé », présente alors une grande diversité d’éléments
chimiques issus de la transformation de l’uranium ou du plutonium initial. Environ un tiers des éléments chimiques de la classification de Mendeleïev sont présents à des degrés d’oxydation et des isotopies différents. Parmi ce vaste ensemble, les actinides « majeurs » représentent la proportion la plus importante avec environ 95% d’uranium et 1% de plutonium en masse pour un combustible UOX déchargé des réacteurs à eau pressurisée. Les actinides « mineurs » (neptunium, américium et curium) sont également produits mais en faible proportion (0,1%). Le reste du combustible irradié est constitué de 4% de produits de fission (PF) comme le technétium, le ruthénium, le zirconium, les lanthanides, etc… La France a opté pour le retraitement des combustibles irradiés sortant des centrales nucléaires par le procédé PUREX mis en œuvre à l’usine de la Hague. Il permet de récupérer avec des rendements quantitatifs l’uranium et le plutonium contenus dans les combustibles irradiés et de les purifier afin de permettre leur réutilisation. Il permet également de conditionner les différents déchets
sous une forme compatible avec un stockage minimisant l’impact sur l’environnement2
(vitrification des produits de fission et des actinides mineurs). L’étape de séparation de l’uranium et du plutonium est basée sur l’extraction liquide-liquide : le combustible irradié est dissous dans l’acide nitrique concentré puis mis en contact avec un solvant organique composé de TBP (tri-n-butylphosphate, Figure 1) dilué dans le TPH (TétraPropylène Hydrogéné). Le TBP possède une forte affinité pour les éléments aux degrés d’oxydation +IV et +VI : il permet la co-extraction du plutonium (IV) et de l’uranium (VI). Ce procédé permet la
récupération quantitative de l’uranium et du plutonium (99,8%) avec des facteurs de
décontamination élevés vis-à-vis des produits de fission et des actinides mineurs (environ 107).
Cependant, il présente certains inconvénients avec notamment le recours à des réactions d’oxydo-réduction pour la partition U/Pu. En effet, elle s’effectue en réduisant le Pu(IV) au degré d’oxydation +III par l’ajout d’U(IV) et d’hydrazinium, dont l’utilisation est fortement contrainte par la réglementation REACh. De plus, le TBP n’est pas complètement incinérable à cause de la présence d’un atome de phosphore.
Dans ce contexte, le CEA développe un nouveau procédé de retraitement des combustibles nucléaires basé sur des molécules extractantes à fonction amide. Parmi les nouveaux
extractants étudiés, les monoamides3-9, de formule générale R
1C(O)NR2R3 (avec Rn des
groupements alkyles), ou les carbamides10-12, de formule générale R
1R2NC(O)NR3R4 (Figure
1), sont particulièrement intéressants car ils permettent l’extraction de l’uranium et du
plutonium avec une forte sélectivité vis-à-vis des produits de fission à forte concentration d’acide nitrique (HNO3 4 M), et surtout, une partition de l’uranium vis-à-vis du plutonium à faible concentration d’acide nitrique, sans « chimie d’oxydo-réduction » donc sans utilisation d’hydrazinium.
Figure 1 : Structure du TBP, des monoamides et carbamides.
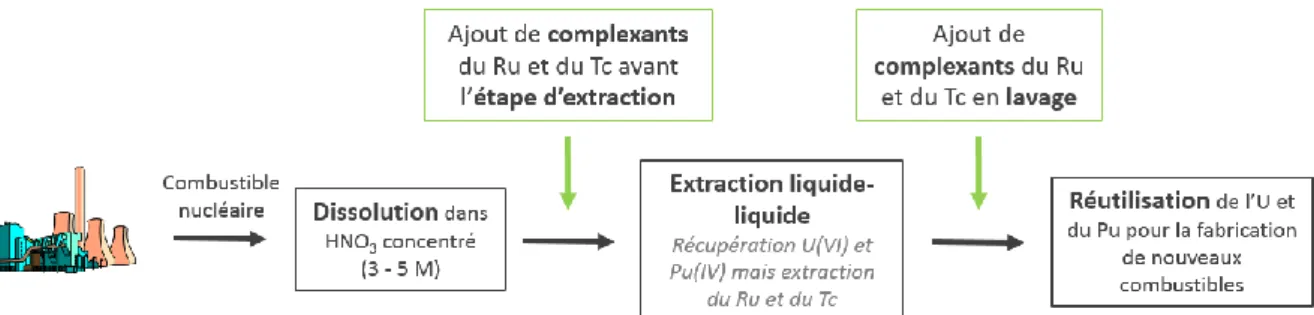
Cependant, les monoamides ou les carbamides, comme le TBP, ne permettent pas d’éviter complètement l’extraction en phase organique de certains produits de fission comme le technétium et le ruthénium. Ainsi, une étape supplémentaire de lavage pour désextraire les produits de fission de la phase organique chargée en uranium et en plutonium est nécessaire afin d’obtenir des facteurs de décontamination pour l’uranium et le plutonium plus satisfaisants. La Figure 2 représente un schéma très simplifié du procédé PUREX. Afin d’éviter l’extraction de ces produits de fission, l’ajout de molécules complexantes en phase aqueuse pour les maintenir sélectivement en milieu acide nitrique ou pour effectuer leur désextraction en phase aqueuse sans ajout de réactifs redox est un axe d’amélioration important (encadrés vert sur la Figure 2).
Figure 2 : Schéma simplifié du procédé PUREX avec les deux options (en vert) pour l’introduction de molécules complexantes dans le procédé.
Trouver un agent masquant pour chacun de ces deux produits de fission est un objectif délicat.
En effet, le technétium est présent sous forme anionique (anion pertechnétate, TcO4-) dans
les solutions de combustibles usés à acidité élevée. Le complexant seléctionné doit donc être
très sélectif de l’anion TcO4- vis-à-vis des ions nitrates, constituant majeur des solutions de
dissolution ([NO3-] 1300 fois supérieure à [TcO4-] à l’étape de co-extraction U/Pu).
En milieu acide nitrique, le ruthénium forme des complexes de ruthénium nitrosyle avec des groupements nitrate, nitreux, hydroxo ou des molécules d’eau. La formule générale de ces
complexes est [𝑅𝑢𝑁𝑂(𝑁𝑂2)𝑥(𝑁𝑂3)𝑦(𝑂𝐻)𝑧(𝐻2𝑂)5−𝑥−𝑦−𝑧]3−𝑥−𝑦−𝑧. Sa spéciation dépend de la
concentration d’acide nitrique, d’acide nitreux, du pH et du temps.
Ainsi, la spéciation du technétium et du ruthénium est imposée par l’acidité élevée du cycle du combustible et doit être abordée de manière très différente pour ces deux éléments. Pour répondre à ces deux problématiques, la méthodologie développée pour cette étude se décompose en plusieurs étapes reportées sur la Figure 3.
Figure 3 : Méthodologie mise au point au cours de la thèse.
Dans un premier temps, les complexants sélectifs du ruthénium et du technétium à tester sont sélectionnés à partir d’une étude approfondie des données de la littérature. Dans un deuxième temps, leur capacité à retenir les deux produits de fission en phase aqueuse est évaluée par des tests d’extraction liquide-liquide dans des conditions proches du procédé (à partir d’un milieu acide nitrique concentré).
Une étude de chimie de coordination est ensuite effectuée pour caractériser les interactions métal-ligand et obtenir une meilleure compréhension des modes d’interaction avec les éléments cibles, pour permettre d’optimiser la structure des complexants.
Une propriété indispensable à toute molécule mise en jeu dans un procédé du cycle du combustible est sa résistance à l’irradiation mais aussi sa stabilité chimique. Pour les complexants les plus intéressants, la stabilité radiolytique et hydrolytique de la molécule organique sera donc évaluée.
Afin de répondre aux enjeux énoncés, ce mémoire s’oriente autour de trois principaux chapitres :
- Une revue bibliographique :
Les notions utiles à la compréhension des travaux réalisés sont présentées et s’articulent autour de trois thèmes : l’extraction liquide-liquide ; le procédé PUREX avec ses points forts, ses limites et le comportement des deux produits de fission ; et enfin les nouveaux solvants avec les données d’extraction disponibles dans la littérature pour le ruthénium et le technétium.
bibliographique, deux catégories de ligands sont apparues intéressantes et ont donc été particulièrement étudiées : l’hydroquinone et la bipyridine.
- Développement de molécules complexantes pour le technétium :
La spéciation du technétium en milieu acide nitrique, ainsi que les complexants et extractants du Tc connus dans la littérature sont présentés. Deux familles de molécules ayant un intérêt ont été analysées : la famille des « polyamines » est étudiée succinctement puis une famille de molécules cages a été largement explorée montrant des résultats très prometteurs pour la reconnaissance du technétium en milieu acide nitrique.
Dans ces travaux de thèse, la différence est faite entre extractants et complexants. Dans cette étude, l’extractant désigne la molécule extractante en phase organique et le complexant représente la molécule ajoutée en phase aqueuse pour complexer le technétium ou le ruthénium.
Les molécules étudiées et les sigles sont référencés dans l’Annexe 1. Afin d’aider à la lecture, les structures des molécules fréquemment rencontrées sont également reportées sur une feuille séparée.
Chapitre 1 :
Revue bibliographique – comportement du
ruthénium et du technétium dans les procédés
I- Retraitement du combustible irradié
Ce chapitre bibliographique présente dans un premier temps les bases de l’extraction
liquide-liquide et du procédé PUREX. Dans un second temps, le technétium et le ruthénium sont introduits avec leur problématique dans le procédé ainsi que leurs mécanismes d’extraction en phase organique. Enfin, des données d’extraction sont présentées avec les monoamides, molécules qui peuvent potentiellement remplacer le TBP dans un nouveau procédé de séparation simplifié et optimisé par rapport au procédé PUREX.
1. RETRAITEMENT DU COMBUSTIBLE IRRADIÉ
1.1. Extraction liquide-liquide
Ce procédé correspond à un transfert de matière depuis une phase liquide aqueuse vers une seconde phase liquide non miscible à la première par contact entre les deux phases. L’extraction liquide-liquide est une technique de séparation utilisée à l’échelle industrielle, dans les domaines aussi variés que l’hydrométallurgie classique, l’industrie nucléaire, la pétrochimie, ou encore l’industrie pharmaceutique. Ce procédé est largement utilisé pour le
retraitement du combustible nucléaire usé. Ce dernier est dissout dans l’acide nitrique
concentré. La solution résultante est composée des actinides d’intérêt à récupérer pour la fabrication de nouveaux combustibles (uranium et plutonium), d’actinides mineurs et de produits de fission. L’extraction liquide-liquide permet de récupérer sélectivement les actinides d’intérêt présents initialement dans la phase aqueuse et produit peu de déchets car le solvant* utilisé peut être recyclé.
Pour obtenir une capacité de traitement conséquente dans les usines de la Hague, toutes les
opérations d’extraction liquide-liquide élémentaires sont mises en œuvre en continu2. Les
contacteurs utilisés pour ces opérations d’extraction sont des appareils dans lesquels les phases aqueuse et organique circulent à contre courant. Cela permet d’obtenir un effet cumulatif de séparation réalisé dans chacun des étages pour récupérer l’uranium et le plutonium avec une très grande pûreté. D’une manière générale, la zone d’extraction comporte une opération d’extraction et une de lavage couplées (Figure 4), la première ayant plutôt la fonction d’assurer un rendement d’extraction correct et la seconde celle d’éliminer les éléments indésirables.
Figure 4 : Opérations d’extraction-lavage dans le procédé PUREX** (chaque rectangle grisé symbolise une opération continue d’extraction liquide-liquide à contre courant nécessitant
I- Retraitement du combustible irradié
Différentes grandeurs sont essentielles pour caractériser et comparer des systèmes extractants :
Le coefficient de distribution (D) : rapport de la concentration d’un élément M entre la phase organique et la phase aqueuse à l’équilibre. Plus le coefficient de distribution est élevé, plus le système extractant est efficace. Dans le cadre de cette étude visant à maintenir le technétium et le ruthénium en phase aqueuse, le but est de minimiser la valeur de leurs coefficients de distribution et atteindre une valeur inférieure à 0,1.
𝐷 =𝐶𝑀
𝑜𝑟𝑔,𝑒𝑞
𝐶𝑀𝑎𝑞,𝑒𝑞
Le facteur de séparation (FSA/B) : rapport des coefficients de distribution de deux éléments A et B mesurés dans les mêmes conditions qui permet d’évaluer la sélectivité d’un système extractant entre deux éléments différents. Plus le facteur de séparation augmente et plus le système extractant est sélectif de l’élément A. Lors de cette étude, le but est d’améliorer le facteur de séparation de l’uranium et du plutonium vis-à-vis du technétium et du ruthénium.
𝐹𝑆𝐴/𝐵 =
𝐷𝐴
𝐷𝐵
Le facteur de décontamination (FD) : rapport de la teneur en impureté ramenée à celle du produit (uranium et plutonium) avant et après traitement. Il donne une mesure de l’efficacité d’une opération de purification ou de séparation. D’après les normes
UNIREP (UNIted REProcessors)2, les facteurs de décontamination doivent être de
l’ordre de 107 pour l’uranium et 108 pour le plutonium, c’est-à-dire des facteurs de décontamination parmi les plus élevés de ceux rencontrés dans l’industrie chimique.
𝐹𝐷 = (𝐶𝑖𝑚𝑝𝑢𝑟𝑒𝑡é𝑠 𝐶 𝑝𝑟𝑜𝑑𝑢𝑖𝑡𝑠 ⁄ )𝑖𝑛𝑖𝑡𝑖𝑎𝑙 (𝐶𝑖𝑚𝑝𝑢𝑟𝑒𝑡é𝑠 𝐶 𝑝𝑟𝑜𝑑𝑢𝑖𝑡𝑠 ⁄ )𝑓𝑖𝑛𝑎𝑙
La décontamination du solvant est donc le fait d’éliminer les impuretés de ce dernier. Dans certains cas, une molécule complexante peut être ajoutée en phase aqueuse
pour empêcher l’extraction de certains éléments en phase organique (palladium)13, ou
pour améliorer certains facteurs de séparation (FSAm/Cm par exemple)14. Ainsi, il est nécessaire d’introduire la constante de formation. Dans un complexe mononucléaire
avec M le métal, L le ligand et MLn le complexe formé, la constante de formation globale
Kn est associée à l’équilibre suivant :
𝑀 + 𝑛𝐿 ⇄ 𝑀𝐿𝑛 et 𝐾𝑛= [𝑀𝐿𝑛] [𝑀][𝐿]𝑛
I- Retraitement du combustible irradié technétium. Cette grandeur permet notamment de comparer le pouvoir complexant des ligands étudiés pour chacun des deux produits de fission.
1.2. Mécanisme d’extraction pour le retraitement des
combustibles nucléaires usés
Il existe deux classes majeures de mécanismes d’extraction liquide-liquide15,16 : l’extraction par
échange d’ions et l’extraction non compensée, appelée aussi extraction par solvatation. Pour le retraitement des combustibles nucléaires usés, l’extraction des actinides d’intérêts se fait par solvatation.
L’extraction avec des réactifs solvatants met en jeu la formation d’un complexe globalement neutre entre l’extractant et le cation métallique à extraire. Dans le domaine du retraitement des
combustibles usés, l’extraction est essentiellement non compensée : le cation métallique Mn+
(uranium ou plutonium) est extrait avec son contre-ion A- (généralement les ions nitrate) par
l’extractant 𝐸̅* présent en phase organique sous la forme d’un complexe neutre. Les extractants solvatants sont principalement des composés possédant des groupements oxygénés, soufrés ou phosphorés donneurs de doublets électroniques comme les éthers, les esters, les cétones, les amides, les sulfoxides ou encore les trialkylphosphates. L’équilibre d’extraction peut s’écrire sous la forme suivante :
𝑀𝑛++ 𝑛𝐴−+ 𝑥𝐸̅ ⇄ 𝑀𝐴̅̅̅̅̅̅̅̅̅̅ 𝑛𝐸𝑥
où 𝐸̅ et 𝑀𝐴̅̅̅̅̅̅̅̅̅̅ représentent respectivement l’extractant et le complexe formé en phase 𝑛𝐸𝑥
organique. La désextraction** de l’élément M se fait généralement en contactant la phase
II- Le procédé PUREX
2. LE PROCÉDÉ PUREX
2.1. Présentation du procédé
2.1.1. Fonctionnement dans les usines de la Hague
Le procédé PUREX2 mis en œuvre dans les usines Orano de la Hague permet le retraitement
des combustibles nucléaires usés. Ce procédé d’extraction liquide-liquide met en œuvre un système extractant à base de TBP (Figure 1) dilué à 30% en volume dans le TPH. Cette molécule possède une forte affinité pour l’U(VI) et le Pu(IV) vis-à-vis des autres éléments présents dans les solutions de combustibles usés. L’extraction de ces deux éléments à partir d’un milieu fortement acide suit un processus d’extraction non compensée selon les équilibres suivants :
𝑈𝑂22++ 2𝑁𝑂3−+ 2𝑇𝐵𝑃̅̅̅̅̅̅ ⇄ 𝑈𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅ 2(𝑁𝑂3)2(𝑇𝐵𝑃)2
𝑃𝑢4++ 4𝑁𝑂
3−+ 2𝑇𝐵𝑃̅̅̅̅̅̅ ⇄ 𝑃𝑢(𝑁𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅ 3)4(𝑇𝐵𝑃)2
Après une période de refroidissement de quelques années qui permet la décroissance de la radioactivité des produits de fission à vie courte, le combustible dit « usé » est composé d’environ un tiers des éléments de la classification périodique (Figure 5). Ces éléments chimiques se retrouvent à des degrés d’oxydation et des isotopies différents, ce qui complexifie le développement de procédés sélectifs.
II- Le procédé PUREX
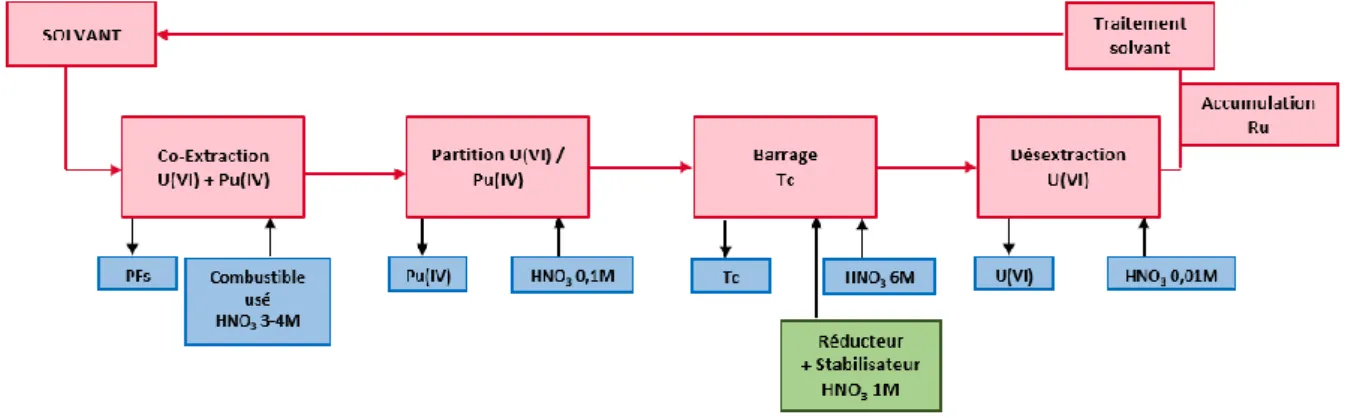
Les principales étapes du procédé PUREX sont (Figure 6)17,18 :
- le cisaillage du combustible afin de permettre sa mise en solution dans un milieu acide
nitrique concentré,
- un premier cycle d’extraction permettant la séparation de l’uranium et du plutonium des
produits de fission et actinides mineurs ; il s’agit de l’étape appelée
extraction
dansla suite du manuscrit,
- un deuxième cycle d’extraction pour désextraire les produits de fission présents en
phase organique ; il s’agit de l’étape appelée
lavage
dans la suite du document,- la séparation U(VI)/Pu(IV) en réduisant le Pu à l’état d’oxydation +III par ajout d’U(IV)
et d’hydrazinium,
- la désextraction d’U(VI) à faible concentration d’acide nitrique,
- la purification de l’uranium et du plutonium dans des cycles d’extraction liquide-liquide
complémentaires et la régénération du solvant,
- la conversion et le conditionnement du plutonium sous forme d’oxyde et de l’uranium
sous forme de nitrate d’uranyle afin de permettre leur réutilisation pour la fabrication de nouveaux combustibles.
Figure 6 : Principe du procédé PUREX (PF = produits de fission, AM = actinides mineurs).
Pour atteindre des facteurs de décontamination élevés, plusieurs cycles d’extraction sont nécessaires pour que l’uranium et le plutonium soient séparés des produits de fission, qui peuvent pour certains, comme le technétium et le ruthénium, être extraits en phase organique. Avant les cycles de purification complémentaires de l’uranium et du plutonium, les facteurs de décontamination sont aux alentours de 105 contre 107 et 108 requis pour l’uranium et le plutonium respectivement.
II- Le procédé PUREX
2.1.2. Avantages et inconvénients
Le système extractant à base de TBP présente de nombreux avantages2 :
- de bonnes propriétés physiques : très faible solubilité du TBP dans la phase aqueuse,
densité et viscosité adéquates (< 10 mPa∙s, limite acceptable pour un procédé),
- une sélectivité importante pour l’U(VI) et le Pu(IV) vis-à-vis de la majorité des produits
de fission et actinides mineurs,
- une capacité de charge en uranium et plutonium élevée (120 g·L-1 pour l’U(VI) et
40 g·L-1 pour le Pu(IV)) sans apparition de troisième phase*,
- une bonne compatibilité avec une opération de désextraction : réversibilité des
complexes formés en phase organique,
- recyclage possible du solvant.
Malgré son emploi à l’échelle industrielle depuis de nombreuses années, ce système
d’extraction a quelques inconvénients parmi lesquels2,9 :
- l’utilisation de chimie redox pour la partition U/Pu en réduisant le Pu(IV) au degré d’oxydation +III par l’ajout d’U(IV) et d’hydrazinium, dont l’utilisation est fortement contrainte par la réglementation REACh,
- la nécessité de plusieurs cycles de séparation et de purification pour atteindre les facteurs de décontamination souhaités,
- l’extraction de certains produits de fission, notamment le zirconium, le technétium et le
ruthénium, qui nécessite de mettre en œuvre des étapes de lavage (ou barrage),
- la dégradation hydrolytique et radiolytique du TBP entraînant une altération de la
sélectivité et la mise en œuvre d’un traitement solvant,
- une molécule pas complètement incinérable : principe CHON** non respecté du fait de
la présence d’un atome de phosphore.
De nombreux éléments sont produits suite à l’irradiation des combustibles. Le technétium et
le ruthénium figurent parmi les éléments les plus problématiques du procédé PUREX.
2.2. Le technétium
Le technétium est le premier radioélément du tableau périodique. Découvert en 1937 par
Perrier et al.19 suite à l’irradiation de molybdène par des neutrons, il est utilisé en médecine
nucléaire pour des diagnostiques ou des applications thérapeutiques. Cependant, il est principalement généré dans les réacteurs nucléaires comme produit de fission de l’uranium
avec un rendement de 6,06%20,21. La teneur en technétium après dissolution d’un combustible
de type UOX est d’environ 0,3 g∙L-1 (environ 4% en masse des produits de fission dans les
solutions de dissolution). Parmi tous les isotopes reportés, trois seulement ont des périodes
radioactives longues : 97Tc (T
II- Le procédé PUREX (T1/2 = 2,1·105 ans). L’isotope 99Tc est le plus abondant dans le combustible usé22. Il se
désintègre en ruthénium 99 en émettant un rayonnement22 β- (Figure 7).
Figure 7 : Schéma de désintégration du 99Mo.
2.2.1. Comportement dans le procédé PUREX
Lors de l’étape de dissolution18, 95% du technétium produit suite à la fission de l’uranium est
dissout dans la solution d’acide nitrique concentré23. Le technétium est alors présent sous
forme d’acide pertechnétique HTcO4 qui se dissocie facilement pour former l’anion
pertechnétate TcO4-. Le technétium peut être extrait soit sous forme d’acide pertechnétique
HTcO4 soit sous forme de complexes mixtes formés avec d’autres cations métalliques
(uranium, plutonium ou zirconium).
2.2.1.1. Extraction de HTcO4 par TBP
L’extraction de l’acide pertechnétique a largement été étudiée dans les conditions du procédé
PUREX24-26 (TBP 30%
vol dans le n-dodecane ; 0 ≤ [HNO3] ≤ 5 M, la concentration de Tc dans
la phase aqueuse initiale n’est pas précisée). Sans uranium en phase aqueuse, les auteurs s’accordent à dire qu’une augmentation de la concentration d’acide nitrique entraine une extraction plus importante du technétium jusqu’à HNO3 1 M. En effet, la concentration de proton est plus importante ce qui favorise l’extraction de l’acide pertechnétique. Puis, les coefficients de distribution du technétium diminuent pour de plus fortes concentrations d’acide nitrique car il y a une compétition entre l’extraction de l’acide nitrique et celle de l’acide pertechnétique (Figure 8).
II- Le procédé PUREX
Figure 8 : Extraction du technétium avec et sans uranium en phase aqueuse24.
L’équilibre à envisager pour l’extraction du technétium en milieu acide nitrique par le TBP en absence d’uranium est le suivant :
𝐻++ 𝑇𝑐𝑂
4−+ 𝑛 𝑇𝐵𝑃̅̅̅̅̅̅ ⇄ (𝐻𝑇𝑐𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅ 1 ≤ 𝑛 ≤ 4 4)(𝑇𝐵𝑃)𝑛
La plupart des auteurs25,27,28 suggèrent qu’en milieu acide nitrique, l’extraction de l’acide pertechnétique se fait avec trois molécules de TBP. Ces données ont été obtenues par la
méthode des pentes*. Le complexe entre l’acide pertechnétique et le TBP n’a pas été
structuralement caractérisé dans la littérature. D’après Pruett29, le complexe se forme par liaisons hydrogène directes avec le TBP et le proton de l’acide pertechnétique ou via des molécules d’eau. Aucun complexe mixte de la forme (𝐻𝑇𝑐𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅ n’est évoqué 4)𝑥(𝐻𝑁𝑂3)𝑦(𝑇𝐵𝑃)𝑛
dans la littérature.
2.2.1.2. Extraction du technétium par le TBP en présence d’uranium En l’absence de technétium, le nitrate d’uranyle est majoritairement extrait par le TBP sous la forme 𝑈𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅. La Figure 8 illustre bien qu’en présence d’uranium, le coefficient de 2(𝑁𝑂3)2(𝑇𝐵𝑃)2
distribution du technétium augmente fortement29. Cet effet peut s’expliquer par un mécanisme
d’extraction différent. C’est le technétium, sous forme d’anion pertechnétate (TcO4-), qui est
co-extrait24,26,27,30-35 en phase organique avec l’uranium en substituant un nitrate dans le complexe neutre formé avec le TBP en phase organique. Dans chaque complexe, un seul
II- Le procédé PUREX
anion nitrate est substitué par un anion pertechnétate8. Ce résultat repose sur la modélisation
des données d’extraction8. La réaction d’extraction mise en jeu est alors :
𝑈𝑂22++ 𝑁𝑂3−+ 𝑇𝑐𝑂4−+ 2 𝑇𝐵𝑃̅̅̅̅̅̅̅̅ ⇄ 𝑈𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅2(𝑁𝑂3)(𝑇𝑐𝑂4)(𝑇𝐵𝑃)2
2.2.1.3. Extraction du technétium par le TBP en présence d’autres cations métalliques
Thibaudeau27 montre que le technétium peut également être co-extrait avec des cations
métalliques au degré d’oxydation +IV, et notamment avec le Pu(IV) et Zr(IV) (Figure 9). L’étude de l’extraction du plutonium par le TBP à partir d’une solution d’acide nitrique met en évidence l’extraction unique de l’espèce 𝑃𝑢(𝑁𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅. D’après ces données d’extraction3)4(𝑇𝐵𝑃)2 27, un anion
pertechnétate remplace un anion nitrate dans le complexe neutre formé en phase organique. Ce résultat repose également sur une modélisation des données d’extraction.
Figure 9 : Coefficient de distribution du technétium en fonction de la concentration de métal (nitrate de plutonium, nitrate de thorium et nitrate de zirconium) (𝑪𝑻𝒄𝒂𝒒,𝒊𝒏𝒊 = 10-4 M, 𝑪
𝑵𝑶𝟑−
𝒂𝒒,𝒊𝒏𝒊
= 4 M, 𝑪𝑯𝒂𝒒,𝒊𝒏𝒊+ = 2,3 M,
CTBP = 30% dans le dodécane, T = 25°C)27.
D’après les résultats reportés sur la Figure 9, le technétium est co-extrait de façon très
prononcée avec le zirconium à HNO3 2,3 M. D’après les différents résultats reportés dans la
littérature, la co-extraction du technétium dépend fortement de l’acidité de la phase aqueuse.
En effet, Jassim36 montre que pour de faibles concentrations d’acide nitrique (C
𝐻𝑁𝑂3 𝑎𝑞,𝑖𝑛𝑖
< 1 M) les coefficients de distribution du technétium sont similaires avec ou sans zirconium car le
zirconium est faiblement extrait. Par contre, Thibaudeau27 et Garraway et al.31, qui ont mené
les études à HNO3 2,3 et 3 M respectivement, indiquent une forte co-extraction du technétium
avec le zirconium.
Dans les conditions d’extraction d’U(VI) et du Pu(IV) dans le procédé PUREX à HNO3 3-4 M,
le coefficient de distribution du technétium est beaucoup plus important en présence de zirconium (encadré bleu sur la Figure 10)27,31. Au-delà de HNO
3 6-7 M, le coefficient de
II- Le procédé PUREX
Figure 10 : Coefficient de distribution du technétium en fonction de la concentration d’acide nitrique, l’encadré bleu correspond aux conditions de co-extraction de l’uranium et du plutonium dans le procédé PUREX (𝑪𝑻𝒄𝒂𝒒,𝒊𝒏𝒊 = 10-4 M et 𝑪
𝒁𝒓 𝒂𝒒,𝒊𝒏𝒊
= 10-2 M, C
TBP = 30% dans dodécane, T = 25°C)27..
Thibaudeau27 montre également que le coefficient de distribution du technétium augmente
avec l’augmentation de la concentration de zirconium (Figure 11). Dans les solutions de combustibles nucléaires usés, la concentration de zirconium est d’environ 0,02 M, soit 1,6 g·L-1.
Figure 11 : Influence de la concentration de zirconium sur l'extraction du technétium (𝑪𝑯𝑵𝑶
𝟑
𝒂𝒒,𝒊𝒏𝒊 = 3 M,
𝑪𝑻𝒄𝒂𝒒,𝒊𝒏𝒊 = 0,28 g·L-1, C
TBP = 30% dans le kérosène, T = 35°C), ajout de nitrate de zirconium.
Les équilibres d’extraction avec le plutonium et le zirconium pris en compte pour modéliser les données d’extraction font intervenir dans les deux cas trois anions nitrate et un anion pertechnétate27 : 𝑍𝑟4++ 3 𝑁𝑂3−+ 𝑇𝑐𝑂4−+ 2𝑇𝐵𝑃̅̅̅̅̅̅ ⇄ 𝑍𝑟(𝑁𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅ 3)3(𝑇𝑐𝑂4)(𝑇𝐵𝑃)2 𝑃𝑢4++ 3 𝑁𝑂3−+ 𝑇𝑐𝑂4−+ 2𝑇𝐵𝑃̅̅̅̅̅̅ ⇄ 𝑃𝑢(𝑁𝑂̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅ 3)3(𝑇𝑐𝑂4)(𝑇𝐵𝑃)2 0,00 0,02 0,04 0,06 0,08 0,10 0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 D(Tc) Caq,ini Zr (M)
II- Le procédé PUREX Dans le procédé PUREX, l’anion pertechnétate, qui a la faculté de former un complexe mixte avec le zirconium, en se substituant à un anion nitrate, est fortement extrait en phase
organique. Lors de la désextraction du zirconium dans l’opération de lavage, le technétium
s’associe alors avec l’uranium et le plutonium et reste ainsi dans la phase organique2.
2.2.2. Problématiques liées au technétium dans PUREX
Lors de la partition U(VI)/Pu(IV), le plutonium est désextrait de la phase organique après sa réduction à l’état d’oxydation +III. Lors de cette étape, de l’uranium (IV) et de l’hydrazinium sont introduits dans le procédé : ils permettent respectivement la réduction et la stabilisation du plutonium au degré d’oxydation +III.
Au cours de cette étape, les problèmes majeurs liés à la présence de grandes quantités de technétium en phase organique sont :
1) l’oxydation d’U(IV) en milieu acide nitrique, où l’anion pertechnétate joue le rôle de catalyseur, selon les équations suivantes :
2 𝑇𝑐𝑂4−+ 3 𝑈4++ 2 𝐻2𝑂 ⇄ 2 𝑇𝑐𝑂2+ 3 𝑈𝑂22++ 4 𝐻+
𝑇𝑐𝑂2+ 𝑁𝑂3−⇄ 𝑇𝑐𝑂4−+ 𝑁𝑂
2) la décomposition de l’hydrazinium37,38 utilisé pour la stabilisation du Pu(III) et d’U(IV). Ce
réactif est utilisé en tant qu’agent anti-nitreux pour éviter l’oxydation du Pu(III) et d’U(IV) par les ions nitreux. Le technétium est réduit sous sa forme tétravalente qui est ensuite
ré-oxydée par l’acide nitrique en TcO4-. Le technétium joue là encore le rôle de catalyseur.
Ainsi, la concentration de TcO4- reste constante alors que l’hydrazinium est complètement
consommé ou que l’U(IV) est oxydé. Ces deux phénomènes dégradent donc les performances à l’étape de partition et nécessitent d’augmenter la concentration de ces deux réactifs introduits (U(IV) et hydrazinium) afin de maintenir des performances équivalentes. Avec l’augmentation du pourcentage de plutonium dans les combustibles de type MOX, le besoin en U(IV) et en hydrazinium augmente, donc le technétium extrait en phase organique devient un problème majeur pour la partition U/Pu.
Pour s’en affranchir, dans le procédé PUREX actuel, le solvant en sortie du lavage PF subit une opération de lavage supplémentaire, dite « lavage Tc », dans laquelle la phase organique chargée est mise en contact avec une solution à forte concentration d’acide nitrique (Figure
12). La grande quantité de contre-ion NO3- favorise le maintien de l’uranium et du plutonium
sous forme de complexes nitratés et non plus de complexes mixtes
(cation-nitrate-pertechnétate). La phase aqueuse effluente qui contient le technétium subit
alors une nouvelle opération d’extraction dite « extraction complémentaire » pour récupérer
l’uranium et le plutonium2 éventuellement désextraits. Enfin, le solvant issu de cette opération
II- Le procédé PUREX
Figure 12 : Schéma du lavage technétium2.
2.3. Le ruthénium
Le ruthénium est un élément unique puisqu’il existe sous tous les états d’oxydation39 compris
entre 0 et +VIII mais aussi à un état d’oxydation –II. Le ruthénium est produit dans les réacteurs nucléaires suite à la fission de l’uranium et du plutonium. Il peut se trouver sous la forme de
sept isotopes naturels et cinq isotopes radioactifs (103Ru et 106Ru avec des périodes
radioactives de 39 jours et 373 jours respectivement ; 97Ru, 105Ru 107Ru ayant des périodes
radioactives inférieures à 3 jours)40. Après une période de refroidissement du combustible en
piscine, le 106Ru est le seul radioisotope à considérer. Il ne représente qu’une faible fraction
du ruthénium total. Le ruthénium est un produit de fission gênant car :
- Il présente un isotope radioactif très irradiant (106Ru) malgré sa faible quantité,
- Une fois extrait en phase organique dans le procédé PUREX, il est très difficile de le
désextraire,
- Sa présence nécessite des cycles d’extraction complémmentaires pour obtenir des
puretés élevées des flux d’uranium et de plutonium,
- L’existence d’un oxyde volatil à température ambiante rend difficile les prévisions de
son comportement dans une usine de retraitement.
La grande faculté à la complexation du ruthénium et la variété de ses degrés d’oxydation font
que cet élément possède une chimie de coordination très riche39.
2.3.1. Étude du ruthénium dans le procédé PUREX
41,42Le ruthénium est présent en quantité importante dans le combustible irradié, et sa teneur dépend de l’enrichissement en isotope fissile, du pourcentage de plutonium pour les
combustibles MOX, du taux de combustion et du temps de refroidissement du combustible41.
Il représente environ 7 à 9% en masse des produits de fission de l’235U et du 239Pu43. Avec
l’augmentation du pourcentage de plutonium dans les combustibles MOX et l’augmentation du temps de séjour des combustibles dans les réacteurs nucléaires, la gestion du ruthénium
devient encore plus contraignante44. Le 106Ru est l’isotope le plus pénalisant dans les procédés
II- Le procédé PUREX
totale, le 106Ru et son descendant direct, le 106Rh, sont responsables d’une forte radioactivité
du combustible : le couple 106Ru-Rh est à l’origine de 80% des émissions γ des effluents des
usines de La Hague.
Dans le procédé PUREX, le ruthénium est présent à chaque étape du retraitement du
combustible irradié. À l’étape de dissolution du combustible usé dans l’acide nitrique45, environ
deux tiers du ruthénium se dissolvent et se retrouvent sous forme de différents complexes. La concentration de ruthénium dissout dans les solutions de dissolution est de l’ordre de 5 mM
(0,5 g∙L-1). Le dernier tiers reste dans les résidus de dissolution à l’état de colloïdes, de boues
ou d’alliages métalliques et enfin 0,02% se volatilisent en tétraoxyde de ruthénium (RuO4) récupéré dans des filtres spécifiques et dédiés.
2.3.1.1. Espèces formées en phase aqueuse
La spéciation du ruthénium en milieu acide nitrique est difficilement prévisible puisqu’elle varie en fonction de la concentration d’acide nitrique, d’acide nitreux ou encore du temps de mise à l’équilibre des solutions. Une fois dissout en milieu acide, le ruthénium forme une espèce de
ruthénium nitrosyle RuNO3+ très stable. Cette entité RuNO3+ forme de nombreux complexes
octaédriques nitratés et/ou nitrés (Figure 13) en phase aqueuse de formule générale [RuNO(NO2)x(NO3)y(OH)z(H2O)5-x-y-z]3-x-y-z qui seront plus largement discutés au chapitre 2.
Figure 13 : Structure du ruthénium nitrosyle.
2.3.1.2. Données d’extraction du ruthénium avec le TBP
Les différentes études de la littérature montrent que l’extraction du ruthénium dépend de l’acidité de la phase aqueuse,46-48. D’après les données publiées par Pruett (Figure 14), le
ruthénium est peu extrait par le TBP et les coefficients de distribution dépendent du temps d’agitation. L’équilibre ne semble pas être atteint après 4 heures d’agitation. Afin de pouvoir comparer les données obtenues au cours de ces travaux avec des données obtenues au CEA, le temps d’agitation pour les tests d’extraction liquide-liquide a été fixé à 1h.
La mesure du coefficient de distribution du ruthénium en fonction de la concentration d’acide nitrique montre qu’un maximum d’extraction est atteint pour une concentration d’acide nitrique
autour de 1 M (DRu = 0,20 pour une heure d’agitation). Ces valeurs sont cohérentes avec la
meilleure décontamination du solvant en ruthénium à l’usine lors de l’étape de « lavage Tc » (effectuée à forte acidité HNO3 5-6 M) et la médiocre décontamination en Ru atteinte au « lavage PF » (autour de HNO3 2 M). Pour HNO3 > 2,5 M, le coefficient de distribution du ruthénium diminue car il y a une compétition entre l’extraction de l’acide nitrique et l’extraction du ruthénium49.
II- Le procédé PUREX
Figure 14 : Coefficients de distribution du ruthénium en fonction de la concentration d’acide nitrique en phase aqueuse et du temps d’agitation (CTBP = 30%vol dans le TPH,
𝑪𝑹𝒖𝑵𝑶(𝑵𝑶
𝟑)𝟑
𝒂𝒒,𝒊𝒏𝒊
= 0,0025 M, T = 25°C)48.
Contrairement au technétium, aucun mécanisme de co-extraction avec l’uranium n’a été mis en évidence. Les coefficients de distribution du ruthénium dépendent de la concentration de TBP libre dans le solvant organique. Ainsi, plus la concentration d’uranium augmente et plus DRu diminue car la concentration d’extractant libre en phase organique diminue8. En effet,
lorsque le solvant est chargé en uranium (112 g·L-1 d’uranium en phase organique), le
coefficient de distribution du ruthénium devient extrêmement faible et inférieur à 5·10-5.
Dans le procédé, le ruthénium est extrait dans les étages où la concentration d’uranium est faible. Une fois extrait dans le solvant, le ruthénium est très difficilement désextrait même après
plusieurs contacts successifs à forte acidité8,50. En effet, même après une heure d’agitation
entre une phase organique chargée en ruthénium et une phase aqueuse à HNO3 5 M, le
coefficent de distribution du ruthénium en lavage est plus de 20 fois supérieur à celui mesuré en extraction. La rétention en phase organique serait due à un changement de spéciation du ruthénium et notamment par la formation de dimères de ruthénium nitrosyle avec des ligands
pontants nitrate ou hydroxo40, ou à la formation de liaison directe Ru-TBP51. Ces espèces sont
supposées être quasiment impossibles à éliminer de la phase organique.
2.3.1.3. Espèces formées en phase organique (TBP 30%vol dans le TPH)
La diversité des espèces de ruthénium présentes en phase aqueuse
([RuNO(NO2)x(NO3)y(OH)z(H2O)5-x-y-z]3-x-y-z , paragraphe 2.3.1.1) rend difficile l’identification de celles extraites en phase organique par le TBP. Les études reportées dans la littérature montrent que :
- les complexes nitratés sont deux fois plus extraits que les complexes nitrés de
II- Le procédé PUREX État de l’art sur la complexation entre le ruthénium et le TBP
Le mode de coordination54 majoritairement décrit dans la littérature suppose que les molécules
de TBP sont liées en seconde sphère de coordination par liaisons hydrogène44,54,55 via les
molécules d’eau ou les ions hydroxo liés au ruthénium (Figure 15)44,54. L’extraction de l’espèce
trinitraté implique alors trois à quatre molécules de TBP liées aux atomes d’hydrogène des
molécules d’eau. Ce nombre important de molécules de TBP nécessaires pour extraire le ruthénium est cohérent avec les faibles coefficients de distribution observés quand le solvant est chargé en uranium. En effet, la quantité d’extractant libre est alors beaucoup plus faible qu’en absence d’uranium.
Les complexes tétranitratés, sous forme d’anions [RuNO(NO3)4(H2O)]- sont extraits sous forme
de paires d’ions avec l’ion H3O+. Cet ion hydronium est solvaté par trois molécules de TBP,
auxquelles viennent s’ajouter deux autres molécules de TBP qui solvatent les deux atomes d’hydrogène du ligand aquo en position axiale du nitrosyle.
Figure 15: Structure des complexes extraits par le TBP et représentation des liaisons hydrogène avec les molécules d’eau ou les ions hydronium.
Des études récentes, menées au CEA par Lefebvre40, confirment l’absence de molécule de
TBP en première sphère de coordination du ruthénium (données infrarouge et EXAFS) même sur des temps longs. Les mesures EXAFS réalisées sur des phases organiques de ruthénium et de TBP dans le TPH montrent que le composé majoritairement extrait est le ruthénium
trinitraté, comme évoqué dans la littérature48,56,57. La sphère de coordination est complétée par
des molécules d’eau.
Modélisation des données d’extraction du ruthénium
Plus récemment, Moeyaert et al.50 ont étudié l’extraction du ruthénium par le TBP (30%
vol dans le TPH) en faisant varier la concentration d’acide nitrique pour une concentration fixe de
ruthénium de 0,51 g·L-1
. La meilleure modélisation des résultats expérimentaux a été obtenue
en considérant la formation des complexes ̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅ 𝑅𝑢𝑁𝑂(𝑁𝑂3)3(𝐻2𝑂)2(𝑇𝐵𝑃)2 et
𝑅𝑢𝑁𝑂(𝑁𝑂3)3(𝐻2𝑂)2(𝑇𝐵𝑃)3
̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅̅ extraits en phase organique avec la participation de deux ou trois
molécules de TBP, ce qui est en accord avec les données obtenues par Lefebvre40.
Lorsque la concentration d’acide nitrique augmente, les coefficients de distribution du
ruthénium augmentent entre 0 et 2,5 M d’acide nitrique puis diminuent. Cette augmentation
est directement corrélée à l’accroissement du pourcentage de complexes de ruthénium